

Abstract
Recent neutron diffraction studies of photoactive yellow protein (PYP) proposed that the H bond between protonated Glu46 and the chromophore [ionized p-coumaric acid (pCA)] was a low-barrier H bond (LBHB). Using the atomic coordinates of the high-resolution crystal structure, we analyzed the energetics of the short H bond by two independent methods: electrostatic pKa calculations and a quantum mechanical/molecular mechanical (QM/MM) approach. (i) In the QM/MM optimized geometry, we reproduced the two short H-bond distances of the crystal structure: Tyr42-pCA (2.50 Å) and Glu46-pCA (2.57 Å). However, the H atoms obviously belonged to the Tyr or Glu moieties, and were not near the midpoint of the donor and acceptor atoms. (ii) The potential-energy curves of the two H bonds resembled those of standard asymmetric double-well potentials, which differ from those of LBHB. (iii) The calculated pKa values for Glu46 and pCA were 8.6 and 5.4, respectively. The pKa difference was unlikely to satisfy the prerequisite for LBHB. (iv) The LBHB in PYP was originally proposed to stabilize the ionized pCA because deprotonated Arg52 cannot stabilize it. However, the calculated pKa of Arg52 and QM/MM optimized geometry suggested that Arg52 was protonated on the protein surface. The short H bond between Glu46 and ionized pCA in the PYP ground state could be simply explained by electrostatic stabilization without invoking LBHB.
