

Abstract
Increased endothelial permeability contributes to the morbidity and mortality associated with chronic inflammatory diseases, including acute lung injury. Cyclic AMP response element-binding protein (CREB) transcriptional factor induces genes that regulate inflammation and vascular remodeling. However, the role of CREB in regulating endothelial barrier function is unknown. Here, we demonstrate that CREB maintains basal endothelial barrier function and suppresses endothelial permeability increase by diverse agonists such as thrombin, lipopolysaccharide, histamine, and VEGF. We show that CREB transcriptionally controls the expression of p190RhoGAP-A, a GTPase-activating protein that inhibits small GTPase RhoA. Impairing CREB function using small interfering RNA or dominant-negative (dn)–CREB mutant (dn-CREB) markedly suppressed p190RhoGAP-A expression, increased RhoA activity, induced actin stress fiber formation, and produced an amplified and protracted increase in endothelial permeability in response to thrombin. Rescuing p190RhoGAP-A expression restored the permeability defect in dn-CREB–transducing endothelial cells. These findings were recapitulated in vivo because dn-CREB expression in mice vasculature increased basal lung microvessel permeability and exaggerated permeability increase induced by thrombin and lipopolysaccharide. Inhibiting RhoA signaling restored endothelial barrier dysfunction in the dn-CREB–expressing lung microvasculature. These results uncover a pivotal role of CREB in regulating endothelial barrier function by restricting RhoA signaling through controlling p190RhoGAP-A expression.
Introduction
The vascular endothelium lining all blood vessels dynamically regulates nutrient supply to underlying tissues and also maintains host-defense and tissue-fluid homeostasis.1 Endothelial monolayer integrity is maintained by the integrated actions of the contractile and interendothelial adhesive forces that couple cells with each other.1–4 However, increased actin-myosin–driven endothelial cell contraction weakens intercellular adhesion, forming minute gaps between endothelial cells, leading to accumulation of protein-rich fluid in the interstitial tissue, a hallmark of tissue inflammation, including acute lung injury.1,2,5,6
Cyclic AMP response element-binding (CREB) protein is a nuclear transcriptional factor that regulates several cellular functions, such as inflammation, cell proliferation, differentiation, adaptation, and survival.7,8 Mice lacking CREB die postnatally within 15 minutes primarily because of impairment of lung function.9,10 However, increased CREB activity has been shown to be associated with pathogenesis of asthma, chronic obstructive pulmonary disease, cognitive memory alteration, and neointima formation.11–14 CREB expression also is induced after endotoxemia or hemorrhage-induced acute lung injury, but its significance remains unclear.15,16 Studies show that various stimuli, including growth factors,7 oxidants,17–19 and G protein–coupled receptors ligands7 induce CREB activity by mediating CREB phosphorylation at serine 133 residue.7,20,21 For example, adenosine by activating adenosine A2 receptor stimulates cAMP/protein kinase A cascade that in turn phosphorylates CREB at serine 133 residue, inducing its transcriptional activity.22,23 Moreover, protein kinase C and MAP kinases as well as Ca2+ calmodulin-dependent kinase can induce CREB activity by phosphorylating it at serine 133 residue.7,21 On being phosphorylated CREB binds to DNA and regulates the transcription of proteins that contains a cAMP response element (CRE) sequence within their promoter.21
The small GTPase RhoA plays a critical role in inducing endothelial cell contraction and thereby in increasing endothelial permeability.1,24,25 RhoA activity is finely regulated by the GTPase-activating proteins (GAPs) that stimulate GTP hydrolysis by GTPases “switching off” the RhoA cycle.26 Studies show that p190RhoGAP (referred to as p190 hereafter) specifically targets RhoA.27,28 Impairment of p190 function leads to constitutive activation of RhoA signaling, leading to persistent increase in endothelial permeability.29,30 Thus, p190, by antagonizing RhoA activity, mitigates the increase in endothelial monolayer permeability. Although signaling mechanisms that regulate p190 function are progressively becoming clear, much less is known about the molecular mechanisms that regulate p190 expression. Interestingly, p190 promoter contains CRE sequence. Thus, we tested the hypothesis that CREB plays an important role in maintaining endothelial barrier function through its ability to transcriptionally control p190 expression.
We interfered with the function of CREB using small interfering RNA (siRNA) or transduced dominant-negative (dn)–CREB mutant (Ser133Ala-CREB mutant) in endothelial cells and in wild type-mice microvasculature to explore the role of CREB in regulating endothelial permeability. Here, we demonstrate p190 as an effector of CREB via which CREB controls RhoA signaling and thereby maintains basal endothelial barrier function and suppresses the persistent increase in endothelial permeability by proinflammatory mediator thrombin as well as lipopolysaccharide (LPS).
Methods
Materials
Human pulmonary arterial endothelial (HPAE) cells and endothelial growth medium (EBM-2) were obtained from Lonza Walkerville. Human α-thrombin was obtained from Enzyme Research Laboratories. The Nucleofactor HCAEC kit and electroporation system were from Amaxa Biosystems. Anti-CREB, anti-RhoA, and HRP-conjugated anti–mouse immunoglobulin G (IgG) antibodies were purchased from Santa Cruz Biotechnology. Anti–phospho-133-CREB antibody was purchased from Cell Signaling Technology, anti–p190RhoGAP-A antibody was purchased from BD Biosciences, and anti–phospho-T850 myosin light chain (MLC) phosphatase 1 (MYPT1) antibody was from Millipore. Alexa-labeled 488 donkey anti–goat secondary antibody and rhodamine-phalloidin were purchased from Invitrogen. CREB siRNA (109994) 5′-GGUGGAAAAUGGACUGGCUtt-3′ was purchased from Ambion.31 Pooled p190RhoGAP-A siRNA and scrambled siRNA with no sequence homology to human genome was purchased from Dharmacon RNA Technologies. T4 polynucleotide kinase was obtained from New England Biolabs. [gamma-32P]ATP (specific activity, 3000 Ci/mmol) was from MP Biomedicals. Ser133Ala-CREB mutant was purchased from Addgene. All of the primers and oligonucleotides were synthesized by Integrated DNA Technologies.
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Illinois. C57BL/6J mice were used as wild-type controls. All experiments were performed on 6- to 8-week-old male mice.
Cell culture and transfection
HPAE cells were cultured as described previously.32 HPAE cells were transfected with siRNA using a Nucleofector device (Amaxa Biosystems) or siRNA transfection reagent (Santa Cruz Biotechnology). cDNA was transduced into HPAE cells using SuperFect (QIAGEN) or Nucleofactor.32,33 We routinely observed 90% siRNA transfection efficiency in endothelial cells,32,33 whereas the efficiency of cDNA lies between 50% and 70%.32,33
Endothelial permeability
Endothelial permeability was determined by measuring the influx of Evans blue–labeled albumin across endothelial monolayer or by assessing changes in transendothelial electrical resistance in real-time, as described previously.27,32
Immunofluorescence
HPAE cells were fixed and incubated with anti–VE-cadherin antibody for 1 hour by Alexa-labeled 488 donkey anti–goat antibody and rhodamine phallodin.27,34 Cells were viewed at room temperature with a 63×/1.2 NA objective and an LSM 510 confocal microscope (Carl Zeiss).
