

Abstract
A previously validated mathematical model of intravascular platelet deposition and tissue factor (TF)-initiated coagulation under flow is extended and used to assess the influence on thrombin production of the activation of factor XI (fXI) by thrombin and of the activation of factor IX (fIX) by fXIa. It is found that the importance of the thrombin-fXIa-fIXa feedback loop to robust thrombin production depends on the concentration of platelets in the blood near the injury. At a near-wall platelet concentration of ∼250,000/μL, typical in vessels in which the shear rate is <200 s−1, thrombin activation of fXI makes a significant difference only at low densities of exposed TF. If the near-wall platelet concentration is significantly higher than this, either because of a higher systemic platelet count or because of the redistribution of platelets toward the vessel walls at high shear rates, then thrombin activation of fXI makes a major difference even for relatively high densities of exposed TF. The model predicts that the effect of a severe fXI deficiency depends on the platelet count, and that fXI becomes more important at high platelet counts.
Introduction
Vascular injury triggers the intertwined processes of platelet deposition and coagulation. These involve many disparate physical and chemical interactions competing with or reinforcing one another in direct and indirect ways. Correctly predicting the overall response of the system under specific conditions is not possible unless these interactions are quantitatively accounted for, and for that mathematical models are a powerful tool. We extend our earlier models (1,2) of platelet deposition and the tissue-factor (TF) pathway of coagulation to include a set of reactions that has received much attention in recent years, activation of factor XI (fXI) to fXIa by thrombin, and activation of factor IX (fIX) to fIXa by fXIa. We are interested in how much these reactions influence thrombin production, and how this influence depends on blood flow parameters, TF level, and the near-wall concentration of platelets.
All blood vessels are lined by endothelial cells that normally work to inhibit platelet deposition and coagulation. Vascular injury exposes the subendothelium (SE) and clot-promoting materials in it. Exposed proteins, e.g., collagen and von Willebrand factor (vWF), mediate platelet adhesion to the SE (3). They also activate the bound platelets causing them to release soluble chemical agonists into the plasma where these can activate other platelets that do not directly contact the SE (3). Activated platelets display high-affinity integrin receptors that serve to anchor interplatelet bridges formed when a molecule of fibrinogen or vWF binds to such receptors on two different platelets. By means of these bridges, a platelet aggregate can be formed on the injured vascular wall (3).
The injury also initiates coagulation by exposing subendothelial TF molecules to the blood (4). The plasma protein fVIIa binds to the exposed TF to form the powerful subendothelial-bound TF:VIIa enzyme complex (see Fig. 1). TF:VIIa activates two inactive plasma proteins (zymogens) into their active enzyme forms, fIX to fIXa and fX to fXa. On their own in the plasma, fIXa and fXa are weak enzymes. However, if fIXa and fXa bind to an activated platelet along with their respective active cofactor molecules, fVIIIa and fVa, they can form powerful enzyme complexes on the platelet's surface (5). The VIIIa:IXa complex (tenase) activates platelet-bound fX to fXa. The Va:Xa complex (prothrombinase) activates platelet-bound prothrombin into the enzyme thrombin (also known as fIIa).
Figure 1.
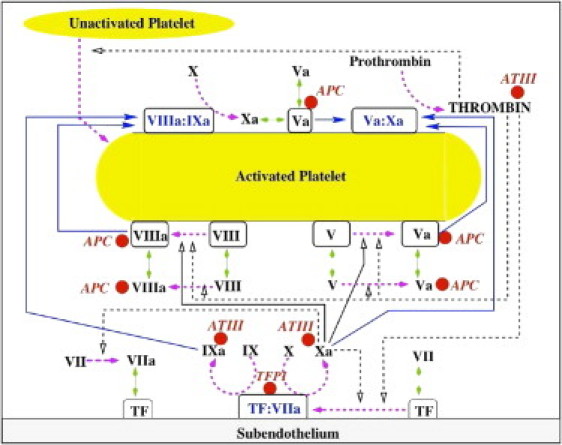
Schematic of coagulation reactions included in our earlier models. (Dashed magenta arrows) Cellular or chemical activation processes. (Blue arrows) Chemical transport in the fluid or on a surface. (Green segments with two arrowheads) Binding and unbinding from a surface. (Rectangular boxes) Surface-bound species. (Solid black lines with open arrows) Enzyme action in a forward direction; (dashed black lines with open arrows) feedback action of enzymes. (Red spheres) Chemical inhibitors.
Thrombin is the critical coagulation enzyme. It converts fibrinogen into fibrin monomers that polymerize to form a gel that mechanically stabilizes platelet aggregates (6). Thrombin contributes powerfully to the production of more thrombin. It activates platelets, and thereby increases the availability of activated platelet surface for the coagulation reactions (3). It activates the cofactor molecules fVIIIa and fVa required for the VIIIa:IXa and Va:Xa complexes on the surfaces of activated platelets (7,8). It activates fVII to fVIIa in plasma and when fVII is bound to TF on the SE (4). As discussed shortly, it also activates fXI to fXIa.
Fig. 1 shows that fXa activates platelet-bound fVIII and fV to their active forms fVIIIa and fVa (7,8). This is the source of these essential cofactor molecules before thrombin becomes available. The figure also shows the targets of the principal chemical inhibitors of coagulation: thrombin, fXa, and fIXa for antithrombin-III (AT-III), fVIIIa, and fVa for activated protein C (APC), and fXa and TF:VIIa for tissue factor pathway inhibitor (TFPI) (9,10). (Note that Fig. 1 does not show the role platelets play in physically inhibiting subendothelial TF:VIIa when they adhere to the SE and thus cover a portion of it (2,11). It also does not show reactions involving fXI. These reactions are described below.)
In the contact pathway, fXIIa activates fXI to fXIa, which, in turn, activates fIX to fIXa (12). This pathway's relevance for in vivo hemostasis is unclear because deficiencies of fXII appear to have no bleeding consequences whereas deficiencies in fXI do (13). The latter vary in severity in ways that do not correlate directly to the degree of fXI deficiency (14), but their existence indicates that fXI plays a role in hemostasis beyond its role in the contact pathway. Because of this, the role of fXI in the TF pathway has received much attention in recent years.
