

This communication reports the integration of nucleobases with small peptides generates a novel kind of nucleopeptides as biocompatible and biostable supramolecular hydrogelators. As a class of molecules that contain both nucleobases and amino acids, nucleopeptides bear considerable biological and biomedical importance.[1] Naturally occurring nucleopeptides, such as willardiine-containing nucleopeptides and peptidyl nucleosides, are antibiotics against microorganisms.[2] A number of unnatural nucleobase containing peptides, such as peptide nucleic acids (PNA), have found successful applications in biology and biomedicine (as an analog of DNA).[3], [4] Such biological significances render nucleopeptides as attractive targets for heterocyclic chemistry and useful molecules for studying biology, which has achieved considerable success.[5] There is, however, little work to use nucleopeptides for developing novel class of materials.[6] Thus, we decide to explore the potential of nucleopeptides to serve as building blocks for biomaterials. Among many possible choices of the types of materials, we chose to generate hydrogels[7] of nucleopeptides for two simple reasons: (i) supramolecular hydrogels, resulted from molecular self-assembly in water that form entangled nanofibers, have exhibited considerable promises for applications in biomedicine because of the inherent biocompatibility and biodegradability associated with the supramolecular nanofibers;[8] (ii) despite their versatility and importance, small nucleopeptides have been hardly explored for hydrogels. Thus, the primary goal of this work is to design, synthesize, and evaluate molecular hydrogelators[7a, 7b, 9] made of nucleopeptides.
Despite the existence of several well-characterized forms of nucleopeptides (chiral nucleopeptides, achiral nucleopseudopeptides, or peptidyl and amino nucleosides),[1b] it is unknown which types of nucleopeptides would be optimal for generating molecular hydrogelators that form nanofibers and hydrogels. Based on that the dipeptide, L-Phe-L-Phe (FF), is able to form nanotube structures[10] and that aromatic rings interact with neighboring nucleobases to stabilize designed DNA structures,[4a] we hypothesize that the conjugation of FF with a nucleobase should lead to a molecular hydrogelator. Such a rationale turns out to be valid. As shown in Scheme 1, the connection of a nucleobase (adenine, guanine, thymine, or cytosine) to the dipeptides segment (FF), affords a novel series of nucleopeptides (1) as hydrogelators that self-assemble in water to form nanofibers and produce hydrogels at the concentration of 2.0 wt% and pH around 5. Molecular mechanics (MM) calculation indicates that the Hoogsteen interactions among nucleobases promote the formation of the nanofibers. The conjugation of a tyrosine phosphate to 1 yields another group of nucleopeptides, precursors 2, which undergo catalytic dephosphorylation to generate hydrogelators 3 that result in supramolecular nanofibers and hydrogels at low concentration (2.0 wt%) and physiological pH. Surprisingly, both 2 and 3 exhibit significant resistance to proteinase K, a powerful digestive enzyme. This result unambiguously confirms the unique advantage of nucleobase. Moreover, circular dichroism (CD) experiment and rheological measurement indicate that the nucleobases of the nucleopeptidic hydrogelators, after self-assembly, are able to interact with the nucleic acids through Waston-Crick H-bonding. Because nucleobases are an important class of biofunctional motifs, this work not only illustrates the first example of nucleopeptides as hydrogelators made by an enzymatic reaction, but also provides a facile way to explore the potential applications of nucleopeptides as biomaterials, which may lead to a new and general platform to examine specific biological functions (e.g., binding to DNA and RNA) of a dynamic supramolecular system that is able to interact with both proteins and nucleic acids.
Scheme 1.