Keywords: low-barrier hydrogen bond, NMR, proton transfer, photoreceptor
Sensing blue light is a prerequisite for organisms to be able to sustain life. Photoactive yellow protein (PYP) serves as a bacterial photoreceptor, in particular, as a sensor for negative phototaxis to blue light (1). The photoactive chromophore of PYP is p-coumaric acid (pCA), which is covalently attached to Cys69 (2). In the PYP ground state, the pCA chromophore is present as a phenolate anion (3–5). Absorption of a blue light photon initiates the trans- cis- isomerization of the pCA region, leading to proton transfer involving the pCA moiety (4, 6). The PYP crystal structure revealed that pCA is H-bonded by protonated Tyr42 and Glu46 (Fig. 1). Tyr42 is further H-bonded by Thr50. Structural analysis suggested that Glu46 is protonated and pCA is ionized in the PYP ground state (7, 8). Remarkably, the distance between the hydroxyl O of Tyr42 and the phenolate O of pCA (OTyr42-OpCA) is 2.49–2.51 Å, and the distance between the carboxyl O of Glu46 and the phenolate O of pCA (OGlu46-OpCA) is 2.54–2.61 Å in most PYP crystal structures at resolution of approximately 1 Å (reviewed in ref. 9). The short distance between Glu46 and pCA is of particular interest because the photoinduced intramolecular proton transfer from protonated Glu46 to ionized pCA occurs in transition to the pB intermediate state during the photocycle (4).
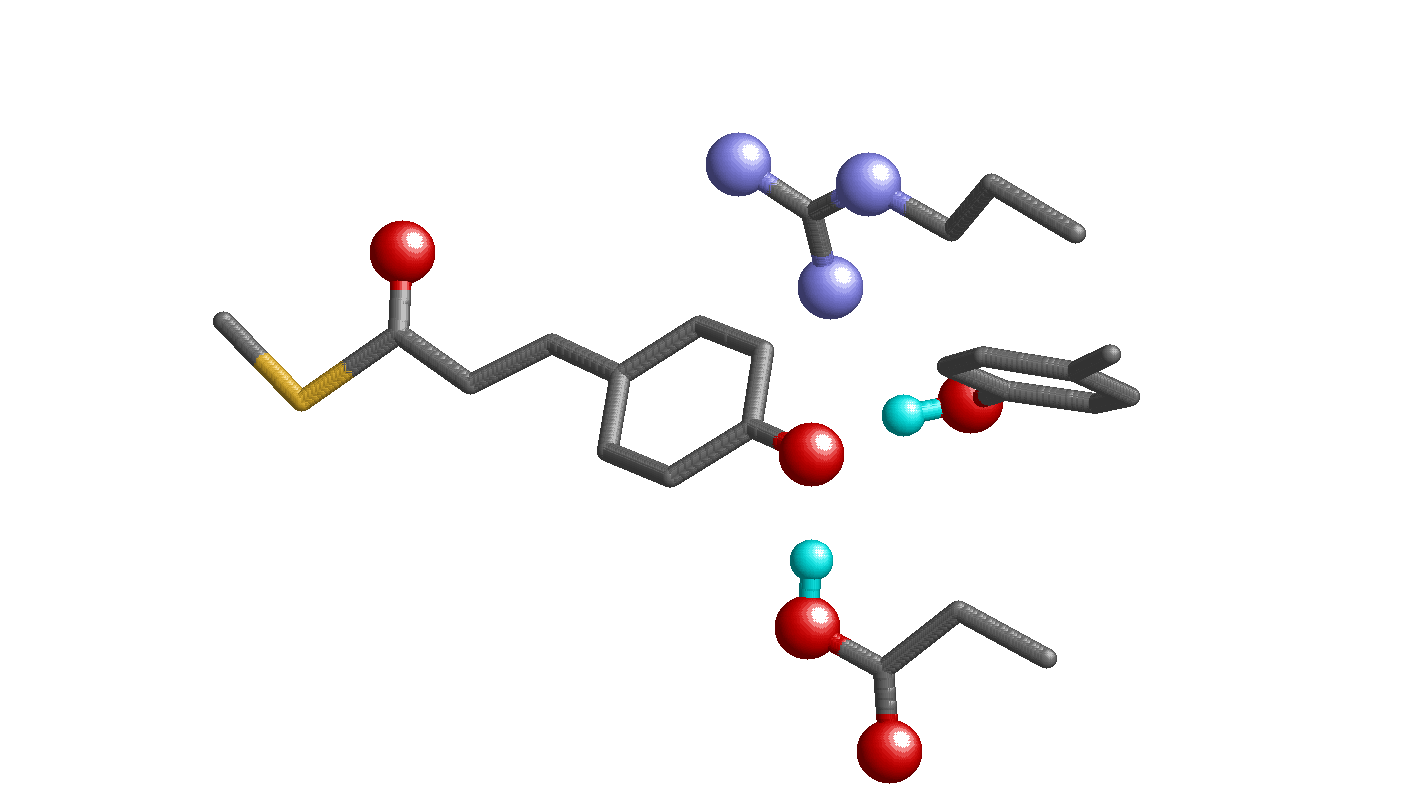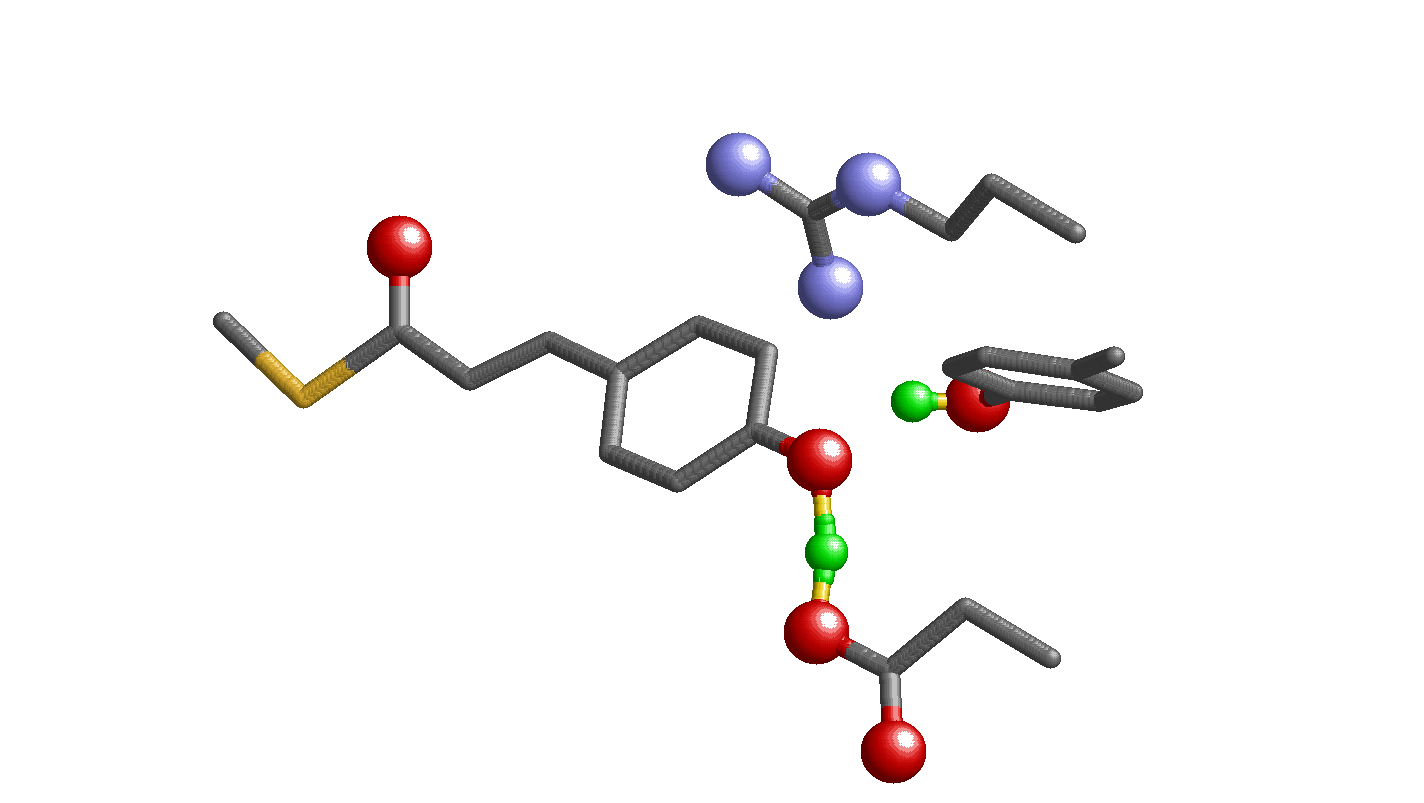
Fig. 1.