RhoA activity
RhoA activity was measured using the GST-rhotekin-Rho binding protein that specifically pulls down activated RhoA, as described previously.27
Western blot analysis
HPAE cells were lysed, and lysates containing equal amounts of protein were resolved by electrophoresis and immunoblotted using appropriate antibodies, as described previously.32,34
G-actin and F-actin ratio
Cells were collected in ice-cold PBS and globular (G)– and filamentous (F)–actin were extracted using buffers, as described previously.35 Equal volumes of G- and F-actin lysates mixed with SDS loading buffer were separated by 10% SDS-PAGE and detected by immunoblotting with an anti-actin antibody.
Quantitative real-time RT-PCR
Total RNA was isolated from HPAE cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA was quantified spectrophotometrically and reverse transcription (RT)–PCR was carried out with a High-Capacity cDNA reverse transcription kit based on the supplier's protocol (AB4368814; Applied Biosystems). cDNA was then used for the PCR amplification using SYBR Green assay kit (AB4309155; Applied Biosystems) and a Prism 7000 Real-Time PCR machine (Applied Biosystems) using the following conditions: 95°C for 10 minutes followed by 40 cycles at 95°C for 15 seconds with extension at 56°C for 1 minute for both p190-A and GAPDH. The PCR amplification run was examined, using the 7300 real-time PCR system–operated SDS Version 1.4 program (Applied Biosystems). The program uses the Delta Rn analysis method (Applied Biosystems). PCR products were separated on 1% agarose gel. Primers used for p190-A were forward (F), AGAAAGAGCCGGTTGGTTCAT and reverse (R), AACATAGCCAAAGAGGCCTTACG according to accession NM_004491.3. Primers used for CREM were F, GCCTGCCTCCTCGCGAACTT and R, TAAACCGCTCCCCTGAGCCG; for ATF2 were F, TGCCCTGTAACCGCCATGCAGA and R, GGCTCTGTACTCTGGTCCGCCA; for Rap1 GTPase were F, GTGTCTCACTGCACCTTCAATGGCA and R, ACGCCTCCTGAACCAAGGACCA; and for p115RhoGEF were F, TCTTCCGGAAAAAGGTGATG and R, GGTCTGTAGCACCTGGCTTC.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from HPAE cells using protocols as described previously.13 Protein–DNA complexes were formed by incubating 5 μg of nuclear protein in a total volume of 20 μL of buffer consisting of 15mM HEPES, pH 7.9, 3mM Tris-HCl, pH 7.9, 60mM KCl, 1mM EDTA, 1mM phenylmethylsulfonyl fluoride, 1mM dithiothreitol, and 4.5 μg of bovine serum albumin, 2 μg of poly(dI-dC), 15% glycerol, and 100 000 cpm of 32P-labeled oligonucleotide probe for 30 minutes at 30°C. The protein–DNA complexes were resolved by electrophoresis on a 4% polyacrylamide gel using 1 × Tris-glycine-EDTA buffer (25mM Tris-HCl, pH 8.5, 200mM glycine, and 0.1mM EDTA). CREB consensus oligonucleotides (5′-AGAGATTGCCTGACGTCAGAGAGCTAG-3′) were used as 32P-labeled oligonucleotide probes to measure CREB DNA-binding activities. Double-stranded oligonucleotides were labeled with [gamma-32P] ATP using the T4 polynucleotide kinase kit (New England Biolabs) following the supplier's protocol.
Chromatin immunoprecipitation assay
Cells were cross-linked in situ by incubating them with 1% formaldehyde at 37°C for 10 minutes, and lysates were collected in ice-cold phosphate-buffered saline. Lysates were resuspended in SDS lysis buffer, followed by sonication (550 sonic dismembrator; Thermo Fisher Scientific) to generate DNA fragments of 500 to 1000 bp. Samples were then diluted in ChIP dilution buffer, precleared with the salmon sperm DNA/protein A agarose-50% slurry, and supernatant was collected after the centrifugation. Supernatant was immunoprecipitated with 10 μg of anti-CREB antibody overnight followed by addition of agarose beads. Immunocomplexes were then retrieved by centrifugation and washed 5 times following the Millipore ChIP assay protocol. The immunoprecipitated DNA was uncross-linked, subjected to proteinase K digestion, and purified using QIAquick columns (QIAGEN). The purified DNA was used as a template for PCR amplification using primers (F, 5′-ATCTCAGCTCACCACAACCACT-3′; R, 5′-CGTGGTAGCTTACGCCTGTAAT-3′) flanking the putative CREB binding site located from −735 and −728 in human p190-A promoter region. The PCR products were resolved on 1.5% agarose gels and visualized by ethidium bromide staining, as described previously.13
Plasmid PGL3-basic-hRhoGap promoter
The human p190Rho-A promoter was cloned from human genomic DNA (BD Biosciences) by PCR amplification using Finnzymes Phusion High-Fidelity DNA polymerase (New England Biolabs). Primers were designed to amplify a fragment of 2951 bp based on the genomic DNA sequence GenBank NT_011109.16. The PCR-amplified fragment spanned a region from −2950 to −1 relative to the translational start site of the human p190RhoGap protein. Shown are the primers sequences that include restriction sites (italicized) that allow for subcloning into PGL3-basic vector: hRGap-Pro-NheI-F, 5′-ATATATGCTAGCGCAggttcagtgcttgcacag-3′ and hRGap-Pro-HindIII-R, 5′-ATATATAAGCTTCGTCGACACATCCTGCCACGA-3′. The amplified human p190RhoGap promoter fragment containing the restriction sites 5′-NheI/HindIII-3′ was digested with NheI and HindIII restriction enzymes (New England Biolabs) and ligated to the same digested restriction sites of the PGL3-basic vector (Promega) using T4 DNA ligase (New England Biolabs). The resulting plasmid PGL3-basic-hRhoGap promoter was verified for DNA quality and integrity through gel analysis and sequencing analysis.
Luciferase assay
HPAE cells were first transfected with scrambled or CREB siRNA, and after 24 hours cells were transduced with p190-A promoter-luciferase construct or pGL3 basic vector using Lipofectamine 2000 reagent (Invitrogen). Cells were left unstimulated or stimulated with thrombin for 6 hours, washed once with ice-cold PBS, and lysed. Luciferase activity was determined using a Promega kit following manufacture's protocols. Values are expressed as relative luciferase units.
Liposomal delivery of cDNA in the mouse lung
Liposomal preparation and delivery of cDNA in the mouse vasculature was performed as described previously.34
LPS treatment
Mice housed in sealed container were exposed to a nebulized solution of lyophilized Escherichia coli LPS in sterile saline (1 mg/mL) for 45 minutes at a driving flow rate (8 L/min) using a small volume nebulizer (Resigard II; Marquest Medical) and killed at the indicated times.32
Assessment of lung microvessel permeability
Evans blue–conjugated albumin (EBA; 20 mg/kg) was injected retro-orbitally 30 minutes before sacrificing the mice to assess vascular leak induced by LPS or protease activating receptor 1 (PAR1) peptide, as described previously.32,34 The extravasated EBA concentration in lung homogenates was calculated against a standard curve (micrograms Evans blue dye per lung). EBA extravasation (EBAE) values were calculated by determining the ratio of EBAE influx in the lung versus plasma.
