

Abstract
Exocyclic ethenobases are highly mutagenic DNA lesions strongly implicated in inflammation and vinyl chloride-induced carcinogenesis. While the alkyladenine DNA glycosylase, AAG (or MPG), binds the etheno lesions 1,N6-ethenoadenine (εA) and 3,N4-ethenocytosine (εC) with high affinity, only εA can be excised to initiate base excision repair. Here, we discover that the human AlkB homolog 2 (ALKBH2) dioxygenase enzyme catalyzes direct reversal of εC lesions in both double- and single-stranded DNA with comparable efficiency to canonical ALKBH2 substrates. Notably, we find that in vitro, the non-enzymatic binding of AAG to εC specifically blocks ALKBH2-catalyzed repair of εC but not that of methylated ALKBH2 substrates. These results identify human ALKBH2 as a repair enzyme for mutagenic εC lesions and highlight potential consequences for substrate-binding overlap between the base excision and direct reversal DNA repair pathways.
Keywords: AAG, AlkB, ALKBH2, DNA repair, ethenocytosine, MPG
1. Introduction
The DNA adducts 1,N6-ethenoadenine (εA) and 3,N4-ethenocytosine (εC), are exocyclic DNA base lesions generated through the reaction of DNA with the aldehyde products of lipid peroxidation, or with metabolites of the industrial agent vinyl chloride [1]. In both bacteria and humans, etheno adducts have been shown to be extremely mutagenic and cytotoxic, leading to miscoding or replication blocks (reviewed in [2]). Significantly, the generation of etheno lesions has been linked to various disease states, including chronic inflammation-associated colon cancer, lung adenocarcinoma and human degenerative disorders [3-5].
Some etheno adducts in the genome can be repaired through the base excision repair (BER) pathway, initiated by excision of the damaged base by a DNA glycosylase (reviewed in [6, 7]). In mammalian cells, the alkyladenine/methylpurine DNA glycosylase, AAG/MPG (hereafter referred to as AAG) represents the primary BER enzyme for a variety of modified base lesions, including 3-methyladenine, hypoxanthine, and εA [8-10]. Recent studies have expanded the substrates recognized by AAG to include 1-methylguanine and 1,N2-ethenoguanine in double-stranded (ds) DNA, as well as hypoxanthine or εA lesions in single-stranded (ss) DNA [11]. Surprisingly however, when AAG binds the εC base lesion in either ss or dsDNA it cannot carry out base excision and instead forms an abortive complex [11-13]. This lack of glycosylic bond cleavage at εC DNA lesions is consistent with the observation that extracts from Aag-null mouse tissues are deficient for excision of εA and hypoxanthine but not deficient for εC excision [9, 10]. Besides being mutagenic, εC lesions can block DNA replication in vitro and are known to indirectly inhibit excision of εA lesions in vivo by hijacking AAG [12]. While weak glycosylase activity on εC substrates has been detected for thymine DNA glycosylase, uracil DNA glycosylase or methyl-CpG-binding protein 4, the primary substrates of these glycosylases are thymine mispaired with guanine, uracil in any base pair or deaminated 5-methylcytosine (i.e. thymine) at CpG sites, respectively [14-18], suggesting that other repair enzymes could be responsible for εC repair.
DNA repair enzymes belonging to the AlkB family of non-heme iron-dependent dioxygenases are now known to repair etheno base lesions through epoxidation of the lipid-derived alkyl chain and its release as glyoxal [19, 20]. In particular, bacterial AlkB directly reverses εA and εC to normal adenine and cytosine in DNA [19, 21]. The human genome encodes several proteins with AlkB dioxygenase motifs, including the eight AlkB homologs (ALKBH1 through ALKBH8) as well as the Fat mass and obesity-associated (FTO) protein (reviewed in [22]). Among the AlkB homologs, ALKBH2 and ALKBH3 have been shown to repair 1-methyladenine (1-meA) and 3-methylcytosine (3-meC) in both DNA and RNA as well as εA lesions in DNA [21, 23-25].
Here, we have purified human ALKBH2 from human cells and discover that it can directly reverse εC lesions in DNA in addition to its known substrates, 1-meA, 3-meC and εA. ALKBH2 displays robust repair activity on εC in both single- and double-stranded DNA at rates comparable to its known methylated substrate, 3-meC. Moreover, we find that AAG specifically inhibits the repair of either εC or εA by ALKBH2 but does not inhibit the repair of other ALKBH2 substrates. These results identify a novel substrate for ALKBH2 and demonstrate potential interactions between AKLBH2 and the base excision repair pathway.
