

Abstract
Current research depicts specific modes of immunity and energy metabolism as being interrelated at the molecular, cellular, organ and organism level. Hence, whereas M2 (alternatively-activated) macrophages dominate insulin-sensitive adipose tissue in the lean, M1-skewed (classically-activated) macrophages accumulate in parallel to adiposity in the obese, and promote inflammation and insulin resistance, that is, meta-inflammation. The latest frontier of immuno-metabolism explores the coregulation of energy metabolism and immune function within hematopoietic cells. M1-skewed macrophages are sustained in edematous, hypoxic tissues by anaerobic glycolysis, whereas mitochondrial biogenesis and respiration dominates in M2 cells. We review the underlying mechanisms and the consequences of the transition from M2 to M1 predominance in adipose tissue, as well as the extracellular signals and transcription factors that control macrophage phenotypes and impose distinct metabolic modes.
Keywords: inflamation, metabolism, macrophages, insulin resistance
1. INSULIN SIGNALS SATIETY
All cells are dependent on the synthesis of ATP to perform energy-expending processes. Formation of ATP from ADP requires energy which is provided by the stepwise enzymatic catabolism of macronutrients—glucose, fatty acids (FA), and amino acids. Cells with functional mitochondria that are not deprived of oxygen can generate great amounts of ATP from the oxidation of glucose and FAs, and of amino acids in certain circumstances. In the absence of mitochondria (e.g., erythrocytes) and during ischemia/hypoxia smaller quantities of ATP are synthesized—through the glycolytic pathway and anaerobic metabolism. The intermittent nature of nutrient availability (prior to the industrial revolution) and the variation in an organism's energy expenditure (for instance, at rest as opposed to during physical exertion or infection) necessitated scenario-dependent metabolic modes of oxidation, storage and distribution of the aforementioned energy substrates [2]. Unraveling the various processes that underlie differential substrate utilization/storage on a cellular organ and whole-body level is ongoing and instrumental in the diagnosis and treatment of metabolic disorders.
The biochemical adaption to a meal—the postprandial state—is one example of a circumscribed metabolic mode. With the rise in plasma concentration of glucose that follows carbohydrate ingestion, cells appropriately switch off pathways of fatty acid, protein, and glycogen catabolism and upregulate their uptake and oxidation of glucose. Presented with a supply of glucose that surpasses immediate energetic requirements, metabolically active cells also respond by synthesizing energy stores: glycogen (by polymerizing glucose), fatty acids (de novo lipogenesis from glucose), and triglycerides (from fatty acids and glycerol). Amino acids are spared from oxidation, being preferentially utilized in the synthesis of proteins. Insulin is the major hormone responsible for orchestrating the postprandial switch in substrate utilization, storage, and prioritization [3].
Insulin is a polypeptide hormone synthesized by a subgroup of pancreatic endocrine cells that reside in the islets of Langerhans, the beta cells. In health, insulin secretion is coupled to the availability of ATP within the beta cell. As in other functioning cells, the availability of energy-providing substrates determines ATP production, and an increase in plasma glucose following ingestion of carbohydrates is the most powerful stimulus for insulin secretion. Circulating insulin binds to its cognate receptor, which is prominently expressed by highly metabolic organs/cells including muscle/myocytes, adipose tissue/adipocytes, and the liver/hepatocytes. The activated insulin receptor transduces insulin's signals via the insulin receptor substrates (IRSs) and the MAPK cascades. Most of insulin's metabolic effects are downstream to the IRSs. Serine protein kinases (e.g., Akt, atypical PKCs, mTOR) are targeted downstream to IRSs such as to modulate the activity (through enzyme phosphorylation status) and expression (through transcription factor phosphorylation) of enzymes that execute insulin's command to “utilize glucose now, build proteins and store energy for the future.” As time passes since the last meal (i.e., the postabsorptive state), the plasma concentration of glucose gradually decreases due to insulin-mediated uptake by muscle and adipose tissue. In response, beta cells secrete less insulin. Nevertheless, insulin continues to regulate metabolism at low, postabsorptive concentrations: it inhibits glucose production by the liver (gluconeogenesis) and the release of free (nonesterified) FAs from adipose tissue (lipolysis) until glucose and insulin concentrations are very low.
2. MACROPHAGE VERSATILITY
Resident and infiltrating macrophages perform an impressive repertoire of functions, in health and in response to injury and infection. Among other roles, macrophages execute phagocytosis and killing of an array of infectious microorganisms, phagocytose tissue debris, apoptotic parenchymal cells and apoptotic neutrophils, orchestrate the repair of wounded tissue, and serve as a bridge between the innate and adaptive arms of immunity. In accordance with their diverse activities, macrophages acquire differential phenotypes, dictated by the form, stage and site of insult [4, 5]. To a certain extent, the phenotypic plasticity of macrophages parallels and cooperates with that of CD4+ T-lymphocytes, which become polarized from a naïve state towards a particular mode of immunity [5].
Macrophage polarization was broadly divided to two opposing phenotypes following the lymphocyte terminology (Th1 and Th2 subsets) and consisting of M1 and M2 (later subdivided to M2a-c) macrophages [6, 7]. Briefly, the M1 designation refers to the “classically-activated” macrophage that emerges during cell-mediated immune responses. Interferon-γ (IFN-γ) and lipopolysaccharide (LPS) prime macrophages to acquire the microbicidal and proinflammatory properties that characterize the M1 phenotype. Macrophage receptors for cytokines and LPS signal through IKK and AP-1 to induce the expression of classic inflammatory mediators, including tumor necrosis factor-α (TNF-α), chemokine (C-C motif) ligand 2 (CCL2/MCP-1), and inducible nitric oxide synthase (iNOS) [8]. M1 macrophages are the first line of defense against intracellular pathogens and they stimulate Th1 polarization of CD4+ lymphocytes. On the other hand, M1 cells can harm bystander parenchymal and immune cells through the generation of microbicidal agents (reactive oxygen species (ROS), proteases, etc.) and further stimulation of other leukocytes. M2 or alternatively-activated macrophages are the counterparts of Th2 lymphocytes and participate in immunity against extracellular parasites, helminthes, and so forth. M2 cells display hyporeactivity to M1-type ligands, such as LPS, and are considered to be anti-inflammatory. Macrophages convert to an M2 phenotype at the later stages of an M1 response. This phenotype switch can take place following the engulfment of apoptotic neutrophils (efferocytosis) as well as other signals [9]. As a result, macrophages initially downregulate their proinflammatory activity and shift their function towards local resolution of inflammation and tissue repair. Activation of macrophage STAT 6 by IL-4 and -13 via their surface receptors, as well as ligand-mediated activation of peroxisome proliferator activating receptors (PPARs) are key signaling pathways mediating M2 skewing and M1 inhibition. Most recently, STAT6 as well as PPARγ and δ were shown to cooperate in regulating the gene transcription pattern specific to M2 [10].
The resolution of inflammation is a regulated series of events. For instance, specialized proresolving lipid mediators (SPM), such as the lipoxins, resolvins, protectins, and maresin, are produced from the same source as the proinflammatory eicosanoids (i.e., omega-3 and omega-6 polyunsaturated fatty acids) by 12/15-lipoxygenase (12/15-LO) to directly promote the termination of inflammation and return to homeostasis. Prostanoids and leukotrienes are synthesized from long-chain polyunsaturated fatty acids early in inflammation, following cleavage of the latter from membrane phospholipids by cPLA2. Recent evidence suggests that the omega-3-type SPMs are derived not from phospholipid fatty acids, but rather from free (nonesterified), omega-3 polyunsaturated fatty acids that transudates from the circulation into edematous tissues while bound to albumin [11].
Macrophages are instrumental in both mediating and responding to the resolution of inflammation, as embodied by the recently described resolution-phase, CD11blow macrophages, now termed Mres (Ariel et al., Front. Immunol. 2011). Recent studies [12–14] have indicated that these macrophages convert from the M2 to the new Mres phenotype in molecular and functional terms. Moreover, these studies highlighted the unique characteristics of this macrophage phenotype and hypothesized that its emergence would prevent exacerbated outcomes of acute and chronic inflammation ([14] and Ariel et al., Front. Immunol. 2011).
3. INSULIN RESISTANCE
Too often, metabolic organs display a pathologic reduction in their responsiveness to insulin, that is, insulin resistance (IR). In such a scenario, muscle and adipose tissue uptake less glucose from the blood after a meal, and hepatocytes inappropriately generate glucose through gluconeogenesis and glycogenolysis. This manifests as postprandial and fasting hyperglycemia, the defining features of IR and Diabetes Mellitus (DM). Whereas hyperglycemia is the most infamous consequence of IR, lipid and protein metabolism is also perturbed, and tissues other than muscle, liver and fat also fail to respond to insulin [15]. Adipocyte IR manifests as excessive release of free (nonesterified) fatty acids (FFAs). IR is detrimental because it results in elevated glucose and FFA concentrations, and also due to impaired function of other insulin-sensitive organs (e.g., endothelial dysfunction secondary to impaired insulin-induced vasodilation). Many acute and chronic disease states are accompanied by IR, including serious infection and trauma, obesity, auto-immune and endocrine disorders, and treatment with certain drugs [16, 17]. Type II diabetes mellitus (DM-II) occurs when beta cells fail to compensate for chronic IR by upregulating the secretion of insulin. The incidence of DM-II is increasing as a result of the obesity epidemic. In addition, a significant proportion of the population displays characteristics of the metabolic syndrome and/or prediabetic states; IR is the underlying pathophysiology in these conditions [16]. Thus, IR is a highly prevalent condition worldwide and across age groups. The clinical consequences of long-standing IR include atherosclerosis, complications of DM and certain cancers, underscoring the relevance of the condition.