Lung weight determination
Left lungs from the same mice used for EBAE were excised and dried in the oven at 60°C overnight for calculation of lung wet/dry ratio, as described previously.32,34
Statistical analysis
Comparisons between experimental groups were made by 1-way ANOVA and paired Student t test. Differences in mean values were considered significant at P < .05.
Results
Impairment of CREB function increases endothelial permeability
We investigated whether HPAE cells depleted of CREB form functional barrier by assessing the decrease in transendothelial electrical resistance (TEER), a measure of endothelial barrier integrity in real time.34 We observed that CREB siRNA significantly reduced endogenous CREB expression at 48 hours after transfection (Figure 1A). CREB siRNA had no effect on the expression of other transcription factors belonging to CREB family, such as ATF2 and CREM (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). We observed that TEER decreased significantly in CREB-depleted endothelial cells, whereas it remained stable in cells transfected with control siRNA (Figure 1B), indicating CREB is required for maintaining endothelial barrier integrity.
Figure 1.

CREB is required for maintaining basal endothelial barrier function and for preventing persistent increases in endothelial permeability postthrombin challenge. (A-C) Depletion of CREB impairs endothelial barrier function. (A) HPAE monolayers were transfected with 2.4 μg of either scrambled (SiSc) or CREB siRNA (SiCREB) for 48 hours. Cell lysates were immunoblotted 48 hours after transfection using anti-CREB antibody to analyze CREB expression. Immunoblot with anti-actin antibody was used as a loading control. (B) Cells plated on gold electrodes were transfected with either SiSc or SiCREB and after 24 hours after transfection, TEER was determined for indicated times. Data represent mean ± SD from 3 experiments performed in duplicates. Asterisk (*) indicates values different from SiSc-transfected endothelial monolayers (P < .05). (C) HPAE monolayers were transfected with SiSc or SiCREB for 48 hours after which cells were simulated with 50nM thrombin, and changes in TEER were recorded overtime. The effects of CREB inhibition on endothelial barrier recovery postthrombin challenge were examined after normalization of TEER values after 48 hours of transfection to 100%. Data represent mean ± SD from 3 experiments performed in duplicates. Asterisk (*) indicates values different from SiSc-transfected endothelial monolayers (P < .05). (D) HPAE cells seeded on Transwell plates were transfected with SiSc or SiCREB and 48 hours after transfection, EBA clearance was determined after without or with thrombin challenge. Data represents the mean ± SD from 3 experiments. Asterisk (*) indicates difference from unstimulated SiSc monolayer, and double asterisk (**) indicates difference from thrombin-stimulated SiSc- or control SiCREB-expressing cells (P < .05). (E-G) Expression of dn-CREB mutant impairs endothelial barrier function. HPAE cells seeded on 6-well plates or gold-plated electrodes were transfected with control cDNA or dn-CREB mutant. After 24 hours after transfection, we determined changes in CREB phosphorylation (E) and TEER in naive monolayer (F) or after stimulation with 50nM thrombin (G) at indicated times. For determining CREB phosphorylation and expression of CREB mutant, cell lysates were immunoblotted with anti–Ser133-CREB antibody anti-CREB antibodies. Immunoblot with anti-actin antibody was performed for protein loading control. The effects of CREB inhibition on endothelial barrier recovery postthrombin challenge were examined after normalization of TEER values after 24 hours after transfection to 100%. Data represent mean ± SD from 3 experiments performed in duplicates. Asterisk (*) indicates values different from SiSc-transfected endothelial monolayers (P < .05). (H-I) CREB knockdown disrupts adherens junctions and increases actin stress fiber formation. HPAE cells expressing SiSc or SiCREB were left unstimulated or stimulated with 50nM thrombin for indicated times, fixed, and immunostained with anti–VE-cadherin antibody and rhodamine phalloidin as described in “Immunofluoresence.” Cells were visualized using an LSM confocal microscope. The images shown are representative of 3 independent experiments. (I) Plot shows time course of fold-change in intercellular gap areas in siSc- or SiCREB-expressing cells after thrombin stimulation. Gap area was quantified using National Institutes of Health ImageJ Version 1.44 software. Asterisk (*) indicates values different from values at time 0 in SiSc-transfected monolayers (P < .05), and double asterisk (**) indicates values different from corresponding thrombin-stimulated SiSc-transfected endothelial monolayers (P < .05). (J) Effect of CREB depletion on actin polymerization. SiSc or SiCREB cells were lysed and centrifuged to separate G- and F-actin. Lysates were then immunoblotted with anti-actin antibody to determine actin polymerization. Numbers indicate densitometric values from 3 individual experiments.
Thrombin, a serine protease released during vascular injury, rapidly induces actin-myosin–driven endothelial contraction, leading to increased endothelial permeability.4,24,34 This is followed by a recovery period during which endothelial contraction is suppressed and interendothelial junctions reseal restoring normal endothelial permeability. Thus, we determined the effect of CREB depletion on thrombin-induced increase in endothelial permeability. We normalized TEER values after 48 hours of transfection in control and CREB siRNA-transfected endothelial monolayers, and changes in TEER were assessed after stimulation with thrombin. As expected, thrombin rapidly decreased TEER in control siRNA-transfected monolayer and subsequently the TEER returned to basal levels within 2 hours (Figure 1C). Inhibition of CREB expression enhanced TEER decrease induced by thrombin, and these responses persisted without recovery to basal level (Figure 1C).
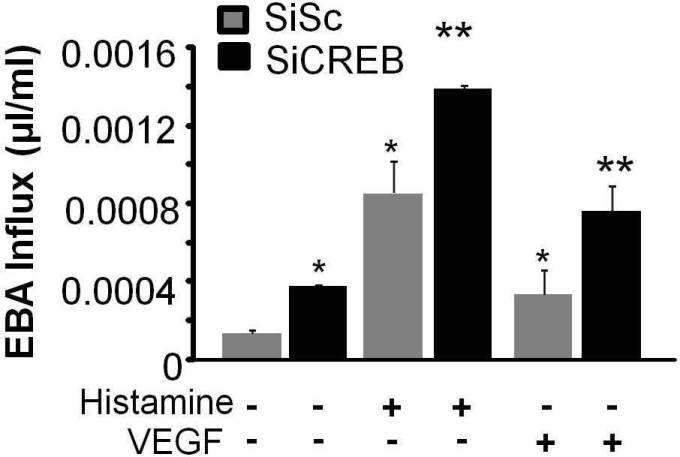
We also determined the effect of CREB knockdown on transendothelial influx of EBA in naive monolayer or after stimulation of monolayer with thrombin. We found that depletion of CREB basally increased transendothelial albumin influx that was further augmented after thrombin stimulation (Figure 1D). Moreover, CREB depletion markedly enhanced barrier disruption after VEGF and histamine (supplemental Figure 3).