Geometry of the photoactive site in PYP. Only the H atom position of the H bonds between Tyr42 and pCA and between Glu46 and pCA are shown by the cyan spheres. (A) Neutron diffraction analysis (PDB ID code 2ZOI). (B) QM/MM optimized structure based on the X-ray crystal structure (PDB ID code 2ZOH). (C) The Glu-pCA model system.
Recently, using the heavy atom coordinates of the PYP X-ray diffraction crystal structure analyzed at 1.25 Å resolution, hydrogen or deuterium atom positions of PYP were assigned in neutron diffraction analysis at 295 K (10). [Note: Both hydrogen (H) and deuterium (D) are called H atom in the present study. Changes in the H-bond donor-acceptor distances due to H/D substitution are subtle, 0.01 Å in NMR studies on PYP (11).] According to the neutron diffraction analysis, an H atom in the OTyr42-OpCA bond was located at 0.96 Å from Tyr42 and was assumed to be an ionic H bond (10). In contrast, in the case of the Glu46–pCA pair, an H atom position was assigned at 1.21 Å from Glu46 and 1.37 Å from pCA, almost at the midpoint of the OGlu46-OpCA bond (2.57 Å) (Fig. 1A). From this unusual H atom position, the H bond between Glu46 and pCA was interpreted as a low-barrier H bond [LBHB (12)] by the authors of ref. 10. LBHB was originally proposed to possess a covalent-bond-like character, thus significantly stabilizing the transition state and facilitating enzymatic reactions (12, 13). To understand the OGlu46-OpCA bond characteristics, the following points should be considered:
-
H-bond length and NMR chemical shift. It was suggested that a strong H-bond results in a more downfield 1H NMR chemical shift. According to the classification of H bonds by Jeffrey (14) or Frey (15), “single-well H bonds” [or “symmetrical H bonds” (16)] are very short typically with O-O distances of 2.4–2.5 Å and display 1H NMR chemical shifts (δH) of 20–22 ppm (15). LBHBs [or “asymmetric H bonds” (16)] are longer, 2.5–2.6 Å with δH of 17–19 ppm (15). “Weak H bonds” are further longer, with δH of 10–12 ppm (15). According to the criteria (15, 16), the OGlu46-OpCA bond is not an LBHB but is more likely to be a single-well H-bond in terms of the H atom position. However, simultaneously, the reported OGlu46-OpCA distance of 2.57 Å (10) is too long for a single-well H bond. Thus, it is not clear whether this protein has an LBHB on the basis of the H-bond geometry.
On the other hand, δH of 15.2 ppm was assigned to protonated Glu46 in NMR studies (11). The value of 15.2 ppm is smaller than that for single-well H bonds [20–22 ppm (15)] or even for LBHB [17–19 ppm (15)].
pKa values. According to Perrin and Nielson (17) or Schutz and Warshel (18), the definition LBHB is vague. Schutz and Warshel (18) concluded that LBHB cannot be defined by only the distance or strength of a H bond, and that only energy-based evaluations can be used to determine H-bond types. In particular, the pKa values of the donor and acceptor moieties are important in determining the energy barrier required for moving an H atom between donor and acceptor moieties (18). In original reports by Frey et al. (13) or Cleland and Kreevoy (12), it was stated that an LBHB may form when the pKa difference between donor and acceptor moieties is nearly zero. Interestingly, it was also speculated by the authors of ref. 10 that the pKa values of Glu46 and pCA would be similar in the PYP ground state. If this were the case, the potential-energy curve of the H bond should resemble the shape of a symmetric potential (18); this would be consistent with the H atom position at the midpoint of the OGlu46-OpCA bond, as reported in the neutron diffraction analysis (10). However, the “similar pKa values of Glu46 and pCA” contradict the protonated Glu46 and deprotonated pCA in the PYP ground state, as suggested in a number of previous studies (3–5).
Covalent-bond-like character. In PYP, Arg52 is located on the protein surface in the chromophore region and it shields the chromophore from the solvent, which, thus far, was considered to be protonated (7) (Fig. 1). In contrast, Arg52 was concluded to be deprotonated by the authors of ref. 10 due to the absence of the corresponding nuclear density. They speculated that ionized pCA was energetically unstable in the hydrophobic chromophore unless a covalent-bond-like LBHB was present. However, it should be noted that several polar groups exist close to the pCA in the PYP chromophore; e.g., Tyr42 and Thr50. In addition, the authors of ref. 10 did not provide any explanation as to how is the existence of deprotonated Arg52 energetically possible on a protein surface where solvation energy is sufficiently available.