The failure of the insulin receptor to propagate its signal despite the interaction with insulin (i.e., IR) is a multifactorial phenomenon. Most commonly, IR is instigated by nutrient overload and/or by inflammation. Proinflammatory cytokines (IL-1β, TNF-α and in certain scenarios, IL-6) and ROS are over-produced in the context of the inflammatory response and activate several serine kinases, particularly IKK and JNK. Phosphorylation of particular IRS serine moieties by the aforementioned kinases precludes IRS tyrosine phosphorylation by the Insulin Receptor's inducible kinase activity [18]. Hence, IRS-dependent signaling is attenuated. Indeed, TNF-α inhibition attenuates inflammation and improves insulin sensitivity in rodent models and patients with rheumatoid arthritis [19]. Serine phosphorylation of IRSs is the generic biochemical inducer of IR across cell types, although cell-specific variations occur when particular serine kinase(s) isoform(s) are involved. Other non-mutually-exclusive mechanisms that underlie defective insulin signaling are activation of SOCS3, an excess of “counterregulatory” mediators that oppose insulin action (e.g., glucagon, epinephrine, cortisol), increased de novo synthesis of DAG and ceramide from excess FAs—which activates Ca++-independent, novel PKC isoforms, and ER stress [15, 20–22].
Obesity, the most common cause of IR, is accompanied by a low-grade, chronic inflammatory response in the circulation and within metabolic organs. Proinflammatory cytokines secreted by recruited and resident macrophages in adipose tissue, the liver, and skeletal muscle are a pivotal link between inflammation and abrogation of insulin signaling in adjacent metabolic cells. The coincidence of weight gain, a chronic, low-grade inflammatory response and IR reflects a causal association and is referred to as meta-inflammation [21]. Adipose tissue has been the focus of meta-inflammation, as described in the following section. The involvement of resident and recruited macrophages in hepatic [23] and skeletal muscle IR is reviewed elsewhere [24].
4. M2 To M1 PHENOTYPE SWITCH IN INSULIN RESISTANCE
Emerging evidence links macrophages phenotype to the degree of organ-specific and whole-body insulin sensitivity, under normal conditions and in disease. In the lean state, the small numbers of macrophages populating adipose tissue are of the M2 phenotype and are associated with tissue-specific and whole body insulin sensitivity. Obesity, in contrast, is associated with accumulation of M1-type macrophages. The M1-to-M2 ratio is inversely related to tissue-specific and whole body insulin sensitivity [25, 26].
Adipocytes are the functional parenchymal cells of adipose tissue. Adipocytes esterify FAs and glycerol to form triglycerides, which are stored in lipid droplets so large that they compress the nucleus and cytoplasm to the periphery. Adipocytes that reside in white adipose tissue (WAT) are dedicated to the storage of FAs when energy substrates are abundant (in insulin-sensitive mammals), and to the release of FFAs to the bloodstream at times of scarcity or when energy requirements are increased. WAT also displays endocrine functions and secretes a battery of adipokines that differentially regulate metabolism, congruent with the body's immediate energy requirements and substrate availability. WAT function is partially determined by its location: subcutaneous and visceral WAT handles FAs and secretes adipokines in a differential manner [27].
Obesity results from a prolonged positive energy balance, that is, an ongoing calorie intake that significantly exceeds energy expenditure. Weight gain in obesity is primarily a consequence of the accumulation of FAs—from the diet or from de novo lipogenesis—stored as triglycerides in WAT. In humans, WAT responds to an excessive supply of fatty acids through hypertrophy—predominantly an increase in lipid droplet size—and to a lesser extent through differentiation of preadipocytes into mature adipocytes (i.e., hyperplasia [28]). The essential role played by adipocytes in metabolic homeostasis is underscored by certain human and rodent lipodystrophies, in which the scarcity of functional WAT culminates in IR, even in the absence of overt obesity [29]. In diet-induced obesity itself, accrual of triglycerides per se is a relatively benign process. However, adipocytes' enzymatic and structural capacity to store FAs is eventually surpassed by an ongoing surplus of calories [30]. In addition, the response of the adipose vasculature and stroma to adipose expansion is often insufficient, leading to hypoxia and overcrowding. As a result of the failure to store excess FAs, the concentration of FFAs (particularly long-chain saturated FAs) and of lipids synthesized from FAs (e.g., diacylglycerol and ceramide) increases to a harmful level. This perturbs adipocyte integrity and function and may lead to apoptosis and necrosis [27, 31, 32]. Lipolysis is no longer inhibited (due to IR) and adipocytes are unable to reesterify FAs, resulting in the spill-over of FFAs into the circulation. Excess circulating free saturated FAs (FSatFAs) are harmful to the vasculature and also undergo ectopic deposition in numerous organs, including liver and muscle. Insulin resistance and lipotoxicity ensue, as the capacity of hepatocytes and myocytes to store triglycerides, is only a fraction of that of adipocytes. Obesity is also associated with differential changes in adipokine secretion, such as a decrease in the insulin-sensitizing and anti-inflammatory adiponectin, and an increase in leptin [22, 27, 32].
In addition to the metabolic and endocrine perturbations that parallel adipose expansion, AT is gradually transformed into an inflamed organ, increasingly populated by proinflammatory myeloid- and lymphoid-derived cells. Effectors of both innate and adaptive immunity are implicated in maintaining an anti-inflammatory environment in the lean, as well as in the coupling of obesity to inflammation, and of the latter to IR [21, 24, 31, 33]. While this paper focuses on M1- and M2-type macrophages in IR, other immune cells that influence an organs' responsiveness to insulin include CD4+ lymphocytes of the Th1, Th2, and Treg phenotypes [34–38] and CD8+ T-cells [39]. Of relevance to our discussion, lymphocytes influence insulin sensitivity in part by differentially modulating the phenotype of macrophages that populate the organ, and macrophage-lymphocyte cross-talk within metabolic tissues may substantiate or negate obesity-associated inflammation and IR [24]. Infiltrating eosinophils [38] also contribute to adipocyte insulin sensitivity in the lean, whereas mast cell accrual is implicated in adipocyte IR in obesity [40]. The coincidence of increased adiposity, AT M1 macrophage accrual and IR, in concert with experiments in which the modulation of AT macrophage recruitment/activation influences adipose and whole-body insulin sensitivity indicates a causal link between AT inflammation and IR [21, 24].
In lean, healthy mammals, macrophages account for approximately 10% of AT cellularity and are interspersed between adipocytes [41]. These cells are of the alternative phenotype, that is, they express CD206, CD301a, and arginase-1 [42, 43]. M2 macrophages may indirectly contribute to adipocyte insulin sensitivity by inhibiting the recruitment and polarization of their M1 counterparts, for example, through IL-10 and IL-1RA (IL-1 receptor antagonist) production [43]. In addition, and in accord with their established role in tissue remodeling, M2 macrophages may facilitate AT expansion in adaption to an increased FA supply. For instance, M2 cells may regulate vascular and stromal modifications to sustain adipocyte hypertrophy and hyperplasia [44] and secrete factors that promote preadipocyte differentiation [33]. There is some evidence in support of the intriguing hypothesis that M2 macrophages directly enhance insulin signaling in adipocytes [25, 45], although the factor(s) that mediate this modulation remain to be identified. The lipolysis that appropriately occurs during weight loss is accompanied by a mild, transient influx of monocytes that mature into M2-type macrophages, which appear to counter excessive lipolysis [45]. The AT chemo-attractant(s) and M2-derived factors that counter lipolysis of weight loss are not clear [46].
An emerging function of AT M2 cells is the clearance of FFAs released from adjacent adipocytes: M2 macrophages upregulate their expression of proteins involved in the uptake (e.g., CD36, a fatty acid translocase and scavenger receptor; [47]), esterification (e.g., DGAT [48]), and oxidation (e.g., LCAD [1]) of FAs. It may be that AT M2 cells thereby extract, catabolize, and/or store free FAs released by adipocytes as neutral triglycerides, while avoiding the formation of large lipid droplets and lipotoxicity that occurs in M1 cells in obesity [49]. A reduction in free FA concentration would attenuate IR and inflammation. Red Eagle and Chawla recently postulated that in weight loss, M2 cells recruited to lipolytic adipocytes take up large quantities of FFAs and transport them as intracellular, neutral lipids to the liver—a process akin to the reverse transport of cholesterol by macrophages—rather than expose the portal and systemic circulation to elevated levels of FFAs [46]. Further study may determine whether AT M2 cells transport FAs in this manner to a quantitatively-substantial degree, in weight loss and/or maintenance of a lean body mass.
In a fascinating experiment, Wu et al. [38] infected mice fed a high-fat diet with a gastrointestinal helminth (Nippostrongylus brasiliensis), which elicits a Th2-type response. Infection resulted in less AT inflammation and attenuation of IR. Parasite infection is uncommon in the industrialized world—due to sanitation and the eradication of malnutrition- and is unlikely to mediate M2 induction in lean AT. In fact, the rarity of parasite infections has putatively contributed to the increase in immune-hypersensitivity diseases (the hygiene hypothesis), possibly extending to obesity-induced inflammation and IR [50].