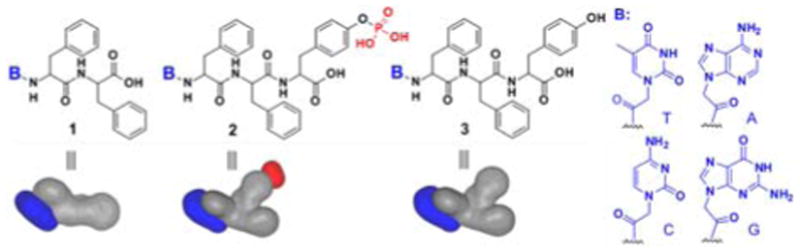
Molecular structures and shapes of the hydrogelators and corresponding precursors based on nucleopeptides.
Fig. 1a shows the typical synthetic route exemplified by the process for making the hydrogelators based on adenine. Following the procedures reported by Nieddu[11] for making nucleobase acetic acids, we first synthesized bis(tert-butyloxycarbonyl) (bis-Boc) protected adenine, (N6-bis-Boc-adenine-9-yl)-acetic acid (4). After being activated by N-hydroxysuccinimide (NHS), 4 reacts with L-Phe to afford 5, which undergoes the same NHS activation and phenylalanine coupling to give the key intermediate 6. Subsequent removal of the Boc-protecting groups with trifluoroacetic acid (TFA) yields the nucleopeptides (1A) in 47% total yield. Encouraged by that 1A, acting as a hydrogelator, self-assembles to form nanofibers with the diameter of 16 nm (Fig. 2a) and results in a hydrogel at the concentration of 2.0 wt% and pH of 5.0, we used the NHS-activated intermediate 6 to react with L-Tyr-phosphate to obtain 7, which forms precursor 2A after the deprotection of the Boc groups. The dephosphorylation process of precursor 2A catalyzed by an enzyme (Fig. 1b) leads to a translucent hydrogel of nucleopeptide 3A (Table 1) at the physiological pH. 31P NMR study confirms that the precursor (2A) completely transforms into the hydrogelator (3A) in 12 hr after the addition of alkaline phosphatase (ALP) (Fig. S2), and the TEM images (Fig. 2) of the negative stained hydrogel of 3A reveals nanofibers with a width of 20 nm, confirming that nanofibers of 3A act as the matrices to sustain the hydrogel (with a storage modulus around 2082 Pa at 2.0 wt%).
Figure 1.
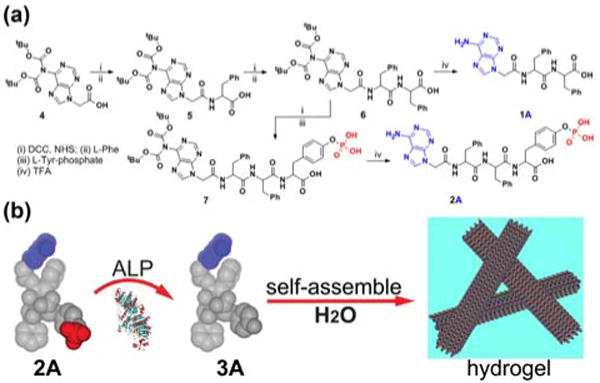
(a) A typical synthetic route of a hydrogelator (1A) and a precursor (2A) based on adenine; (b) illustration of the dephosphorylation process catalyzed by alkaline phosphatase (ALP) that converts 2A to 3A and results in nanofibers and a hydrogel.
Figure 2.
