

Abstract

The stereospecific synthesis of aryloxy and amino substituted E- and Z-ethyl-3-acrylates is of interest because of their potential in the polymer industry and in medicinal chemistry. During work on a copper-catalyzed cross-coupling reaction of E- and Z-ethyl-3-iodo-acrylates with phenols and N-heterocycles, we discovered a very simple (non-metallic) method for the stereospecific synthesis of aryloxy and amino substituted acrylates. To study this long standing problem on the stereoselectivity of aryloxy and amino substituted acrylates, a series of O- and N-substituted nucleophiles was allowed to react with E- and Z-ethyl-3-iodo-acrylates. Screening of different bases indicated that DABCO (1,4-diazabicyclo[2.2.2]octane) afforded successful conversion of E- and Z- ethyl-3-iodoacrylates into aryloxy and amino substituted ethyl acrylates in a stereospecific manner. Herein are the details of this DABCO-mediated stereospecific synthesis of aryloxy and amino substituted E- or Z-acrylates.
1. Introduction
Vinyl ethers are important intermediates in organic and medicinal chemistry and as raw materials for the polymer industry.2–5 Indeed, vinyl aryl ethers constitute useful intermediates in a wide range of reactions; for example, cycloadditions, cyclopropanations, metathesis reactions, natural product analogues, and polymers.2 N-Vinyl amines are widely used in the preparation of polymeric dyes, catalysts and ion-exchange resins, while the vinyl group can also act as an efficient protecting group of phenol derivatives.1 Vinyl aryl ether moieties are found in numerous biologically active molecules, for example 1-phenoxy-3-triazolyl-1-hexene derivatives are plant growth regulators.2 In particular, acrylate ester derivatives are used as the base acrylic monomer in a wide range of coatings, adhesives, as well as finishes for paper, leather and textiles. In addition they are used in floor and wood polishes, acrylic resins, and powder coatings. Acrylic acid derived polymers are widely used in everyday life including super adsorbent polymers (SAPs).6 Recent reports indicate that classes of alkoxy acrylates are potently active against drug resistant strains of tuberculosis7–8 and N-azolyl acrylates inhibit CRM1(Chromosome Region Maintenance)-mediated nucleocytoplasmic transport, a selective congener which inhibits the HIV-1 production in latently infected cells.9
RESULTS AND DISCUSSION
Synthesis of aryloxy and amino substituted E-Acrylates from Ethyl-Z-3-iodoacrylates
The last decade has shown significant progress in the development of Cu-catalyzed cross-coupling processes and other methods10, 11 for the synthesis of aryl ethers, aryl thioethers, aryl amines, and their respective vinyl analogues of synthetic, polymeric, and biological importance.10–33 The use of copper-catalysis in the formation of C-N, C-O and C-S bonds including aryl and vinyl substituted derivatives covering a broad spectrum of amines, ethers, and thioethers, in good to excellent yields has been reported.7–8,36 It has now been shown that the formation of vinylic C-N and C-O bonds by a Cu-catalytic method7 is ineffective with the functionalized ethyliodoacrylate substrates, for both phenols and N-heterocycles as nucleophiles.1 These products obtained from an ethyl-Z-3-iodoacrylate substrate, presumably, resulted from the conjugate addition, which produced only the E- isomers of the desired products of ethers or amines from an ethyl-Z-3-iodoacrylate instead of giving the previously reported7 copper mediated stereospecific cross-coupled Z- products at the 40 °C optimized reaction temperature. This previous result reported from our laboratory with ethyl-Z-3-iodoacrylate employing a copper catalytic method for the synthesis of aryloxy and amino substituted Z-acrylates yielded exclusively the E-isomers (as shown in Table 1)1 and were inadvertently assigned as the Z-isomers.7–8 The coupling constants for arylthio substituted Z-acrylates were in the range of 10–12 Hz according to the literature.35–36 In this case the coupling constants for aryloxy and amino substituted E-acrylates were also in the range of 10–12 Hz, which led us to assign the E-isomers as Z-isomers when compared to the arylthio derivatives of Z-acrylates.1, 7–8, 36 Hence, in order to correct the error due to miss-assignment in the previous communications7, 8 as well as develop an efficient method for the stereospecific synthesis of phenoxy- and amino- E- and Z-ethylacrylates, it was decided to investigate the reaction conditions step-by-step.
Table 1.
Trans acrylate ethers and amines from the starting cis acrylates, are believed to be from a conjugate addition
 | |||||
|---|---|---|---|---|---|
| Entry | Product | % Yield | Entry | Product | Yield (%) |
| 1 | 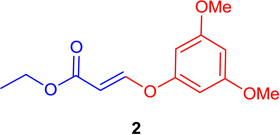 |
98 | 8 | 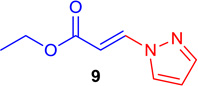 |
92 |
| 2 | 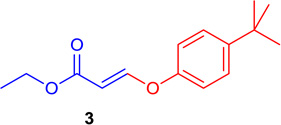 |
93b | 9 | 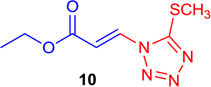 |
96 |
| 3 | 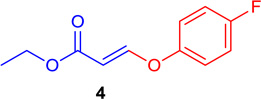 |
84 | 10 | 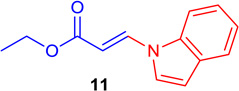 |
93c |
| 4 | 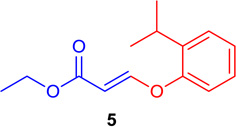 |
95 | 11 | 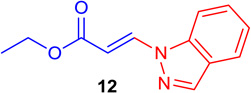 |
84 |
| 5 | 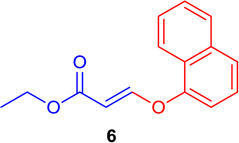 |
96 | 12 | 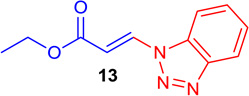 |
84c |
| 6 | 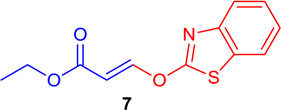 |
92b | 13 | 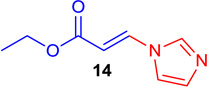 |
96c |
| 7 | 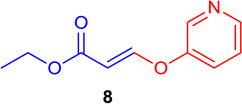 |
86b | 14 | 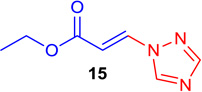 |
92c |
Isolated yields, the average of at least two runs.
The reaction was carried out at 40 °C and was stirred for 2 h.
The reaction was carried out at 40 °C and was stirred for 3 h.
The Role of a Base for the Synthesis of aryloxy and amino substituted E-Acrylates from Z-Ethyl-iodo-acrylate
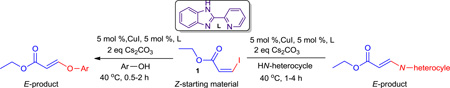
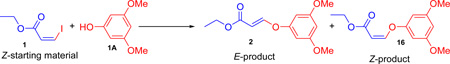
To investigate this reaction a series of experiments were performed and the results are summarized in Scheme 1. The optimized reaction conditions with a copper catalyst in combination with an inorganic base, Cs2CO3, at 40 °C was applied to ethyl-Z-3-iodoacrylate1, 7–8 with a phenol or an amine yielded only the E-products 2 and 9, respectively. With a copper catalyst in the presence of Cs2CO3 at room temperature, this yielded mixtures of E- and Z-isomers in an approximately 3:2 ratio for both aryloxy substituted acrylates (2, 16) and amino substituted acrylates (9, 22). The use of Cs2CO3, without a copper catalyst, at room temperature yielded the same results obtained with a copper catalyst, as mentioned. At 0 °C the reaction did not proceed with or without the catalyst. The copper catalyst was clearly not necessary in this process.7
Scheme 1.