M2 skewing of AT macrophages in lean rodents maintaining their weight is attributed to the local secretion of the Th2 cytokines, IL-4 and IL-13, albeit at concentrations lower than those accompanying overt parasitic infection (Figure 1). The cellular source of these interleukins appears to be lean, “un-stressed” adipocytes [25], recruited eosinophils [38], and unique “innate Th2 lymphocytes” [35]. AT IL-10 production may also contribute to M2 recruitment and polarization [51]. Interestingly, insulin stimulates in vitro Th2 skewing of CD4+ lymphocytes—including a switch from IFN-γ to IL-4 secretion—possibly through ERK phosphorylation [52]. It is, therefore, tempting to hypothesize that in the lean and insulin sensitive state, insulin promotes triglyceride storage (rather than FFA release) in a dual manner: directly, through inhibition of adipocyte lipases and indirectly, by maintaining adjacent macrophages in the alternative mode.
Figure 1.
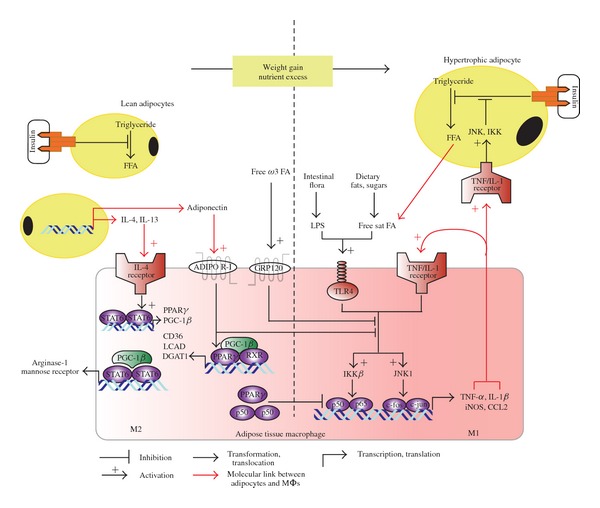
Interdependence of adipocyte and macrophage functional modes in the lean and obese states. The left side of the diagram depicts the insulin-sensitive adipocyte and M2-type adipose tissue macrophage characteristic of adipose tissue in the lean state. Adipocytes are of a normal size and are sensitive to insulin's signal to minimize the cleavage of fatty acids from triglyceride stores (lipolysis). Consequently, local and circulating concentrations of FFA are maintained below harmful levels. Healthy adipocytes (as well as other cell types not shown here) are a source of M2-inducing cytokines—IL-4 and IL-13, which signal through STAT6 phosphorylation to induce transcription of PPARs, their coactivator, PGC-1β, and 12/15-LO. PGC-1β promotes the assembly of the STAT6 transcription complex, thereby amplifying the expression of signature M2 proteins, such as arginase-1 and the pattern recognition/endocytic receptor CD206. Induction of 12/15-LO further enhances the M2 phenotype and insulin sensitivity by synthesizing 15-HETE (from arachidonic acid), 17S-HDHA, and RvD1 (the latter two from DHA). RvD1 is a specialized proresolution mediator and appears to act on its cognate receptor (not shown) expressed on macrophages and adipocytes to promote the M2 phenotype and to induce adiponectin secretion, respectively. Indeed, healthy adipocytes produce and secrete greater amounts of adiponectin than in the obese, and this adipokine exerts insulin sensitizing and anti-inflammatory effects, locally and systemically. Adiponectin signaling inhibits M1-skewing, enhances PPAR-γ activity, and increases oxidative metabolism in adipose tissue macrophages. The ligand binding domain of PPAR-γ recognizes oxygenated metabolites of polyunsaturated fatty acids which are consumed in the context of a healthful diet, including 15-HETE and 17S-HDHA. Activated PPAR-γ inhibits NF-κB-dependent gene expression. It also forms a heterodimer with RXR to induce proteins involved in the uptake (e.g., CD36), esterification (e.g., DGAT), and oxidation (e.g., LCAD) of FFAs released from adjacent adipocytes. Consequently, the concentration of FFAs does not reach toxic levels. Free, extracellular DHA contributes to M2-skewing by activating a novel surface receptor (GRP120). During weight gain, adipocytes undergo hypertrophy in an attempt to store excess FAs as neutral lipids, that is, triglyceride. Unfortunately, the excess supply of FAs eventually surpasses the capacity of adipocytes and adipose tissue to store lipids, leading to an increase in FFA concentrations. The reduced sensitivity to insulin that accompanies obesity also increases FA release by allowing lipolysis of triglycerides. In conjunction with LPS derived from the abnormal intestinal flora that populates the obese, and with M1/Th1-type cytokines, free saturated FAs activate macrophage IKK and JNK1, thereby inducing the M1 immune program. M1 macrophages produce IL-1β and TNFα, which increase and activate adipocyte IKK and JNK to block insulin signaling. As a result, the number of M1 macrophages parallels the expansion of adipose tissue, exacerbating inflammation and insulin resistance.
The intracellular pathways leading to the alternative activation of macrophages are gradually being deciphered. Signaling through the IL-4 receptor-associated Janus kinases, IL-4 and IL-13 mediate the rapid tyrosine phosphorylation of STAT6. Phosphorylated STAT6 dimerizes and translocates to the nucleus where it transactivates the expression of the lipid-sensing nuclear factors PPAR-γ and PPAR-δ, and their coactivator, the PPAR-γ coactivator-1beta (PGC-1β). Interestingly, PGC-1β serves as a coactivator for the STAT6 dimer as well, thereby amplifying the expression of mouse M2-defining molecules, such as CD206 and arginase-1 [10, 33, 53].
The PPARs form heterodimers with RXR (retinoid X receptor) and upon binding a lipid or synthetic ligand they mediate gene expression through trans-activation. In general and in the context of the lean AT macrophage, PPARs are instrumental in determining the metabolic mode of M2 macrophages, and in inhibiting the M1 phenotype [26, 53]. Manipulation of PPAR-γ activity in macrophages provides evidence of its link to AT M2 skewing and insulin sensitivity. For example, thiazolidinediones are synthetic PPAR-γ agonists that attenuate IR in patients. This has been attributed to the PPAR-γ-dependent maturation of subcutaneous adipocytes, which are associated with greater insulin sensitivity than visceral adipocytes [27]. Notwithstanding the beneficial redistribution of adipose tissue, it appears that macrophage PPAR-γ is also necessary for the thiazolidinediones' insulin-sensitizing effect [54]. Indeed, administration of a thiazolidinedione attenuated the AT M1-to-M2 ratio, in conjunction with improved insulin sensitivity [51, 55]. In contrast, disruption of PPAR-γ in myeloid cells impairs whole-body and AT M2 maturation and activation, and predisposes mice to development of diet-induced obesity and IR [55]. A fraction of activated PPAR-γ undergoes SUMOylation, without forming a heterodimer, and trans-represses the p50–p65 (NF-κB) heterodimer [56]—an M1-dictating transcription factor. In contrast to p50–p65, the p50–p50 homodimer—induced, for example, by IL-10, promotes M2 skewing and M1 inhibition [57].
PPAR-γ binds polyunsaturated fatty acids that have been transformed by lipoxygenases and may also be activated by the adipokine, adiponectin. Adiponectin is unique among the adipokines in that it exerts insulin-sensitizing and anti-inflammatory effects, and in that its secretion is gradually reduced as adipose tissue expands. In the lean state, adiponectin binds to its two cognate receptors expressed by metabolically active cells, as well as monocytes and macrophages. As a result of signaling through PPAR-γ and AMP-dependent kinase (AMPK) stimulation, adiponectin inhibits the M1 and promotes the M2 phenotype [58–60]. Adiponectin also induces ceramidase in macrophages, thereby attenuating ceramide's proinflammatory effects [61]. Adiponectin's M2-skewing and M1-opposing properties and the inverse correlation between adiposity and adiponectin concentrations constitute an important link between body weight, macrophage phenotype, and insulin responsiveness.
Whereas a diet rich in saturated fatty acids is detrimental to cardiometabolic health, consumption of omega-3 polyunsaturated fatty acids (PUFAs: e.g., EPA and DHA, which are present in significant amounts in oily fish) is inversely associated with cardiovascular and metabolic disease [62]. Indeed, omega-3 PUFAs are essential nutrients, in virtue of their oxygenated metabolites that potently modulate multiple cellular processes. EPA and DHA undergo enzymatic transformation by 12/15-LO and other enzymes in the production of SPMs (such as Resolvin E1, Resolvin D1, and Protectin D1) and of intermediate metabolites that are potent PPAR ligands [23, 63]. IL-4 and IL-13 induce 12/15-LO expression in murine and human monocytes/macrophages [64–68] thereby promoting the production of SPM and PPAR-ligands from DHA. Emerging evidence suggests that metabolism of long-chain omega-3 PUFAs by the 12/15-LO pathway provides mediators that support M2-skewing of AT macrophages and stimulate adiponectin production. Interestingly, these mediators also attenuate hepatic steatosis (excessive accumulation of triglycerides in hepatocytes) and Kupffer cell activation in nonalcoholic fatty liver disease, the hepatic component of the metabolic syndrome [23, 69]. Specifically, DHA administration attenuated inflammation [70] in AT and the liver of obese rodents and reduced hepatic accumulation of triglycerides [71, 72]. AT concentrations of DHA-derived SPMs, RvD1 and PD1, and of 17S-HDHA (an intermediate metabolite in the 12/15-LO-dependent synthesis of SPMs) were increased by DHA ingestion, and 17S-HDHA was shown to potently activate PPAR-γ and mimic the therapeutic effects of rosiglitazone. A study in mice that are transgenic for fat-1, an enzyme that converts omega-6 to omega-3 PUFAs, confirmed the benefit conferred by omega-3 PUFAs in obesity-induced AT perturbations. These mice displayed less obesity-induced IR and AT M1 accumulation than their wild-type controls [73].