In other studies, we transduced dn-CREB mutant (Ser133Ala-CREB mutant) in HPAE cells and quantified the effect of impairment of CREB function on TEER. As expected, dn-CREB suppressed basal CREB phosphorylation (Figure 1E). We observed that transduction of dn-CREB mutant also disrupted basal barrier function (Figure 1F) and led to persistent increase in the duration of endothelial permeability in response to thrombin (Figure 1G), recapitulating the above-mentioned findings in CREB knockdown cells.
Next, we determined the morphology of CREB-depleted endothelial monolayer by staining for adherens junction (AJ) marker VE-cadherin as well as assessed actin fibers. We found that CREB depletion resulted in AJ disruption and actin stress fiber formation, leading to interendothelial gap formation (Figure 1H-I). Thrombin exaggerated AJs disruption and actin stress fiber formation in CREB-depleted monolayer, and these changes persisted even after 120 minutes postthrombin challenge, resulting in formation of numerous opened gaps (Figure 1H-I). However, in control siRNA-transfected HPAE cells, thrombin induced transient disruption of AJs and actin stress fiber formation, and as a result, interendothelial gaps were not apparent at 120 minutes (Figure 1G-I). CREB depletion had no effect on actin polymerization because amount of G- versus F-actin remained the same (Figure 1J). These findings along with above-mentioned TEER data suggest that CREB is required for maintaining a normal endothelial barrier and that it prevents amplified and protracted increase in endothelial permeability by inflammatory mediators such as thrombin.
CREB maintains endothelial barrier function by down- regulating RhoA signaling
RhoA plays a critical role in inducing actin-stress fiber formation and endothelial permeability.27,34 We therefore reasoned whether impairment of CREB function increased endothelial permeability by up-regulating RhoA signaling. As shown in Figure 2, in control siRNA-transfected cells, RhoA activity was barely detectable in unstimulated endothelial monolayers (Figure 2A-B). Thrombin induced a 4-fold activation of RhoA that returned to basal level in 30 minutes (Figure 2A-B). However, in CREB-depleted monolayers, RhoA activity was increased to 3-fold without thrombin stimulation (Figure 2A-B). Thrombin augmented RhoA activity by 12-fold, and this activity remained elevated to 6-fold until 30 minutes (Figure 2A-B).
Figure 2.

CREB negatively regulates RhoA signaling. (A) Effects of CREB depletion on RhoA (top row and panel B) or myosin phosphatase 1 (MYPT1; second row and panel C) activities. HPAE cells transfected with either SiSc or SiCREB were left unstimulated or stimulated with 50nM thrombin for indicated times after which they were lysed to measure RhoA activity. MYPT1 activity was determined by immunoblotting with anti–phospho-381 MYPT antibody. Top row shows RhoA activity as indicated by the amount of RBD-bound RhoA, whereas third row shows total amount of RhoA protein in whole cell lysate. Bottom row shows immunoblot of CREB using anti-CREB antibodies. (B-C) Plots show mean + SD of fold-increase in RhoA (B) or MYPT1 (C) activities from multiple experiments over value at time 0 (n = 4). Asterisk (*) indicates values different from unstimulated SiSc group (P < .05), and double asterisk (**) indicates values different from corresponding thrombin-stimulated, SiSc-transfected monolayers (P < .05).
An important consequence of RhoA signaling is the phosphorylation of the Rho kinase substrate MYPT1, which prevents dephosphorylation of MLC and thereby sustains actin-myosin contraction.1,4 Therefore, we determined MYPT1 phosphorylation in CREB-depleted cells. Consistently, we observed that MYPT1 was basally phosphorylated in CREB-depleted cells and that the phosphorylation increased further after thrombin stimulation (Figure 2A,C). Depletion of CREB had no effect on endogenous levels of RhoA, because the total amount of RhoA was similar in control and CREB siRNA-transfected cells (Figure 2A).
CREB suppresses RhoA activity by controlling p190-A expression and thereby establishes endothelial barrier
Guanosine exchange factors (GEFs) and GAPs regulate RhoA activity. GEFs increases RhoA activity by inducing GDP-GTP exchange on RhoA.1,25 On the contrary, GAPs antagonizes RhoA activity specifically by increasing GTPase activity of RhoA.36–38 We have shown previously that p115RhoGEF and p190-A play a critical role in regulating RhoA activity.27 We therefore tested the possibility that CREB controls RhoA signaling via regulating the expression of either p190-A or p115RhoGEF. In addition, we assessed the effect of CREB depletion on the expression of VE-cadherin, β-catenin, p120-catenin, and FAK proteins because these proteins form endothelial barriers and interact with upstream regulators of RhoA, such as p190-A.1,37 We found that CREB deficiency did not alter the expression levels of FAK, p120-catenin, or β-catenin (Figure 3A). However, knockdown of CREB markedly suppressed p190-A expression (Figure 3A). Quantitative PCR analysis of p190-A in CREB-depleted or dn-CREB–transducing cells showed that impairment of CREB function decreased p190-A mRNA by 70% (Figures 3B-C) while sparing the expression of p115RhoGEF (supplemental Figure 2), thus identifying p190-A as the transcriptional target of CREB.
Figure 3.

CREB depletion suppresses p190-A expression. (A) Effect of CREB knockdown on p190-A protein expression. HPAE cells expressing SiSc or SiCREB were lysed and immunoblotted with indicated antibodies to determine their expression. Numbers indicate densitometric values under each condition taking SiSc as 100%. (B-C) Effect of impairment of CREB function on p190-A mRNA expression. HPAE cells transfected with either SiCREB for 48 hours (B) or dn-CREB mutant for 36 hours (C) were lysed, and RNA was extracted. mRNA was analyzed by quantitative RT-PCR using primers specific for the p190-A. GAPDH expression was used as a paired control. Asterisk (*) indicates values different from SiSc-transfected monolayers (P < .05).
To determine that loss of p190-A expression was responsible for impairment of endothelial barrier function in dn-CREB–transducing cells, we overexpressed wild-type p190-A in dn-CREB–expressing cells and determined TEER and interendothelial gaps formation after without or with thrombin stimulation. We found that rescuing p190-A expression (Figure 4A inset) markedly suppressed interendothelial gaps formation in CREB knockdown cells (Figure 4A) and thereby restored basal barrier function as well as facilitated the reformation of barrier after thrombin challenge (Figure 4B-C). In addition, we determined that decreased p190-A expression is sufficient to impair barrier function seen in CREB knockdown cells. Thus, we depleted p190-A in HPAE cells using siRNA and determined TEER in response to thrombin. We found that ∼ 70% reduction in p190-A expression (similar to that induced by CREB knockdown; Figure 4D inset) impaired basal endothelial barrier function as seen in CREB knockdown cells (Figure 4D-E). Furthermore, thrombin persistently increased endothelial permeability in these monolayers (Figure 4E). Collectively, these findings demonstrate that CREB maintains endothelial barrier function by controlling p190-A expression, which dampens RhoA signaling.
Figure 4.