Thrombin can activate fXI to fXIa (15,16) and fXIa can activate fIX to fIXa (17,18) in the plasma and on activated platelets' surfaces (13,19). This provides a path to fIXa production that augments that by TF:VIIa. Factor XIa activation of fIX increases thrombin production at low [TF] (13,20,21), it allows continued production of fIXa after TF:VIIa is inhibited by TFPI or by coverage of the injury by adherent platelets, and it may provide a burst of thrombin production leading to thrombin-activatable fibrinolysis inhibitor activation and consequent inhibition of fibrinolysis (22). Most in vitro studies of the role of fXI in the TF pathway were done in static closed systems (no flow) (21–23) with soluble TF (von dem Borne et al. (22) used surface-bound TF) and many were done without platelets. It is difficult to extrapolate meaningful quantitative results from such studies to an in vivo situation in which production of thrombin in response to vascular injury involves a complex biochemical network coordinated with the activation and deposition of platelets on the vessel wall, and occurs under flow. Ex vivo studies have shown thrombus growth is significantly different with fXI and with antibody-inhibited-fXI (24), but are not able to provide detailed quantitative data about the dynamics of the protein concentrations that underlie these observations. Even under more-controlled conditions in vitro, the useful information that can be extracted from experiments involving coagulation, platelet deposition, and flow (25) is quite limited.
Our earlier models (1,2) are based on the reaction network pictured in Fig. 1, a simple description of platelet deposition, activation, and participation in coagulation reactions, and a simple treatment of flow-mediated transport of proteins and platelets to or from the vicinity of the injury. Studies of these models revealed a sharp threshold in thrombin production in response to different densities of exposed TF and showed how the threshold [TF] and the concentration of thrombin produced were modified by changes in the flow shear rate. They showed that the reduced thrombin production that occurs with hemophilias A and B and severe thrombocytopenia are natural consequences of the kinetics of the reaction system. This work also led us to propose that as platelets adhere to the SE, they cover (i.e., pave-over) and so physically inhibit subendothelial enzymes including TF:VIIa. Our hypothesis was given experimental support (11), and the predicted threshold and its dependence on shear rate were seen experimentally (25). Our studies indicated that, for small injuries, flow-mediated dilution is a much more potent inhibitor than TFPI and APC, and highlighted the role of flow and platelet deposition in establishing the TF threshold. Support for this prediction comes from comparison between results in studies of coagulation and platelet deposition under flow where a TF threshold is seen (25), and others without flow and platelet deposition, in which no TF threshold is observed (26).
In Fogelson and Tania (1), platelet deposition shut down TF:VIIa activity within a few minutes. Only during that time-window was fIXa produced. For a wide range of [TF], most of the fIXa and fXa produced by TF:VIIa was carried away by the flow. A small amount bound to activated platelets, where the fXa activated fVIII. Only if TF:VIIa produced sufficient amounts of fIXa and fXa, was enough VIIIa:IXa complex formed on the adhering platelets' surfaces to allow substantial fXa production to continue after platelets covered the SE. We found this to be the key to whether robust thrombin generation occurred. In this article, we explore whether a second path to fIXa production (by fXIa) changes the dynamics seen in Fogelson and Tania (1) and Kuharsky and Fogelson (2).
Simulations with the new model show that thrombin-activated fXIa can indeed lead to robust thrombin production at low [TF] that, without fXI, would initiate only small amounts of thrombin production. In these cases, a burst in thrombin production is seen 15–20 min after the initial injury, rather than after 6–7 min as with high [TF]. More surprising is the model's prediction that the concentration of platelets in the blood near the injury has a strong impact on how much the availability of fXI influences thrombin production. If the near-wall concentration of platelets is high because the bulk platelet count is high, or because, at high shear rates, the red blood cells' motion leads to a redistribution of platelets toward the vessel walls (27,28), then thrombin-catalyzed fXI activation and fXIa-catalyzed fIX activation are important even at relatively high levels of exposed TF. The essential reason for this behavior is that higher platelet counts lead to faster coverage of the SE by adherent platelets, which causes faster shutdown of TF:VIIa activity. Hence, the second path to fIXa production becomes more important. If sensitivity to fXI deficiency depends on the platelet count, this might help explain the variations in bleeding tendencies seen with fXI deficiency.
Methods
We assume that the reactions depicted in Fig. 1 occur in a reaction zone above a small patch of exposed SE (Fig. 2 A). Initially, the reaction zone does not include platelets and so its height (1–2 μm) is determined by the height of the boundary layer from which a chemical in the blood flowing past the injury can reach the SE by diffusion before vanishing downstream. The height of the reaction zone increases to account for the changing volume of platelets in the thrombus. Adjacent to the reaction zone, in the direction perpendicular to the flow, is an endothelial zone (Fig. 2 B) with height equal to that of the reaction zone and width dependent on the flow shear rate and protein diffusion coefficients (1). Thrombin can diffuse from the reaction zone into the endothelial zone, bind to TM, and produce APC, which may then diffuse into the reaction zone.
Figure 2.
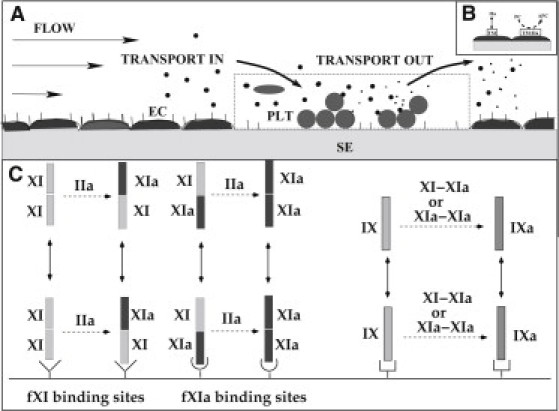
Schematic of (A) reaction zone and (B) endothelial zone. (C) Schematic of reactions involving fXI or fXIa. (Left) Dimers fXI-fXI and fXI-fXIa can bind (unbind) to (from) the fXI-specific binding sites, whereas fXI-fXIa and fXIa-fXIa can bind (unbind) to (from) the fXIa-specific binding sites. Thrombin can activate fXI to fXIa in solution and on an activated platelet. Only the dangling-end of a bound dimer can be activated by thrombin or can activate fIX. (Right) fXI-fXIa and fXIa-fXIa dimers can activate fIX to fIXa in solution or on an activated platelet.
