

Abstract
The mechanism of umpolung amide synthesis was probed by interrogating potential sources for the oxygen of the product amide carbonyl that emanates from the α-bromo nitroalkane substrate. Using a series of 18O-labeled substrates and reagents, evidence is gathered to advance two pathways from the putative tetrahedral intermediate. Under anaerobic conditions, a nitro-nitrite isomerization delivers the amide oxygen from nitro oxygen. The same homolytic nitro-carbon fragmentation can be diverted by capture of the carbon radical intermediate with oxygen gas (O2) to deliver the amide oxygen from O2. This understanding was used to develop a straightforward protocol for the preparation of 18O-labeled amides in peptides by simply performing the umpolung amide synthesis reaction under an atmosphere of 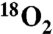 .
.
Preparative needs in amide synthesis, particularly those in the peptide regime, continue to drive new methods for their construction (1–15). The increasing success of peptide therapeutics, as well as continuing biophysical studies of peptides, have generally outpaced the scope of purely chemical methods for their preparation on scale (16, 17). Native chemical ligation has teamed with solid phase peptide synthesis to target an increasing list of complex problems, whereas α-keto acid-based methods (18, 19) and umpolung amide synthesis (UmAS) (20) are beginning to target others. Historically, improvements to dehydrative amide synthesis have received the lion’s share of effort, and this is justified by the broad inventory of carboxylic acids available commercially and synthetically (21). Aside from the need to greatly generalize any one of these developments, it remains a challenge to routinely incorporate chiral nonnatural amino acids. Site-selective labeling of amide oxygen, despite its power as a technique for the spectroscopic study of protein structure (22), also remains quite difficult beyond the use of methods that label the precursor carboxylic acids through a combination of acid and H218O (23–26).
We recently reported a unique amide synthesis based on the reaction of α-halo nitroalkanes and amines, using a halonium activating agent and alkaline conditions (20). This basic reaction also holds promise in more complex settings such as peptide synthesis, and the nascent principles for its use in enantioselective peptide homologation with aryl glycine amino acids were outlined. Although the rapid and complete formation of an N-halamine (Fig. 1, 2), and its competency in the coupling was established, we were unable to directly observe the putative “tetrahedral intermediate” (Fig. 1, 3) that constitutes a logical species en route to amide, particularly because it bears the proper oxidation state at carbon, requiring only a formal hydrolysis to provide amide product (Fig. 1, 4). Although we have failed to observe this intermediate directly, we remained interested in how it might evolve to amide and began a reverse-mechanistic approach to explore the various possibilities. Our ulterior motive was practical: The ability to site-selectively label amide oxygens with the 18O isotope remains challenging in many contexts, and methods remain limited in number (23–25).
Fig. 1.

Experiments reveal that water is not the source of amide oxygen in umpolung amide synthesis.
Results and Discussion
Our first hypothesis was that this intermediate simply hydrolyzed to amide by either ionization of bromide or nitrite to an intermediate azomethine (e.g., Scheme 1, 6). Should this be the case, the use of H218O would provide the 18O-labeled amide. In the event, we observed the formation of only 16O-labeled amide (Table 1, entry 1), suggestive of a pathway to amide that might be more complex. The experiments that followed attempted to isolate the possible sources of oxygen: 1) residual water, 2) the nitro functionality in the α-halo nitroalkane substrate, and 3) potassium carbonate.
Scheme 1.

Observations using 18O-labeling experiments and corresponding anaerobic and aerobic pathways from tetrahedral intermediate to amide
Table 1.
18O-labeling study of amide synthesis from α-bromo nitroalkanes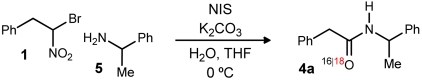
| Entry* | NO2 label (% N ) ) |
H2O (%18O) | Atmosphere | Amide (% 18O) | Δ18O | Yield† (%) |
| 1‡ | 0 | > 99 | open | < 1 | < 1 | 75 |
| 2‡ | 82 | 0 | open | 17 | −65 | 76 |
| 3§ | 82 | 0 | open | 49 | −33 | 70 |
| 4§ | 82 | > 99 | open | 49 | −33 | 70 |
| 5§ | 82 | 0 | Ar | 66 | −16 | 70 |
| 6 | 0 | 0 |
 ¶ ¶
|
83 | +83 | 68 |
*Reactions employed one equivalent of α-bromo nitroalkane (0.2 M in THF), with N-iodosuccinimide (one equivalent) added as the final reagent at 0 °C. “Open” atmosphere refers to use of a static atmosphere provided by a cap or septum. Other variations used a balloon of the indicated gas. Isotopic distribution determined by high resolution mass spectrometry.