Restoration of p190RhoGAP expression rescues endothelial barrier defect in CREB-impaired endothelial cells. (A) HPAE cells were cotransduced with control cDNA plus GFP-tagged p190-A cDNA or dn-CREB mutant along with green fluorescent protein (GFP)-tagged p190-A cDNA. After 24 hours, cells were lysed, and lysates were immunoblotted with anti-GFP and anti-p190-A antibodies to determine protein expression (inset). In parallel, cells were stimulated with thrombin for indicated times, fixed, and stained with anti–VE-cadherin antibody and viewed under confocal microscope. Intracellular gap areas were quantified using National Institutes of Health ImageJ Version 1.44 software. The values shown are representative of 3 independent experiments. Asterisk (*) indicates values different from unstimulated control vector-expressing cells (P < .05), and double asterisk (**) indicates values different from corresponding thrombin-stimulated control vector-transducing monolayers (P < .05). Pound (#) indicates values different from corresponding thrombin-stimulated dn-CREB–transducing monolayers (P < .05). (B-C) HPAE cells cotransducing indicated cDNA were stimulated without (B) or after thrombin stimulation (C), and TEER was determined at indicated times. Data represent mean ± SD of percentage of decrease in TEER. Asterisk (*) indicates values different from value at 24 hours (P < .05), and double asterisk (**) indicates values different from dn-CREB vector-transducing monolayers with and without treatment of thrombin (P < .05). (D-E) Effect of p190-A knockdown on endothelial monolayer permeability. HPAE cells were transfected with 2.4 μg of SiSc or p190-A siRNA for 48 hours, after which changes in TEER were determined without (D) or after thrombin stimulation (E). Inset, cell lysates were immunoblotted 48 hours after transfection using anti–p190-A antibody to analyze p190-A expression. Immunoblot with anti–β-actin antibody was used as a loading control. Plot shows mean ± SD from 3 experiments performed in duplicates. Asterisk (*) indicates values different from corresponding SiSc-transfected endothelial monolayers (P < .05).
CREB induces p190RhoGAP promoter activity
Phosphorylation at serine 133 residue and nuclear translocation determine CREB activity.8,20,21 We first determined whether CREB activity is altered in association with disruption of endothelial barrier function by thrombin. CREB phosphorylation was determined in cell lysates using anti–phospho-serine 133 antibody. Nuclear extracts containing equal amount of protein were analyzed by EMSA for CREB DNA-binding activity using radiolabeled CREB consensus oligonucleotide probe. We observed that CREB is basally phosphorylated and thrombin induced a 4-fold increase in CREB phosphorylation within 5 minutes, which further increased to 6-fold at 30 minutes and remained elevated around this level even at 1 hour (Figure 5A). Thrombin also induced an increase in CREB DNA-binding activity above basal level within 1 hour that persisted at 2 hours (Figure 5B). These findings suggest that CREB is induced during alteration of endothelial barrier function.
Figure 5.

CREB is required for inducing p190-A promoter activity. (A) Thrombin activates CREB. HPAE cells were stimulated with thrombin (50nM) for the indicated times, and phosphorylation of CREB was determined by immunoblotting using Ser133-phosphospecific antibody. Membrane was reprobed with anti-CREB antibody to normalize for protein loading. Data represent mean ± SD from 3 individual experiments. Asterisk (*) indicates significant increase in phosphorylation above time 0 (P < .05). (B) Thrombin induces CREB nuclear localization. HPAE cells stimulated with thrombin for indicated times were lysed, and nuclear extracts were prepared as described in “Electrophoretic mobility shift assay.” Nuclear extracts containing equal amount of proteins were analyzed for protein-CRE DNA-binding activity using 32P-labeled consensus CRE oligonucleotide as a probe. (C) Effect of CREB knockdown on p190-A promoter expression. Nuclear extracts from cells transducing control vector or dn-CREB mutant were used for ChIP assay using monoclonal anti-CREB antibodies, and the resulting DNA fragments were subjected to PCR amplification using primers spanning the CREB consensus sequences from human p190-A. (D) Depletion of CREB suppresses p190-A promoter activity. HPAE cells expressing Sc or SiCREB for 24 hours were transfected with p190-A luciferase promoter construct. Cells were then left unstimulated or stimulated with thrombin for 6 hours, and the luciferase activities were determined. Asterisk (*) indicates values different from values from SiSc group at time 0 (P < .05), and double asterisk (**) indicates value different from SiSc cells after without or with thrombin stimulation.
Transfac analysis of human p190-A promoter revealed the presence of a putative CREB-binding motif spanning from −735 to −728 bp. To address the possibility that CREB transcribes p190-A by binding to CRE site on p190-A promoter, we performed a ChIP assay. ChIP analysis revealed that CREB directly binds to p190RhoGAP promoter, whereas it failed to bind p190-A promoter in HPAE cells transducing dn-CREB mutant (Figure 5C).
We also determined the functional role of CREB interaction with the p190RhoGAP promoter in inducing p190-A promoter activity. To do this, we a generated p190-A promoter luciferase reporter plasmid and cotransduced this plasmid in HPAE cells without or with CREB siRNA. These cells were stimulated with thrombin for 6 hours after which luciferase activity was determined. As shown in Figure 5D, basal promoter activity was suppressed by 5-fold in CREB-depleted HPAE cells. Moreover, thrombin increased luciferase activity by 2-fold in cells transducing control vector, but it failed to increase the luciferase activity in CREB-depleted cells (Figure 5D). These findings demonstrate that CREB controls p190-A expression by directly inducing its promoter activity.
CREB is required for restoring lung vascular barrier function
We have shown that thrombin increases lung vascular barrier permeability by activating protease activating receptor 1 (PAR1) that resolves naturally within the next 2 to 3 hours.34,39 We also have shown that nebulized endotoxin, LPS disrupts lung vascular barrier function that resolves within 24 hours.32 Thus, we used these mice models of lung injury to address the in vivo relevance of CREB activation in regulating endothelial barrier function via p190-A. We conjugated dn-CREB mutant with liposome and injected retro-orbitally into mouse vasculature.35 Mice receiving vector served as a control. After 48 hours after transfection, these mice were challenged with PAR1 peptide (1 mg/kg),34,39 and the lungs were harvested at indicated times. In other studies, mice transducing vector or dn-CREB mutant were allowed to inhale LPS for 45 minutes as described in “LPS treatment” and, after 4 and 24 hours, these mice were sacrificed. We determined EBAE in the lung parenchyma and lung wet-dry weight ratio to assess the effect of impaired CREB function on lung vascular barrier function. Injection of PAR1 peptide (intravenously) increased edema formation and lung vascular permeability in mice receiving control vector or CREB mutant at 30 minutes (Figure 6A-C). Lung vascular barrier function recovered at 150 minutes in vector-expressing lungs (Figure 6A-C). However, mice lungs transducing dn-CREB mutant showed increased vascular leak under basal conditions (Figure 6A-C). PAR1 activation further augmented lung vascular dysfunction, but this did not resolve in 150 minutes (Figure 6A-C). Likewise, we observed that LPS-induced increase in lung vascular permeability returned to near normal levels within 24 hours in vector-expressing lungs, but these responses persisted in dn-CREB mutant–expressing lungs (Figure 6D-E). These findings indicate that CREB plays a critical role in maintaining lung vascular function and for opposing barrier dysfunction by the 2 diverse inflammatory agents PAR1 and LPS.