We assume that in the reaction zone, each species is uniformly distributed and is described by its concentration. Platelets are 1), unactivated, unattached, and free to move with the fluid, or 2), activated and adherent to the SE, or 3), activated and bound to other platelets in the thrombus. In cases 2 and 3, the platelets are stationary. Proteins are distinguished by their chemical identity and by whether they are in the plasma, bound to the SE, or bound to an activated platelet. Only those in the plasma move with the fluid; the others are stationary while they are bound to a surface. For a protein to get from the SE to a platelet's surface, it must unbind from the SE, move through the plasma, and then bind to a receptor on an activated platelet. While in the plasma, it may be carried downstream by the flow.
Advective and diffusive transport of each fluid-phase species into or out of the reaction zone occurs at a rate proportional to 1), the difference in the species' concentration in the bulk fluid and in the reaction zone, and 2), the value of a mass transfer coefficient. The latter depends on the flow shear rate, the species' diffusion coefficient, and the size of the injured patch (2).
Platelets in the bulk fluid are not activated. Platelets can be activated by contact with the SE, by exposure to thrombin in the plasma, or by contact with other activated platelets. The last is used as a surrogate for activation by chemical agonists (ADP and TXA2) released by platelets that the model does not track.
Surface binding sites for coagulation proteins play a prominent part in the model. We assume that fVII and fVIIa compete for the TF binding sites on the SE. Each of the pairs, fIX/fIXa, fX/fXa, fV/fVa, and fVIII/fVIIIa, has their distinct binding sites on the surfaces of activated platelets for which the proteins in that pair compete. There are also binding sites specific for fIXa. In the endothelial zone, thrombin can bind to TM. In a change from Fogelson and Tania (1) and Kuharsky and Fogelson (2), in which prothrombin and thrombin were assumed to share platelet binding sites, here we assume that they have separate sites. This agrees with evidence available as of this writing that thrombin is released from the platelet surface upon its activation (29), and that the mechanisms of prothrombin and thrombin binding to the platelet are different (see the Supporting Material).
Our assumptions about protein interactions, other than the new ones involving fXI and fXIa, follow. Further discussion of them including citations to the literature can be found in Kuharsky and Fogelson (2):
-
1.
fVII and fVIIa can bind to subendothelial TF. fXa can activate fVII in plasma and when fVII is bound to TF. fXa can bind to TF:VII directly from the plasma.
-
2.
fIX and fX can be activated by the TF:VIIa complex on the SE. They compete to bind to TF:VIIa directly from plasma. fX can also be activated by the platelet-bound VIIIa:IXa complex.
-
3.
Prothrombin can be activated to thrombin by the platelet-bound Va:Xa complex.
-
4.
fV and fVIII can be activated by thrombin in plasma and by thrombin and fXa on the surfaces of activated platelets.
-
5.
The chemical inhibitors are AT-III, APC, and TFPI. Because [AT-III] is high, we assume it acts in a first-order manner to permanently inactivate plasma fIXa, fXa, and thrombin. APC is produced in the endothelial zone by the TM-thrombin complex. It can bind to platelet-bound fVa and fVIIIa to permanently inactivate them. TFPI in the plasma must first bind to fXa, and then the TFPI:fXa complex must bind to the TF:VIIa complex to inhibit it.
-
6.
When a platelet adheres to the SE, it physically blocks the activity of the TF:VIIa complexes on the patch of SE to which it has adhered.
To this model, we have added reactions in which thrombin activates fXI to fXIa, and ones in which fXIa activates fIX to fIXa. We assume that each of these reactions can take place in plasma or on an activated platelet's surface. We also have added reactions describing fXI and fXIa binding to and unbinding from the surfaces of activated platelets.
That fXI circulates as a dimer may be essential for some of its functions in coagulation (30,31). Because of the slow rate of activation of fXI by thrombin, dimers in which only one of the halves has been activated are common (32,33). We track three versions of the dimer in the plasma, which we denote fXI-fXI, fXI-fXIa, and fXIa-fXIa, with no, one, or both halves activated, respectively. The half-activated dimer is an active enzyme (33) and we assume it activates fIX at the same rate as that for each half of a fully activated dimer. We assume that there are 1500 binding sites for fXI and 250 distinct ones for fXIa on each activated platelet (34,35), and that the fXI dimers bind to these sites as depicted in Fig. 2 C, a scenario similar to that suggested in Gailani et al. (30). We denote the four types of the bound dimer as Plt-fXI-fXI, Plt-fXI-fXIa, Plt-fXIa-fXI, and Plt-fXIa-fXIa. In the first two, an unactivated half-dimer is bound to a fXI-specific site; in the latter two, an activated half-dimer is bound to a fXIa-specific site. We assume that the dimer half that is bound to a platelet is not available for other reactions. Only the dangling ends of the dimers are available for reactions with thrombin or fIX. We use kcat = 1.3 (10)−4 s−1 and KM = 50 nM (15) for activation of fXI by thrombin in plasma and on platelets; kcat = 0.21 s−1and KM = 0.2 μM for fXIa activation of fIX (30) in plasma and on platelets (36); KD = 10 nM for fXI binding with platelets (37); and KD = 1.7 nM for fXIa binding with platelets (35).