†Isolated yields.
‡1.2 equivalents of amine used.
§Five equivalents of amine used to replace potassium carbonate base.
¶97% enriched 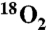 gas used.
gas used.
We were mindful that the reaction proceeds to amide—albeit at a slower rate—despite attempts to thoroughly remove water, suggesting that the nitro oxygen might play a role. We prepared doubly 18O-labeled nitroalkane (82% 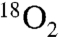 , Fig. 1, 1) [see SI Appendix for complete details. The doubly labeled nitroalkane (Fig. 1, 1) was prepared (67.7%
, Fig. 1, 1) [see SI Appendix for complete details. The doubly labeled nitroalkane (Fig. 1, 1) was prepared (67.7% 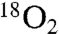 , 29.1% 18O/16O) from labeled nitrite] (27), and using standard conditions for amide formation the amide product was produced with a detectable level of isotope incorporation (17%; Table 1, entry 2). This corresponded to a high level of label loss (
, 29.1% 18O/16O) from labeled nitrite] (27), and using standard conditions for amide formation the amide product was produced with a detectable level of isotope incorporation (17%; Table 1, entry 2). This corresponded to a high level of label loss (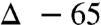 ). Suspecting a possible role for potassium carbonate as an unlabeled oxygen source, this experiment was again performed, but with the amine in excess to replace exogenous potassium carbonate base. This experiment led to diminished but still significant label loss (
). Suspecting a possible role for potassium carbonate as an unlabeled oxygen source, this experiment was again performed, but with the amine in excess to replace exogenous potassium carbonate base. This experiment led to diminished but still significant label loss (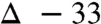 ; Table 1, entry 3). We continued this series with excess amine (no carbonate), and reinvestigated the possible role of water but found no observable change in the level of isotope conservation (Table 1, entry 4). Finally, the role of atmospheric oxygen was examined by degassing the solvent and using an argon atmosphere for the reaction. This variation provided a high degree of isotope conservation from nitroalkane to amide product (
; Table 1, entry 3). We continued this series with excess amine (no carbonate), and reinvestigated the possible role of water but found no observable change in the level of isotope conservation (Table 1, entry 4). Finally, the role of atmospheric oxygen was examined by degassing the solvent and using an argon atmosphere for the reaction. This variation provided a high degree of isotope conservation from nitroalkane to amide product (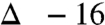 ; Table 1, entry 5).
; Table 1, entry 5).
The outline in Scheme 1 provides a mechanistic picture consistent with these observations. An alternative to our original hypothesis that the tetrahedral intermediate hydrolyzes to amide by ionization of either bromide (Scheme 1, 6) or nitrite (not shown) is homolysis of the tetrahedral intermediate to aminomethyl radical (Scheme 1, 7) and nitrogen dioxide radical (28). Under anaerobic conditions, this radical pair can recombine with isomerization to form nitrite (Scheme 1, 8). Nitrosyl transfer to water under the basic conditions then leads to its collapse to amide (Scheme 1, 10). This mechanistic pathway is consistent with the high level of isotope conservation in the key anaerobic experiment (Table 1, entry 5). Support of this proposal is found in the ability of tertiary nitro compounds to isomerize to nitrite reversibly (29–36). The combination of alkyl, amino, and bromo groups may favor the nitrite isomer (Scheme 1, 8), or alternatively, its subsequent hydrolysis may provide an irreversible step forward to amide.