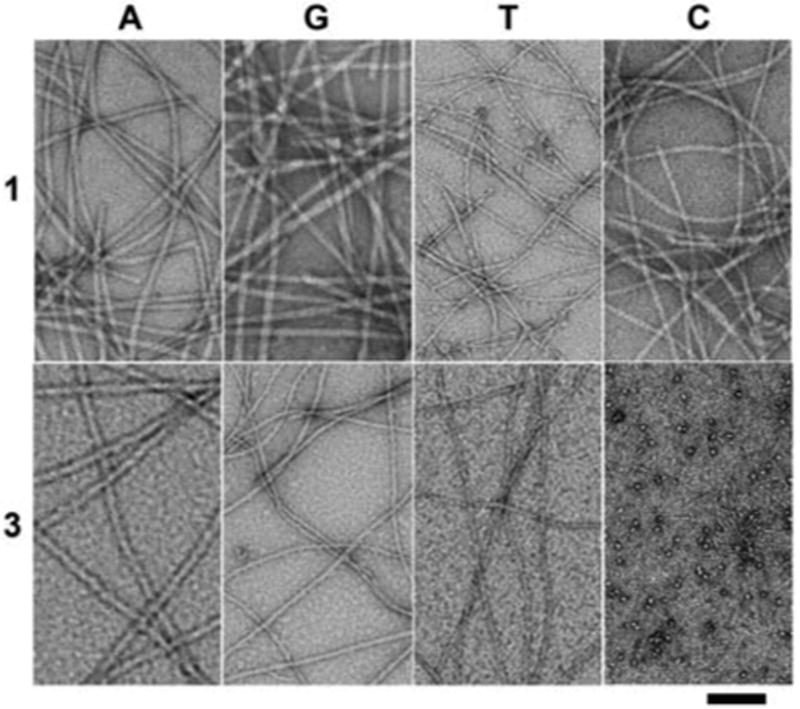
Transmission electron micrograph (TEM) of the hydrogels formed by 1A, 1G, 1T, 1C, 3A, 3G, 3T and the solution of 3C (scale bar = 100 nm).
Table 1.
The conditions and properties of the nucleopeptidic hydrogelators and corresponding supramolecular nanofibers and hydrogels.
| Sample | 1A | 1G | 1T | 1C | 3A | 3G | 3T | 3C |
|---|---|---|---|---|---|---|---|---|
| wt % | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 |
| pH | 5.0 | 5.0 | 5.0 | 5.0 | 7.4 | 7.4 | 7.4 | 7.4 |
| Optical images |

|

|

|

|

|

|

|

|
| Width of nanofibers (nm) | 16 | 15 | 9 | 10 | 20 | 14 | 95 | 5a |
| Critical strain (%) | 1.0 | 0.8 | 1.2 | 0.6 | 0.4 | 2.0 | 8.0 | - |
| G′ (pa) | 8090 | 12613 | 6346 | 26 | 2082 | 682 | 2.9 | - |
| IC50 (μM) | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
These thin nanofibers have low quantity and coexist with nanoparticles, thus fail to result in a hydrogel
The formation of the nanofibers and the hydrogel of 1A or 3A indicates that the direct attachment of a purine or pyrimidine base to a small peptide is a valid approach to design hydrogelator of nucleopeptides. To examine the generality of this approach, we used the synthetic procedures similar to Fig. 1a to produce the nucleopeptides consisting of other nucleobases (G, T, or C) and examined their capability to form nanofibers and hydrogels. As revealed by TEM (Fig. 2), hydrogelators 1G, 1T and 1C self-assemble to form nanofibers with the width of 15, 9, and 10 nm, respectively, and the nanofibers entangle to trap water and result in the hydrogels (Table 1) at the concentration of 2.0 wt% and slight acid condition (pH 5.0).
Like 2A, precursors 2G and 2T, at 2.0 wt% and pH 7.4, upon the addition of ALP (10 U), turn into hydrogelators 3G and 3T, respectively. This enzymatic conversion leads to the formation of nanofibers 3G and 3T and results in the corresponding hydrogels shown in Table 1. TEM reveals that the diameters of the nanofibers of 3G (14 nm) and 3T (9 nm) are similar to those of the nanofibers of 1G and 1T, respectively. At the concentration of 2.0 wt% and pH 7.4, 3C self-assembles to afford both nanoparticles (11 nm) and short, thin nanofibers (4 nm in diameter and about 200 nm long), but fails to form well-defined nanofiber networks to provide effective matrices that warrant a hydrogel of 3C.