Obese adults treated with omega-3 fatty acids display higher adiponectin concentrations [74–76], plausibly mediated in part by metabolism to RvD1 and RvE1, as administration of these SPMs increases adiponectin secretion in obese rodents [72, 77]. RvD1, at the nanomolar range, also reduced the M1-to-M2 ratio in AT, at least in part through adiponectin induction. Interestingly, DHA and RvD1 also promoted the emergence of proresolving macrophages [14, 78]. It was recently confirmed that RvD1 enhanced nonphlogistic phagocytosis and M2 marker expression by macrophages residing in the stroma-vascular compartment of obese AT [70]. Furthermore, intraperitoneal administration of DHA induced the emergence of a low-expressing CD11b/F4/80 subset of macrophages in the adipose tissue of obese mice [70]. This suggests that the Mres-like macrophages that emerge during resolution of zymosan-induced inflammation [14] may also be induced therapeutically in the context of obesity-induced inflammation. The beneficial impact of DHA at high concentrations on M2 skewing and M1 inhibition of AT macrophages—and as a result on AT function—is also mediated through direct stimulation of the macrophage cell-surface receptor, GPR120 [79]. Two recent reviews have addressed the pleiotropic effects of omega-3 FA consumption and conversion to SPMs in ameliorating inflammation and insulin resistance in adipose tissue and the liver [23, 69]. A clinical trial (NCT00760760) is presently assessing whether a similar beneficial response occurs (i.e., a reduction in AT inflammation) in morbidly obese adults ingesting fish oil capsules.
In summary, in the lean and the treated obese, several exogenous and endogenous factors target intersecting pathways in AT macrophages to inhibit classic activation and to induce M2 polarization (Figure 1). There is ample evidence the M2 phenotype is causatively associated with adipocyte insulin sensitivity.
In contrast to the lean state and to weight loss, obesity is accompanied by a robust influx of monocytes [80] which transform into M1 macrophages that induce adipocyte IR [81], recruit additional immune cells, and exacerbate inflammation [82]. In addition to secreting cytokines and reactive oxygen species (ROS) that antagonize insulin signaling, proinflammatory macrophages appear to inhibit adipogenesis through presentation of Wnt5a, thereby curtailing the adaptive increase in cellular depots for FAs [83]. The magnitude of AT macrophage accumulation during weight gain is remarkable, reaching 40% of AT cellularity. These staggering statistics exemplify the concept of an inflamed adipose tissue. In contrast to macrophages recruited to the AT in weight loss, the macrophages detected in the obese continue to reside in expanded subcutaneous and visceral WAT and maintain their proinflammatory status unless weight loss is achieved [84, 85] and/or physical activity is increased [85].
The inflamed adipose tissue of obesity resembles an organ chronically infected by an intracellular organism, despite the absence of any pathogen. This raises the question of the identity of cells and molecules that attract monocytes to AT and impose the transformation to M1 macrophages. In 1993, Hotamisligil et al. [86] demonstrated that the proinflammatory cytokine TNF-α was expressed by AT of obese, but not lean rodents, and that TNF-α induced adipocyte IR through serine phosphorylation of IRSs by IKK. TNF-α also stimulates the NH2-terminal kinase (JNK) pathway, a stress kinase that normally phosphorylates the c-Jun component of the activator protein 1 (AP-1; c-jun-c-fos). JNK also phosphorylates serine residues on the IRS, thereby inhibiting insulin signaling in adipocytes in concert with IKK (Figure 1 and [18]). These data and subsequent studies led to a paradigm switch in the perception of AT and the adipocyte as a tissue/cell of limited biologic repertoire beyond FA storage and release. In addition to the prototypical M1-inducing cytokine, TNF-α, the chemokine CCL2 and its cognate receptor, CCR2 are also upregulated in the adipose tissue of obese subjects and rodents [41, 87–89]. Indeed, selective deletion of CCR2 in myeloid cells reduced the M1 prevalence of AT macrophages in the obese [89, 90].
As regards the cellular source of AT cytokines and chemokines, there is some evidence that hypertrophic, FFA-stimulated, and hypoxic adipocytes themselves are a source of these cytokines and chemokines [89]. In addition, adipocyte apoptosis may attract macrophages [26]. Whether this could be quantitatively accountable for monocyte influx is not clear and many authors believe that neither adipocyte apoptosis nor secretion of cytokines/chemokines by adipocytes can fully account for macrophage influx and M1 specialization [91].
Adipocyte necrosis is increased in the context of obesity. The observation that the vast majority of infiltrating macrophages encircle necrotic adipocytes to form crown-like structures in obese rodents and humans highly suggests that necrosis-associated factors chemo-attract monocytes [92, 93]. As previously mentioned, monocytes acquire an M1 phenotype and secrete cytokines and ROS that interfere with insulin signaling in adjacent adipocytes. As a result, lipolysis of triglycerides is not inhibited by insulin. In turn, excessive release of FFA targets macrophage signaling to promote the proinflammatory output (as detailed below). The paracrine cross-talk between adipocytes and macrophages thereby evolves into a partially self-sustaining cycle of “M1-driven inflammation → adipocyte IR → increased FFA → M1-driven inflammation” that serves also to recruit more monocytes (see Figure 1 and discussion below) [94]. M1 macrophages themselves are probably the major source of AT cytokines and chemokines in established obesity.
In infection, the activation of the TLR4 is central to the development of the M1 phenotype. Similar to other TLRs, TLR4 detects specific PARPs (pathogen-associated recognition patterns). Macrophage TLR4 preferentially ligates LPS released from the outer membrane of Gram-negative bacteria (GNB). TLR4 signaling elicits ROS generation and nuclear translocation of canonical NF-κB, the p50–p65 heterodimer. Indeed, GNB septicemia [17] and systemic administration of LPS [95, 96] induce an insulin-resistant state. Furthermore, LPS infusion impaired insulin signaling and induced proinflammatory cytokine and chemokine expression in human subcutaneous AT [95, 96]. These findings suggest that LPS may mediate M1 activation in obese AT. Whereas Gram-negative bacteremia is incapacitating and cannot account for IR in the general population, emerging evidence now depicts obesity and DM as states of “metabolic endotoxemia,” that is, mildly elevated concentrations/activity of flora-derived LPS detectable in the circulation. The mechanisms underlying the mild endotoxemia include enhanced intestinal translocation of LPS from intestinal microbes [97] and impaired LPS neutralization [98]. Indeed, the vast population of microorganisms residing in the intestine is emerging as a major regulator of gastrointestinal, metabolic, enteroendocrine, and immune function [50]. Obesity and DM are accompanied by an increase in LPS-containing microbiota in the gut [99]. This impairs epithelial barrier function, precipitating the translocation of LPS, macrophage infiltration of AT, and induction of adipose IR in a CD14-dependent manner [100, 101]. A high-fat meal also facilitates absorption of intestinal LPS and promotes low-grade systemic [102] and AT [101, 103]. The intestinal flora can be safely manipulated through intake of probiotics and prebiotics such as to augment insulin sensitivity [104]. We hypothesize that certain probiotic strains may ameliorate the increased M1-to-M2 ratio of AT macrophages, thereby attenuating IR.
In addition to LPS, FSatFAs (particularly lauric (C12:0), myristic (C14:0), and palmitic acids (C16:0)) are ligands for TLR4 and TLR2. FSatFAs (but not free polyunsaturated FAs) activate these receptors in monocytes, eliciting NF-κB-responsive gene transcripts in an MyD88-dependent manner [105–107]. The detection of an endogenous ligand (FSatFA) by the macrophage pattern recognition receptors (PRR), TLR4/TLR2, is in accordance with the role of macrophages (and other sentinels of the immune system) in recognizing “danger signals” even in the absence of “nonself” entities [108]. A plausible mechanistic explanation for the ability of TLR4/2 to be activated by LPS and by FSatFA is that the latter mimics the Lipid A moiety of Gram-negative bacteria (e.g., E. coli and S. typhimurium) LPS, which contains several modules of lauric and myristic acid [109]. Of note, the stimulatory effect of FSatFAs on TLR4 is not limited to immune cells. TLR4 is constitutively expressed by a diverse range of cells, including epithelial and endothelial cells, hepatocytes, myocytes, and adipocytes. Palmitic acid (C16:0), for example, stimulated IKK and induced IL-6 and TNF-α expression in both adipocytes and endothelial cells in a TLR4-dependent fashion. Also of interest is that the long-chain omega-3 FA, DHA blocks the activation of TLR4/2 by LPS or FSatFA, at least in vitro [105].