In the model, the concentration of each protein satisfies an ordinary differential equation (ODE) that includes terms for each reaction in which that species is involved and a term describing the species' transport if it is in the fluid. A complete description of the model is given in the Supporting Material. Here, we use two of the new equations to illustrate the types of reactions that the model includes:
Here, = [fXI-fXIa], = [Plt-fXI-fXIa], and = [Plt-fXIa-fXI]. Ten processes directly change and . fXI-fXIa is transported by the fluid (A) and can bind to the platelet binding sites for fXIa (B) or fXI (C) and unbind from these sites. It can bind to fluid-phase fIX (concentration z9) to form a transient complex on the way to activating fIX to fIXa (D). It is produced by thrombin's activation of fXI-fXI (E), binds with thrombin (e2) on the way to being fully activated by it (F), and is inactivated by AT-III (G). Plt-fXI-fXIa is formed when fXI-fXIa binds to platelet binding sites for fXI (H) or by activation of Plt-fXI-fXI () by platelet-bound thrombin ( (I)), and it forms a complex with platelet-bound fIX () on the way to activating it (J). The values and are the concentrations of unoccupied platelet binding sites for fXI and fXIa, respectively.
The model's ODEs are solved using DLSODE (38). For each simulation, we specify the plasma concentrations of all zymogens and enzymes (typically only [fVIIa] is nonzero) and platelets, the rate constants for all reactions, the numbers of each type of binding site on an activated platelet, the dimensions of the injury, the flow velocity near the injured wall, the diffusion coefficients for all fluid-phase species, and the density of exposed TF. The simulation outputs are the concentration of every zymogen and enzyme within the reaction and endothelial zones every 1 s from initiation of the injury until the completion of the simulation, as well as the concentrations of unactivated platelets and platelets attached either directly to the SE or to other platelets.
Results
Uniform platelet concentration
We performed a series of simulations distinguished only by the [TF] exposed in the injury. The model equations were solved for 20 min and we recorded all concentrations at 10 min and 20 min post injury for each [TF]. We did this for normal [fXI] (NfXI = 30 nM) and with no fXI. Fig. 3 shows the total thrombin concentration [fIIa] (the concentration of all species that include the active thrombin enzyme) at these times for a simulation with a flow shear rate of 100 s−1. For each curve, there is a small interval of [TF] over which [fIIa] increases sharply from values (≤10 pM) at which thrombin has little physiological effect to values (≥10 nM) at which it has a profound effect. We refer to the center of this interval as the threshold; it is ∼3 fmol/cm2 at 20 min and just under 5 fmol/cm2 at 10 min. For shear rate 50 s−1, the results (not shown) are very similar with a maximum [fIIa] ∼4% higher and the threshold shifted slightly to the left. For shear rate 500 s−1 (not shown), the curves are again similar, but the upstroke in the curves is shifted to the right by 2–3 fmol/cm2 and the maximum [fIIa] is ∼4% less than at shear rate 100 s−1.
Figure 3.
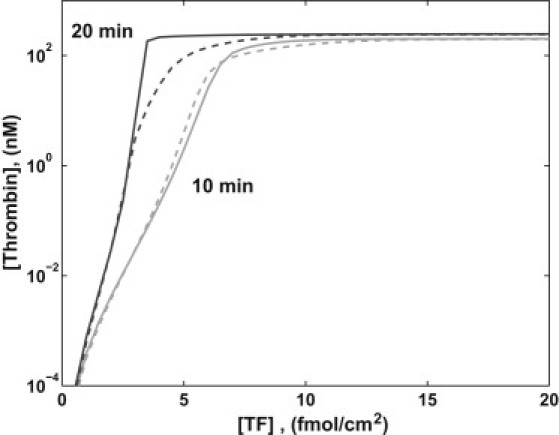
[fIIa] at 10 min and 20 min versus [TF] with normal [fXI] (solid) and no fXI (dashed) with near-wall platelet count 250,000/μL and shear rate 100 s−1.
From Fig. 3, we see that without fXI, [fIIa] has a threshold dependence on [TF], and that the threshold [TF] varies somewhat depending on the flow shear rate, consistent with results in Kuharsky and Fogelson (2). The threshold level is lower if one considers [fIIa] at 20 min rather than at 10 min. With NfXI, for sufficiently low [TF] there is no significant change in [fIIa] at 10 and 20 min, and in these cases, [fIIa] is subpicomolar. For high [TF], a high [fIIa] develops by 10 min and remains high at 20 min, and the fXI reactions have little effect on [fIIa] achieved. There is an interval of [TF] for which the fXI reactions lead to a substantial increase in [fIIa] at 20 min. No similar interval of [TF] exists for which a significant increase occurs in [fIIa] at 10 min, so the effect of fXI appears late.
To understand why it takes >10 min before significant additional amounts of thrombin are produced with NfXI, we look at the timecourse of several species' concentrations in simulations, one with NfXI and one without fXI, performed with [TF] = 3.5 fmol/cm2, shear rate 100 s−1, and near-wall platelet count of 250,000/μL. In both cases, [fIIa] builds slowly, reaching 1 nM only after >800 s. In Fig. 4 A, we see that with NfXI, the growing [fIIa] is accompanied by a slow rise in the concentration of the platelet-bound fXI-thrombin complex, paralleled with delays, by a slow rise in [Plt-fXI-fXIa] (other fXIa species also form but at much lower concentrations) and in the concentration of the complex of Plt-fXI-fXIa with Plt-fIX. [Plt-fXI-fXIa] rises slowly because [fIIa] does and because the kcat for thrombin-catalyzed activation of fXIa is small. Starting at ∼850 s, the rates of increase of [Plt-fXI-fXI:fIIa], [Plt-fXI-fXIa], and [Plt-fXI-fXIa:fIX] (see Fig. 4 B) successively increase at a rapid rate as thrombin binds to Plt-fXI-fXI and half-activates it, and then the resulting Plt-fXI-fXIa dimer binds to Plt-fIX on the way to activating it. Starting at ∼500 s, the effect of Plt-fXI-fXIa becomes increasingly apparent, as the decline in [Plt-fIXa] slows and then reverses with NfXI, while it continues to drop without fXI (Fig. 4 B). The upswing in [Plt-fIXa] and the increase of [Plt-fVIIIa] (with and without fXI) lead to a rapid increase in the concentrations of the tenase complex and the complex of tenase and Plt-fX (Fig. 4 C) starting at ∼900 s. This is followed in quick succession (Fig. 4 D) by rapid increases in [Plt-fXa], [Plt-Va:Xa], and [fIIa] so that from ∼1000 s on, there is substantially more thrombin with fXI than without.