The Role of a Base for the Synthesis of aryloxy and amino substituted E-Acrylates from Z-Ethyl-iodo-acrylate
The use of inorganic bases Cs2CO3, K3PO4 and K2CO3 at elevated temperature (40 °C) yielded exclusively E-isomers of ethers and amines from the Z-acrylate exclusively, but at room temperature they gave mixtures of Z- and E-isomers, indicating the lack of stereospecificity. The results presented in Scheme 1 suggested that in the presence of an inorganic base, a conjugate addition took place between the acrylate iodide and nucleophiles1 instead of a copper-catalyzed cross-coupling process.7
Optimization of Base-Mediated Stereospecific Synthesis of aryloxy and amino substituted Acrylates from Ethyl-3-iodoacrylate
These findings prompted us to investigate the long-standing problem of stereoselectivity with ethyl-Z-3-iodoacrylate from different approaches. Since it was now known that the reaction proceeded without a copper catalyst1 and only a base was essential, it was decided to screen different organic bases, such as strong bases (HMDS, LiHMDS), nucleophilic hindered bases (DABCO and DBU), the unhindered nucleophilic base (DMAP), and a commonly used simple amine base (Et3N). The results indicated that a nucleophilic hindered base, DABCO, efficiently promoted the conversion of ethyl- Z-3-iodoacrylates into aryloxy and amino substituted Z-acrylates in a stereospecific fashion. The authors are unaware of any published reports for such a DABCO-mediated stereospecific addition of O- or N-nucleophiles to ethyliodoacrylates for the stereospecific synthesis of biologically important aryloxy and amino substituted acrylates.
As mentioned earlier, various inorganic bases (Cs2CO3, K2CO3, K3PO4) were screened with and without a copper catalyst at ambient and elevated (40 °C) temperatures. None of the conditions were effective in controlling the stereochemistry of the ether and amine products. The prototypical base employed by us and others for the synthesis of vinyl ethers and amines from vinyl iodide substrates was Cs2CO3.36 Treatment of 1 with the inorganic bases K2CO3 or K3PO4 with the nucleophile 1A produced almost the same results as with Cs2CO3, either with or without a copper catalyst. The results obtained with Cs2CO3 under various conditions are summarized in Table 2, entries 1–7. None of these conditions provided the desired stereospecificity and in most cases yielded mixtures of the Z- and E-isomers 16 and 2 in a 2:3 ratio. We next screened the DBU, DABCO, DMAP, Et3N, HMDS, LiHMDS in various solvents at room temperature. The results are summarized in Table 1. With DBU, DMAP and Et3N in DMF either with or without a copper catalyst the process did not proceed (Table 2, entries 24–29). However, it was found that treatment of 1 with 1A and 2 equivalents of DABCO in DMF in the absence of a copper catalyst at room temperature for 1 hour afforded exclusively Z-isomer 16 in nearly quantitative yield with full retention of stereochemistry (Table 2, entry 15). The use of 1.1 equiv of DABCO under the same conditions required longer reaction times to finish and resulted in somewhat lower yield (Table 2, entry 16). The use of a catalytic amount of DABCO did not provide full conversion of the starting material (Table 1, entries 17–19) into product. To summarize, the optimized conditions for the stereospecific conversion of 1 to 16 were: 1 equivalent of 1 when stirred for 1 hour with 1.5 equivalents of 1A and 2 equiv of DABCO in DMF at room temperature afforded Z-isomer 16 stereospecifically.
Table 2.
Stereospecific cross coupling of ethyl-Z-3-iodoacrylate with 3,5-dimethoxyphenol
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | eq. base | solvent | temp. (°C) | time (h) | yield (%)a | Z/E |
| 1 | CuI, L | 2.0 Cs2CO3 | DMF | 40 | 0.5 | 95 | 0:100 |
| 2 | CuI, L | 2.0 Cs2CO3 | DMF | rt | 0.5 | 94 | 40:60 |
| 3 | CuI | 2.0 Cs2CO3 | DMF | rt | 0.5 | 95 | 40:60 |
| 4 | L | 2.0 Cs2CO3 | DMF | rt | 0.5 | 95 | 40:60 |
| 5 | - | 2.0 Cs2CO3 | DMF | 40 | 0.5 | 94 | 0:100 |
| 6 | - | 2.0 Cs2CO3 | DMF | rt | 0.5 | 95 | 40:60 |
| 7 | - | 2.0 Cs2CO3 | toluene | rt | 6 | 88 | 70:30 |
| 8 | - | 2.0; K2CO3 | DMF | rt | 0.5 | 91 | 40:60 |
| 9 | - | 2.0; K2CO3 | toluene | rt | 6 | 75 | 65:35 |
| 10 | - | 2.0; K3PO4 | DMF | rt | 0.5 | 93 | 40:60 |
| 11 | - | 2.0; K3PO4 | toluene | rt | 10 | 60 | 65:35 |
| 12 | - | 2.0; HMDS | DMF | rt | 6 | 10 | 100:0 |
| 13 | - | 2.0; HMDS | toluene | rt | 15 | 10 | 100:0 |
| 14 | - | 2.0; LiHMDS | DMF | rt | 6 | 0 | N/A |
| 15 | - | 2.0 DABCO | DMF | rt | 1.0 | 95 | 100:0 |
| 16 | - | 1.1 DABCO | DMF | rt | 6.0 | 84 | 100:0 |
| 17 | - | 0.5 DABCO | DMF | rt | 2.5 | 50 | 100:0 |
| 18 | - | 0.2 DABCO | DMF | rt | 2.5 | 20 | 100:0 |
| 19 | - | 0.1 DABCO | DMF | rt | 2.5 | 10 | 100:0 |
| 20 | CuI, L | 2.0 DABCO | DMF | rt | 0.5 | 95 | 100:0 |
| 21 | CuI, L | 2.0 DABCO | toluene | rt | 12 | 87 | 100:0 |
| 22 | CuI | 2.0 DABCO | DMF | rt | 0.5 | 95 | 100:0 |
| 23 | CuI | 2.0 DABCO | toluene | rt | 12 | 84 | 100:0 |
| 24 | - | 2.0 DMAP | DMF | rt | 6 | 0 | N/A |
| 25 | CuI, L | 2.0 DMAP | DMF | rt | 6 | 0 | N/A |
| 26 | - | 2.0 Et3N | DMF | rt | 6 | 0 | N/A |
| 27 | CuI, L | 2.0 Et3N | DMF | rt | 6 | 0 | N/A |
| 28 | - | 2.0 DBU | DMF | rt | 6 | 0 | N/A |
| 29 | CuI, L | 2.0 DBU | DMF | rt | 6 | 0 | N/A |
The starting aryl vinyl halides contained ~ 3–9 % Z-isomer; this resulted in ~3–9 % of the cis-isomer, included in the overall yield.
2.4 Application of DABCO-Mediated Stereospecific Synthesis of Aryloxy and Amino substituted Z-Acrylates from Ethyl-Z-3-iodoacrylate
With optimized conditions in hand, it was decided to determine the scope of this process. To ensure the coupling reaction proceeded in a regio- and stereospecific fashion, the coupling reaction was investigated using 1 with various electron-poor and electron-rich substituted aromatic phenols and N-heterocycles. Examination of the results (summarized in Table 3) indicated that the DABCO-mediated system worked well when the Z-vinyl iodide was subjected to the coupling with aryl- or heterocyclic-substituted phenols (Table 3, entries 1–6). Electron-rich, electron poor, and ortho-substituted hindered aromatic phenols gave excellent yields with full retention of stereochemistry (16–19) when the process was carried out at room temperature for 1–2 hours. As expected, the 3-hydroxypyridine substrate (Table 3, entry 6) gave the corresponding vinyl ether 21, but this reaction took 3 hours to go to completion. This demonstrated that the scope of the reaction can be extended to the synthesis of heterocyclic substituted vinyl ethers in excellent yields.
Table 3.