The plasma concentration of FSatFAs is typically elevated in diet-induced obesity, due to increased intake, de novo lipogenesis (of palmitic acid), and resistance to insulin's anti-lipolytic effect. Infusion of lipids (triglyceride emulsion) and heparin (which activates lipoprotein lipase) elevates FFA concentrations and induces IR in healthy adults [110]. This experimental model recapitulates the FSatFA-induced IR of obesity, but does not directly implicate TLR4 activation, since FSatFAs may interfere with insulin signaling through TLR4-independent mechanisms, such as an increase in DAG and induction of Endoplasmatic Stress [20, 22]. The evidence implicating TLR4 in FSatFA-induced IR arises from studies in Tlr4−/− mice and cells, demonstrating protection from obesity-induced AT inflammation and IR compared with their wild-type counterparts [111, 112]. One may still argue that FSatFA-mediated adipose IR and inflammation is elicited via adipocyte, rather than macrophage TLR4 signaling. The confirmation that FSatFAs drive M1-skewing in obese AT was provided by demonstrating that Tlr4 knockdown confined to hematopoietic cells attenuates the increase in AT M1 versus M2 markers [113] and the detection of a subpopulation of AT-infiltrating macrophages that are classically activated by FSatFAs in obesity [114].
Interestingly, FSatFAs uptake into classically activated macrophages may be increased in IR, and this appears to be associated with impaired insulin signaling in macrophages (which express the insulin receptor) and elevated expression of CD36, compared with the insulin-sensitive state [115]. It appears that whereas AT M2 cells (that also express increased levels of CD36) are equipped with enzymes that store or consume SatFAs, M1 cells preferentially depend on glycolysis for energy. As a result, M1 cells in obese AT accumulate large lipid droplets and are exposed to the deleterious effects of excess saturated FAs [49]. A recent study demonstrated that upon uptake, SatFAs serve as a substrate for ceramide synthesis in macrophages [116], a fate shared by metabolic organs exposed to nutrient excess [20, 22]. In the absence of sufficient adiponectin-driven ceramidase activity [61], ceramide activates the Nlrp3 inflammasome, which induces caspase-1-mediated processing and activation of IL-1α [116]. In addition, deletion of CD36 in hematopoietic-derived cells alone was suffice to attenuate adipose IR and macrophage accumulation that results from consuming a high-fat diet [117].
In short, saturated FAs classically activate AT macrophages through activation of PRR directly, through activation of cell-surface TLRs and indirectly, through uptake and conversion to ceramide, which activates the intracellular NLRP3.
Proinflammatory cytokines (TNF-α, IL-1β) and TLRs signal through IKK and JNK. The latter kinases induce IR by serine phosphorylation of IRSs and by activating their downstream proinflammatory transcription factors, NF-κB and AP-1. Cell-specific knockout experiments have established that activation of IKKβ and JNK1 in monocyte macrophages is sufficient to induce obesity-induced IR and inflammation in metabolic organs, including adipose tissue [118, 119]. These findings further corroborate the centrality of M1 signaling in obesity-induced inflammation, and IR.
Activation of M1 cells by proinflammatory cytokines and FSatFAs has been discussed in detail (summarized in part in Figure 1). In infection, IFN-γ secreted across the immune synapse forged by Th1-polarized CD4+ lymphocytes and antigen-presenting macrophages contributes directly to M1 skewing. Several studies not elaborated on here have demonstrated that IFN-γ-secreting, Th1 lymphocytes participate in M1 specialization, inflammation and IR in obese AT [34, 36].
An important caveat to the M1-M2 dichotomy is that in vivo, macrophages generally acquire a mixed phenotype along the M1-M2 axis [4, 5]. This also applies to macrophages that increasingly populate expanding adipose. For instance, an increase in CD11c+ macrophages displaying a mixture of M1 (e.g., exhibiting enhanced IL-1β expression) and M2 (demonstrating enhanced arignase-1 and suppressed iNOS expression) characteristics was observed in AT from mice fed a high-fat diet [120]. Other studies have also demonstrated the presence of various mixed phenotypes in obese AT and have called into question the strict depiction of M1 cells as increasing in number and inducing IR and of M2 cells as insulin sensitizers but dwindling in number as AT expands [44, 120]. Clearly some of the discrepancies may be attributed to the different diets (e.g., 30% versus 60% fat), mouse species, and methods employed in the many studies investigating this issue. Indeed, recent expert opinion maintains that the M1/M2 paradigm is a reasonable starting point to conceptualize and investigate macrophages' involvement in AT function. Nevertheless, a more accurate depiction necessarily includes several mixed phenotypes associated with human and rodent obesity [33].
5. IMMUNO-METABOLISM: METABOLIC MODES OF M1, M2, AND Mres CELLS
Historically, immunity and energy metabolism are considered to be distinct capabilities, subserved by different cell-types and differentially regulated to respond to an array of departures from homeostasis. More recently, specific modes of immunity and energy metabolism are being interrelated at the molecular, cellular, organ and organism level. The coupling of AT responsiveness to insulin to the phenotype of local macrophages is a striking example of immune and metabolic cross-talk. The exciting interdisciplinary field of immuno-metabolism focuses on the coregulation of hematopoietic cell types' specific immune function, and their mode of ATP generation [121]. It appears that in the course of the inflammatory response, macrophages switch their metabolic mode in conjunction and in cooperation with the timely transition from the M1 to the M2, and eventually to the Mres phenotype.
The dependence of neutrophils and macrophages on glycolysis, rather than mitochondrial ATP production, even in normoxia, was first demonstrated close to a century ago [122]. Since then, the hypoxia-inducible factor (HIF-1) transcriptional complex has emerged as a critical regulator of cellular and systemic responses to low oxygen levels. Oxygen tension is low in inflamed, edematous tissues, curtailing oxidative metabolism that could provide ATP for the energy-requiring functions of phagocytes recruited to a site of infection: migration, protein synthesis, and so forth. Hypoxia allows the HIF-1α subunit to be translocated to the nucleus, where it forms a dimer with HIF-1β. The HIF heterodimer binds hypoxic-response elements in the promoter region of genes that promote phagocyte energy generation, inflammatory and bactericidal activities, and survival. The HIF-regulated genes function together to orchestrate the metabolic shift to anaerobic glycolysis: increased insulin-independent uptake of glucose (GLUT-1), glycolysis to pyruvate (e.g., M-type pyruvate kinase), and pyruvate conversion to lactate (lactate dehydrogenase) rather than its entry into the Krebs cycle (pyruvate dehydrogenase (PDH) kinase 1; PDK1) [122]. HIF-1α is intimately associated with classic macrophage activation: TLR signaling to NF-κB upregulates macrophage HIF-1α expression [123], HIF-1α induces the synthesis of M1-type cytokines and mediators [124], and the levels of HIF-1α increase during the differentiation of blood monocytes into tissue macrophages [125]. Hif1−/− macrophages and rodents produce less lactate and ATP and exhibit impairment of tissue extravasation and intracellular killing of phagocytosed microorganisms, both ATP-requiring processes [126]. Thus, HIF-1α is instrumental in macrophage (and neutrophil) metabolic adaption to the challenge of performing ATP-dependent immune functions despite the low ambient oxygen that characterizes acutely inflamed tissues. HIF-1α also defers programmed neutrophil apoptosis [127], a signal for efferocytosis by macrophages and the consequent transition to M2 and Mres phenotypes [14]. Interestingly, M1 stimulating factors, such as LPS and IFN-γ, enhanced glycolysis in macrophages independently of HIF-1 (through the induction of a more active phosphofructokinase (PFK) isoform), yet M2 stimulants failed to elicit such a response [128, 129].
The intense burst of anaerobic metabolism in acute-phase macrophages has been likened to that occurring in “fast-twitch” muscle fibers that allow us to sprint short distances. Applying this analogy, the later stages of inflammation and tissue remodeling necessitate an endurance mode of activity, supported by on-going aerobic metabolism. We previously described how Th2-type cytokines, PPAR-γ, and PGC-1β cooperate to induce the molecular machinery that drives the immune functions and metabolism in M2 macrophages. In a sense, the metabolic capacity of AT M2 cells to uptake, esterify, and oxidize FSatFA constitutes a unique immune function, that is, the removal of FSatFA so as to avoid M1 activation and IR. Furthermore, M2 cells that arise as inflammation subsides may undergo a gradual increase in tissue oxygen tension, allowing the utilization of FA to generate ATP. Hence, induction of enzymes involved in FA oxidation (stimulation of macrophages with IL-4 increased the rate of β-oxidation by ~200%) would provide energy for M2 cells to reestablish tissue homeostasis [1]. Pharmacologic inactivation of oxidative metabolism (fatty acid oxidation, oxidative phosphorylation, or mitochondrial respiration) markedly reduced the IL-4-induced expression of arginase-1, dectin-1, and the mannose receptor, but not LPS- and IFN-γ-induced production of M1 markers. This suggests tight coupling of the metabolic and immune functions of M2 macrophages. In contrast to the metabolic mode of M1 cells (anaerobic glycolysis) that supports the latter's phenotype-specific immune function through provision of ATP, inhibition of oxidative metabolism in macrophages stimulated by Th2 cytokines had little impact on ATP levels. Further studies are needed to determine how oxidative metabolism is coupled to M2 immune function [1].