Because the reaction of oxygen with carbon radicals is often at, or near, the rate of diffusion (37–39), we hypothesized that putative aminomethyl radical (Scheme 1, 7) might be intercepted by 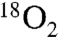 . A solution of unlabeled nitroalkane (Fig. 1, 1) was subjected to the standard reaction conditions and an
. A solution of unlabeled nitroalkane (Fig. 1, 1) was subjected to the standard reaction conditions and an 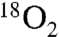 atmosphere (balloon), and a high degree of isotope incorporation into the amide product was observed (
atmosphere (balloon), and a high degree of isotope incorporation into the amide product was observed (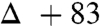 ; Table 1, entry 6). This is suggestive of an aerobic pathway that diverts from Scheme 1, 7, and the intermediate peroxide (Scheme 1, 9) can fragment to amide and the elements of bromonium nitrate. Together, each pathway explains why water is not necessarily required as a reagent but may be a helpful additive by promoting nitrite hydrolysis (anaerobic) or formation of nitrate or (potassium) hypobromite (aerobic).
; Table 1, entry 6). This is suggestive of an aerobic pathway that diverts from Scheme 1, 7, and the intermediate peroxide (Scheme 1, 9) can fragment to amide and the elements of bromonium nitrate. Together, each pathway explains why water is not necessarily required as a reagent but may be a helpful additive by promoting nitrite hydrolysis (anaerobic) or formation of nitrate or (potassium) hypobromite (aerobic).
In an effort to devise a direct preparation of 18O-labeled amides, we developed an experimental preparation that favors the aerobic pathway to amide. Two key modifications were made: 1) solutions of the reactants were degassed by freeze-pump-thaw cycles, and 2) the final cycle was finished by drawing  into the head space of the vial. To minimize overall cost, a vial was used to maintain a small volume without unduly limiting the solution’s contact with the labeled oxygen atmosphere. (The SI Appendix provides a description of the experiment as well as a picture of the experimental setup.) Application of this protocol to a series of couplings provided labeled amides (Table 2, 11a–f) with a high degree of enrichment (65–93%; Table 2), as well as site-selectivity (Table 2, entries 4–6). Although it is likely that higher levels of incorporation could be achieved using techniques to further enrich the solvent with labeled oxygen, or a vessel pressurized by oxygen, our goal was to establish the efficiency of incorporation using an inexpensive, easily reproduced experimental setup.
into the head space of the vial. To minimize overall cost, a vial was used to maintain a small volume without unduly limiting the solution’s contact with the labeled oxygen atmosphere. (The SI Appendix provides a description of the experiment as well as a picture of the experimental setup.) Application of this protocol to a series of couplings provided labeled amides (Table 2, 11a–f) with a high degree of enrichment (65–93%; Table 2), as well as site-selectivity (Table 2, entries 4–6). Although it is likely that higher levels of incorporation could be achieved using techniques to further enrich the solvent with labeled oxygen, or a vessel pressurized by oxygen, our goal was to establish the efficiency of incorporation using an inexpensive, easily reproduced experimental setup.