Figure 4.
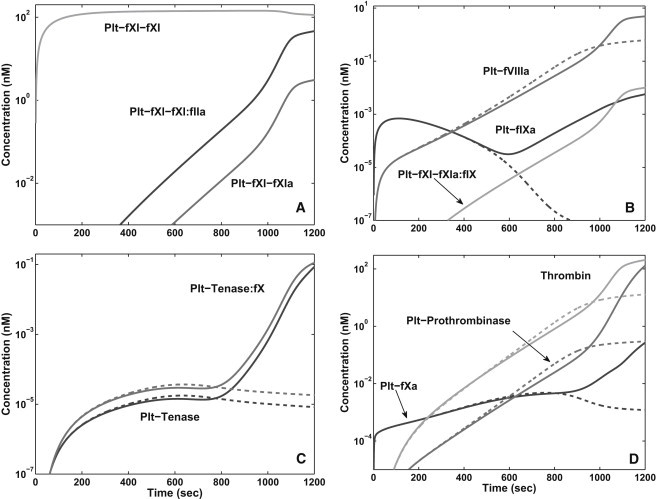
[TF] = 3.5 fmol/cm2, shear rate 100 s−1, and near-wall platelet count 250,000/μL. Various concentrations versus time with normal [fXI] (solid) and no fXI (dashed).
It is noteworthy that, up until the time that the effect of fXIa really takes hold, there is actually a little more thrombin (and platelet-bound fVa, fVIIIa, tenase, and prothrombinase) without fXI than with it. This happens because fXI competes with fV and fVIII for thrombin and so slows their activation by it. Fig. 4 shows that a significant fraction of the total thrombin is, in fact, bound to fXI on platelets.
Platelet near-wall excess
For the simulations just reported, we assumed that the concentration of platelets supplied to the reaction zone by transport was 250,000/μL. This is reasonable if the platelets are distributed uniformly across the vessel lumen, which is the case for shear rates <∼200 s−1. At higher shear rates, the platelet concentration is enhanced within a few microns of the vessel walls and is reduced over the central part of the lumen. The platelet near-wall excess (NWE) concentration can be 2–8-fold higher than at the vessel's center (27,28). A reasonable way to account for the platelet NWE in the simulations is to supply nonactivated platelets to the reaction zone at a higher concentration that is based on experimental observations (27,28).
The effect of the platelet NWE on thrombin production in simulations without fXI is shown in Fig. 5 A. [fIIa] is significantly less when the near-wall platelet count is 750,000/μL, three times the normal uniform count. This reduction can be traced to the fact that the SE is covered by platelets much faster at the higher platelet count. Hence, production of fIXa and fXa by TF:VIIa drops off more rapidly. The formation of Plt-VIIIa:IXa requires the simultaneous presence of Plt-fVIIIa (activated initially by fXa) and fIXa (1), and these are both substantially reduced with the higher platelet count. Consequently, there is less and slower formation of prothrombinase and less production of thrombin.
Figure 5.
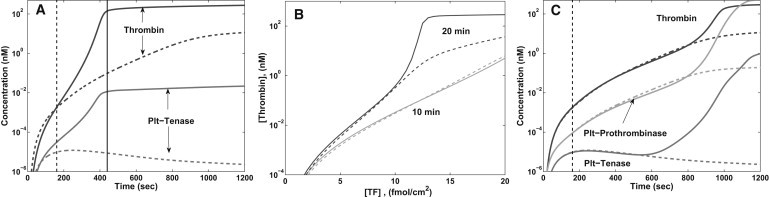
[TF] = 15 fmol/cm2, shear rate 500 s−1. (A) No fXI and near-wall platelet count (#/μL) 250,000 (solid) or 750,000 (dashed). Plots of [Plt-VIIIa:IXa] and [fIIa] versus time. (B and C) Near-wall platelet count 750,000. (B) [fIIa] versus [TF] after 10 min and 20 min with NfXI (solid) and with no fXI (dashed). (C) [Plt-VIIIa:IXa], [Plt-Va:Xa], and [fIIa] versus time with NfXI (solid) and with no fXI (dashed). (A and C, vertical lines) When platelets covered 99% of the SE.
Adding fXI provides another pathway to fIXa production. Fig. 5 B shows thrombin production in simulations with various [TF], shear rate 500 s−1, and with near-wall platelet count 750,000/μL. [fIIa] at 10 min is changed little by the fXI reactions. There is a gradual increase in [fIIa] at 10 min with [TF]; [fIIa] = 1 nM occurs only for [TF] above ∼17 fmol/cm2, and no threshold dependence is exhibited. Without fXI, [fIIa] at 20 min again increases only gradually with [TF] and no threshold is apparent. In contrast, with NfXI, [fIIa] is >100-fold higher for [TF] of 12–13 fmol/cm2 than at 10 fmol/cm2. Thus, the presence of fXI restores the threshold nature of thrombin production and leads to substantially more thrombin for above-threshold [TF] than for below-threshold [TF], and for the same [TF] in the absence of fXI. Fig. 5 C shows how [Plt-VIIIa:IXa], [Plt-Va:Xa], and [fIIa] vary in time for shear rate 500 s−1, [TF] = 15 fmol/cm2, and platelet count 750,000/μL with and without fXI. We see that the effect of fXI on the final amount of thrombin produced can be strongly influenced by the concentration of platelets.