Figure 6.

Effect of impairment of CREB function on lung microvessel permeability. (A-E) Expression of dn-CREB mutant in lungs exaggerate pulmonary edema formation. Mice were injected retro-orbitally with liposome encapsulating either vector or dn-CREB mutant. After 48 hours after transfection, either PAR1 agonist (1 mg/kg) or control peptide was injected intravenously into mice (A-C), or mice were exposed to nebulized LPS (D-E). Lung vascular permeability was determined by quantifying EBAE or wet-dry weight ratio at the indicated times. (A) Representative images of PAR1-induced Evans blue accumulation in the lungs-transducing vector or dn-CREB mutant. (B-E) Plot shows mean ± SEM of changes in EBAE and wet-dry weight ratio after PAR1 peptide administration or LPS exposure in control and dn-CREB-transdcucing lungs (n = 4). Asterisk (*) indicates values different from control vector group value at time 0, and double asterisk (**) indicates values different from corresponding PAR1 peptide treated control vector group (P < .05). (F) Control vector or dn-CREB–transducing lungs were harvested after treatment with either control peptide or PAR1 peptide at indicated times. Lung lysates were immunoblotted with the indicated antibodies to determine PAR1-induced phosphorylation of CREB and MYPT1. Immunoblot with anti-CREB antibody was used to confirm the overexpression of CREB in dn-CREB mutant lungs. (G-H) Plot shows mean ± SEM of fold-increase in CREB phosphorylation and MYPT1 activity in lungs-transducing dn-CREB mutant. Asterisk (*) indicates significance from its control vector group at time 0 (P < .05), and double asterisk (**) indicates significance from the corresponding PAR1 peptide–treated control vector mice group (P < .05).
We also assessed the effect of CREB impairment on p190RhoGAP function in the lungs. Consistent with findings in endothelial cells, we observed that transduction of dn-CREB impaired p190-A expression, leading to increased MYPT1 phosphorylation under basal condition that increased further after PAR1 activation (Figure 6F-G). PAR1 activation significantly increased CREB phosphorylation in lungs expressing control vector (Figure 6H). However, dn-CREB–expressing lungs showed diminished CREB phosphorylation, confirming the expression of dn-CREB mutant in the lungs (Figure 6H).
Inhibition of RhoA signaling reverses lung vascular barrier dysfunction occurring secondary to impairment of CREB function
To address whether inhibition of RhoA signaling would restore endothelial barrier function in lungs transducing dn-CREB mutant, we inhibited Rho kinase by injecting Y-27632 into mice (10 mg/kg body) 15 minutes before the PAR1 peptide infusion in mice transducing control vector or dn-CREB mutant. We observed that inhibition of RhoA restored albumin leakage and edema formation in mutant-expressing lungs to the level seen in control lungs after without or with PAR1 activation (Figure 7A-B). To compare the effectiveness of RhoA inhibition in restoring lung edema formation in vector and CREB mutant–expressing lungs, we recalculated findings in Figure 7A-B, taking 0-minute values in vector-expressing lungs as 100%. Data show a pronounce effect of RhoA inhibition in mitigating basal as well as PAR1 induced increase in lung edema formation in mutant-expressing lungs (Figure 7C-D). Similar findings were observed in endothelial cells transducing dn-CREB mutant where inhibition of Rho kinase attenuated persistent increase in endothelial permeability by thrombin (data not shown). Thus, CREB prevents lung vascular dysfunction via p190-A–mediated suppression of RhoA signaling.
Figure 7.

Inhibition of RhoA signaling restores endothelial barrier function in dn-CREB lungs. Mice transducing control vector or dn-CREB mutant were given with either normal saline (vehicle) or Rho kinase inhibitor Y-27632 (10 mg/kg body weight mice) intravenously for 15 minutes. Mice were then injected with either control peptide or PAR1 peptide for the indicated times. Evans blue was injected into the mice 30 minutes before they were euthanized. Lung vascular permeability measurements were determined by quantifying transendothelial albumin flux of EBA (A) or lung wet-dry ratio (B). Data represent mean ± SEM from 3 individual experiments. Asterisk (*) indicates values different from control vector group at 0 time point, and double asterisk (**) indicates values different from values in corresponding PAR1 peptide–treated control group (P < .05). Pound (#) indicates values different from dn-CREB mutant lungs (P < .05). (C-D) Mean + SD of percentage of change in inhibition of lung vascular permeability (C) and lung wet-dry weight ratio (D) by Y-27632. Values shown in panels A and B were recalculated taking time 0 value in vector-expressing lungs as 100%. Asterisk (*) indicates values different from control vector group.
Discussion
Our results identified a crucial role of CREB in maintaining basal endothelial monolayer integrity and in restoring normal endothelial barrier function after increase in endothelial permeability by the inflammatory mediators thrombin, VEGF, and histamine as well as the endotoxin LPS. We specifically demonstrate that CREB transcriptionally controls p190-A expression that antagonizes RhoA signaling, thereby preventing disruption of endothelial monolayer integrity and long-lasting increase in endothelial permeability in response to inflammatory agonists. The increased RhoA signaling secondary to the loss of CREB and p190-A function was a decisive factor in disrupting endothelial barrier integrity and in amplifying endothelial permeability increase in response to thrombin because normal barrier function was restored by either rescuing p190-A expression or by inhibiting the activity of RhoA effector Rho kinase in CREB-impaired cells and lungs.
Impairment of endothelial barrier function is a crucial factor in the pathogenesis of several diseases arising because of tissue inflammation and hypoxia.1,22,23,40 In the lung, increased microvessel endothelial permeability leads to protein-rich alveolar edema and severely impairs oxygenation, leading to the life-threatening disease acute lung injury.2,6 It is well established that actin-myosin–driven endothelial cell contraction primarily determines endothelial permeability increase induced by inflammatory mediators such as thrombin.3,4,41 However, whether CREB directly influences the formation of functional endothelial barrier and whether it is necessary for altering endothelial permeability in response to inflammatory mediators is unknown. We showed that impairment of CREB function increased stress fiber formation under naive conditions consequently disrupting endothelial barrier disruption. Moreover, the proinflammatory mediator thrombin, which generally induces a reversible increase in endothelial permeability,27,32,34,42 produced an amplified and protracted increase in endothelial permeability in CREB-impaired cells. Histamine and VEGF also induced exaggerated increase in endothelial permeability in CREB-depleted endothelial cells. We show that thrombin enhanced actin stress fiber formation in CREB-depleted cells, which prevented resealing of AJs. Hence, lungs transducing dn-CREB mutant showed persistent edema formation after in response to PAR1 activation. We also showed that impairment of CREB function prevented resolution of lung edema in a model of LPS-induced acute lung injury. Thus, these findings demonstrate that CREB function is required to establish endothelial barrier and to mitigate long-lasting increase in endothelial permeability by diverse edemagenic agents.