DABCO mediated stereospecific cross coupling of ethyl-Z-3-iodoacrylate 1
 | |||||
|---|---|---|---|---|---|
| Entry | Product | Yield (%)a | Entry | Product | Yield (%)a |
| 1 | 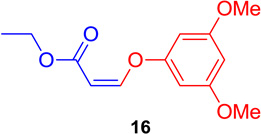 |
97 | 8 | 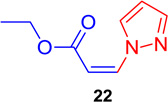 |
90 |
| 2 | 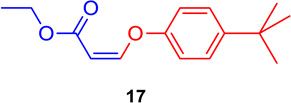 |
91 | 9 | 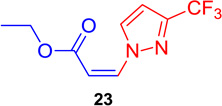 |
93 |
| 3 | 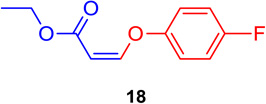 |
82 | 10 | 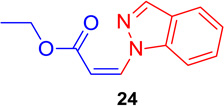 |
89 |
| 4 | 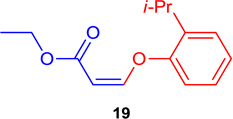 |
93 | 11 | 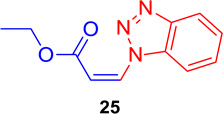 |
91b |
| 5 | 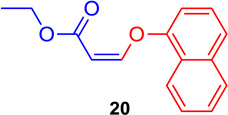 |
91 | 12 | 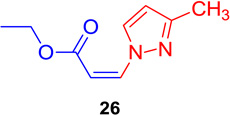 |
75 |
| 6 | 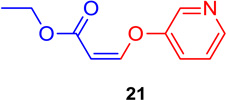 |
92 | 13 | 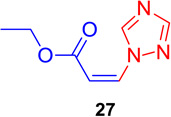 |
92b |
Isolated yields, the average of at least two runs.
The reaction was carried out at rt and was stirred for 3 h.
Encouraged by these results, the DABCO-mediated reaction conditions were employed for the coupling of 1 with various N-heterocycles, including pyrazole, indazole, benzotriazole, and triazoles (Table 3, entries 8–13). Interestingly, 1 gave the desired Z-vinyl amines (22–27, Table 3, entries 8–13), but required somewhat longer reaction times as compared to oxygen nucleophiles. No reaction between 1 and indole took place, presumably because of the unique character of the indole N-H bond (pKa = 19). The pKas of the nucleophiles that worked well in this study ranged from 9.5–15 for both phenols and N-heterocycles.
2.5 Application of DABCO-Mediated Stereospecific Synthesis of Aryloxy and Amino substituted E-Acrylates from Ethyl-E-3-iodoacrylate
To investigate whether or not the stereochemistry of aryloxy and amino substituted acrylates was retained regardless of the E- or Z-configuration of the starting ethyliodoacrylate, a series of experiments was conducted which employed the DABCO-mediated reaction conditions using ethyl E-3-iodoacrylate 29 in the presence of the same phenols and N-heterocycles, as shown previously with 1 (Table 3). The results of the DABCO-mediated coupling between 29 with various phenols and N-heterocycles are summarized in Table 4. Examination of the results indicated that reaction of 29 with nucleophiles proceeded in a stereospecific fashion and yielded exclusively the aryloxy substituted E-acrylates with electron-rich, electron poor, and ortho substituted, as well as heterocyclic substituted aromatic phenols in excellent yields (2–6) over a 1–3 hour period (Table 4 entries 1–5). To test the hypothesis and to further extend the scope of the reaction, E-isomer 29 was subjected to the optimized reaction conditions with N-heterocycles, including pyrazole, indazole, triazole, and benzotriazole (Table 4, entries 9–15). As expected, 29 afforded the desired amino substituted E-acrylates 9–15 in excellent yield when stirred at room temperature for a 2–3 hour period.
Table 4.
DABCO mediated stereospecific cross coupling of ethyl-Z-3-iodoacrylate with phenols and N-heterocycles
 | |||||
|---|---|---|---|---|---|
| Entry | Product | Yield (%) | Entry | Product | Yield (%) |
| 1 | 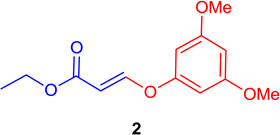 |
98 | 6 | 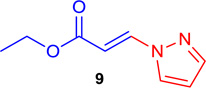 |
92 |
| 2 | 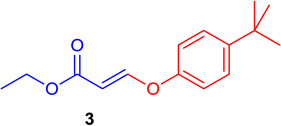 |
93b | 7 | 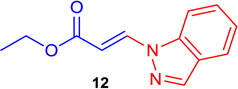 |
96b |
| 3 | 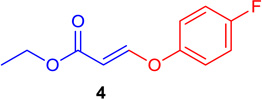 |
84 | 8 | 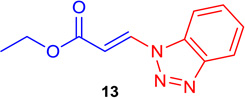 |
93 |
| 4 | 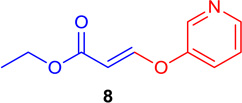 |
95b | 9 | 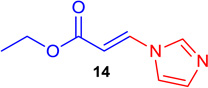 |
84b |
| 5 | 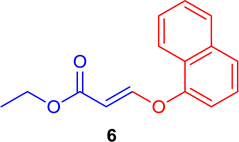 |
96 | 10 | 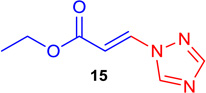 |
84b |
Isolated yields, the average of at least two runs.
The reaction was carried out at rt and was stirred for 3 h.
A Potential Mechanism for the Stereospecificity
Key points that were observed from the experiments:
Ethyliodoacrylate substrates reacted with oxygen and nitrogen nucleophiles in stereospecific fashion in the absence of a copper catalyst.
The hindered, nucleophilic base DABCO was required to control the stereospecificity.
At least 1.1 equivalents of DABCO was necessary for efficient conversion.
On the other hand, the stereospecific nature of sulfur substitution was unusual (Scheme 2, 30), and gave the reported arylthio derivatives of Z-acrylates stereospecifically, in contrast to most oxygen and nitrogen nucleophiles (2, 16 and 9, 22).1, 7
Scheme 2.

The Cs2CO3-mediated stereospecific formation of arylthio substituted Z-acrylates from ethyl-Z-iodoacrylate.
Hence, it was speculated that an addition-elimination mechanism may be involved in these types of substitution reactions with oxygen and nitrogen nucleophiles.
In the case of thiolate or DABCO, addition to Z- or E-ethyl-3-iodo-acrylate 1 or 29 would lead to 33 or 34, respectively (Scheme 3). In order to give distinct products by this mechanism, loss of iodide must be faster than rotation about the C-C bond to interconvert 33 and 34. Consequently, these may be transition states rather than intermediates, which is reasonable, given the leaving group ability of iodide.37 Both product isomers are stable to the reaction conditions, so that loss of stereochemistry in the earlier cases (Scheme 1; 2, 16 and 9, 22) was not due to a stereospecific reaction followed by interconversion. The other possible distinction between these reactions involves enolate stereochemistry, which will be discussed below.
Scheme 3.

Schematic representationa of possible mechanistic pathways of ethyliodoacrylate with thiols or DABCO
a In the case of ethyl-3-iodo-acrylate starting material, no considerable preference for the s-cis or the s-trans conformation is expected. b Rotation along the single bond in 33 and 34 (indicated by red color) must be slower than elimination of iodide.
If the rate of iodide loss were the only determinant of stereospecific substitution, then DABCO must function as a base, rather than a nucleophilic catalyst. This was in question because the other amine bases were found to be much less effective at mediating substitution (Table 2, entries 16–21). The nucleophilic catalytic behavior of DABCO in the process was proven as follows:
Treatment of E- and Z-ethyl-3-iodoacryate 1 or 29 with a stoichiometric amount of DABCO was monitored by 1H NMR spectroscopy. Examination of the data in Figure 1 indicates that each acrylate reacts to form a distinct product, 31 and 32 respectively, and the proton NMR coupling constants indicate that ethyl-Z-3-iodoacrylate (Figure 1, 1) gave the intermediate Z-adduct (Figure 1, 31) and ethyl-E-3-iodoacrylate (Figure 1, 29) gave the E-adduct (Figure 1, 32). Subsequent treatment of these intermediates with nucleophile stereospecifically led to the product (see Figure 1). While reaction of iodoacrylates plausibly proceeds through a single transition state, substitution of these ammonium ions almost certainly does not. Presumably intermediates 35 and 37 (Scheme 4) have a sufficient lifetime that they could interconvert with their respective rotamers before elimination. While it is certainly true that a barrier exists to slow interconversion of rotamers, it is not expected to be sufficient to cause the observed results. Thus the observed stereospecific substitution of both 31 and 32 requires some other distinction between their intermediates than the rotamer formed.
Figure 1.

Experimental evidence for the formation of a stereospecific acrylate –DABCO adduct
Scheme 4.