Fig. 6 shows further evidence that the platelet count influences the sensitivity of the coagulation system to variations in [fXI]. For shear rates 100 s−1 and 500 s−1, simulations were carried out with several different platelet counts and [fXI] equal to 100%, 10%, and 1% of its normal plasma level. For each set of parameter values, [fIIa] at 20 min is plotted against [TF]. Fig. 6 A shows four sets of plots for platelet counts 125,000–500,000/μL. For each [fXI], the threshold level of TF increases with platelet count, because the faster coverage of the SE at higher platelet count necessitates higher [TF] to produce the same thrombin concentrations. For each platelet count, there is little difference between the curves for NfXI and 10% NfXI, but for 1% NfXI the reduction in thrombin production grows steadily with the platelet count. For platelet count 125,000/μL, there is a narrow range of [TF] for which thrombin production for 10% NfXI is approximately twice that for 1% NfXI. For platelet count 500,000/μL, there is a broader range of [TF] for which thrombin production is substantially more (by close to 100-fold in some cases) for 10% NfXI than for 1% NfXI. It is also useful to consider for fixed [TF], say 3 fmol/cm2, what the interplay between platelet count and [fXI] implies about thrombin production. For this [TF], reduction of [fXI] to 10% or 1% of normal has little effect for platelet count 125,000/μL, a major impact for 250,000/μL, and very little effect for 375,000/μL. Without knowing the platelet count, the effect of a particular deficiency of fXI cannot be predicted.
Figure 6.
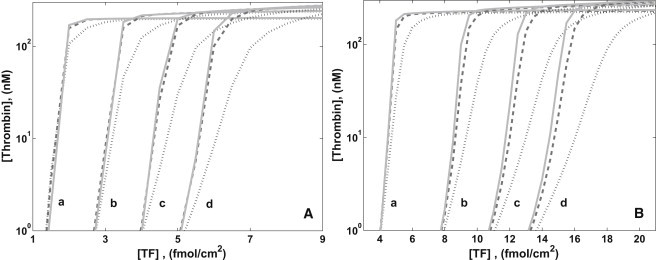
[fIIa] at 20 min versus [TF] for [fXI] = 30 nM (solid), 3 nM (dashed), and 0.3 nM (dotted). (A) Shear rate 100 s−1. Platelet counts (#/μL) (a) 125,000, (b) 250,000, (c) 375,000, and (d) 500,000. (B) Shear rate 500 s−1. Platelet counts (a) 250,000, (b) 500,000, (c) 750,000, and (d) 1,000,000.
For a shear rate 500 s−1 at which a platelet NWE occurs, we looked at higher platelet counts from 250,000/μL to 1,000,000/μL. Fig. 6 B shows that the curves shift to the right as the near-wall platelet count increases. Further, the width of the interval of [TF] for which the thrombin production at 1% NfXI is much less than at 10% NfXI grows significantly with increasing platelet count. At higher platelet counts, there are intervals of [TF], albeit narrow ones, for which a 10% NfXI leads to substantially less thrombin production than for NfXI.
Discussion
Using a mathematical model of intravascular clotting in which coagulation occurs in tandem with platelet deposition and under flow, we explored the effect of thrombin-activated fXIa on thrombin production during TF-initiated coagulation. Our main result is the prediction that the sensitivity of thrombin production to the plasma [fXI] depends strongly not only on the level of exposed TF but also on the concentration of platelets. A high concentration of platelets in the blood within a few microns of the vessel wall increases the importance of fXI in the production of thrombin.
Based primarily on in vitro experiments without flow or platelets, it is believed that thrombin-activated fXIa plays a significant role in thrombin production only at low [TF]. The results depicted in Fig. 3 are consistent with this view and suggest that, even accounting for platelet deposition and flow, this view is correct. Further explorations show that the story may be more interesting.
Because platelet deposition physically inhibits its activity, TF:VIIa remains active for a length of time that depends on the near-wall platelet count and on the flow speed near the injury. Because high shear rates lead to higher near-wall platelet counts (28) and thus to faster platelet deposition, thrombin production that relies solely on TF:VIIa to generate fIXa is much reduced at higher shear rates (500 s−1 and above) even at high [TF] (15 fmol/cm2). As seen in Fig. 5, simply increasing the near-wall platelet count from 250,000 to 750,000/μL caused a significant drop in [fIIa] at both 10 min and 20 min. In another simulation with the higher platelet count, but with NfXI, [fIIa] at 20 min was substantially higher. The presence of the fXI reactions neutralized the effect of the threefold-higher platelet count on [fIIa] at 20 min.
Fluid-phase fXIa is inhibited by potent plasma proteins (including C1-inhibitor) and by protease-nexin 2 released from activated platelets (32), whereas platelet-bound fXIa is protected from these inhibitors (39,40). These inhibitors are not included in our model, so it may overpredict fXIa concentrations in the fluid. This should have little impact on the results because flow-mediated removal of plasma-phase species from the reaction zone is itself a potent regulator of local concentrations. The dominant fXIa species, by far, in the simulations is Plt-fXI-fXIa, which is the only form of fXIa in the model that at no time during its formation requires the enzyme to be free in the plasma.
Some people with fXI deficiency bleed excessively when they suffer trauma to tissues with high fibrinolytic activity (14,41). A rationale for this is that fXI is required for high levels of thrombin production after clot formation (i.e., a thrombin burst), which, in turn, is required to activate the fibrinolytic inhibitor thrombin-activatable fibrinolysis inhibitor-a (13,22). Although our model includes neither fibrin formation nor fibrinolysis, it does make predictions about the conditions under which a burst of thrombin production will occur. Our results suggest that the bleeding that fXI-deficient people experience in lysis-prone tissues may be more severe if their platelet count is also elevated. This counterintuitive result arises because of the platelets' dual role; in addition to providing the surfaces on which thrombin production occurs, the platelets also play an anticoagulant role by covering the TF exposed on the vascular wall.
Below-normal [fXI] protects against venous thrombosis (42) and some forms of arterial thrombosis (41), whereas above-normal [fXI] increases the risk of these diseases (43,44). Arterial thrombosis is primarily associated with rupture or erosion of atheroma and the consequent exposure of TF and other clot-promoting materials. Because our model indicates an important role for fXI under high shear conditions even for relatively high [TF], reduced fXI may lead to lower thrombin production in these situations. How venous thrombosis is initiated is much less clear; it may involve the adhesion of TF-bearing microparticles to activated endothelial cells (45). It seems reasonable that the level of TF expression in these situations will often be less than with overt vascular wall trauma. Also unclear is whether the concept of adherent platelets paving-over the damaged vascular wall is directly relevant, but the observation that platelet attachment to vWF released by activated endothelial cells may be an essential component of venous thrombosis (46) suggests that it may be. In any case, the model's prediction that fXI deficiency reduces thrombin generation for low TF exposures under venous flow conditions and normal platelet counts is consistent with a protective effect. In our simulations, above-normal [fXI] does not increase thrombin production. This suggests that mechanisms other than those under study here are the reasons for increased risk of thrombosis with elevated fXI.