2. Materials and Methods
2.1 Expression and purification of ALKBH proteins from human cells
The coding region for full-length human ALKBH2 (NM_001001655) was PCR amplified and cloned into a modified version of pcDNA3.1 (Invitrogen) [26, 27] for expression as an N-terminal triple FLAG-streptavidin binding peptide fusion protein. ALKBH2Δ (AB277859), missing amino acid residues 94-261 and replaced with a different protein isoform of 94-157 amino acid residues, was cloned by RT-PCR from total RNA extracted from HeLa human cervical carcinoma cells. All constructs were verified by DNA sequencing.
Empty pcDNA3.1-FLAG vector or pcDNA3.1-FLAG-ALKBH2 protein expression constructs (20 μg) were transiently transfected by calcium phosphate precipitation into 293T human embryonic kidney cells followed by cellular extract production as previously described [27]. Whole cell extract from transiently transfected cells (1 mg of total protein) was rotated with 10 μL of FLAG M2 antibody resin (Sigma) for 2 h at 4° C in wash buffer (20 mM HEPES at pH 7.9, 2 mM MgCl2, 0.2 mM EGTA, 10% glycerol, 1 mM DTT, 0.1 mM PMSF, 0.1% NP-40) with 150 mM NaCl. Resin was washed extensively using the same buffer. Bound samples were eluted with two sequential volumes of wash buffer containing 100 ug/mL of 3×FLAG peptide (Sigma). The elutions were subject to a second round of purification on streptavidin-conjugated resin (Pierce, Thermo Scientific) for 1 h at 4° C followed by three rinses in wash buffer and two rounds of elution with wash buffer containing 1 mM biotin. Protein samples were immediately flash frozen in LN2 and stored at -80° C.
2.2. Protein and DNA analysis
Cellular extracts and purified protein samples were fractionated on 4-12% Bis-Tris polyacrylamide gels (Invitrogen) followed by silver staining (SilverQuest Kit, Invitrogen) or transfer to nitrocellulose membrane for immunoblotting. Primary monoclonal mouse antibodies against the FLAG tag (F1804, Sigma) were detected using IR Dye-conjugated secondary antibodies (Rockland). Immunoblots were scanned using direct infrared fluorescence via the Odyssey System (LI-COR Biosciences).
2.3 Protein mass spectrometry
Protein identification was performed by the Biopolymers and Proteomics Facility of the David H. Koch Institute for Integrative Cancer Research as previously described [27]. Briefly, processed peptide samples were analyzed by chromatography on an Agilent Model 1100 Nanoflow HPLC system coupled with electrospray ionization on a Thermo Electron Model LTQ Ion Trap mass spectrometer. Protein identification through tandem mass spectra correlation was performed using SEQUEST [28].
2.4. Restriction enzyme-mediated oligonucleotide demethylase repair assays
Oligonucleotide repair assays were performed essentially as described [27, 29]. Radiolabeled DNA oligonucleotides (100 fmol) containing 1-meA, 3-meC, εA or εC lesions (Eurogentec, Supplementary Table 2) were incubated at 25°C for 1 hour in a total volume of 50 uL containing 50 mM Tris pH 8.0, 2 mM ascorbic acid, 0.5 mM α-ketoglutarate, 40 uM FeSO4 and 10 uL of purified enzyme (∼50 ng protein) or buffer followed by heat inactivation at 70°C for 15 min. For dsDNA substrates, radiolabeled oligonucleotides were pre-annealed with an equimolar amount of complementary DNA prior to reaction. For ssDNA substrates, a 2-fold excess of complementary oligonucleotide was hybridized to form dsDNA after heat inactivation. An aliquot of each reaction was digested with DpnII (New England Biolabs) for 1 h at 37°C followed by electrophoresis on 7M urea/18% denaturing polyacrylamide gels for 1 hour at 500V. Gels were visualized using a Cyclone Phosphorimager (Perkin Elmer).