Table 2.
18O-labeled amide synthesis using aerobic UmAS*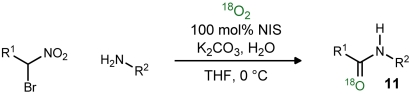
| Entry | Amide product | Amide 18O (%)† | Yield (%) | |
| 1 | 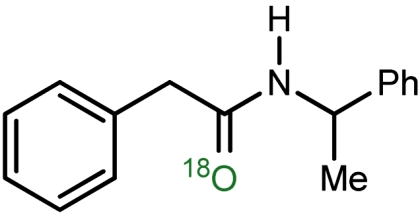 |
a | 79 | 79 |
| 2 | 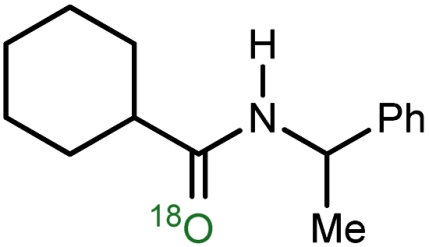 |
b | 65 | 81 |
| 3 | 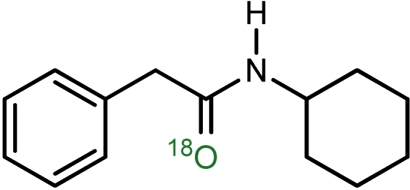 |
c | 79 | 78 |
| 4 | 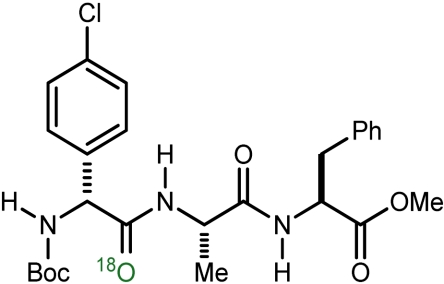 |
d | 93 | 58 |
| 5 | 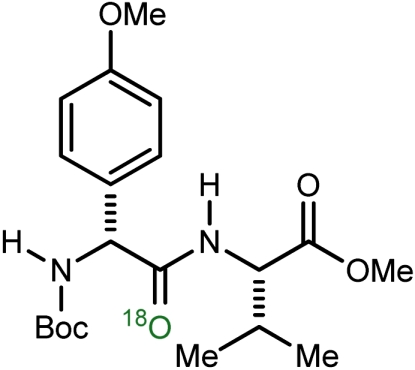 |
e | 76 | 65 |
| 6 | 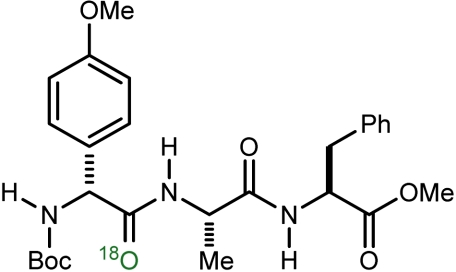 |
f | 88 | 49 |
*See SI Appendix for complete experimental details for introduction of a static atmosphere of labeled oxygen (97% 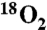 ).
).
†Enrichment determined by mass spectrometry. Analysis by 13C NMR was consistent with incorporation. See SI Appendix for complete details.
Conclusions
In summary, we have identified two pathways to amide product from the putative tetrahedral intermediate formed in UmAS. The pathway is highly dependent on the nature of the atmosphere above the solution. When degassed and stirred under argon, the nitro oxygen serves as the primary source of oxygen in the amide product. If the solution is instead stirred under an atmosphere of oxygen, nearly all of the amide oxygen can result from gaseous oxygen. This mechanistic picture also rationalizes our earlier observation that amide is still produced when measures are taken to use anhydrous conditions for UmAS. In this case, particularly with the anaerobic variant, the isomerization to nitrite can still occur, and amine (or carbonate) can serve the role of nitrosyl acceptor. Aside from the clearer picture this study provides for further reaction optimization and development, we have demonstrated that 18O-labeled amides can be prepared conveniently using UmAS by the straightforward supply of 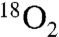 as an atmosphere during an otherwise standard coupling with amine.
as an atmosphere during an otherwise standard coupling with amine.
Supplementary Material
ACKNOWLEDGMENTS.
We would like to thank Libin Xu and Ned Porter (Vanderbilt University) for helpful discussions and Ahmad Al-Mestarihi and Brian Bachmann for initial access to  . We are grateful to Amgen for financial support of this work. Chiral nonracemic nitroalkane donors were prepared with the support of the National Institutes of Health (GM 084333). J.P.S. was supported by a National Science Foundation Predoctoral Fellowship.
. We are grateful to Amgen for financial support of this work. Chiral nonracemic nitroalkane donors were prepared with the support of the National Institutes of Health (GM 084333). J.P.S. was supported by a National Science Foundation Predoctoral Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1113553108/-/DCSupplemental.