The pathways regulating M2 immune and metabolic modes also intersect to provide PPAR-γ (Figures 1 and 2) and PPAR-δ with a ligand they require in order to initiate transcription. Th2 cytokines induce 15-LO in M2 cells in a STAT6-dependent manner [65, 66, 68]. Furthermore, engulfed apoptotic neutrophils also induce 12/15-LO expression by macrophages [9, 14]. 15-HETE isomers and 17S-HDHA are intermediate metabolites in the 12/15-LO-dependent synthesis of SPM from the long-chain polyunsaturated FA, AA, EPA, and DHA [65, 66, 68, 71, 130]. Whereas SPM act on G-protein coupled receptors in a autocrine and paracrine manner, 15-HETE isomers 17S-HDHA are intracrine mediators, potently binding to both PPAR-γ [53, 71] and PPAR-δ [130]. Furthermore, PPAR-δ is responsible for enhancing the expression of macrophage receptors for apoptotic cells, in addition to boosting aerobic metabolism and mitochondrial biogenesis. Lipids derived from engulfed apoptotic cells also activate PPAR-δ, thereby reinforcing efferocytosis and coupling it to aerobic metabolism [131], until satiety is attained.
Figure 2.
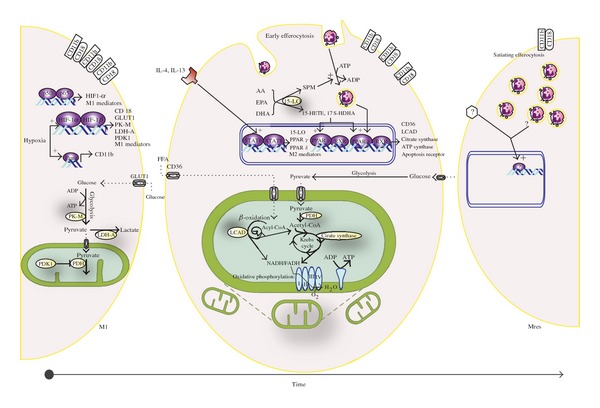
Coregulation of immune phenotype and metabolic mode in macrophages. Recruited macrophages display striking phenotype plasticity in the course of acute, spontaneously resolving inflammation. Upon exudation into edematous, hypoxic tissue, macrophages initially acquire the proinflammatory and microbicidal M1 phenotype and depend on anaerobic glycolysis for ATP. Pathogen-associated molecular patterns, Th1-type cytokines (not shown), and hypoxia-sensitive transcription factors (HIF-1 and Pur, see text for details) cooperate to induce the expression both of M1 mediators (e.g., cytokines, iNOS, CD11b/CD18) and of the proteins/enzymes that perform anaerobic glycolysis. The insulin-independent glucose transporter (GLUT 1) facilitates glucose uptake; pyruvate kinase catalyzes pyruvate synthesis, lactate dehydrogenase produces lactate from pyruvate and generates NAD+ (required for further glycolysis). PDH-kinase inhibits pyruvate dehydrogenase, thereby diverting pyruvate away from the Krebs cycle, and towards lactate. With the clearance of microbes and ablation of edema, specialized proresolution mediators (SPM), Th2-type cytokines, and the uptake of apoptotic neutrophils (early efferocytosis) produce a phenotype switch to the M2 macrophage. IL-4 and IL-13 activate STAT6 transcriptive activity, inducing not only M2 signatures (e.g., nonopsonic endocytic receptors) but also two PPAR isoforms and 12/15-LO. The latter synthesizes SPMs—which promote further nonphlogistic recruitment and efferocytosis by macrophages—and intermediate metabolites (15-HETE and 17S-HDHA) that are necessary for PPAR activation. Lipids from engulfed neutrophils also provide PPARδ ligands. The PPARs in turn orchestrate mitochondrial biogenesis and the metabolic shift to aerobic metabolism of glucose and particularly fatty acids. PPARδ also reinforces efferocytosis by inducing receptors for apoptotic cells. Upregulated metabolic proteins/enzymes include fatty acid translocator CD36, enzymes that catalyze β-oxidation of fatty acids (e.g., LCAD-Long Chain Acyl CoA Dehydrogenase), Krebs cycle enzymes (e.g., citrate synthase), pyruvate dehydrogenase, which enables glycolysis-derived pyruvate to enter the Krebs cycle, and constituents of the Electron Transport Chain that perform oxidative phosphorylation. Inhibition of these metabolic pathways hampers M2 function [1]. Handling of fatty acids by adipose tissue M2 constitutes a unique metabolic function in that cells up-take, catabolize or esterify (not shown) fatty acids released by adipocytes, thereby constraining the level of these potentially harmful molecules within a normal range. On-going efferocytosis “satiates” the macrophage that ceases cell engulfment and promotes the resolution of inflammation, that is, phenotype switching to Mres (CD11blow). The metabolic mode and signaling cascades that regulate immune and metabolic functions of Mres remain to be elucidated.
As evidence of the existence and uniqueness of Mres gathers, it is tempting to speculate that the emergence of this phenotype is regulated by distinct pathways and transcription factors. Furthermore, considering that CD11blow cells are destined to egress through the lymphatic system—where oxygen tension is low—a switch from the oxidative mode of M2 cells to oxygen-independent metabolism seems plausible. Even if this is the case, it is unlikely that the transition to glycolysis is mediated through HIF-1α, as this would be presumably associated with enhanced TLR-NF-κB signaling. Rather, we found Mres cells to be hypo-responsive to LPS and other TLR ligands [14]. We, therefore, speculate that a transcription factor(s) other than HIF-1 and PPAR-γ respond to resolution-promoting signals to mediate the metabolic and resolution phase functions of Mres. Identifying such a factor would open a new avenue for combating relentless inflammation.
6. FUTURE DIRECTIONS
The association of improper nutrition, obesity, and inflammatory leukocyte phenotypes in the immune system, and particularly in fat tissue macrophages, highlights the role of macrophages in the development of metabolic disorders, like diabetes mellitus and insulin resistance. Moreover, the dominance of M2-like macrophages in lean fat stores and the differential metabolic pathways expressed in M1 and M2 macrophages suggests an intimate crosstalk between adipose tissue macrophages and their surrounding adipocytes. The identification of a new macrophage phenotype, that is, associated with resolution of acute, spontaneously-resolving inflammation and that may be induced (through DHA administration) in chronic, metabolic-associated inflammation, suggests that these macrophages might play beneficial regulatory roles in additional diseases characterized by an inflammatory response of excessive intensity or duration. Further studies are required to determine whether this is indeed the case and whether Mres are directly involved in controlling adipocyte function in the lean state and during weight loss. Moreover, it will be important to determine whether the broad change in macrophage metabolic pathways during polarization and conversion between phenotypes is essential for these phenotype switches or is merely a consequence of the adaptation of the macrophage to the new functions and environments it has taken upon itself.
ACKNOWLEDGMENTS
This work was supported by grants from the Israel Science Foundation (Grant no. 534/09), the Nutricia Research Foundation, and the Marc Rich Foundation (to A. Ariel). A. Ariel is a recipient of the young scientist award from Teva Pharmaceuticals Ltd.
ABBREVIATIONS
- IR:
Insulin resistance
- PKC:
Protein kinase C
- IRS:
Insulin receptor substrate
- AT:
Adipose tissue
- WAT:
White adipose tissue
- ATM:
Adipose tissue macrophage
- mTOR:
Mammalian target of rapamycin
- FA:
Fatty acid
- FFA:
Free fatty acid
- FSatFA:
Free saturated fatty acid
- DM:
Diabetes mellitus
- Mres:
Resolution-promoting macrophage
- IFN-γ:
Interferon-γ
- LPS:
Lipopolysaccharide
- TNF-α:
Tumor necrosis factor-α
- CCL2:
Chemokine (C-C motif) ligand 2
- MCP-1:
Monocyte chemotactic Protein 1
- EPA:
Eicosapentaenoic acid
- DHA:
Docosahexaenoic acid
- PPAR:
Peroxisome-proliferator activating receptor
- PGC:
Peroxisome proliferator-activated receptor-gamma coactivator
- DGAT:
Diacyl-glycerol acyl-transferase
- LCAD:
Long chain acyl CoA dehydrogenase
- Nlrp3:
Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3
- PFK:
Phosphofructokinase
- SPM:
Specialized proresolving lipid mediators
- 12/15-LO:
12/15-lipoxygenase
- 17S-HDHA:
17S-hydroxy-DHA
- PUFA:
Polyunsaturated fatty acids
- cPLA2:
Cytosolic phospholipase A2.