Kinetic analysis experiments were carried out under single-turnover conditions using an excess of ALKBH2 enzyme (10 pmol) to the labeled DNA oligonucleotide substrate (100 fmol). An aliquot of the reaction mixture was removed at 1, 2, 3, 5, 10, 15, 30, 45 and 60 min for heat inactivation at 70°C for 15 min. The amount of cleaved product to uncleaved substrate was determined using OptiQuant (Packard Instruments) and normalized against the signal in the buffer control reaction. Enzymatic rate constants were determined as described previously [11] using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA) by fitting the single-turnover kinetic data into the One Phase Exponential Association equation: y=ymax(1-e-kt) where y is the amount of substrate cleaved at any particular time point, ymax is the maximum amount of cleaved substrate, t is time, and kobs is the observed rate constant. Quantification represents the average of at least three independent experiments.
2.5 AAG protein studies
For AAG inhibition studies, increasing amounts of AAG protein were pre-mixed with ALKBH2 before addition to each repair reaction at the indicated concentration. Reactions were incubated at 25°C for 1 hour before heat inactivation and PAGE analysis as described above. Purification and analysis of full-length AAG, AAGΔ80 and AAG E125Q protein samples used in this study were previously described [11]. Escherichia coli single stranded DNA binding protein (SSB02200, Epicentre Biotechnologies) and bovine serum albumin (New England Biolabs) was tested at 200 nM as specificity controls.
3. Results
3.1 Expression and purification of human ALKBH2
Previous studies with human ALKBH proteins have utilized recombinant human proteins purified from Escherichia coli [23, 25, 30]. Here, we overexpressed and purified ALKBH2 from human embryonic kidney cells to reproduce the biochemical properties of human AlkB proteins from their physiological source. In addition, we cloned and purified an ALKBH2 variant (ALKBH2Δ) encoded by an alternatively spliced transcript with a frameshift deletion of the C-terminal dioxygenase domain [31]. Tagged ALKBH2 proteins were tandem affinity purified from whole cell extract along with a parallel mock purification from cells transfected with empty vector alone (Fig. 1A-B). Polyacrylamide gel fractionation and silver staining of the purifications confirmed that the tagged ALKBH2 or ALKBH2Δ was the predominant protein band in each of the purifications with very few copurifying polypeptides (Fig. 1C). The mock and ALKBH2 samples were also analyzed by liquid chromatography-mass spectrometry for the identification of all proteins present in the protein preparations (Supplementary Table 1). Amongst the copurifying proteins, we detected only the heat shock proteins, common non-specific contaminants in affinity purifications. Importantly, we did not detect the presence of any other repair enzyme in any of the purifications besides the ALKBH2 protein.
Figure 1.
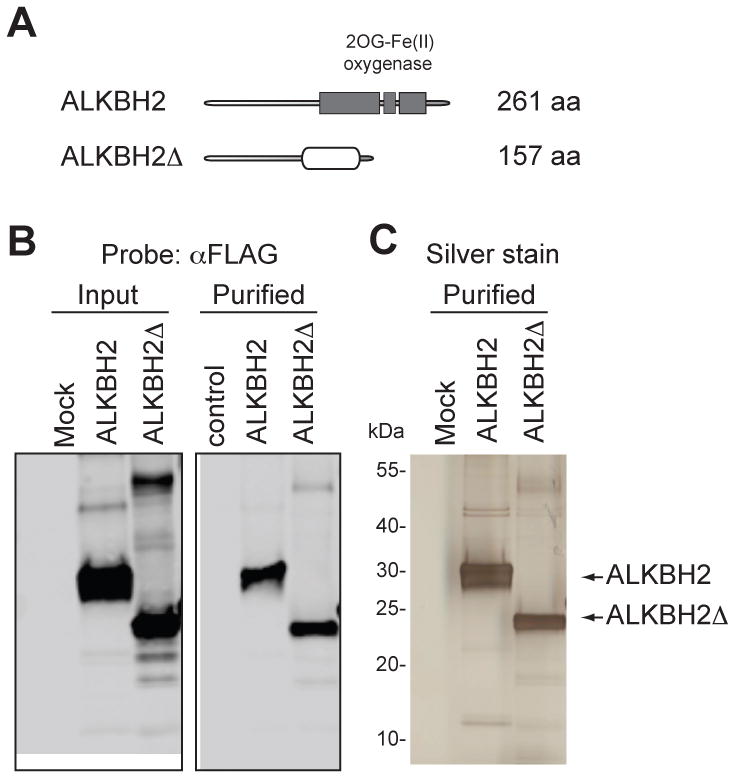
(A) Schematic of full-length human AlkB homolog 2 (ALKBH2) and splice variant (ALKBH2Δ). The 2-oxoglutarate, Fe(II) dioxygenase domain is noted with shaded boxes and the alternative sequence of ALKBH2Δ is denoted with a clear oval. (B) Immunoblot analysis of input and purified samples from 293T cells transfected with ALKBH2 expression constructs. Mock represents a control purification from 293T cells transfected with empty vector (C) Protein profile of purified ALKBH2 complexes by silver stain analysis. Arrows indicate the tagged ALKBH2 proteins.