We measured the rheological properties of the hydrogels to gain further insight on their characteristics. As shown in Table 1, among them, the hydrogel of 1G exhibits the highest storage modulus (12. 6 KPa), the hydrogels of 1A and 1T possess relatively high storage moduli of 8.1 KPa and 6.3 KPa, respectively, and the hydrogel of 1C has the lowest storage modulus (26 Pa). The storage moduli of the hydrogels of 3G and 3T are 682 Pa and 2.9 Pa, respectively, indicating that the hydrogel of 3T possesses much weaker mechanical strength than those of the hydrogels 3A and 3G (Table 1). The relatively high storage moduli of hydrogels of 1A, 1G, 3A, and 3G may stem from that purine bases favor the formation of Hoogsteen base pair,[12] in addition to strong π-π interaction of purine nucleobases that contain two fused five- and six-member heterocyclic rings. Moreover, the lower storage moduli of the hydrogels of 3 than those of the corresponding hydrogels of 1 suggest that the presence of tyrosine may reduce the efficiency of the non-covalent interactions required for the stabilization of self-assembled nanostructures, thus resulting in a relatively weak viscoelastic property of those hydrogels.
Furthermore, the addition of an oligomeric deoxyadenosine (A10) to the hydrogel of 1T or 3T results in a more stable hydrogel (Figs. S3, S6), as demonstrated by the increase of storage modulus (G') from 6.3 KPa (of hydrogel 1T) to 14.3 KPa (of the hydrogel of 1T and A10), or from 2.9 Pa (of hydrogel 3T) to 12.0 Pa (of the hydrogel of 3T and A10) (Fig. S6). This result suggests that Waston-Crick interactions between the self-assembly of 1T (or 3T) and A10 favor molecular aggregation and enhance the mechanical strength of the hydrogels. To further examine Waston-Crick H-bonding between complementary nucleobases among the hydrogelators, we use hydrogelators of 1T and 1A (or 3T and 3A) to prepare a mixed hydrogel and find that the increase of the storage modulus (G′) from 6.3 KPa (of hydrogel 1T) to 18 KPa (of the hydrogel of 1T and 1A), or from 2.9 Pa (of hydrogel 3T) to 150 Pa (of the hydrogel of 3T and 3A). The mixed hydrogel of the mismatched nucleobases (i.e., (1T and 1G) or (1T and 1C)) exhibits, however, little increase of the storage moduli (Fig. S8) in comparison to that of hydrogel 1T. These results indicate that these nucleopeptidic hydrogelators preserve Waston-Crick interaction of the nucleobases.
We used circular dichroism (CD) to study the superstructures of these nanofibers of self-assembled nucleopeptides in the gel phase. The hydrogels of 1 all give a common feature of β-sheet structure according to the CD spectra with a positive peak near 195 nm and a negative peak around 210 nm (Fig, S3), suggesting these nucleopeptides arrange into β-sheet-like configurations. The hydrogels of 3A, 3G, and 3T display the common feature of the CD spectra with a positive peak near 195 nm and a negative peak around 210 nm, also suggesting that the nucleopeptides adopt β-sheet-like configurations. The CD spectrum of 3C solution exhibits a positive peak near 203 nm and a negative peak around 215 nm, which red-shifts regarding to those in typical β-sheet. The red-shifted β-sheet signal likely associates with a twisted structure as opposed to the standard planar β-sheet, agreeing with the fact that the increase in β-sheet twisting causes disorder and results in the short nanofibers and the nanoparticles, which leads to weak mechanical strength.[13] Overall, the signals of β-sheet (i.e., transitions at 195 nm∼225 nm) of 1 are stronger than those of 3, following the trends that the storage moduli of 1 are larger than those of 3. The CD signals with broad bands around 300 nm among the hydrogels 1 and 3 likely originate from the formation of mesophases of hydrogelators because they locate far from the chromophoric absorption region (ca. 270 nm) of the hydrogelators (Fig. S4).