In the present study, we investigated how the formation of the short H bond between Glu46 and pCA was energetically favored in the 1.25-Å PYP crystal structure, using a large-scale quantum mechanical/molecular mechanical (QM/MM) approach. We also performed pKa calculations by solving the linear Poisson–Boltzmann (LPB) equation and considered the protonation states of all titratable sites of PYP (electrostatic calculation). Note that in general, electrostatic and QM/MM calculations give consistent results; e.g., (19).
Results and Discussion
H-Bond Distances in PYP.
The QM/MM geometry optimization resulted in an OTyr42-OpCA distance of 2.50 Å and an OGlu46-OpCA distance of 2.57 Å, which are in agreement with the distances of 2.52 and 2.57 Å in neutron diffraction analysis (10), respectively (Table 1). Note that the corresponding distances are 2.50 and 2.59 Å in another X-ray diffraction analysis at a resolution of 1.00 Å (9), respectively. The H atom in the OTyr42-OpCA bond was located at a distance of 1.01 Å from Tyr42 rather than pCA, in agreement with neutron diffraction studies (10).
Table 1.
H-bond distances in optimized geometries in the PYP protein environment and model systems (in Å)
In the OGlu46-OpCA bond, neutron diffraction studies (10) suggested that the H atom is located at a distance of 1.21 and 1.37 Å from Glu46 and pCA, respectively. In contrast, the present QM/MM studies suggested that the H atom is at a distance of 1.00 and 1.58 Å from Glu46 and pCA, respectively, irrespective of the consistency in the OGlu46-OpCA distance (Movie S1). In agreement with the present QM/MM result, deprotonated pCA and protonated Glu46 were observed in experimental studies (3–5). Hence, to explain the short OGlu46-OpCA distance of 2.57 Å, it is not prerequisite to locate an H atom near the midpoint between OGlu46 and OpCA. According to Frey (15), an essential requirement for a short H-bond is that the proton lies inline with the donor and acceptor atoms; the OGlu46-H-OpCA angles are 167.9° in the neutron diffraction studies (10) and 169.7° in the QM/MM geometry (Table S1), essentially the same.
The OTyr42-OpCA bond possessed an asymmetric double-well potential (17) (Fig. 2A), which agrees with the conclusion from the neutron diffraction study that the short H bond between Tyr42 and pCA was not a LBHB (10). The energy value at 0.96 Å from OTyr42, which is the corresponding H atom position in neutron diffraction studies (10), was only approximately 0.9 kcal/mol higher than that at 1.01 Å for the energy minimum in the QM/MM geometry; i.e., essentially the same (Fig. 2A). The energy near the OpCA moiety is higher than that in the OTyr42 moiety (i.e., near energy minimum), which indicates that the pKa of Tyr42 is higher than that of pCA. The OGlu46-OpCA bond also possessed an asymmetric double-well potential, also corresponding to a classical H bond (Fig. 2B). There is no energy minimum near 1.21 Å from OGlu46, and the energy is approximately 5 kcal/mol higher than that at 1.00 Å. Thus, we did not essentially observe differences in the potential-energy profile between OTyr42-OpCA and OGlu46-OpCA. The asymmetric potential curves obtained for Tyr42-pCA and Glu46-pCA cannot be classified to those of LBHB (18).
Fig. 2.

Energy profiles along the proton transfer coordinate for H-bond donor-acceptor pairs (A) Tyr42-pCA, (B) Glu46-pCA in the PYP protein environment, and (C) Glu-pCA in the model system (Fig. 1C).
We also analyzed the potential-energy profile by altering the OGlu46-OpCA distance. An OGlu46-OpCA distance where the H atom is located nearly at the midpoint of OGlu46-OpCA was obtained at approximately 2.3 Å (Fig. 3). The potential-energy curve with OGlu46-OpCA = 2.32 Å resembles that of a single-well potential, but is not yet completely symmetric [i.e., the minimum is not at the center of the OGlu46-OpCA bond (18)] due to the originally larger pKa value of Glu46 with respect to pCA in the PYP environment (3–5). Obviously, even in this case the OGlu46-OpCA bond is unlikely to satisfy the condition of LBHB. As suggested by Schutz and Warshel (18), identification of LBHB with a single minimum potential can be valid only if the minimum is at the center of the OGlu46-OpCA bond. More importantly, the energy minimum with OGlu46-OpCA = 2.32 Å is obviously energetically higher than that with OGlu46-OpCA = 2.57 Å. Hence, the common case of asymmetric single minimum H bond is not LBHB but a standard H bond where the pKa difference between the donor and acceptor moieties is large; this has been already demonstrated by Schultz and Warshel (18). Thus, unless the pKa values of the donor and acceptor moieties are already similar in the original geometry, the resulting energy minimum of the bond is affected more by the moiety whose pKa value is lower, and as a result, the bond becomes energetically unstable before decreasing the OGlu46-OpCA distance.
Fig. 3.

Dependence of the potential-energy profiles at OGlu46-OpCA = 2.32, 2.57, and 2.77 Å. The red arrow indicates the energy difference from the energy minimum obtained at OGlu46-OpCA of 2.57 Å.