Schematic representation of probable mechanistic pathways of ethyl-3-iodo-acrylate with oxygen and nitrogen nucleophiles
There is another distinction besides rotamers that is possible: enolate stereochemistry. Nucleophilic attack on Z-ammonium acrylate 31 (Scheme 4) could proceed so as to allow close approach of the ammonium species with the nascent oxyanion of 35. This requires formation of the E-enolate (assuming an ester with a higher CIP priority than the oxyanion). In contrast, the E-ammonium acrylate 32 cannot bring the nascent oxyanion of 37 into close proximity to the ammonium ion, so E-enolate is not favored. Charge repulsion between anionic nucleophile and the forming oxyanion may explain Z-enolate geometry in 37, which also would allow better solvation of oxyanion. Note that s-cis versus s-trans conformational preference of 31 and 32 is likely to be small, transition state stability should overwhelm minor preferences. Rotation about the single bond interconverts 37 and 38; however, 37, with the smaller H eclipsing the OEt group, is preferred over 38. Intermediates 35 and 37 are distinct, and would remain so throughout their reactions: E-enolate 35 leading to the corresponding Z-product through the reaction pathway that retains closest approach of oppositely charged atoms, and Z-enolate 37 leading to the corresponding E-product through the pathway of lowest steric repulsion with H eclipsing OEt. Intermediate 37 has a larger distance between charged atoms and little variation depending on the rotamer.
This mechanism provides a self-consistent picture, but does not answer all questions. Among others, the direct thiolate substitution, and the initial formation of ammonium acrylate intermediates cannot be controlled by these factors. We fall back on the extremely rapid loss of β iodide from the enolate, faster than rotameric interconversion. With smaller nucleophiles, bond making proceeds to a different extent in the transition state and rotation competes with elimination, even with the iodide leaving group.
This is our preferred explanation, but other possibilities remain. Despite our efforts to exclude them, adventitious transition metal catalysts may be present. A mechanism that does not involve loss of double bond integrity is possible via SRN1. Electron transfer to acrylate would form a radical anion that loses iodide, and reaction of the resulting vinyl radical with nucleophile forms a radical anion that can transfer an electron to form product and propagate the reaction. The iodoacrylates would cleave more readily as the radical anion, but the ammonium acrylate intermediates could accept an electron more readily. While this mechanism is mentioned, we consider it less likely because of the range of nucleophiles that function in this process.
SUMMARY
In summary, an efficient DABCO-mediated stereospecific synthesis of aryloxy and amino substituted ethyl acrylates has been achieved in good to excellent yields. The DABCO-mediated system tolerates a wide range of functional groups while employing a wide applicability. The generality and simplicity of the system permits open air reactions without special precautions or metals.1 This stereospecific process should have wide applicability in the polymer and resin industries. The optimized conditions remain as illustrated for the synthesis of Z-isomer 16 (Table 2, entry 15). Treatment of 1 with 1A and 2 equiv of DABCO in DMF at room temperature for 1 hour provided Z-isomer 16 in nearly quantitative yield.
EXPERIMENTAL SECTION
General considerations
The N,N-dimethylformamide (DMF, anhydrous, 99.8% purity), cesium carbonate (99.98% purity), DABCO, phenols, and N-heterocyclic compounds were used as received without further purification. Silica gel (230–400 mesh) chromatography was utilized for purification of the products. 1H and 13C NMR spectra were obtained on a 300 MHz or 500 MHz NMR instrument with chemical shifts reported relative to TMS.
General Procedure A: DABCO-Mediated Synthesis of Aryloxy and Amino substituted Acrylates from Ethyl-Z-3-iodo-acrylate and Ethyl-E-3-iodo-acrylate (Table 3 & 4)
An oven dried round bottom flask containing a magnetic stir bar was sealed with a rubber septum and then evacuated and backfilled with argon while cooling to rt. The round bottom flask was then charged with anhydrous DABCO (2.0 eq), E- or Z-ethyl-3-iodo-acrylate (1.0 eq) and dry DMF (2 mL). The solution which resulted was stirred for 5–10 min at rt. The appropriate phenol or N-heterocycle (0.75 mmol, 1.5 equiv) in 0.5 mL of dry DMF was added to the reaction mixture through a rubber septum and the mixture stirred for an additional 0.5–3 h at rt depending on the structure of the substrate. The reaction mixture was then filtered through a pad of silica gel to remove insoluble residues. The pad of silica gel was washed with ethyl acetate and hexane (40:60, 100 mL). The combined filtrate was washed with brine (5 × 50mL), dried (Na2SO4) and concentrated in vacuo on a rotary evaporator. The concentrated crude oil was purified by flash column chromatography on silica gel using the eluent (2–20 %) ethyl acetate and hexane (depending on the substrate) to obtain the pure products (81–98%).
General Procedure B: Cu-Catalyzed Cross-Coupling of Z-Ethyl-2-iodo-acrylate l with Phenols and N-heterocycles (Table 1)
An oven dried round bottom flask containing a magnetic stir bar was sealed with a rubber septum and evacuated and backfilled with argon (the sequence was repeated three times) while cooling to rt. The round bottom flask was then charged with anhydrous cesium carbonate (2.0 equiv), copper (I) iodide (5 mol %), L (5 mol %) and dry DMF (2 mL). The solution which resulted was stirred for 5–10 min at rt. The reaction mixture turned a light green color within 3–5 min. The reaction vessel was evacuated and backfilled with argon once more before adding the N-heterocycle or phenol. The appropriate N-heterocycle or phenol (0.75 mmol, 1.5 equiv) was added to the reaction mixture through a rubber septum and the mixture stirred for an additional 5 min at rt. Then, Z-ethyl-3-iodo-acrylate 1 (0.5 mmol, 1.0 equiv) of choice was added in a minimum amount of dry DMF to the resulting reaction mixture through a rubber septum. The contents of the reaction mixture were heated from rt to 40 °C to 80°C for 0.5–4 h depending on the substrate. The reaction mixture was then cooled to rt and filtered through a pad of silica gel to remove insoluble residues. The pad of silica gel was washed with ethyl acetate and hexane (40:60) (100 mL). The combined filtrate was washed with brine (5 × 50mL), dried (Na2SO4) and concentrated in vacuo on a rotatory evaporator. The concentrated crude oil was purified by flash column chromatography on silica gel using the eluent ethyl acetate and hexane to obtain the pure product (81–95%). With oxygen and nitrogen nucleophiles this gave a mixture of Z- and E-0 products. With sulfur nucleophiles it gave the desired Z-isomer, but this is not a copper-mediated process.
Characterization data for products shown in Tables 1 & 4
(E)-Ethyl 3-(3,5-dimethoxy-phenoxy)-acrylate (Tables 1 & 4; 2)
General procedure A was followed (rt, 1 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 3,5-dimethoxyphenol (101.8 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5 % EtOAc in hexane) provided pure E-ether 2 (108.8 mg, 0.431 mmol, 98% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.78 (1H, d, J = 12.2 Hz), δ 6.30 (1H, t, J =2.2Hz), δ 6.25-6.24 (2H, m), δ 5.59 (1H, d, J = 12.2 Hz), δ 4.22 (2H, q, J = 3.6 Hz, J = 10.7 Hz), δ 3.80 (6H, s), δ 1.31 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 167.1, 161.6, 158.4, 157.5, 102.3, 96.9, 96.5, 60.0, 55.4, 14.2 ppm. HRMS (EI), calcd. for C13H16O5 252.0998; found 252.1007.
(E)-Ethyl 3-(4-tert-butyl-phenoxy)-acrylate (Tables 1 & 4; 3)
General procedure A was followed (rt, 3 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), tert-butylphenol (99.1 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 3 (101.6 mg, 0.409 mmol, 93% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.81 (1H, d, J = 12.2 Hz), δ 7.43-7.38 (2H, m), δ 7.04-6.99 (2H, m), δ 5.54 (1H, d, J = 12.2 Hz), δ 4.21 (2H, q, J = 3.6Hz, J = 10.7Hz); δ 1.34 (9H, s), δ 1.30 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 166.2, 158.5, 152.6, 147.2, 126.5, 117.4, 101.6, 60.1, 35.3, 32.6, 15.5 ppm. HRMS (ESI), calcd. for C15H21O3 (M + H)+:249.1491; found 249.1502.