Similar to other nucleobase-containing small molecules that bind with nucleic acids through Waston-Crick interaction,[14] hydrogelator 1T or 3T also binds to oligomeric deoxyadenosine (e.g., A10), which results in the distinctive changes in the CD spectra. For example, comparing to the CD of hydrogel 1T, the CD of the 1T-A10 mix gel (Fig. S5) exhibits the decreased ellipticity of positive band at 192, 228 nm and negative bands around 205, 247 and 287 nm regions. The CD spectrum of 3T-A10 mix gel (Fig. S5) shows that the addition of A10 both changes the intensity of bands at 195, 205 nm and creates a new band at 303 nm that can be resulted from the conformational change of self-assembled structures of 3T induced by A10.[14a-c] In addition, comparing to the solution of 1T or 2T, the CD spectra of the mix solution of A10 with hydrogelator 1T (or precursor 2T) shows slight changes of the band shape, indicating the relatively weak interaction in the solution state (Fig. S5).
We also used molecular mechanical (MM) calculation to evaluate non-covalent interactions[15] and simulate the width of the nanofibers of 1. As shown in Fig. 3a, the simulated widths of the nanofibers are 15 nm, 16 nm, 9 nm and 11 nm for nucleopeptides 1A, 1G, 1T and 1C, respectively, which correlate well with the trend in the experimental observation (Fig. 2). According to simulation, the thicker width of nanofibers in the hydrogels of 1A and 1G are likely resulted from the formation of Hoogsteen base pair[12, 16] by adenine or guanine nucleobases (Figs. S10 and S11). In addition, MM calculation supports the formation of β-sheet like structure.
Figure 3.
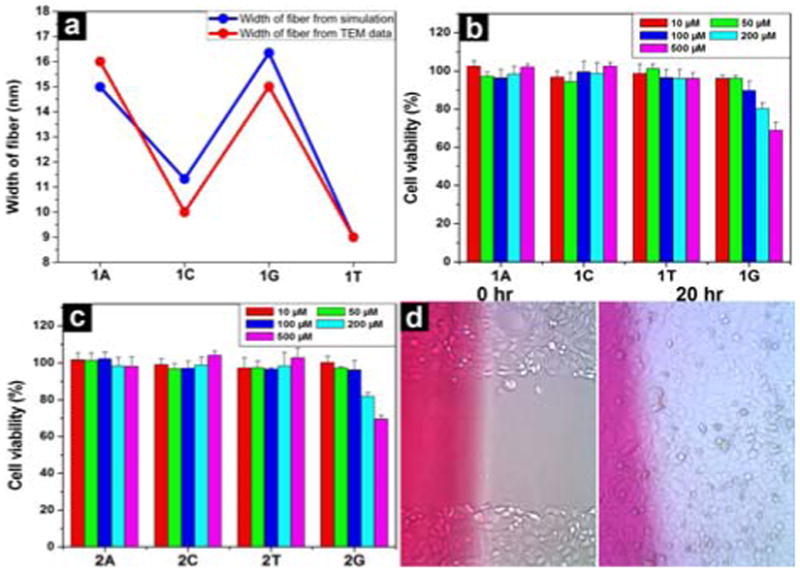
(a) A comparison of the width of fibers of hydrogels 1A, 1C, 1G and 1T based on transmission electron micrographs (in red) and molecular mechanical calculations (in blue). 72 hr cell viability test of (b) 1A, 1C, 1T and 1G; (c) 2A, 2C, 2T and 2G. (d) Optical images of HeLa cells on the surface 0 h and 20 h after the creation of scratch-wound in the presence of hydrogel 3T (by adding 27.7 mM of 3T in the media).