H-Bond Distances in Model Systems.
To investigate the contributions of the PYP environment to the H-bond length, we performed geometry optimizations for model systems that comprised only side chains; i.e., Tyr-pCA or Glu-pCA. The Tyr-pCA model yielded a slightly longer OTyr-OpCA distance (2.57 Å; Table 1) than in PYP (2.50 Å). In contrast, the OGlu-OpCA distance in the Glu-pCA model was 2.52 Å, shorter than that of 2.57 Å in PYP. The obtained energy curve in the model system (Fig. 2C) was that of an asymmetric double-well potential but less asymmetric than that in the PYP protein environment (Fig. 2B); the shorter donor acceptor distance and the less asymmetric potential-energy curve were due to the absence of the protein environment, as already demonstrated by Warshel et al. (20, 21). Hence, in the PYP active site, a delocalized charge distribution over the OGlu-OpCA bond (i.e., low-barrier for H atom movement, Fig. 2C) is energetically less favorable because a concentrated charge (i.e., due to localization of the H atom, Fig. 2B) can interact more strongly with the PYP protein dipoles.
pKa Values of Glu46 and pCA in PYP.
Schutz and Warshel (18) concluded that the determination of the pKa values for the donor and acceptor moieties is the clearest way of examining the LBHB proposal. From the unusual H atom position of the Glu46-pCA bond, the authors of ref. 10 speculated that the pKa values of Glu46 and pCA would be similar in the PYP ground state, contributing to the LBHB formation. In contrast, we calculated the pKa values of Glu46 and pCA to be 8.6 and 5.4, respectively (Table 2); these values are in agreement with a number of experimental studies that attributed the two pKa values near approximately nine and approximately six to those of Glu46 and pCA, respectively (3–5). Hence, there is little basis of emphasizing the equal pKa values for Glu46 and pCA in the PYP ground state.
Table 2.
Calculated pKa values
| WT | R52A | |||||
| Glu46 | pCA | Arg52 | Glu46 | pCA | ||
| pKa | protein | 8.6 | 5.4 | 13.7 | 8.1 | 5.9 |
| water (reference) | 4.4 | 8.8* | 12.0 | 4.4 | 8.8* | |
| pKa shift (water → protein) | total | 4.2 | –3.4 | 1.7 | 3.7 | –2.9 |
| (i) protein van der Waals volume† | 8.1 | 4.7 | –4.7 | 8.0 | 4.4 | |
| (ii) atomic charge | –3.9 | –8.1 | 6.4 | –4.3 | –7.3 |
*See ref. 29.
†The protein van der Waals volume (i.e., the space obtained by merging the volumes of the van der Waals spheres of all protein atoms) that decreases the availability of solvation energy.
The protein charge of the entire PYP contributes to decreasing pKa(Glu46) by 3.9 (Table 2). The key components that decrease pKa(Glu46) are Thr50 (by 1.9), Arg52 (by 1.8), and Tyr42 (by 1.5) (Table S2). Arg52 was protonated in the PYP ground state (Table 2; explicitly discussed later), and positively charged Arg52 facilitates deprotonation of Glu46. Notably, Thr50 and Tyr42 are not charged groups, but they have essentially the same influence on pKa(Glu46) as the positively charged Arg52 does (Table S2), implying the importance of protein dipoles in stabilizing the charged group in the protein inner core.
The influence of the protein van der Waals volume on pKa(Glu46) is larger than that of the protein charge (Table 2). The protein van der Waals volume (i.e., the space obtained by merging the volumes of the van der Waals spheres of all protein atoms) is the main part that provides the so-called “protein hydrophobicity” to the charged group; the protein van der Waals volume prevents access of water molecules to Glu46 and, thus, decreases the availability of solvation energy. In particular, the loss of solvation energy (22) destabilizes the deprotonated states for acidic residues, increasing the pKa values.
A slightly smaller contribution of the protein van der Waals volume to pKa(pCA) upshift (by 4.7) with respect to pKa(Glu46) upshift (by 8.1) was due to the fact that Glu46 is completely buried in the protein (7). In contrast, the protein atomic charge plays a major role in decreasing pKa(pCA) (Table 2). Arg52 decreases pKa(pCA) by 2.9 in the wild-type PYP (Table S3). Although Tyr42 and Thr50 are not charged groups, they also significantly contribute to decreasing pKa(pCA) by 2.3 and 1.5, respectively. See SI Results and Discussion for the pKa values of the R52A mutant.
Protonation State of Arg52.
One of the backgrounds to propose the Glu46-pCA bond as LBHB was the interpretation of Arg52 as being deprotonated in the neutron diffraction analysis (10). Arg52 has two H-bond partners, the backbone carbonyl O atoms of Thr50 and Tyr98 (Fig. S1). The Nη1 atom of Arg52 appears to have only a single nuclear density toward Thr50 according to ref. 10. In contrast, Arg52 was proposed to be protonated in a previous X-ray diffraction study (7) because the residue is exposed to the solvent. Hence, even if the basic residues are in the neighborhood of Arg52, the electrostatic influence on Arg52 will be considerably shielded by bulk water.