References
- 1.Bode JW. Emerging methods in amide- and peptide-bond formation. Curr Opin Drug Discov Devel. 2006;9:765–775. [PubMed] [Google Scholar]
- 2.Saxon E, Armstrong JI, Bertozzi CR. A “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- 3.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 4.Saxon E, et al. Investigating cellular metabolism of synthetic azidosugars with the Staudinger ligation. J Am Chem Soc. 2002;124:14893–14902. doi: 10.1021/ja027748x. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson BL, Kiessling LL, Raines RT. Staudinger ligation: A peptide from a thioester and azide. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 6.Dawson PE, Muir TW, Clarklewis I, Kent SBH. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 7.Cho SH, Yoo EJ, Bae L, Chang S. Copper-catalyzed hydrative amide synthesis with terminal alkyne, sulfonyl azide, and water. J Am Chem Soc. 2005;127:16046–16047. doi: 10.1021/ja056399e. [DOI] [PubMed] [Google Scholar]
- 8.Cassidy MP, Raushel J, Fokin VV. Practical synthesis of amides from in situ generated copper(I) acetylides and sulfonyl azides. Angew Chem Int Ed Engl. 2006;45:3154–3157. doi: 10.1002/anie.200503805. [DOI] [PubMed] [Google Scholar]
- 9.Gunanathan C, Ben-David Y, Milstein D. Direct synthesis of amides from alcohols and amines with liberation of H2. Science. 2007;317:790–792. doi: 10.1126/science.1145295. [DOI] [PubMed] [Google Scholar]
- 10.Nordstrom LU, Vogt H, Madsen R. Amide synthesis from alcohols and amines by the extrusion of dihydrogen. J Am Chem Soc. 2008;130:17672. doi: 10.1021/ja808129p. [DOI] [PubMed] [Google Scholar]
- 11.Yoo WJ, Li CJ. Highly efficient oxidative amidation of aldehydes with amine hydrochloride salts. J Am Chem Soc. 2006;128:13064–13065. doi: 10.1021/ja064315b. [DOI] [PubMed] [Google Scholar]
- 12.Gao J, Wang GW. Direct oxidative amidation of aldehydes with anilines under mechanical milling conditions. J Org Chem. 2008;73:2955–2958. doi: 10.1021/jo800075t. [DOI] [PubMed] [Google Scholar]
- 13.Chan WK, Ho CM, Wong MK, Che CM. Oxidative amide synthesis and N-terminal α-amino group ligation of peptides in aqueous medium. J Am Chem Soc. 2006;128:14796–14797. doi: 10.1021/ja064479s. [DOI] [PubMed] [Google Scholar]
- 14.Li XC, Danishefsky SJ. New chemistry with old-functional groups: On the reaction of isonitriles with carboxylic acids—A route to various amide types. J Am Chem Soc. 2008;130:5446–5447. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao Y, Li X, Danishefsky SJ. Thio FCMA intermediates as strong acyl donors: A general solution to the formation of complex amide bonds. J Am Chem Soc. 2009;131:12924–12926. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groner B, editor. Peptides as Drugs: Discovery and Development. Weinheim: Wiley-VCH; 2009. [Google Scholar]
- 17.Bray BL. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat Rev Drug Discov. 2003;2:587–593. doi: 10.1038/nrd1133. [DOI] [PubMed] [Google Scholar]
- 18.Carrillo N, Davalos EA, Russak JA, Bode JW. Iterative, aqueous synthesis of β3-oligopeptides without coupling reagents. J Am Chem Soc. 2006;128:1452–1453. doi: 10.1021/ja057706j. [DOI] [PubMed] [Google Scholar]
- 19.Bode JW, Fox RM, Baucom KD. Chemoselective amide ligations by decarboxylative condensations of N-alkylhydroxylamines and α-ketoacids. Angew Chem Int Edit. 2006;45:1248–1252. doi: 10.1002/anie.200503991. [DOI] [PubMed] [Google Scholar]
- 20.Shen B, Makley DM, Johnston JN. Umpolung reactivity in amide and peptide synthesis. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valeur E, Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem Soc Rev. 2009;38:606–631. doi: 10.1039/b701677h. [DOI] [PubMed] [Google Scholar]