To verify the biocompatibility of the hydrogelators, we added hydrogelator 1 or precursor 2 into the culture of HeLa cells and measured the proliferation of the cells. According to the MTT assay shown in Fig. 3, after being incubated with the 500 μM of hydrogelator (1A, 1T, or 1C) or the precursor (2A, 2T, or 2C) for 72 hr, the cell viability remain at 100%. Although the cell viability decreases slightly when they are incubated with 500 μM of 1G or 2G for 72 hr, the value of IC50 is still > 500 μM. These results prove that nucleopeptides 1, 2, and 3 are biocompatible. We also used a simple wound-healing assay[17] to examine the capability of the nanofibers and hydrogels of 3 to serve as a material for maintain cell-matrix interaction. As shown in Fig. 3d, the presence of the hydrogel of 3T in cell culture has little inhibitory effect on the migration of cells, further supporting the biocompatibility of 3.
Besides biocompatibility, biostability is also an essential requisite for a biomaterial. Thus, we examine the stability of hydrogelators by incubating them with proteinase K, a powerful protease that hydrolyzes a wide range of peptidic substrates and cleavage the peptide bond adjacent to the carboxyl group of aliphatic and aromatic amino acids with blocked alpha amino groups.[18] As shown in Fig. 4, more than 85 % of 2T, 3T, or 3A, more than 70 % of 2A or 3C, and above 50% of 2C remain after 24 hrs of the incubation with proteinase K; more than 40% of 2G or 3G remains after 4 hrs of the incubation with proteinase K. Although less that 10% of 1T or Nap-FFY[19] remains after 4 hrs of the incubation with proteinase K (Fig. S13), the excellent or fair resistance to enzymatic digestion, exhibited by the nucleopeptides 2 and 3, confirms the unique advantage of the nucleobases. Because of their high resistance to proteases, the hydrogel formed by hydrogelator 3T, 3A, or 3C promises to serve as new biomaterials for applications that require long-term biostability. In addition, this result suggests that the incorporation of nucleobase may be an effective approach for improving the biostability of other small peptidic hydrogelators.
Figure 4.
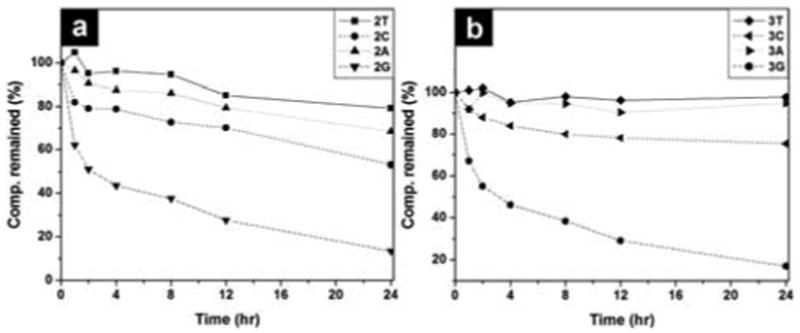
The time-dependent course of the digestions of hydrogelators of (a) 2T, 2C, 2G, 2A, and (b) 3T, 3A, 3G and compound 3C by proteinase K.
In conclusion, this work not only demonstrates the generation of a new type of hydrogelators based on the conjugates of nucleobases and short peptides that self-assemble in water to afford supramolecular hydrogels upon a pH- or enzymatic trigger, but also introduces a new, simple, and general approach for developing soft, biocompatible materials from nucleopeptides. Since it is easy to incorporate other bioactive peptides or molecular recognition motifs[20] with nucleobases, this work provides a facile way to explore the new applications of nucleopeptides as functional biomaterials.