(E)-Ethyl 3-(4-fluorophenoxy)-acrylate (Tables 1 & 4; 4)
General procedure A was followed (rt, 3 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 4-fluorophenol (74 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 4 (77.8 mg, 0.370 mmol, 84% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.75 (1H, d, J = 12.2 Hz), δ 7.12-7.02 (4H, m), δ 5.52 (1H, d, J = 12.2 Hz), δ 4.21 (2H, q, J = 3.6 Hz, J = 10.7 Hz), δ 1.30 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 161.3, 159.2, 119.7, 119.6, 116.6, 116.3, 102.1, 60.0, 14.2 ppm. HRMS (EI), calcd. for C11H11O3F: 210.0692; found 210.0690.
(E)-Ethyl 3-(2-isopropylphenoxy)-acrylate (Table 1; 5)
General procedure B was followed (rt, 6 h or 40 °C, 30 min). Z-vinyl iodide 1 (100 mg, 0.44 mmol), CuI (4.2 mg, 0.022 mmol), Cs2CO3 (260 mg, 0.80 mmol), L (4.3 mg, 0.022 mmol), 2-isopropylphenol (119.9 mg, 0.88 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 5 (97.9 mg, 0.418 mmol, 95% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.80 (1H, d, J = 12.3 Hz), δ 7.34-7.30 (1H, m), δ 7.26-7.16 (2H, m), δ 7.02-6.99 (1H, m), δ 5.46 (1H, d, J = 12.3), δ 4.20 (2H, q, J = 3.6 Hz, J = 10.7 Hz), δ 3.22 (1H, hep), δ 1.35-1.23 (9H, m) ppm. 13C NMR (75 MHz, CDCl3): δ 167.3, 160.3, 152.9, 139.2, 127.0, 125.5, 118.5, 101.2, 59.9, 27.0, 22.8, 14.2 ppm. HRMS (ESI), calcd. for C14H19O3 (M + H)+: 235.1334; found 235.1334.
(E)-Ethyl 3-(naphtalen-1-yloxy) acrylate (Tables 1 & 4; 6)
General procedure A was followed (rt, 3 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), naphthalene-1-ol (95.2 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 6 (102.3 mg, 0.422 mmol, 96% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.12-8.09 (1H, m) δ 7.97 (1H, d, J = 12.2 Hz), δ 7.92-7.87 (1H, m), δ 7.73-7.70 (1H, m), δ 7.63-7.56 (2H, m), δ 7.46 (1H, t, J = 7.8 Hz), δ 7.17-7.15 (1H, m), δ 5.65 (1H, d, J = 12.2 Hz), δ 4.25 (2H, q, J = 6.0 Hz J = 15.4 Hz) δ 1.31 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 167.1, 159.6, 151.7, 134.6, 127.7, 126.8, 126.4, 125.7, 125.4, 125.0, 121.3, 112.7, 102.4, 60.0, 14.2 ppm. HRMS (ESI), calcd. for C15H15O3 (M + H)+:243.1021; found 243.1013.
(E)-Ethyl 3-(benzo[d]thiazol-2-yloxy) acrylate (Table 1; 7)
General procedure B was followed (40 °C 30 min-2 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), CuI (4.2 mg, 0.022 mmol), Cs2CO3 (260 mg, 0.80 mmol), L3 (4.3 mg, 0.022 mmol), benzothiazol-2-ol (133.0 mg, 0.88 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (10% EtOAc in hexane) provided 7 (100.9 mg, 0.405 mmol, 92% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 14.3 Hz), δ 7.48-7.36 (3H, m), δ 7.31-7.26 (1H, m), δ 6.95 (1H, d, J = 14.3 Hz), δ 4.30 (2H, q, J = 3.6 Hz, J = 10.7 Hz), δ 1.36 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 168.8, 167.1, 134.8, 133.5, 126.8, 124.7, 122.9, 122.0, 111.5, 110.4, 60.7, 14.2 ppm. HRMS (ESI), calcd. for C12H12NO3S (M + H)+: 250.0538; found 250.0540.
(E)-Ethyl 3-(pyridin-3-yloxy) acrylate (Tables 1 & 4; 8)
General procedure A was followed (rt, 3 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), pyridine-3-ol (62.8 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (15% EtOAc in hexane) provided 8 (73.0 mg, 0.378 mmol, 86% yield) as a colorless oil.
1H NMR (300 MHz, CDCl3): δ 8.48 (2H, m), δ 7.78 (1H, d, J = 12.2 Hz), δ 7.46-7.41 (1H, m), δ 7.37-7.33 (1H, m), δ 5.62 (1H, d, J = 12.2 Hz), δ 4.22 (2H, q, J = 3.6 Hz, J = 10.7 Hz), δ 1.30 (3H, t, J = 7.1) ppm. 13C NMR (75 MHz, CDCl3): δ 166.6, 157.8, 152.2, 146.2, 140.7, 125.1, 124.2, 103.6, 60.2, 14.2 ppm. HRMS (ESI), calcd. for C10H12NO3 (M + H)+: 194.0817; found 194.0819.
(E)-Ethyl 3-(1H-pyrazol-1-yl) acrylate (Tables 1 & 4; 9)
General procedure A was followed (rt, 2 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 1H-pyrazole (34 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 9 (67.3 mg, 0.405 mmol, 92% yield) as a white solid. 1H NMR (300 MHz, CDCl3): δ 8.03 (1H, d, J = 13.9 Hz), δ 7.74 (1H, s, br), δ 7.68 (1H, d, J = 2.5 Hz), δ 6.46 (1H, m), δ 6.39 (1H, d, J = 13.9 Hz), δ 4.29 (2H, q, J = 7.1), δ 1.32 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 166.4, 143.3, 139.4, 129.8, 108.9, 105.7, 60.5, 14.1 ppm; HRMS (EI), calcd. for C8H10N2O2: 166.0742; found 166.0716.
E-Ethyl 3-(5-(methylthio)-1H-tetrazol-1-yl) acrylate (Table 1; 10)
General procedure B was followed (40 °C, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), CuI (4.2 mg, 0.022 mmol), Cs2CO3 (73 mg, 0.88 mmol), L (4.3 mg, 0.022 mmol), 5-(methylsulfanyl)-1H-tetraazole (76.6 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 10 (77 mg, 0.36 mmol, 81 % yield) as a yellowish semisolid. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 14.1 Hz, HC(N)=CH), δ 6.77 (1H, d, J = 14.1 Hz HC=CH(N)), δ 4.34 (2H, q, J = 7.1 Hz, H2CCH3), δ 1.37 (3H, t, J = 7.1 Hz, H3C-CH2) ppm; HRMS (EI), calcd. for C7H10N4O2S: 214.0524; found: 214.0515.
(E)-Ethyl 3-(1H-indol-1-yl) acrylate (Table 1; 11)
General procedure B was followed (40°C, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), CuI (4.2 mg, 0.022 mmol), Cs2CO3 (73 mg, 0.88 mmol), L (4.3 mg, 0.022 mmol), indole (77.3 mg, 0.66 mmol) and DMF (2.0 mL)) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 11 (88 mg, 0.41 mmol, 93 % yield) as an off-white solid. 1H NMR (300 MHz, CDCl3): δ 8.33 (1H, d, J = 14 Hz), δ 7.64 (d, 1H, J = 3.3Hz), δ 7.61 (1H, m), δ 7.41 (1H, d, J = 3.5 Hz), δ 7.35 (1H, m), 7.25 (1H, m), δ 6.75 (1H, d, J = 3.5 Hz), δ 6.00 (1H, d, J = 14 Hz), δ 4.31 (2H, q, J = 7.1 Hz), δ 1.37 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 167.3, 137.0, 136.0, 129.7, 123.8, 123.4, 122.3, 121.4, 109.9, 108.6, 100.5, 103.2, 60.2, 14.3 ppm. Elemental analysis calcd. for C13H13NO2·0.27 H2O: C, 70.95; H, 6.20; N, 6.36; found: C, 70.95; H, 6.10; N, 6.32.