Our predictions can be tested in a number of ways: One could do studies in an in vitro flow chamber with a surface coated with collagen and TF (as in Okorie et al. (25)), using normal and fXI deficient blood, and controlling the platelet count by adding washed platelets to the perfusate. Experiments could also be done using fXI knockout mice (47). With clinical data tabulating [fXI], platelet count, and bleeding behavior, one could determine whether patients with fXI deficiency and platelet count in the low-normal range bled less than those with fXI deficiency and platelet count in the high-normal range. Such studies could validate the predictions made with the model and establish whether platelet count provides an independent, clinically relevant risk factor for bleeding in fXI-deficient people.
In the model presented, we assume that each chemical or platelet species is well mixed in the reaction zone. We treat flow and transport in a simple way and allow for little influence of the deposited platelets on the delivery of proteins and platelets to the reaction zone. These are reasonable approximations for small spatial domains and small thrombi. For larger injuries and for larger thrombi that grow substantially into the vessel lumen, and to account more fully for interaction between growing thrombi and the flow, a more complex spatial-temporal model is required. For a larger thrombus, the relative importance of fIXa generated by TF:VIIa and that generated by platelet-bound fXIa may shift toward the latter because of the greater number of platelets in the thrombus. We have made significant progress in developing a model that can address such issues (48).
The results of this article suggest a general principle that the higher the bulk platelet count, the more important thrombin-activated fXIa is in further thrombin production. In vessels where the shear rate is sufficiently high to cause a high near-wall concentration of platelets to develop, this feedback mechanism seems to be critical to a strong response to vascular injury, even at relatively high TF exposures.
Acknowledgments
The authors thank P. N. Walsh and J. P. Keener for helpful discussions.
This work was supported by National Science Foundation grants DMS-0540779 and DMS-0943760 and by National Institute of General Medical Sciences grant R01-GM090203.
Supporting Material
References
- 1.Fogelson A.L., Tania N. Coagulation under flow: the influence of flow-mediated transport on the initiation and inhibition of coagulation. Pathophysiol. Haemost. Thromb. 2005;34:91–108. doi: 10.1159/000089930. [DOI] [PubMed] [Google Scholar]
- 2.Kuharsky A.L., Fogelson A.L. Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys. J. 2001;80:1050–1074. doi: 10.1016/S0006-3495(01)76085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savage B., Ruggeri Z. Platelet thrombus formation in flowing blood. In: Michelson A., editor. Platelets. Elsevier; New York: 2007. pp. 359–376. [Google Scholar]
- 4.Nemerson Y. The tissue factor pathway of blood coagulation. Semin. Hematol. 1992;29:170–176. [PubMed] [Google Scholar]
- 5.Mann K.G., Nesheim M.E., Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 6.Blombäck B. Fibrinogen structure, activation, polymerization and fibrin gel structure. Thromb. Res. 1994;75:327–328. doi: 10.1016/0049-3848(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 7.Lollar P., Knutson G.J., Fass D.N. Activation of porcine factor VIII:C by thrombin and factor Xa. Biochemistry. 1985;24:8056–8064. doi: 10.1021/bi00348a033. [DOI] [PubMed] [Google Scholar]
- 8.Monkovic D.D., Tracy P.B. Activation of human factor V by factor Xa and thrombin. Biochemistry. 1990;29:1118–1128. doi: 10.1021/bi00457a004. [DOI] [PubMed] [Google Scholar]
- 9.van 't Veer C., Mann K.G. Regulation of tissue factor initiated thrombin generation by the stoichiometric inhibitors tissue factor pathway inhibitor, antithrombin-III, and heparin cofactor-II. J. Biol. Chem. 1997;272:4367–4377. doi: 10.1074/jbc.272.7.4367. [DOI] [PubMed] [Google Scholar]
- 10.Esmon C.T. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem. 1989;264:4743–4746. [PubMed] [Google Scholar]
- 11.Hathcock J.J., Nemerson Y. Platelet deposition inhibits tissue factor activity: in vitro clots are impermeable to factor Xa. Blood. 2004;104:123–127. doi: 10.1182/blood-2003-12-4352. [DOI] [PubMed] [Google Scholar]
- 12.Bouma B.N., Griffin J.H. Human blood coagulation factor XI. Purification, properties, and mechanism of activation by activated factor XII. J. Biol. Chem. 1977;252:6432–6437. [PubMed] [Google Scholar]
- 13.von dem Borne P.A., Meijers J.C., Bouma B.N. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995;86:3035–3042. [PubMed] [Google Scholar]
- 14.Asakai R., Chung D.W., Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N. Engl. J. Med. 1991;325:153–158. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- 15.Gailani D., Broze G.J., Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 16.Naito K., Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J. Biol. Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 17.Gailani D., Renné T. Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007;27:2507–2513. doi: 10.1161/ATVBAHA.107.155952. [DOI] [PubMed] [Google Scholar]
- 18.Baird T.R., Walsh P.N. The interaction of factor XIa with activated platelets but not endothelial cells promotes the activation of factor IX in the consolidation phase of blood coagulation. J. Biol. Chem. 2002;277:38462–38467. doi: 10.1074/jbc.M205902200. [DOI] [PubMed] [Google Scholar]
- 19.Oliver J.A., Monroe D.M., Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler. Thromb. Vasc. Biol. 1999;19:170–177. doi: 10.1161/01.atv.19.1.170. [DOI] [PubMed] [Google Scholar]
- 20.Cawthern K.M., van 't Veer C., Mann K.G. Blood coagulation in hemophilia A and hemophilia C. Blood. 1998;91:4581–4592. [PubMed] [Google Scholar]