References
- 1.Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metabolism. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Botham KM. Bioenergetics: the role of ATP. In: Granner DK, editor. Harper’s Illustrated Biochemistry. chapter 11. New York, NY, USA: McGraw-Hill; 2006. pp. 88–94. [Google Scholar]
- 3.Rasmussen H, Zawalich KC, Ganaesan S, Calle R, Zawalich WS. Physiology and pathophysiology of insulin secretion. Diabetes Care. 1990;13(6):655–666. doi: 10.2337/diacare.13.6.655. [DOI] [PubMed] [Google Scholar]
- 4.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature Immunology. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 6.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annual Review of Immunology. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 8.Nathan C. Points of control in inflammation. Nature. 2002;420(6917):846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 9.Freire-de-Lima CG, Yi QX, Gardai SJ, Bratton DL, Schiemann WP, Henson PM. Apoptotic cells, through transforming growth factor-β, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. Journal of Biological Chemistry. 2006;281(50):38376–38384. doi: 10.1074/jbc.M605146200. [DOI] [PubMed] [Google Scholar]
- 10.Szanto A, Balint BL, Nagy ZS, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33(5):699–712. doi: 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasuga K, Yang R, Porter TF, et al. Rapid appearance of resolvin precursors in inflammatory exudates: novel mechanisms in resolution. Journal of Immunology. 2008;181(12):8677–8687. doi: 10.4049/jimmunol.181.12.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bannenberg GL, Chiang N, Ariel A, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. Journal of Immunology. 2005;174(7):4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 13.Bystrom J, Evans I, Newson J, et al. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. 2008;112(10):4117–4127. doi: 10.1182/blood-2007-12-129767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schif-Zuck S, Gross N, Assi S, Rostoker R, Serhan CN, Ariel A. Saturated-efferocytosis generates pro-resolving CD11blow macrophages: modulation by resolvins and glucocorticoids. European Journal of Immunology. 2011;41(2):366–379. doi: 10.1002/eji.201040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Defronzo RA. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallagher EJ, LeRoith D, Karnieli E. The metabolic syndrome-from insulin resistance to obesity and diabetes. Endocrinology and Metabolism Clinics of North America. 2008;37(3):559–579. doi: 10.1016/j.ecl.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Losser MR, Damoisel C, Payen D. Bench-to-bedside review: glucose and stress conditions in the intensive care unit. Critical Care. 2010;14(4):p. 231. doi: 10.1186/cc9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Science’s STKE. 2005;2005(268):p. pe4. doi: 10.1126/stke.2682005pe4. [DOI] [PubMed] [Google Scholar]
- 19.Wasko MC, Kay J, Hsia EC, Rahman MU. Diabetes mellitus and insulin resistance in patients with rheumatoid arthritis: risk reduction in a chronic inflammatory disease. Arthritis Care & Research. 2011;63(4):512–521. doi: 10.1002/acr.20414. [DOI] [PubMed] [Google Scholar]
- 20.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 22.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. The Lancet. 2010;375(9733):2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Claria J, González-Périz A, López-Vicario C, Rius B, Titos E. New insights into the role of macrophages in adipose tissue inflammation and fatty liver disease: modulation by endogenous omega-3 fatty acid-derived lipid mediators. doi: 10.3389/fimmu.2011.00049. Front. Immun.. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology. 2011;11(2):98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 25.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metabolism. 2008;7(6):485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annual Review of Pathology. 2011;6:275–297. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blüher M. Adipose tissue dysfunction in obesity. Experimental and Clinical Endocrinology and Diabetes. 2009;117(6):241–250. doi: 10.1055/s-0029-1192044. [DOI] [PubMed] [Google Scholar]
- 28.Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 29.Chehab FF. Minireview: obesity and lipodystrophy—where do the circles intersect? Endocrinology. 2008;149(3):925–934. doi: 10.1210/en.2007-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—an allostatic perspective. Biochimica et Biophysica Acta. 2010;1801(3):338–349. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Gustafson B. Adipose tissue, inflammation and atherosclerosis. Journal of Atherosclerosis and Thrombosis. 2010;17(4):332–341. doi: 10.5551/jat.3939. [DOI] [PubMed] [Google Scholar]
- 32.Lionetti L, Mollica MP, Lombardi A, Cavaliere G, Gifuni G, Barletta A. From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutrition, Metabolism and Cardiovascular Diseases. 2009;19(2):146–152. doi: 10.1016/j.numecd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 33.Morris DL, Singer K, Lumeng CN. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Current Opinion in Clinical Nutrition and Metabolic Care. 2011;14(4):341–346. doi: 10.1097/MCO.0b013e328347970b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(7):1304–1310. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- 35.Moro K, Yamada T, Tanabe M, et al. Innate production of TH 2 cytokines by adipose tissue-associated c-Kit+ Sca-1+ lymphoid cells. Nature. 2010;463(7280):540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 36.Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-γ, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circulation Research. 2008;103(5):467–476. doi: 10.1161/CIRCRESAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nature Medicine. 2009;15(8):921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu D, Molofsky AB, Liang HE, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine. 2009;15(8):914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nature Medicine. 2009;15(8):940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. Journal of Clinical Investigation. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lumeng CN, Delproposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57(12):3239–3246. doi: 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeyda M, Stulnig TM. Adipose tissue macrophages. Immunology Letters. 2007;112(2):61–67. doi: 10.1016/j.imlet.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Bourlier V, Zakaroff-Girard A, Miranville A, et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation. 2008;117(6):806–815. doi: 10.1161/CIRCULATIONAHA.107.724096. [DOI] [PubMed] [Google Scholar]
- 45.Kosteli A, Sugaru E, Haemmerle G, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. Journal of Clinical Investigation. 2010;120(10):3466–3479. doi: 10.1172/JCI42845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Red Eagle A, Chawla A. In obesity and weight loss, all roads lead to the mighty macrophage. Journal of Clinical Investigation. 2010;120(10):3437–3440. doi: 10.1172/JCI44721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bassaganya-Riera J, Misyak S, Guri AJ, Hontecillas R. PPAR γ is highly expressed in F4/80hi adipose tissue macrophages and dampens adipose-tissue inflammation. Cellular Immunology. 2009;258(2):138–146. doi: 10.1016/j.cellimm.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koliwad SK, Streeper RS, Monetti M, et al. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. Journal of Clinical Investigation. 2010;120(3):756–767. doi: 10.1172/JCI36066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prieur X, Mok CYL, Velagapudi VR, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. 2011;60(3):797–809. doi: 10.2337/db10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Musso G, Gambino R, Cassader M. Obesity, diabetes, and gut microbiota: the hygiene hypothesis expanded? Diabetes Care. 2010;33(10):2277–2284. doi: 10.2337/dc10-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujisaka S, Usui I, Bukhari A, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58(11):2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viardot A, Grey ST, Mackay F, Chisholm D. Potential antiinflammatory role of insulin via the preferential polarization of effector T cells toward a T helper 2 phenotype. Endocrinology. 2007;148(1):346–353. doi: 10.1210/en.2006-0686. [DOI] [PubMed] [Google Scholar]
- 53.Chawla A. Control of macrophage activation and function by PPARs. Circulation Research. 2010;106(10):1559–1569. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hevener AL, Olefsky JM, Reichart D, et al. Macrophage PPARγ is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. Journal of Clinical Investigation. 2007;117(6):1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature. 2005;437(7059):759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porta C, Rimoldi M, Raes G, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(35):14978–14983. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lovren F, Pan Y, Quan A, et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. American Journal of Physiology. 2010;299(3):H656–H663. doi: 10.1152/ajpheart.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization link between the metabolic and innate immune activity of full-length adiponectin. Journal of Biological Chemistry. 2011;286(15):13460–13469. doi: 10.1074/jbc.M110.204644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohashi K, Parker JL, Ouchi N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. Journal of Biological Chemistry. 2010;285(9):6153–6160. doi: 10.1074/jbc.M109.088708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holland WL, Miller RA, Wang ZV, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nature Medicine. 2011;17(1):55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106(21):2747–2757. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- 63.Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends in Immunology. 2007;28(4):176–183. doi: 10.1016/j.it.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 64.Ariel A, Li PL, Wang W, et al. The docosatriene protectin D1 is produced by TH2 skewing promotes human T cell via lipid raft clustering. Journal of Biological Chemistry. 2005;280(52):43079–43086. doi: 10.1074/jbc.M509796200. [DOI] [PubMed] [Google Scholar]
- 65.Heydeck D, Thomas L, Schnurr K, et al. Interleukin-4 and -13 induce upregulation of the murine macrophage 12/15-lipoxygenase activity: evidence for the involvement of transcription factor STAT6. Blood. 1998;92(7):2503–2510. [PubMed] [Google Scholar]
- 66.Huang JT, Welch JS, Ricote M, et al. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400(6742):378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 67.Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. Journal of Clinical Investigation. 1993;92(3):1572–1579. doi: 10.1172/JCI116738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nassar GM, Morrow JD, Roberts LJ, Lakkis FG, Badr KF. Induction of 15-lipoxygenase by interleukin-13 in human blood monocytes. Journal of Biological Chemistry. 1994;269(44):27631–27634. [PubMed] [Google Scholar]
- 69.González-Périz A, Clària J. Resolution of adipose tissue inflammation. The Scientific World Journal. 2010;10:832–856. doi: 10.1100/tsw.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Titos E, Rius B, González-Périz A, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. Journal of Immunology. 2011;187(10):5408–5418. doi: 10.4049/jimmunol.1100225. [DOI] [PubMed] [Google Scholar]