Electrostatics: The calculated pKa value of Arg52 was 13.7, suggesting that Arg52 is undoubtedly protonated (Table 2). Although the PYP protein volume that comprises the van der Waals radii of the PYP residues partly covers Arg52 and contributes to decreasing the pKa value by 4.7, the pKa(Arg52) was more upshifted mainly by the ionized pCA (by 2.2 in pKa) and backbone regions (namely carbonyl group) at Tyr98 (by 2.0 in pKa, NArg52-OTyr98 = 2.98 Å), Thr50 (by 1.0 in pKa, NArg52-OThr50 = 2.93 Å), and Val66 (by 0.7 in pKa, NArg52-OVal66 = 3.66 Å; Table S4). In contrast, Lys60 is the residue that most significantly contributes to Arg52 deprotonation, but decreases the Arg52 pKa value by only 0.4 (Table S4), which is located more than 7 Å away from Arg52. Thus, we could not find any reasonable mechanism to favor deprotonation of Arg52 based on the geometry of the PYP crystal structure (10).
QM/MM: We also independently performed QM/MM calculations to carefully evaluate the protonation state of Arg52. When Arg52 was deprotonated as suggested in ref. 10, the NArg52-OThr50 distance of 2.93 Å in the original crystal structure was significantly increased to 3.78 Å, which does not form H bonds (Table 3 and Fig. S1). The corresponding distance with protonated Arg52 was 3.14 Å. The NArg52-OTyr98 distance of 2.98 Å in the original crystal structure remains unchanged in the QM/MM geometry optimized in the presence of protonated Arg52 (2.99 Å), whereas it was increased to 3.31 Å in the presence of deprotonated Arg52 (Table 3). The resulting rmsd of the optimized side chain heavy atoms of Arg52 relative to the crystal structure [Protein Data Bank (PDB) ID code 2ZOH) was 0.143 Å for protonated Arg52 but 0.349 Å for deprotonated Arg52, the latter being above the uncertainty at a resolution of approximately 1 Å. Clearly, the positive charge of protonated Arg52 is prerequisite to the strong interaction with the two backbone carbonyl groups, as deprotonated Arg52 is too weak to maintain the H-bond distance in particular with Thr50.
Table 3.
Comparison of the H-bond donor-acceptor distances of Arg52 between the crystal structure (10) and the QM/MM optimized geometry with protonated /deprotonated Arg52 (in Å)
| Crystal | QM/MM (protein) | ||
| X-ray | Protonated | Deprotonated | |
| Arg52-Thr50 (Nη1-OCO) | 2.93 | 3.14 | 3.78 |
| Arg52-Tyr98 (Nη2-OCO) | 2.98 | 2.99 | 3.31 |
| Rmsd | — | 0.143 | 0.349 |
OCO = backbone carbonyl O atom. Rmsd of the optimized side chain heavy atoms of Arg52 with respect to those of the crystal structure. —; not applicable.
The two independent results of the pKa and QM/MM calculations demonstrated that Arg52 is highly likely to be protonated. The LBHB concept that was employed to explain the stabilization of the ionized pCA in the absence of protonated Arg52 (10) may need to be reevaluated. If protonated Arg52 is the case for the PYP ground state, employment of the LBHB concept will be less relevant.
Conclusions
We reproduced the two short H-bond donor-acceptor distances for Tyr42-pCA (2.50 Å) and Glu46-pCA (2.57 Å) using the QM/MM calculations for the entire PYP. Tyr42 was protonated and pCA was ionized. In the OGlu46-OpCA bond, a H atom was located at approximately 1 Å from Glu46, and Glu46 was obviously protonated in the presence of deprotonated pCA (Table 1). The potential-energy profile of the OGlu46-OpCA bond revealed an asymmetric double-well potential (17). Furthermore, there was no energy minimum near the midpoint of the OGlu46-OpCA bond or the pCA moiety (Fig. 2B). We also calculated the pKa values for Glu46 and pCA to be 8.6 and 5.4, respectively (Table 2). Ionized pCA was electrostatically stabilized by protonated Arg52 (by 2.9 in pKa), and the short H bond of Tyr42 (by 2.3) and dipole of Thr50 (by 1.5) (Table S3). Obviously, the negative charge on ionized pCA is not a bare charge but already stabilized by the short H bond of Tyr42. The calculated pKa value of Arg52 on the protein surface was 13.7, suggesting that Arg52 was protonated in the geometry of the crystal structure (10) as in previous structural studies (7). QM/MM geometries also resulted in a significantly smaller rmsd with protonated Arg52 (0.14 Å) than with deprotonated Arg52 (0.35 Å) relative to the original crystal structure (Table 3). The NMR chemical shift of 15.2 ppm (11) for the OGlu46-OpCA bond is smaller than that for single-well H bonds [20–22 ppm (15)] or even for LBHB [17–19 ppm (15)]. Indeed, the H bond was not explicitly concluded to be an LBHB in NMR studies (11). Thus, together with the potential-energy curve of the OGlu46-OpCA bond (Figs. 2 and 3), there is no necessity to stabilize ionic pCA by LBHB. The short H-bond distance of 2.57 Å for the OGlu46-OpCA bond can be simply explained by electrostatic interaction without invoking the LBHB concept.