- 21.Keularts I.M., Zivelin A., Béguin S. The role of factor XI in thrombin generation induced by low concentrations of tissue factor. Thromb. Haemost. 2001;85:1060–1065. [PubMed] [Google Scholar]
- 22.von dem Borne P.A., Cox L.M., Bouma B.N. Factor XI enhances fibrin generation and inhibits fibrinolysis in a coagulation model initiated by surface-coated tissue factor. Blood Coagul. Fibrinolysis. 2006;17:251–257. doi: 10.1097/01.mbc.0000224843.33216.5f. [DOI] [PubMed] [Google Scholar]
- 23.Butenas S., van 't Veer C., Mann K.G. Evaluation of the initiation phase of blood coagulation using ultrasensitive assays for serine proteases. J. Biol. Chem. 1997;272:21527–21533. doi: 10.1074/jbc.272.34.21527. [DOI] [PubMed] [Google Scholar]
- 24.Tucker E.I., Marzec U.M., Hanson S.R. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okorie U.M., Denney W.S., Diamond S.L. Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: amplification of 100 fM circulating tissue factor requires flow. Blood. 2008;111:3507–3513. doi: 10.1182/blood-2007-08-106229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatterjee M.S., Denney W.S., Diamond S.L. Systems biology of coagulation initiation: kinetics of thrombin generation in resting and activated human blood. PLOS Comput. Biol. 2010;6:9. doi: 10.1371/journal.pcbi.1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tilles A.W., Eckstein E.C. The near-wall excess of platelet-sized particles in blood flow: its dependence on hematocrit and wall shear rate. Microvasc. Res. 1987;33:211–223. doi: 10.1016/0026-2862(87)90018-5. [DOI] [PubMed] [Google Scholar]
- 28.Woldhuis B., Tangelder G.J., Reneman R.S. Concentration profile of blood platelets differs in arterioles and venules. Am. J. Physiol. 1992;262:H1217–H1223. doi: 10.1152/ajpheart.1992.262.4.H1217. [DOI] [PubMed] [Google Scholar]
- 29.Kamath P., Krishnaswamy S. Fate of membrane-bound reactants and products during the activation of human prothrombin by prothrombinase. J. Biol. Chem. 2008;283:30164–30173. doi: 10.1074/jbc.M806158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gailani D., Ho D., Walsh P.N. Model for a factor IX activation complex on blood platelets: dimeric conformation of factor XIa is essential. Blood. 2001;97:3117–3122. doi: 10.1182/blood.v97.10.3117. [DOI] [PubMed] [Google Scholar]
- 31.Wu W., Sinha D., Walsh P.N. Factor XI homodimer structure is essential for normal proteolytic activation by factor XIIa, thrombin, and factor XIa. J. Biol. Chem. 2008;283:18655–18664. doi: 10.1074/jbc.M802275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley J., McEwan P.A., Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith S.B., Verhamme I.M., Gailani D. Characterization of novel forms of coagulation factor XIa: independence of factor XIa subunits in factor IX activation. J. Biol. Chem. 2008;283:6696–6705. doi: 10.1074/jbc.M707234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baglia F.A., Jameson B.A., Walsh P.N. Identification and characterization of a binding site for platelets in the Apple 3 domain of coagulation factor XI. J. Biol. Chem. 1995;270:6734–6740. doi: 10.1074/jbc.270.12.6734. [DOI] [PubMed] [Google Scholar]
- 35.Miller T.N., Sinha D., Walsh P.N. A catalytic domain exosite (Cys527-Cys542) in factor XIa mediates binding to a site on activated platelets. Biochemistry. 2007;46:14450–14460. doi: 10.1021/bi701310x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh P.N., Sinha D., Bradford H. Functional characterization of platelet-bound factor XIa: retention of factor XIa activity on the platelet surface. Blood. 1986;68:225–230. [PubMed] [Google Scholar]
- 37.Greengard J.S., Heeb M.J., Griffin J.H. Binding of coagulation factor XI to washed human platelets. Biochemistry. 1986;25:3884–3890. doi: 10.1021/bi00361a022. [DOI] [PubMed] [Google Scholar]
- 38.Hindmarsh A. ODEPACK, a systematized collection of ODE solvers. In: Stepleman R.S., editor. Scientific Computing, Vol. 1, IMACS Transactions on Scientific Computation. North-Holland; Amsterdam, The Netherlands: 1983. pp. 55–65. [Google Scholar]
- 39.Scandura J.M., Zhang Y., Walsh P.N. Progress curve analysis of the kinetics with which blood coagulation factor XIa is inhibited by protease nexin-2. Biochemistry. 1997;36:412–420. doi: 10.1021/bi9612576. [DOI] [PubMed] [Google Scholar]
- 40.Walsh P.N. Roles of platelets and factor XI in the initiation of blood coagulation by thrombin. Thromb. Haemost. 2001;86:75–82. [PubMed] [Google Scholar]
- 41.Seligsohn U. Factor XI deficiency in humans. J. Thromb. Haemost. 2009;7(Suppl 1):84–87. doi: 10.1111/j.1538-7836.2009.03395.x. [DOI] [PubMed] [Google Scholar]
- 42.Salomon O., Steinberg D.M., Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb. Haemost. 2011;105:269–273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 43.Meijers J.C., Tekelenburg W.L., Rosendaal F.R. High levels of coagulation factor XI as a risk factor for venous thrombosis. N. Engl. J. Med. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 44.Doggen C.J., Rosendaal F.R., Meijers J.C. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–4051. doi: 10.1182/blood-2005-12-023697. [DOI] [PubMed] [Google Scholar]
- 45.Mackman N., Tilley R.E., Key N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 46.Brill A., Fuchs T.A., Wagner D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–1407. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schumacher W.A., Luettgen J.M., Seiffert D.A. Inhibition of factor XIa as a new approach to anticoagulation. Arterioscler. Thromb. Vasc. Biol. 2010;30:388–392. doi: 10.1161/ATVBAHA.109.197178. [DOI] [PubMed] [Google Scholar]
- 48.Leiderman K., Fogelson A.L. Grow with the flow: a spatial-temporal model of platelet deposition and blood coagulation under flow. Math. Med. Biol. 2011;28:47–84. doi: 10.1093/imammb/dqq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.