(E)-Ethyl 3-(1H-indazol-1-yl) acrylate (Tables 1 & 4; 12)
General procedure A was followed (rt, 2 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 1H-indazole (78 mg, 0.66 mmol)and DMF (2.0 mL)) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 12 (80 mg, 0.37 mmol, 84% yield) as a white solid. 1H NMR (300 MHz, CDCl3): δ 8.38 (1H, d, J = 13.7 Hz), δ 8.22 (s, 1H), δ 7.79 (1H, d, J = 8 Hz), δ 7.69 (1H, d, J = 8.4 Hz), δ 7.54 (1H, t, J = 7.5 Hz), δ 7.32 (1H, t, J = 7.5 Hz), δ 6.55 (1H, d, J = 13.7 Hz), δ 4.32 (2H, q, J = 7.1 Hz), δ 1.37 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 167.2, 139.5, 138.9, 136.5, 128.2, 125.5, 123.1, 121.5, 109.5, 103.2, 60.3, 14.1 ppm. Elemental analysis calcd. for C12H12N2O2: C, 66.65; H, 5.59; N, 12.96; found: C, 66.51; H, 5.65; N, 12.85.
(E)-Ethyl 3-(1H-benzo[d][1,2,3]triazol-1-yl) acrylate (Tables 1 & 4; 13)
General procedure A was followed (rt, 2 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 1H-1,2,3-benzotriazole (59 mg, 0.66 mmol) and DMF (2.0 mL)) were stirred to obtain the crude product. Column chromatography on silica gel (5 % EtOAc in hexane) provided 13 (80.3 mg, 0.37 mmol, 84 % yield) as a white crystalline solid. 1H NMR (300 MHz, CDCl3): δ 8.55 (1H, d, J = 14.3 Hz), δ 8.17 (d, 1H, J = 8.3 Hz), δ 7.77 (1H, d, J = 8.3 Hz), δ 7.66 (1H, t, J = 7.4 Hz), δ 7.50 (1H, t, J = 7.4 Hz), δ 6.79 (1H, d, J = 14.3 Hz), δ 4.36 (2H, q, J = 7.1 Hz), δ 1.39 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 165.8, 146.5, 135.0, 131.4, 129.2, 125.3, 120.7, 110.0, 108.1, 60.9, 14.1 ppm. HRMS (EI), calcd. for C11H11N3O2: 217.0851; found: 217.0832.
(E)-Ethyl 3-(1H-imidazol-1-yl) acrylate (Tables 1 & 4; 14)
General procedure A was followed (rt, 2 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 1H-imidazole (34 mg, 0.66 mmol) and DMF (2.0 mL)) were stirred to obtain the crude product. Column chromatography on silica gel (10 % EtOAc in hexane) provided 14 (70 mg, 0.42 mmol, 96% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.93 (1H, d, J = 14.2 Hz), δ 7.81 (1H, s), δ 7.26 (1H, s), δ 7.20 (1H, s), δ 6.10 (1H, d, J = 14.2 Hz), δ 4.30 (2H, q, J = 7.1 Hz), δ 1.33 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 166.1, 137.7, 136.2, 131.6, 116.1, 107.1, 60.8, 14.1 ppm. HRMS (EI), calcd. for C8H10N2O2: 166.0742; found: 166.0769.
(E)-Ethyl 3-(1H-1,2,4-triazol-1-yl) acrylate (Tables 1 & 4; 15)
General procedure A was followed (rt, 2 h). E-vinyl iodide 29 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 1H-1,2,4-triazole (45.6 mg, 0.66 mmol) and DMF (2.0 mL)) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 15 (67.6 mg, 0.40 mmol, 92% yield) as a white solid. 1H NMR (300 MHz, CDCl3): δ 8.35 (1H, s), 8.08 (1H, s), 8.03 (1H, d, J = 13.8 Hz), δ 6.63 (1H, d, J = 13.8 Hz), δ 4.31 (2H, q, J = 7.1 Hz), δ 1.35 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 165.5, 153.5, 144.5, 134.9, 110.4, 61.0, 14.1 ppm. HRMS (EI), calcd. for C7H9N3O2: 167.0694; found: 167.0666.
Characterization data for products shown in Table 3
(Z)-Ethyl 3-(3,5-dimethoxyphenoxy) acrylate (16)
General procedure A was followed (rt, 1 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 3,5-dimethoxyphenol (101.8 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 16 (108.8 mg, 0.431 mmol, 98% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 6.89 (1H, d, J = 7.0 Hz), δ 6.30 (3H, s), δ 5.17 (1H, d, J = 7.0 Hz), δ 4.23 (2H, q, J = 10.7 Hz, J = 3.6 Hz), δ 3.80 (6H, s), δ 1.33 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 161.5, 158.7, 153.5, 100.1, 96.8, 96.1, 59.8, 55.4, 14.2 ppm. HRMS (EI), calcd. for C13H16O5: 252.0998; found: 252.1007.
(Z)-Ethyl 3-(4-tert-butylphenoxy) acrylate (17)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 4-tert-butylphenol (99.2 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 17 (101.6 mg, 0.409 mmol, 93% yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.40 (2H, d, J = 8.7 Hz), δ 7.06 (2H, d, J = 8.7 Hz), δ 6.88 (1H, d, J = 7.2 Hz), δ 5.15 (1H, d, J = 7.0 Hz), δ 4.23 (2H, q, J = 10.7 Hz, J = 3.6 Hz); δ 1.35-1.30 (12H, m) ppm. 13C NMR (75 MHz, CDCl3): δ 164.8, 155.0, 154.5, 147.7, 126.6, 117.1, 99.6, 59.9, 34.4, 31.4, 14.3 ppm. HRMS (ESI), calcd. for C15H21O3 (M + H)+: 249.1491; found: 249.1490.
(Z)-Ethyl 3-(4-fluorophenoxy) acrylate (18)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 4-fluorophenol (74.0 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 18 (77.8 mg, 0.370 mmol, 84 % yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.11-7.04 (4H, m), δ 6.81 (1H, d, J = 7.0 Hz), δ 5.18 (1H, d, J = 7.0 Hz), δ 4.23 (2H, q, J = 10.7 Hz, J = 3.6 Hz), δ 1.33 (3H, t, J = 7.1) ppm. 13C NMR (75 MHz, CDCl3): δ 164.5, 161.1, 157.8, 154.1, 119.1, 116.5, 100.2, 59.9, 14.2 ppm. HRMS (EI), calcd. for C11H11O3F: 210.0692; found: 210.0690.
(Z)-Ethyl 3-(2-isopropyl-phenoxy) acrylate (19)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), 2-isopropylphenol (90 mg, 0.66 mmol) and DMF (2.0 mL)) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 19 (97.9 mg, 0.418 mmol, 95 % yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.33-7.30 (1H, m), δ 7.25-7.16 (2H, m), δ 6.98 (1H, d, J = 1.2 Hz), δ 6.87 (1H, d, J = 6.9 Hz), δ 5.15 (1H, d, J = 6.9 Hz), δ 4.25 (2H, q, J = 10.7 Hz, J = 3.6Hz), δ 3.40 (1H, hep), δ 1.36-1.21 (9H, m) ppm. 13C NMR (75 MHz, CDCl3): δ 165.0, 154.9, 154.6, 139.0, 127.0, 125.0, 116.8, 99.6, 59.9, 27.5, 22.8, 14.4 ppm. HRMS (ESI), calcd. for C14H19O3 (M + H)+: 235.1334; found: 235.1326.
(Z)-Ethyl 3-(naphtalen-1-yloxy) acrylate (20)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), naphthalene-1-ol (95.2 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (5% EtOAc in hexane) provided 20 (102.3 mg, 0.422 mmol, 96 % yield) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.39-8.36 (1H, m), δ 7.88-7.85 (1H, m), δ 7.67 (1H, d, J = 8.2 Hz), δ 7.60-7.55 (2H, m), δ 7.42 (1H, t, J = 7.8 Hz), δ 7.11-7.08 (2H, m), δ 5.28 (1H, d, J = 6.9 Hz), δ 4.31 (2H, q, J = 10.7 Hz J = 3.6Hz), δ 1.39 (3H, t, J = 7.1 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 164.9, 153.8, 153.0, 134.5, 127.4, 126.9, 126.3, 125.9, 125.3, 124.4, 121.9,110.4, 100.3, 59.9, 14.4 ppm. HRMS (ESI), calcd. for C15H15O3 (M + H)+: 243.1021; found: 243.1013.