Computational Methods
Initial Coordinates and Atomic Partial Charges.
The atomic coordinates of PYP were taken from the X-ray structure of the wild-type PYP at 1.25-Å resolution (PDB ID code 2ZOH) (10). H atoms were generated and energetically optimized with CHARMM (23), whereas the positions of all heavy atoms were fixed. Atomic partial charges of the amino acids were adopted from the all-atom CHARMM22 (24) parameter set. The atomic charges of pCA (Table S5) were determined by fitting the electrostatic potential in the neighborhood of these molecules using the restrained electrostatic potential (RESP) procedure (25). The electronic wave functions were calculated after the optimization of the geometry with the density functional theory (DFT) module in JAGUAR (26) (B3LYP/LACVP**+).
QM/MM Calculations.
We employed the so-called electrostatic embedding QM/MM scheme and used the Qsite (27) program code as performed in previous studies (19). We employed the restricted DFT method with the B3LYP functional and LACVP**+ basis sets. The geometries were refined by constrained QM/MM optimization; the coordinates of the heavy atoms in the surrounding MM region were exactly fixed to the original X-ray coordinates, whereas those of H atoms in the MM region were optimized with the OPLS2005 force field. (i) To investigate the energetics of the H-bond network, pCA with the covalently bonded Cys69 and all H-bond partner residues (i.e., Tyr42, Glu46, and Thr50) were considered as the QM region whereas other residues were approximated by the MM force field (Table S1). The potential-energy profile of the H bond was obtained as follows: first, we prepared for the QM/MM optimized geometry without constraints, and we used the resulting geometry as the initial geometry. Next, we moved the H atom from the H-bond donor atom (OD) to the acceptor atom (OA) by 0.05 Å, optimized the geometry by constraining either the OD-H and H-OA distances (Fig. 2) or the OD-H and OD-OA distances (Fig. 3), and calculated the energy of the resulting geometry. This procedure was repeated until the H atom reached the OA atom. After obtaining the stable geometry of the QM fragment, we determined the electrostatic potential (ESP) charges for the anionic state of the [Tyr42, Glu46, Thr50, and pCA] system (Table S1). (ii) To carefully evaluate the protonation state of Arg52, we defined the side chains of Arg52 and Met100 and the carbonyl backbone groups of Thr50 and Tyr98 as the QM region and the remaining protein residues as the MM region (Tables S6 and S7).
Protonation Pattern and pKa.
The present computation is based on the electrostatic continuum model created by solving the LPB equation with the MEAD program (28). We used identical computational conditions and parameters (e.g., ref. 19). The calculated difference in the pKa value of the protein relative to the reference system was added to the known reference pKa value. Experimentally measured pKa values employed as references are 8.8 for pCA (29). The ensemble of the protonation patterns was sampled by the Monte Carlo (MC) method with Karlsberg (30). The dielectric constants were set to ϵp = 4 inside the protein and ϵw = 80 for water. All computations were performed at 300 K, pH 7.0, and an ionic strength of 100 mM. The LPB equation was solved using a 3-step grid-focusing procedure at resolutions of 2.5 Å, 1.0 Å, and 0.3 Å. The MC sampling yielded the probabilities [protonated] and [deprotonated] of the two protonation states of a molecule. The pKa value was evaluated using the Henderson–Hasselbalch equation. A bias potential was applied to obtain an equal amount of both protonation states ([protonated] = [deprotonated]), yielding the pKa value as the resulting bias potential.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the JST PRESTO program (H.I.), Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) in Japan (21770163 to H.I. and 22740276 to K.S.), Special Coordination Fund for Promoting Science and Technology of MEXT (H.I.), Takeda Science Foundation (H.I.), Kyoto University Step-up Grant-in-Aid for young scientists (H. I.), and Grant for Basic Science Research Projects from The Sumitomo Foundation (H. I.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1113599108/-/DCSupplemental.