Supplementary Material
Acknowledgments
This work was partially supported by NIH (R01CA142746), NSF (DMR 0820492), a HFSP grant (RGP0056/2008), and start-up grant from Brandeis University. The images were taken at Brandeis EM facilities.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Roviello GN, Musumeci D, Bucci EM, Pedone C. Mol Biosyst. 2011;7:1073. doi: 10.1039/c0mb00214c. [DOI] [PubMed] [Google Scholar]; b) Roviello GN, Benedetti E, Pedone C, Bucci EM. Amino Acids. 2010;39:45. doi: 10.1007/s00726-010-0567-6. [DOI] [PubMed] [Google Scholar]
- 2.a) Azzam ME, Algranat Id. Proc Natl Acad Sci USA. 1973;70:3866. doi: 10.1073/pnas.70.12.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hector RF, Zimmer BL, Pappagianis D. Antimicro Agents Chemother. 1990;34:587. doi: 10.1128/aac.34.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Itaya M, Yamaguchi I, Kobayashi K, Endo T, Tanaka T. J Biochem. 1990;107:799. doi: 10.1093/oxfordjournals.jbchem.a123128. [DOI] [PubMed] [Google Scholar]
- 3.a) Nielsen PE, Egholm M, Berg RH, Buchardt O. Science. 1991;254:1497. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]; b) Haaima G, Lohse A, Buchardt O, Nielsen PE. Angew Chem Int Ed. 1996;35:1939. [Google Scholar]
- 4.a) Kool ET. Acc Chem Res. 2002;35:936. doi: 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]; b) Pradeepkumar PI, Hobartner C, Baum DA, Silverman SK. Angew Chem Int Ed. 2008;47:1753. doi: 10.1002/anie.200703676. [DOI] [PubMed] [Google Scholar]; c) Kool ET, Morales JC, Guckian KM. Angew Chem Int Ed. 2000;39:990. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Nielsen PE. Chem Biodivers. 2010;7:786. doi: 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]
- 6.a) Iwaura R, Yoshida K, Masuda M, Ohnishi-Kameyama M, Yoshida M, Shimizu T. Angew Chem, Int Ed. 2003;42:1009. doi: 10.1002/anie.200390257. [DOI] [PubMed] [Google Scholar]; b) Shimizu T, Iwaura R, Masuda M, Hanada T, Yase K. J Am Chem Soc. 2001;123:5947. doi: 10.1021/ja010201i. [DOI] [PubMed] [Google Scholar]
- 7.a) Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]; b) Yang ZM, Gu HW, Fu DG, Gao P, Lam JK, Xu B. Adv Mater. 2004;16:1440. [Google Scholar]; c) Lee KY, Mooney DJ. Chem Rev. 2001;101:1869. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]; d) Choi SW, Zhang Y, Xia YN. Angew Chem Int Edit. 2010;49:7904. doi: 10.1002/anie.201004057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Kiyonaka S, Sada K, Yoshimura I, Shinkai S, Kato N, Hamachi I. Nat Mater. 2004;3:58. doi: 10.1038/nmat1034. [DOI] [PubMed] [Google Scholar]; b) Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]; c) Toledano S, Williams RJ, Jayawarna V, Ulijn RV. J Am Chem Soc. 2006;128:1070. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]; d) Ulijn RV, Woolfson DN. Chem Soc Rev. 2010;39:3349. doi: 10.1039/c0cs90015j. [DOI] [PubMed] [Google Scholar]; e) Valery C, Paternostre M, Robert B, Gulik-Krzywicki T, Narayanan T, Dedieu JC, Keller G, Torres ML, Cherif-Cheikh R, Calvo P, Artzner F. Proc Natl Acad Sci USA. 2003;100:10258. doi: 10.1073/pnas.1730609100. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wada A, Tamaru S, Ikeda M, Hamachi I. J Am Chem Soc. 2009;131:5321. doi: 10.1021/ja900500j. [DOI] [PubMed] [Google Scholar]; g) Micklitsch CM, Knerr PJ, Branco MC, Nagarkar R, Pochan DJ, Schneider JP. Angew Chem Int Ed. 2011;50:1577. doi: 10.1002/anie.201006652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) George M, Weiss RG. Acc Chem Res. 2006;39:489. doi: 10.1021/ar0500923. [DOI] [PubMed] [Google Scholar]; b) Terech P, Weiss RG. Chem Rev. 1997;97:3133. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]; c) Wang QG, Yang ZM, Zhang XQ, Xiao XD, Chang CK, Xu B. Angew Chem Int Ed. 2007;46:4285. doi: 10.1002/anie.200700404. [DOI] [PubMed] [Google Scholar]; d) Yang Z, Liang G, Guo Z, Xu B. Angew Chem Int Ed. 2007;46:8216. doi: 10.1002/anie.200701697. [DOI] [PubMed] [Google Scholar]; e) Li XM, Li JY, Gao YA, Kuang Y, Shi JF, Xu B. J Am Chem Soc. 2010;132:17707. doi: 10.1021/ja109269v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Gorbitz CH. Chem Eur J. 2001;7:5153. doi: 10.1002/1521-3765(20011203)7:23<5153::aid-chem5153>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]; b) Gorbitz CH. Chem Commun. 2006:2332. doi: 10.1039/b603080g. [DOI] [PubMed] [Google Scholar]; c) Gorbitz CH. Chem Eur J. 2007;13:1022. doi: 10.1002/chem.200601427. [DOI] [PubMed] [Google Scholar]
- 11.Porcheddu A, Giacomelli G, Piredda I, Carta M, Nieddu G. Eur J Org Chem. 2008:5786. [Google Scholar]
- 12.a) Araki K, Yoshikawa I. Low Molecular Mass Gelators: Design, Self-Assembly, Function. Vol. 256. Springer-Verlag Berlin; Berlin: 2005. p. 133. [Google Scholar]; b) Davis JT. Angew Chem, Int Ed. 2004;43:668. doi: 10.1002/anie.200300589. [DOI] [PubMed] [Google Scholar]
- 13.Manning MC, Illangasekare M, Woody RW. Biophys Chem. 1988;31:77. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]; b) N. W. Sreerama, R. W, (Ed.: N. Berova, Nakanashi, K., Woody, R. W.), Wiley-VCH, New York, 2000, pp. p 601.
- 14.a) Arigon J, Prata CAH, Grinstaff MW, Barthelemy P. Bioconjugate Chem. 2005;16:864. doi: 10.1021/bc050029y. [DOI] [PubMed] [Google Scholar]; b) Dong GM, Zhang LR, Zhang LH. Helv Chim Acta. 2003;86:3516. [Google Scholar]; c) Godeau G, Bernard J, Staedel C, Barthelemy P. Chem Commun. 2009:5127. doi: 10.1039/b906212b. [DOI] [PubMed] [Google Scholar]; d) Iwaura R, Hoeben FJM, Masuda M, Schenning A, Meijer EW, Shimizu T. J Am Chem Soc. 2006;128:13298. doi: 10.1021/ja064560v. [DOI] [PubMed] [Google Scholar]; e) Janssen PGA, Vandenbergh J, van Dongen JLJ, Meijer EW, Schenning A. J Am Chem Soc. 2007;129:6078. doi: 10.1021/ja0711967. [DOI] [PubMed] [Google Scholar]
- 15.Mayo SL, Olafson BD, Goddard WA. J Phys Chem. 1990;94:8897. [Google Scholar]
- 16.Sivakova S, Rowan SJ. Chem Soc Rev. 2005;34:9. doi: 10.1039/b304608g. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez LGWX, Guan JL. In: Cell Migration: Developmental Methods and Protocols. Guan JL, editor. Vol. 294. Humana Press Inc.; Totowa, NJ: 2004. p. 23. [Google Scholar]
- 18.a) Bromme D, Peters K, Fink S, Fittkau S. Arch Biochem Biophys. 1986;244:439. doi: 10.1016/0003-9861(86)90611-9. [DOI] [PubMed] [Google Scholar]; b) Liang GL, Yang ZM, Zhang RJ, Li LH, Fan YJ, Kuang Y, Gao Y, Wang T, Lu WW, Xu B. Langmuir. 2009;25:8419. doi: 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- 19.a) Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Kuang Y, Gao YA, Xu B. Langmuir. 2011;27:529. doi: 10.1021/la1020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider HJ. Angew Chem Intl Ed. 2009;48:3924. doi: 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.