CREB belongs to a family of cAMP response genes.7 It is well established that increased cAMP concentration strengthens endothelial barrier function and prevents the increases in endothelial permeability in response to several agonists such as thrombin, LPS, and hypoxia.1,43,44 For example, up-regulated adenosine B2 receptor by inducing cAMP generation suppresses hypoxia-induced vascular leak.45,46 Adenosine B2 receptor contains hypoxia-inducible factor binding elements in the promoter region.45,46 Because CREB can regulate hypoxia-inducible factor expression47 our findings raise the possibility that CREB play a general role in mitigating endothelial barrier dysfunction during inflammation as well as oxygen stress.17,23,46
The small GTPases RhoA and Rac1 play a key role in regulating endothelial permeability.25,27,44 Because RhoA induces actin stress fiber formation and endothelial permeability increase,1 we speculated that CREB-regulated endothelial permeability by regulating RhoA expression. However, we showed that impairment of CREB function did not alter RhoA expression. Notably, we showed that CREB function was required not only for suppressing basal RhoA activity but also for attenuating thrombin-induced RhoA activity. Inhibition of RhoA signaling reversed endothelial permeability defect in CREB-impaired cells and lungs, demonstrating CREB-regulated endothelial barrier by antagonizing RhoA activity. Baumer et al showed that Rac1 activity significantly contributes to mitigate the increase in endothelial permeability by thrombin and LPS.43 However, our findings demonstrate that inhibition of RhoA signaling was sufficient to restore basal endothelial barrier function in CREB knockdown cells under naive conditions and after thrombin challenge, indicating CREB specifically alters RhoA signaling.
RhoA is known to cycle between the active (GTP-bound) and inactive (GDP-bound) states.26,48 Rho activation state is regulated by the opposing actions of the GEFs and the GAPs. The P115 RhoGEF proteins mediate the activation of RhoA in response to thrombin by stimulating the release and exchange of GDP for GTP.26 However, GAP proteins inactivate RhoA by increasing intrinsic RhoA-GTPase activity, converting RhoA-GTP into RhoA-GDP form.25,26 p190RhoGAP family consisting of p190-A and p190-B isoforms primary accounts for the total RhoGAP activity.49 Interestingly, p190 specifically inactivates RhoA.38 Several studies showed that p190-A prevents the increase in endothelial permeability induced by thrombin27,28 and endotoxin.29,30 Our findings implied that CREB transcriptionally controls an upstream regulator of RhoA whose function is to dampen RhoA activity. Therefore, we posited that a functionally important relationship exists between CREB activity and p115RhoGEF or p190-A expression that regulates RhoA signaling to maintain endothelial barrier function. Hence, we focused on determining the role of CREB in regulating p115RhoGEF and p190-A expression. We showed that impairment of CREB function lead to depletion of p190-A protein. However, CREB depletion had no effect on expression of p115RhoGEF expression. Moreover, depletion of CREB did not alter the expression of VE-cadherin, β-catenin, p120-catenin, or FAK, all which play a fundamental role in regulating endothelial permeability.1 Thus, CREB specifically targets p190-A. Decreased p190-A expression helps to explain amplified RhoA activity, consequently inducing MLC phosphatase activity and actin stress fiber formation in CREB impaired endothelial cells and mice lungs, which leads to amplified and protracted increase in endothelial permeability by thrombin and LPS. Consistently, we showed that restoring p190RhoGAP expression in dn-CREB mutant–transfected cells reestablished the endothelial barrier function toward the levels seen in control cells. Also, we showed that siRNA-induced knockdown of p190RhoGAP recapitulated endothelial barrier defect seen in CREB-depleted cells. These findings are consistent with the notion that CREB and p190RhoGAP operate in a linear pathway, where p190 acts downstream of CREB in dampening RhoA signaling and thereby in establishing endothelial barrier function and limiting protracted increase in endothelial barrier dysfunction post thrombin challenge.
Several mechanisms such as protein–protein interactions, phospholipid modification, subcellular translocation, and proteolytic degradation regulate p190-A activity.50 For example, phosphorylation of p190-A by cSrc and FAK induced its GAP activity.27,37 p190 interaction with p120-RasGAP, Rnd3, and p120-catenin was crucial for p190 GAP activity.37 We showed that CREB transcriptionally controls p190RhoGAP expression. We identified CRE site in the p190-A promoter where CREB binds to induce p190-A promoter activity. Consistently, depletion of CREB markedly suppressed p190RhoGAP promoter activity. We also showed that thrombin increased CREB activity as well as p190-A promoter activity. However, thrombin did not induce p190-A promoter activity in CREB-depleted cells. Thus, our results demonstrate that transcriptional control of the p190-A expression by CREB was another regulatory mechanism of p190-A activity and thereby maintenance of endothelial barrier function.
In conclusion, our findings provided new insight into the physiologic roles of CREB. CREB down-regulates RhoA via maintaining the expression of p190-A and thereby helps to maintain endothelial barrier function under basal conditions and prevent long-lasting increases in endothelial permeability by inflammatory mediators. CREB functional interactions with RhoA via p190 have significant implications in vivo under physiologic and pathologic conditions.
Supplementary Material
Acknowledgments
The authors acknowledge Dr Asrar B. Malik for critiques during the progress of this work. They thank Dr Michael Holinstat (Thomas Jefferson University, Philadelphia, Pennsylvania) for Rap1 beads and Dr Sarah J. Parson (University of Virginia School of Medicine, Charlottesville) for p190-A cDNA and Marc Montminy (Salk Institute for Biologic Studies) and Addgene (Cambridge, Massachusetts) for dn-CREB mutant.
This work was supported by National Institutes of Health grants HL71794, HL84153, and PO1HL060678.
Footnotes
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.R.C. and D.M. designed research, analyzed and interpreted data, and wrote the manuscript; and K.R.C., M.T., and T.S. performed the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dolly Mehta, Department of Pharmacology, The University of Illinois, College of Medicine, 835 S Wolcott Ave, Chicago, IL 60612; e-mail: dmehta@uic.edu.