References
- 1.Sprenger WW, Hoff WD, Armitage JP, Hellingwerf KJ. The eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J Bacteriol. 1993;175:3096–104. doi: 10.1128/jb.175.10.3096-3104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baca M, et al. Complete chemical structure of photoactive yellow protein: novel thioester-linked 4-hydroxycinnamyl chromophore and photocycle chemistry. Biochemistry. 1994;33:14369–14377. doi: 10.1021/bi00252a001. [DOI] [PubMed] [Google Scholar]
- 3.Kim M, Mathies RA, Hoff WD, Hellingwerf KJ. Resonance Raman evidence that the thioester-linked 4-hydroxycinnamyl chromophore of photoactive yellow protein is deprotonated. Biochemistry. 1995;34:12669–12672. doi: 10.1021/bi00039a024. [DOI] [PubMed] [Google Scholar]
- 4.Xie A, Hoff WD, Kroon AR, Hellingwerf KJ. Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein. Biochemistry. 1996;35:14671–14678. doi: 10.1021/bi9623035. [DOI] [PubMed] [Google Scholar]
- 5.Demchuk E, et al. Protonation states and pH titration in the photocycle of photoactive yellow protein. Biochemistry. 2000;39:1100–13. doi: 10.1021/bi991513p. [DOI] [PubMed] [Google Scholar]
- 6.Pan D, Philip A, Hoff WD, Mathies RA. Time-resolved resonance raman structural studies of the pB′ intermediate in the photocycle of photoactive yellow protein. Biophys J. 2004;86:2374–2382. doi: 10.1016/S0006-3495(04)74294-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borgstahl GE, Williams DR, Getzoff ED. 1.4 Å structure of photoactive yellow protein, a cytosolic photoreceptor: Unusual fold, active site, and chromophore. Biochemistry. 1995;34:6278–6287. doi: 10.1021/bi00019a004. [DOI] [PubMed] [Google Scholar]
- 8.Getzoff ED, Gutwin KN, Genick UK. Anticipatory active-site motions and chromophore distortion prime photoreceptor PYP for light activation. Nat Struct Biol. 2003;10:663–668. doi: 10.1038/nsb958. [DOI] [PubMed] [Google Scholar]
- 9.Anderson S, Crosson S, Moffat K. Short hydrogen bonds in photoactive yellow protein. Acta Crystallogr D Biol Crystallogr. 2004;60:1008–1016. doi: 10.1107/S090744490400616X. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi S, et al. Low-barrier hydrogen bond in photoactive yellow protein. Proc Natl Acad Sci USA. 2009;106:440–444. doi: 10.1073/pnas.0811882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sigala PA, Tsuchida MA, Herschlag D. Hydrogen bond dynamics in the active site of photoactive yellow protein. Proc Natl Acad Sci USA. 2009;106:9232–7. doi: 10.1073/pnas.0900168106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cleland WW, Kreevoy MM. Low-barrier hydrogen bonds and enzymic catalysis. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 13.Frey PA, Whitt SA, Tobin JB. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science. 1994;264:1927–30. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 14.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford: Oxford University Press; 1997. [Google Scholar]
- 15.Frey PA. In: Isotope Effects in Chemistry and Biology. Kohen A, Limbach H.-H, editors. Boca Raton, FL: CRC; 2006. pp. 975–993. [Google Scholar]
- 16.Frey PA. Strong hydrogen bonding in molecules and enzymatic complexes. Magn Reson Chem. 2001;39:S190–S198. [Google Scholar]
- 17.Perrin CL, Nielson JB. “Strong” hydrogen bonds in chemistry and biology. Annu Rev Phys Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 18.Schutz CN, Warshel A. The low barrier hydrogen bond (LBHB) proposal revisited: The case of the Asp…His pair in serine proteases. Proteins. 2004;55:711–723. doi: 10.1002/prot.20096. [DOI] [PubMed] [Google Scholar]
- 19.Saito K, et al. Distribution of the cationic state over the chlorophyll pair of photosystem II reaction center. J Am Chem Soc. 2011;133:14379–14388. doi: 10.1021/ja203947k. [DOI] [PubMed] [Google Scholar]
- 20.Warshel A, Weiss RM. An empirical valence bond approach for comparing reactions in solutions and in enzymes. J Am Chem Soc. 1980;102:6218–6226. [Google Scholar]
- 21.Warshel A, Papazyan A, Kollman PA. On low-barrier hydrogen bonds and enzyme catalysis. Science. 1995;269:102–106. doi: 10.1126/science.7661987. [DOI] [PubMed] [Google Scholar]
- 22.Kato M, Pisliakov AV, Warshel A. Barrier for proton transport in aquaporins as a challenge for electrostatic models: the role of protein relaxation in mutational calculations. Proteins. 2006;64:829–844. doi: 10.1002/prot.21012. [DOI] [PubMed] [Google Scholar]
- 23.Brooks BR, et al. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 24.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 25.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 26.Jaguar v. 7.5. New York, NY: Schrödinger, LLC; 2008. [Google Scholar]
- 27.Qsite v. 5.6. New York, NY: Schrödinger, LLC; 2010. [Google Scholar]
- 28.Bashford D, Karplus M. pKa’s of ionizable groups in proteins: Atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 29.Kroon AR, et al. Spectral tuning, fluorescence, and photoactivity in hybrids of photoactive yellow protein, reconstituted with native or modified chromophores. J Biol Chem. 1996;271:31949–31956. doi: 10.1074/jbc.271.50.31949. [DOI] [PubMed] [Google Scholar]
- 30.Rabenstein B, Knapp EW. Calculated pH-dependent population and protonation of carbon-monoxy-myoglobin conformers. Biophys J. 2001;80:1141–1150. doi: 10.1016/S0006-3495(01)76091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}