(Z)-Ethyl 3-(pyridin-3-yloxy) acrylate (21)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (100 mg, 0.44 mmol), DABCO (98.7 mg, 0.88 mmol), pyridine-3-ol (62.8 mg, 0.66 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (15% EtOAc in hexane) provided 21 (73.0 mg, 0.378 mmol, 86 % yield) as a colorless oil.
1H NMR (300 MHz, CDCl3): δ 8.49-8.42 (2H, m), δ 7.45-7.41 (1H, m), δ 7.33-7.29 (1H, m), δ 6.83 (1H, d, J = 6.9Hz), δ 5.25 (1H, d, J = 6.9Hz), δ 4.21 (2H, q, J = 3.5 Hz, J = 10.7 Hz), δ 1.30 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 164.3, 153.5, 152.7, 146.0, 140.1, 124.8, 124.2, 101.8, 60.1, 14.3 ppm. HRMS (ESI), calcd. for C10H12NO3 (M + H)+: 194.0817; found: 194.0821.
(Z)-Ethyl 3-(1H-pyrazol-1-yl) acrylate (22)
General procedure A was followed (rt, 1 h). Z-vinyl iodide 1 (90 mg, 0.4 mmol), DABCO (90 mg, 0.8 mmol), 1H-pyrazole (40.5 mg, 0.6 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (10% EtOAc in hexane) provided pure Z-vinyl amine 22 (60 mg, 90 % yield) as a colorless liquid. 1H NMR (300 MHz, CDCl3): δ 9.13 (1H, d, J = 2.7 Hz), δ 7.68 (1H, d, J = 1.5 Hz), δ 7.32 (1H, d, J = 11.1 Hz), δ 6.59 (1H, t, J = 2.1 Hz), δ 5.44 (1H, d, J = 11.1 Hz), δ 4.24 (2H, q, J = 7.2 Hz), δ 1.34 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 165.0, 142.2, 137.8, 133.0, 108.6, 102.2, 60.6, 14.2 ppm; HRMS (ESI) calcd. for C8H11N2O2 (M + H)+: 167.0821; found: 167.0830.
(Z)-Ethyl 3-(3-(trifluoromethyl)-1H-pyrazol-1-yl) acrylate (23)
General procedure A was followed (rt, 1 h). Z-vinyl iodide 1 (90 mg, 0.4 mmol), DABCO (90. mg, 0.8 mmol), 3-(trifluoromethyl) pyrazole (81.8 mg, 0.6 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (10% EtOAc in hexane) provided pure Z-vinyl amine 23 (87 mg, 93 % yield) as a white solid; mp 36.1-37.1 °C. 1H NMR (300 MHz, CDCl3): δ 9.11 (1H, m), δ 7.27 (1H, d, J = 11.1 Hz), δ 6.65 (1H, d, J = 3.0 Hz), δ 5.62 (1H, d, J = 11.1 Hz), δ 4.26 (2H, q, J = 7.2), δ 1.34 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 164.4, 145.0, 144.5, 136.7, 134.7, 122.5, 119.0, 106.3, 106.2, 61.0, 14.1 ppm; Anal. calc. for C9H9N2O2F3·0.07 CH3(CH2)4CH3: C, 47.12; H, 4.19; N, 11.66; found: C, 47.18; H, 3.90; N, 11.54; HRMS (ESI) calcd. for C9H10N2O2F3 (M + H)+: 235.0694; found: 235.0717.
(Z)-Ethyl 3-(1H-indazol-1-yl) acrylate (24)
General procedure A was followed (rt, 2 h). Z-vinyl iodide 1 (90 mg, 0.4 mmol), DABCO (90. mg, 0.8 mmol), indazole (70.5 mg, 0.6 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (15% EtOAc in hexane) provided pure Z- vinyl amine 24 (77 mg, 89 % yield) as a colorless liquid. 1H NMR (300 MHz, CDCl3): δ 8.18 (1H, s), δ 7.76 (1H, dd, J1 = 8.1 Hz, J1 = 1.2 Hz), δ 7.46 (2H, m), δ 7.33 (1H, d, J = 9.9 Hz), δ 7.29 (1H, m), δ 5.70 (1H, d, J = 9.9), δ 4.28 (2H, q, J = 7.2 Hz), δ 1.26 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 166.1, 139.5, 137.4, 129.7, 127.5, 124.8, 122.5, 121.4, 109.9, 106.7, 60.8, 14.1 ppm; HRMS (ESI) calcd. for C12H13N2O2 (M + H)+: 217.0977; found: 217.1004.
(Z)-Ethyl 3-(1H-benzo[d][1,2,3]triazol-1-yl) acrylate (25)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (90 mg, 0.4 mmol), DABCO (90. mg, 0.8 mmol), 1H-benzotriazole (71.3 mg, 0.6 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (20 % EtOAc in hexane) provided pure Z-vinyl amine 25 (79 mg, 91 % yield) as a yellowish brown liquid.
1H NMR (300 MHz, CDCl3): δ 8.13 (1H, d, J = 9.0 Hz), δ 7.58 (t, 2H, J = 7.8 Hz), δ 7.46 (2H, t, J = 7.8 Hz), δ 6.10 (1H, d, J = 9.0 Hz), δ 4.20 (2H, q, J = 7.2 Hz), δ 1.14 (3H, t, J = 7.2 Hz) ppm; 13C NMR (75 MHz, CDCl3): δ 164.3, 145.6, 132.4, 129.2, 128.3, 124.6, 120.3, 114.4, 110.4, 61.3, 13.8 ppm; HRMS (ESI) calcd. for C11H12N3O2 (M + H)+: 218.0930; found: 218.0931.
(Z)-Ethyl 3-(3-methyl-1H-pyrazol-1-yl) acrylate (26)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (90 mg, 0.4 mmol), DABCO (90. mg, 0.8 mmol), 3-methylpyrazole (49.5 mg, 0.6 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (10% EtOAc in hexane) provided pure Z-vinyl amine 26 (54 mg, 75 % yield) as a light yellow liquid. 1H NMR (300 MHz, CDCl3): δ 9.04 (1H, d, J = 2.7 Hz), δ 7.22 (1H, d, J = 11.1 Hz), δ 6.23 (1H, d, J = 2.7 Hz), δ 5.33 (1H, d, J = 11.1), δ 4.23 (2H, q, J = 7.2 Hz), δ 1.33 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 165.2, 151.9, 137.7, 139.9, 109.2, 100.7, 60.4, 14.2, 13.4 ppm; HRMS (ESI) calcd. for C9H13N2O2 (M + H)+: 181.0977; found: 181.0975.
(Z)-Ethyl 3-(1H-1,2,4-triazol-1-yl) acrylate (27)
General procedure A was followed (rt, 3 h). Z-vinyl iodide 1 (90 mg, 0.4 mmol), DABCO (90. mg, 0.8 mmol), 1,2,4-triazole (41.3 mg, 0.6 mmol) and DMF (2.0 mL) were stirred to obtain the crude product. Column chromatography on silica gel (20 % EtOAc in hexane) provided pure Z-vinyl amine 27 (61.5 mg, 92 % yield) as a colorless liquid. 1H NMR (300 MHz, CDCl3): δ 9.68 (1H, s), δ 8.01 (1H, s), δ 7.26 (1H, d, J = 11.1 Hz), δ 5.71 (1H, d, J = 11.1 Hz), δ 4.26 (2H, q, J = 7.2 Hz), δ 1.33 (3H, t, J = 7.2 Hz) ppm. 13C NMR (75 MHz, CDCl3): δ 164.3, 152.0, 146.7, 133.5, 107.5, 61.2, 14.1 ppm; HRMS (ESI) calcd. for C7H10N3O2 (M + H)+: 168.0773; found: 168.0783.