- 22.Fang C, Senes A, Cristian L, DeGrado WF, Hochstrasser RM. Amide vibrations are delocalized across the hydrophobic interface of a transmembrane helix dimer. Proc Natl Acad Sci USA. 2006;103:16740–16745. doi: 10.1073/pnas.0608243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponnusamy E, Jones CR, Fiat D. Synthesis of O-18 isotope labeled amino-acids and dipeptides and its effect on C-13 NMR. J Labelled Compd Rad. 1987;24:773–778. [Google Scholar]
- 24.Charette AB, Chua P. Thiolysis and hydrolysis of imino and iminium triflates: Synthesis of secondary and tertiary thioamides and O-18-labeled amides. Tetrahedron Lett. 1998;39:245–248. [Google Scholar]
- 25.Seyfried MS, Lauber BS, Luedtke NW. Multiple-turnoverisotopic labeling of Fmoc- and Boc-protected amino acids with oxygen isotopes. Org Lett. 2010;12:104–106. doi: 10.1021/ol902519g. [DOI] [PubMed] [Google Scholar]
- 26.Ponnusamy E, Fiat D, Jones CR. O-18 isotope labeling and its effect on C-13 chemical-shifts of the peptide-bond. Int J Pept Protein Res. 1986;28:542–545. doi: 10.1111/j.1399-3011.1986.tb03290.x. [DOI] [PubMed] [Google Scholar]
- 27.Yang CC, Goldberg IH. Synthesis of 1-([O-18(2)]-2-nitro-1-imidazolyl)-3-methoxy-2-propanol ([O-18(2)]-misonidazole) J Labelled Compd Rad. 1989;27:423–434. [Google Scholar]
- 28.Astolfi P, Panagiotaki M, Greci L. New insights into the reactivity of nitrogen dioxide with substituted phenols: A solvent effect. European J Org Chem. 2005;14:3052–3059. [Google Scholar]
- 29.Hochstein W, Schollkopf U. Lone pair rearrangements .13. mechanism of nitroalkane-alkyl nitrite rearrangement. Justus Liebigs Annalen der Chemie. 1978;11:1823–1834. [Google Scholar]
- 30.Hartshorn MP, Robinson WT, Wright GJ, Cheng LY. N-15-labeling study of the rearrangement of R-2,4-dichloro-T-6-hydroxy-3,6-dimethyl-2,T-5-dinitrocyclohex-3-enone to 4-chloro-C-6-hydroxy-3,6-dimethyl-R-5-nitro-cyclohex-3-ene-1,2-dione. Aust J Chem. 1989;42:1569–1578. [Google Scholar]
- 31.Amin MR, Dekker L, Hibbert DB, Ridd JH, Sandall JPB. Evidence for a reversible nitro nitrito rearrangement following ipso-attack in nitration. J Chemical Society-Chemical Communications. 1986;9:658–659. [Google Scholar]
- 32.Nguyen NV, Baum K. Preparation of 1,2-dibromodinitroethylene and 1,1-dibromodinitroethylene. Tetrahedron Lett. 1992;33:2949–2952. [Google Scholar]
- 33.Baum K, et al. Synthesis and properties of 1,2-difluorodinitroethylene. J Org Chem. 1991;56:537–539. [Google Scholar]
- 34.Bergman J, Brimert T. Synthesis and reactions of some dinitrodiazoquinones. Tetrahedron. 1999;55:5581–5592. [Google Scholar]
- 35.Ketari R, Foucaud A. Reactions of triphenylphosphine and trialkyl phosphite-silver nitrate complexes with positive halogen compounds. Synthesis of α-nitro nitriles. J Org Chem. 1981;46:4498–4501. [Google Scholar]
- 36.Tzeng D, Baum K. Reactions of hexanitroethane with alcohols. J Org Chem. 1983;48:5384–5385. [Google Scholar]
- 37.Maillard B, Ingold KU, Scaiano JC. Rate constants for the reactions of free radicals with oxygen in solution. J Am Chem Soc. 1983;105:5095–5099. [Google Scholar]
- 38.Pratt DA, Mills JH, Porter NA. Theoretical calculations of carbon-’oxygen bond dissociation enthalpies of peroxyl radicals formed in the autoxidation of lipids. J Am Chem Soc. 2003;125:5801–5810. doi: 10.1021/ja034182j. [DOI] [PubMed] [Google Scholar]
- 39.Lalevee J, Graff B, Allonas X, Fouassier JP. Aminoalkyl radicals: Direct observation and reactivity toward oxygen, 2,2,6,6-tetramethylpiperidine-N-oxyl, and methyl acrylate. J Phys Chem A. 2007;111:6991–6998. doi: 10.1021/jp071720w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.