References
- 1.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 2.Minshall RD, Malik AB. Transport across the endothelium: regulation of endothelial permeability. Handb Exp Pharmacol. 2006;1:107–144. doi: 10.1007/3-540-32967-6_4. [DOI] [PubMed] [Google Scholar]
- 3.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163(3):510–522. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- 4.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 5.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med. 2009;11(30):e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichiki T. Role of cAMP response element binding protein in cardiovascular remodeling: good, bad, or both? Arterioscler Thromb Vasc Biol. 2006;26(3):449–455. doi: 10.1161/01.ATV.0000196747.79349.d1. [DOI] [PubMed] [Google Scholar]
- 8.Persengiev SP, Green MR. The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis. 2003;8(3):225–228. doi: 10.1023/a:1023633704132. [DOI] [PubMed] [Google Scholar]
- 9.Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci U S A. 1998;95(8):4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bleckmann SC, Blendy JA, Rudolph D, Monaghan AP, Schmid W, Schutz G. Activating transcription factor 1 and CREB are important for cell survival during early mouse development. Mol Cell Biol. 2002;22(6):1919–1925. doi: 10.1128/MCB.22.6.1919-1925.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiappara G, Chanez P, Bruno A, et al. Variable p-CREB expression depicts different asthma phenotypes. Allergy. 2007;62(7):787–794. doi: 10.1111/j.1398-9995.2007.01417.x. [DOI] [PubMed] [Google Scholar]
- 12.Mroz RM, Holownia A, Chyczewska E, et al. Cytoplasm-nuclear trafficking of CREB and CREB phosphorylation at Ser133 during therapy of chronic obstructive pulmonary disease. J Physiol Pharmacol. 2007;58(Suppl 5):437–444. [PubMed] [Google Scholar]
- 13.Chava KR, Karpurapu M, Wang D, et al. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2009;29(6):809–815. doi: 10.1161/ATVBAHA.109.185777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reusch JE, Klemm DJ. Cyclic AMP response element-binding protein in the vessel wall: good or bad? Circulation. 2003;108(10):1164–1166. doi: 10.1161/01.CIR.0000084296.45158.50. [DOI] [PubMed] [Google Scholar]
- 15.Abraham E, Arcaroli J, Shenkar R. Activation of extracellular signal-regulated kinases, NF-kappa B, and cyclic adenosine 5′-monophosphate response element-binding protein in lung neutrophils occurs by differing mechanisms after hemorrhage or endotoxemia. J Immunol. 2001;166(1):522–530. doi: 10.4049/jimmunol.166.1.522. [DOI] [PubMed] [Google Scholar]
- 16.Shenkar R, Abraham E. Hemorrhage induces rapid in vivo activation of CREB and NF-kappaB in murine intraparenchymal lung mononuclear cells. Am J Respir Cell Mol Biol. 1997;16(2):145–152. doi: 10.1165/ajrcmb.16.2.9032121. [DOI] [PubMed] [Google Scholar]
- 17.Leonard MO, Howell K, Madden SF, et al. Hypoxia selectively activates the CREB family of transcription factors in the in vivo lung. Am J Respir Crit Care Med. 2008;178(9):977–983. doi: 10.1164/rccm.200712-1890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barlow CA, Shukla A, Mossman BT, Lounsbury KM. Oxidant-mediated cAMP response element binding protein activation: calcium regulation and role in apoptosis of lung epithelial cells. Am J Respir Cell Mol Biol. 2006;34(1):7–14. doi: 10.1165/rcmb.2005-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichiki T, Tokunou T, Fukuyama K, Iino N, Masuda S, Takeshita A. Cyclic AMP response element-binding protein mediates reactive oxygen species-induced c-fos expression. Hypertension. 2003;42(2):177–183. doi: 10.1161/01.HYP.0000079791.26014.04. [DOI] [PubMed] [Google Scholar]
- 20.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2(8):599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 21.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 22.Eltzschig HK, Ibla JC, Furuta GT, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198(5):783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson LF, Eltzschig HK, Ibla JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200(11):1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Nieuw Amerongen GP, Draijer R, Vermeer MA, van Hinsbergh VW. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res. 1998;83(11):1115–1123. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- 25.Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87(2):243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 26.Takai Y, Kaibuchi K, Sasaki T, Tanaka K, Shirataki H, Nakanishi H. Rho small G protein and cytoskeletal control. Princess Takamatsu Symp. 1994;24:338–350. [PubMed] [Google Scholar]
- 27.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem. 2006;281(4):2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- 28.Harrington EO, Newton J, Morin N, Rounds S. Barrier dysfunction and RhoA activation are blunted by homocysteine and adenosine in pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1091–1097. doi: 10.1152/ajplung.00421.2003. [DOI] [PubMed] [Google Scholar]
- 29.Mammoto T, Parikh SM, Mammoto A, et al. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007;282(33):23910–23918. doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- 30.Birukova AA, Zebda N, Cokic I, et al. p190RhoGAP mediates protective effects of oxidized phospholipids in the models of ventilator-induced lung injury. Exp Cell Res. 2011;317(6):859–872. doi: 10.1016/j.yexcr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem. 2007;282(13):9482–9491. doi: 10.1074/jbc.M700067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tauseef M, Kini V, Knezevic N, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103(10):1164–1172. doi: 10.1161/01.RES.0000338501.84810.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knezevic N, Roy A, Timblin B, et al. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol. 2007;27(18):6323–6333. doi: 10.1128/MCB.00523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein betagamma subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J Exp Med. 2009;206(12):2761–2777. doi: 10.1084/jem.20090652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein IK, Predescu DN, Sharma T, Knezevic I, Malik AB, Predescu S. Intersectin-2L regulates caveola endocytosis secondary to Cdc42-mediated actin polymerization. J Biol Chem. 2009;284(38):25953–25961. doi: 10.1074/jbc.M109.035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12(9):2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wildenberg GA, Dohn MR, Carnahan RH, et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127(5):1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 38.Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113(2):147–158. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 39.Vogel SM, Gao X, Mehta D, et al. Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol Genomics. 2000;4(2):137–145. doi: 10.1152/physiolgenomics.2000.4.2.137. [DOI] [PubMed] [Google Scholar]
- 40.Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med. 2004;30(9):1702–1714. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- 41.Minami T, Sugiyama A, Wu SQ, Abid R, Kodama T, Aird WC. Thrombin and phenotypic modulation of the endothelium. Arterioscler Thromb Vasc Biol. 2004;24(1):41–53. doi: 10.1161/01.ATV.0000099880.09014.7D. [DOI] [PubMed] [Google Scholar]
- 42.Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW, 2nd, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol. 1986;128(1):96–104. doi: 10.1002/jcp.1041280115. [DOI] [PubMed] [Google Scholar]
- 43.Baumer Y, Spindler V, Werthmann RC, Bunemann M, Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol. 2009;220(3):716–726. doi: 10.1002/jcp.21819. [DOI] [PubMed] [Google Scholar]
- 44.Schlegel N, Baumer Y, Drenckhahn D, Waschke J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit Care Med. 2009;37(5):1735–1743. doi: 10.1097/CCM.0b013e31819deb6a. [DOI] [PubMed] [Google Scholar]
- 45.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111(4):2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kvietikova I, Wenger RH, Marti HH, Gassmann M. The transcription factors ATF-1 and CREB-1 bind constitutively to the hypoxia-inducible factor-1 (HIF-1) DNA recognition site. Nucleic Acids Res. 1995;23(22):4542–4550. doi: 10.1093/nar/23.22.4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Settleman J. Getting in shape with Rho. Nat Cell Biol. 2000;2:E7–9. doi: 10.1038/71390. [DOI] [PubMed] [Google Scholar]
- 49.Matheson SF, Hu KQ, Brouns MR, Sordella R, VanderHeide JD, Settleman J. Distinct but overlapping functions for the closely related p190 RhoGAPs in neural development. Dev Neurosci. 2006;28(6):538–550. doi: 10.1159/000095116. [DOI] [PubMed] [Google Scholar]
- 50.Naoe H, Araki K, Nagano O, et al. The anaphase-promoting complex/cyclosome activator Cdh1 modulates Rho GTPase by targeting p190 RhoGAP for degradation. Mol Cell Biol. 2010;30(16):3994–4005. doi: 10.1128/MCB.01358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}