- 71.González-Périz A, Planagumà A, Gronert K, et al. Docosahexaenoic acid (DHA) blunts liver injury by conversion to protective lipid mediators: protectin D1 and 17S-hydroxy-DHA. FASEB Journal. 2006;20(14):2537–2539. doi: 10.1096/fj.06-6250fje. [DOI] [PubMed] [Google Scholar]
- 72.González-Périz A, Horrillo R, Ferré N, et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by ω-3 fatty acids: a role for resolvins and protectins. FASEB Journal. 2009;23(6):1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White PJ, Arita M, Taguchi R, Kang JX, Marette A. Transgenic restoration of long-chain n-3 fatty acids in insulin target tissues improves resolution capacity and alleviates obesity-linked inflammation and insulin resistance in high-fat-fed mice. Diabetes. 2010;59(12):3066–3073. doi: 10.2337/db10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Itoh M, Suganami T, Satoh N, et al. Increased adiponectin secretion by highly purified eicosapentaenoic acid in rodent models of obesity and human obese subjects. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(9):1918–1925. doi: 10.1161/ATVBAHA.106.136853. [DOI] [PubMed] [Google Scholar]
- 75.Kratz M, Swarbrick MM, Callahan HS, Matthys CC, Havel PJ, Weigle DS. Effect of dietary n-3 polyunsaturated fatty acids on plasma total and high-molecular-weight adiponectin concentrations in overweight to moderately obese men and women. American Journal of Clinical Nutrition. 2008;87(2):347–353. doi: 10.1093/ajcn/87.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krebs JD, Browning LM, McLean NK, et al. Additive benefits of long-chain n-3 polyunsaturated fatty acids and weight-loss in the management of cardiovascular disease risk in overweight hyperinsulinaemic women. International Journal of Obesity. 2006;30(10):1535–1544. doi: 10.1038/sj.ijo.0803309. [DOI] [PubMed] [Google Scholar]
- 77.Hellmann J, Tang Y, Kosuri M, Bhatnagar A, Spite M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB Journal. 2011;25(7):2399–2407. doi: 10.1096/fj.10-178657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krishnamoorthy S, Recchiuti A, Chiang N, et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1660–1665. doi: 10.1073/pnas.0907342107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nishimura S, Manabe I, Nagasaki M, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. Journal of Clinical Investigation. 2008;118(2):710–721. doi: 10.1172/JCI33328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lumeng CN, DeYoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56(1):16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- 82.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bilkovski R, Schulte DM, Oberhauser F, et al. Adipose tissue macrophages inhibit adipogenesis of mesenchymal precursor cells via wnt-5a in humans. International Journal of Obesity. 2011;35(11):1450–1454. doi: 10.1038/ijo.2011.6. [DOI] [PubMed] [Google Scholar]
- 84.Cancello R, Henegar C, Viguerie N, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54(8):2277–2286. doi: 10.2337/diabetes.54.8.2277. [DOI] [PubMed] [Google Scholar]
- 85.Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exercise Immunology Review. 2010;16:105–118. [PubMed] [Google Scholar]
- 86.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 87.Bruun JM, Lihn AS, Pedersen SB, Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. Journal of Clinical Endocrinology and Metabolism. 2005;90(4):2282–2289. doi: 10.1210/jc.2004-1696. [DOI] [PubMed] [Google Scholar]
- 88.Dahlman I, Kaaman M, Olsson T, et al. A unique role of monocyte chemoattractant protein 1 among chemokines in adipose tissue of obese subjects. Journal of Clinical Endocrinology and Metabolism. 2005;90(10):5834–5840. doi: 10.1210/jc.2005-0369. [DOI] [PubMed] [Google Scholar]
- 89.Jiao P, Chen Q, Shah S, et al. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: involvement of nuclear factor-κB and c-Jun NH2-terminal kinase pathways. Diabetes. 2009;58(1):104–115. doi: 10.2337/db07-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ito A, Suganami T, Yamauchi A, et al. Role of CC chemokine receptor 2 in bone marrow cells in the recruitment of macrophages into obese adipose tissue. Journal of Biological Chemistry. 2008;283(51):35715–35723. doi: 10.1074/jbc.M804220200. [DOI] [PubMed] [Google Scholar]
- 91.Li P, Lu M, Nguyen MTA, et al. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. Journal of Biological Chemistry. 2010;285(20):15333–15345. doi: 10.1074/jbc.M110.100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of Lipid Research. 2005;46(11):2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 93.Murano I, Barbatelli G, Parisani V, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. Journal of Lipid Research. 2008;49(7):1562–1568. doi: 10.1194/jlr.M800019-JLR200. [DOI] [PubMed] [Google Scholar]
- 94.Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor α. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(10):2062–2068. doi: 10.1161/01.ATV.0000183883.72263.13. [DOI] [PubMed] [Google Scholar]
- 95.Agwunobi AO, Reid C, Maycock P, Little RA, Carlson GL. Insulin resistance mid substrate utilization in human endotoxemia. Journal of Clinical Endocrinology and Metabolism. 2000;85(10):3770–3778. doi: 10.1210/jcem.85.10.6914. [DOI] [PubMed] [Google Scholar]
- 96.Mehta NN, McGillicuddy FC, Anderson PD, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59(1):172–181. doi: 10.2337/db09-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. American Journal of Physiology. 2007;292(3):E740–E747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- 98.Gubern C, López-Bermejo A, Biarnés J, Vendrell J, Ricart W, Fernández-Real JM. Natural antibiotics and insulin sensitivity: the role of bactericidal/permeability-increasing protein. Diabetes. 2006;55(1):216–224. [PubMed] [Google Scholar]
- 99.Cani PD, Neyrinck AM, Fava F, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50(11):2374–2383. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- 100.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 101.Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 102.Ghanim H, Abuaysheh S, Sia CL, et al. Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. 2009;32(12):2281–2287. doi: 10.2337/dc09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakarai H, Yamashita A, Nagayasu S, et al. Adipocyte-macrophage interaction may mediate LPS-induced low-grade inflammation: potential link with metabolic complications. Innate Immunity. 2011;17(6) doi: 10.1177/1753425910393370. [DOI] [PubMed] [Google Scholar]
- 104.Andreasen AS, Larsen N, Pedersen-Skovsgaard T, et al. Effects of Lactobacillus acidophilus NCFM on insulin sensitivity and the systemic inflammatory response in human subjects. British Journal of Nutrition. 2010;104(12):1831–1838. doi: 10.1017/S0007114510002874. [DOI] [PubMed] [Google Scholar]
- 105.Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through toll-like receptors. Molecules and Cells. 2006;21(2):174–185. [PubMed] [Google Scholar]
- 106.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. Journal of Biological Chemistry. 2001;276(20):16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 107.Lee JY, Ye J, Gao Z, et al. Reciprocal modulation of toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. Journal of Biological Chemistry. 2003;278(39):37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- 108.Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. Journal of Pathology. 2008;214(2):161–178. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nature Reviews Microbiology. 2005;3(1):36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 110.Jensen CB, Storgaard H, Holst JJ, Dela F, Madsbad S, Vaag AA. Insulin secretion and cellular glucose metabolism after prolonged low-grade Intralipid infusion in young men. Journal of Clinical Endocrinology and Metabolism. 2003;88(6):2775–2783. doi: 10.1210/jc.2002-021430. [DOI] [PubMed] [Google Scholar]
- 111.Tsukumo DML, Carvalho-Filho MA, Carvalheira JBC, et al. Loss-of-function mutation in toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 112.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. Journal of Clinical Investigation. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saberi M, Woods NB, de Luca C, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metabolism. 2009;10(5):419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nguyen MTA, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. Journal of Biological Chemistry. 2007;282(48):35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- 115.Liang CP, Han S, Okamoto H, et al. Increased CD36 protein as a response to defective insulin signaling in macrophages. Journal of Clinical Investigation. 2004;113(5):764–773. doi: 10.1172/JCI19528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nicholls HT, Kowalski G, Kennedy DJ, et al. Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes. 2011;60(4):1100–1110. doi: 10.2337/db10-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arkan MC, Hevener AL, Greten FR, et al. IKK-β links inflammation to obesity-induced insulin resistance. Nature Medicine. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 119.Solinas G, Vilcu C, Neels JG, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metabolism. 2007;6(5):386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 120.Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet—induced obesity in mice. Diabetes. 2010;59(5):1171–1181. doi: 10.2337/db09-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mathis D, Shoelson SE. Immunometabolism: an emerging frontier. Nature Reviews Immunology. 2011;11(2):81–83. doi: 10.1038/nri2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nature Reviews Immunology. 2009;9(9):609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rius J, Guma M, Schachtrup C, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008;453(7196):807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Peyssonnaux C, Datta V, Cramer T, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. Journal of Clinical Investigation. 2005;115(7):1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Oda T, Hirota K, Nishi K, et al. Activation of hypoxia-inducible factor 1 during macrophage differentiation. American Journal of Physiology. 2006;291(1):C104–C113. doi: 10.1152/ajpcell.00614.2005. [DOI] [PubMed] [Google Scholar]
- 126.Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Elks PM, Van Eeden FJ, Dixon G, et al. Activation of hypoxia-inducible factor-1α (hif-1α) delays inflammation resolution by reducing neutrophil apoptosis and reverse migration in a zebrafish inflammation model. Blood. 2011;118(3):712–722. doi: 10.1182/blood-2010-12-324186. [DOI] [PubMed] [Google Scholar]
- 128.Rodríguez-Prados JC, Través PG, Cuenca J, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. Journal of Immunology. 2010;185(1):605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 129.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. Journal of Experimental Medicine. 1983;158(3):670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Naruhn S, Meissner W, Adhikary T, et al. 15-Hydroxyeicosatetraenoic acid is a preferential peroxisome proliferator-activated receptor β/δ agonist. Molecular Pharmacology. 2010;77(2):171–184. doi: 10.1124/mol.109.060541. [DOI] [PubMed] [Google Scholar]
- 131.Mukundan L, Odegaard JI, Morel CR, et al. PPAR-Δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nature Medicine. 2009;15(11):1266–1272. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]