(Z)-Ethyl 3-(4-tert-butylphenylsulfanyl) acrylate (30) (Scheme 2)
To a suspension of Cs2CO3 (286.3 mg 0.88 mmol) in 5 mL DMF vinyl iodide 1 (100 mg, 0.44 mmol) and 4-tert-butyl-benzenethiol (88 mg, 0.53 mmol) were added respectively at rt. The reaction mixtures were stirred either for 30 minutes at 45°C or at room at room temperature for 2 h to obtain 30 (114 mg, 98% yield) as a colorless oil. Column chromatography solvent (2–3% EtOAc in hexane) provided pure 30. The 1HNMR, 13CNMR and HRMS data for compound 30 is in agreement according to the reported literature.36
1H NMR (300 MHz, CDCl3): δ 7.43 (4H, q, J = 13.4, 8.5 Hz), δ 7.28 (1H, d, J = 10.1 Hz), δ 5.90 (1H, d, J = 10.1 Hz), δ 4.27 (2H, q, J = 14.3, 7.1 Hz), δ 1.34 (12H, t, J = 7.9 Hz); 13C NMR (75 MHz, CDCl3): δ 166.5, 151.5, 150.4, 132.6, 131.0,126.3, 112.9, 60.2, 34.5, 31.1, 14.3. HRMS (ESI) (M + H)+, Calcd. for C15H21O2S 265.1262; Found 265.1259.
Supplementary Material
Acknowledgement
We thank Dr. Douglas Stafford, Director, Milwaukee Institute for Drug Discovery, for technical advice and the NIH and the Research Growth Initiative of the University of Wisconsin-Milwaukee for financial support. We also wish to thank Professor Wim Dehaen (Department of Chemistry, University of Leuven, Belgium) who pointed out our mistake in the assignments in tables 2 and 4 in reference 7 to us.
Footnotes
Supporting Information Available: Copies of 1H NMR and 13C NMR are provided in the Supporting Information sections. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.This work was presented previously in preliminary fashion; see: Kabir MS, Namjoshi OA, Verma R, Asad MSA, Lorenz M, Cook JM. Abstracts of Papers, 241st ACS National Meeting; Anaheim, CA, United States. 2011. ORGN-188.
- 2.Taillefer M, Ouali A, Renard B, Spindler J-F. Chem. Eur. J. 2006;12:5301. doi: 10.1002/chem.200501411. [DOI] [PubMed] [Google Scholar]
- 3.Bao W, Xin Lv YL. Synthesis. 2008;12:1911. [Google Scholar]
- 4.Ouali A, Laurent R, Caminade A-M, Majoral J-P, Taillefer M. J. Am. Chem. Soc. 2006;128:15990. doi: 10.1021/ja066505s. [DOI] [PubMed] [Google Scholar]
- 5.Kaddouri H, Vicente V, Ouali A, Ouazzani F, Taillefer M. Angew. Chem. Int. Ed. 2009;48:333. doi: 10.1002/anie.200800688. [DOI] [PubMed] [Google Scholar]
- 6.US outlook for acrylic acids and derivatives with forecasts to 2006 and 2011, Freedonia Industry Study, # 1488.
- 7.Kabir MS, Lorenz M, Namjoshi OA, Cook JM. Org. Lett. 2010;12:464–467. doi: 10.1021/ol9026446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kabir MS, Namjoshi OA, Verma R, Polanowski R, Krueger SM, Sherman D, Rott MA, Schwan WR, Monte A, Cook JM. Bioorg. Med. Chem. 2010;18:4178. doi: 10.1016/j.bmc.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 9.Neck VN, Pannecouque C, Vanstreels E, Stevens M, Dehaen W. Bioorg. Med. Chem. 2008;16:9487. doi: 10.1016/j.bmc.2008.09.051. [DOI] [PubMed] [Google Scholar]
- 10.Monnier F, Taillefer M. Angew. Chem. Int. Ed. 2009;48:6954. doi: 10.1002/anie.200804497. (Minireview) and references cited therein. [DOI] [PubMed] [Google Scholar]
- 11.Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;42:5400. doi: 10.1002/anie.200300594. (Review) and references cited therein. [DOI] [PubMed] [Google Scholar]
- 12.Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;115:5558. [Google Scholar]
- 13.Kunz K, Scholz U, Ganzer D. Synlett. 2003:2428. [Google Scholar]; (e) Beletskaya IP, Cheprakov AV. Coord. Chem. Rev. 2004;248:2337. [Google Scholar]
- 14.Fraln R, Kikeji D. Synthesis. 2006:2271. [Google Scholar]
- 15.Corbet JP, Mignani G. Chem. Rev. 2006;106:2651. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]
- 16.Kienle M, Dubakka SR, Brade K, Knochel P. Eur. J. Org. Chem. 2007:4166. [Google Scholar]
- 17.Carril M, SanMartin R, Dominguez E . Chem. Soc. Rev. 2008;37:639. doi: 10.1039/b709565c. [DOI] [PubMed] [Google Scholar]
- 18.Shafir A, Lichtor PA, Buchwald SL. J. Am. Chem. Soc. 2007;129:3490. doi: 10.1021/ja068926f. [DOI] [PubMed] [Google Scholar]
- 19.Altman RA, Buchwald SL. Org. Lett. 2007;9:643. doi: 10.1021/ol062904g. [DOI] [PubMed] [Google Scholar]
- 20.Jiang L, Job GE, Klapars A, Buchwald SL. Org. Lett. 2003;5:3667. doi: 10.1021/ol035355c. [DOI] [PubMed] [Google Scholar]
- 21.Gujadhur RK, Bates CG, Venkataraman D. Org. Lett. 2001;3:4315. doi: 10.1021/ol0170105. [DOI] [PubMed] [Google Scholar]
- 22.Harada H, Thalji RK, Bergman RG, Ellman JA. J. Org. Chem. 2008;73:6772. doi: 10.1021/jo801098z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nordmann G, Buchwald SL. J. Am. Chem. Soc. 2003;125:4978. doi: 10.1021/ja034809y. [DOI] [PubMed] [Google Scholar]
- 24.Chemler SR, Fuller PH. Chem Soc. Rev. 2007;36:1153. doi: 10.1039/b607819m. [DOI] [PubMed] [Google Scholar]
- 25.For a highlight on recent major developments in C-C, C-N and C-N coupling please see: Evano G, Blanchard N, Toumi M. Chem. Rev. 2008;108:3054. doi: 10.1021/cr8002505..
- 26.Monnier F, Taillefer M. Angew. Chem. Int. Ed. 2008;47:3096. doi: 10.1002/anie.200703209. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y-G, Chen H-H. Org. Lett. 2006;8:5609. doi: 10.1021/ol062339h. [DOI] [PubMed] [Google Scholar]
- 28.Pan X, Cai Q, Ma D. Org. Lett. 2004;6:1809. doi: 10.1021/ol049464i. [DOI] [PubMed] [Google Scholar]
- 29.Smith AB, III, Duffey MO, Basu K, Walsh SP, Suennemann HW, Frohn M. J. Am. Chem. Soc. 2008;130:422. doi: 10.1021/ja078293k. [DOI] [PubMed] [Google Scholar]
- 30.Nicolaou KC, Leung GYC, Dethe DH, Guduru R, Sun YP, Lim CS, Chen DY-K. J. Am. Chem. Soc. 2008;130:10019. doi: 10.1021/ja802803e. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez M, Buchwald SL. Org. Lett. 2007;9:973. doi: 10.1021/ol062978s. [DOI] [PubMed] [Google Scholar]
- 32.Shen R, Lin CT, Bowman EJ, Bowman BJ, Proco JA., Jr J. Am. Chem. Soc. 2003;125:7889. doi: 10.1021/ja0352350. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Shao N, Huryk R, Palani A, Aslanian R, Seidel-Dugan C. Org. Lett. 2009;11:867. doi: 10.1021/ol802772s. [DOI] [PubMed] [Google Scholar]
- 34. Cesati RR, III, Dwyer G, Jones RC, Hayes MP, Yalamanchili P, Casebier DS. Org. Lett. 2007;9:5617. doi: 10.1021/ol7025729.. Other contributors were cited in the aforementioned referenced reviews and minireviews.
- 35.Bates CG, Saejueng P, Doherty MQ, Venkataraman D. Org. Lett. 2004;6:5005. doi: 10.1021/ol0477935. [DOI] [PubMed] [Google Scholar]
- 36.Kabir MS, Lorenz M, Van Linn ML, Namjoshi OA, Ara S, Cook JM. J. Org. Chem. 2010;75:3626. doi: 10.1021/jo1004179. [DOI] [PubMed] [Google Scholar]
- 37.Jencks WP. Acc. Chem. Res. 1980;13:161–169. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.