3.2 Ethenocytosine is a novel substrate of ALKBH2
The purified proteins were tested for DNA repair activity using a previously described oligonucleotide cleavage assay that is based on the fact that the presence of a DNA base lesion in the DpnII restriction site blocks cleavage by the restriction enzyme and repair of the lesion restores a cleavable restriction site [29]; DpnII cleavage produces a smaller, lower migrating product (Fig 2). As expected, full-length ALKBH2 displayed repair activity on 1-meA, 3-meC and εA, known substrates of human AlkB proteins (Fig. 3A)[21, 23-25, 29]. In contrast, the ALKBH2Δ variant was inactive for repair on any substrate tested in this study, consistent with truncation of the catalytic motifs critical for AlkB dioxygenase activity (Fig. 3A). The extent of repair for 1-meA and 3-meC lesions by ALKBH2 was greater than that of εA in either ds- or ssDNA, consistent with previous results (Fig. 3A, B) [19, 24].
Figure 2.

Schematic of restriction enzyme-mediated oligonucleotide demethylase assay and lesions tested in this study. Purified ALKBH2 variants were incubated with a 49-mer DNA oligonucleotide containing the indicated lesion followed by digestion with the methylation-sensitive restriction enzyme, DpnII, and gel electrophoresis. Single-stranded DNA substrates were annealed with a complementary oligonucleotide after heat inactivation of ALKBH2 prior to restriction digestion with DpnII.
Figure 3.
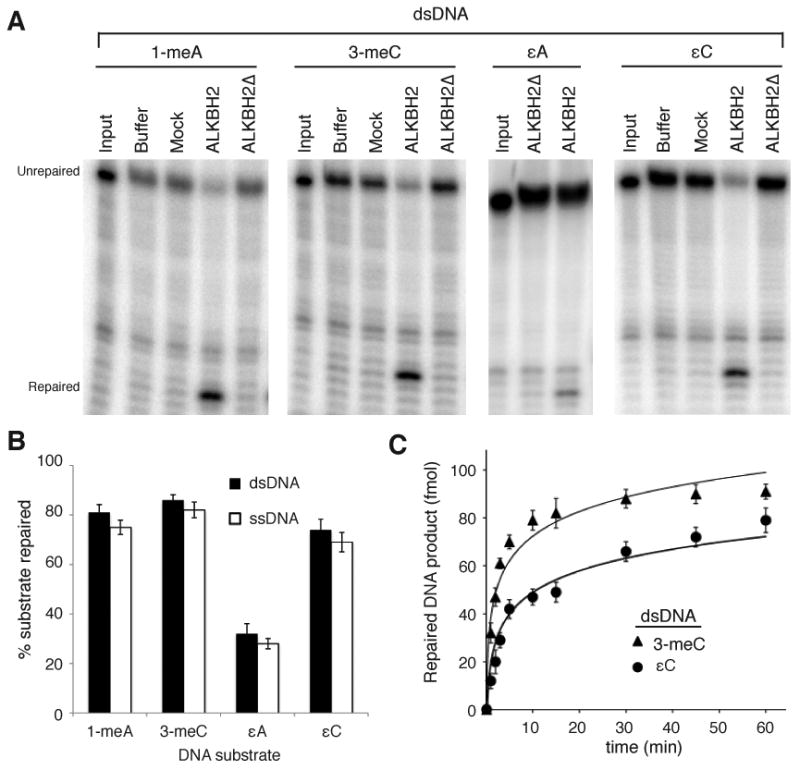
Human ALKBH2 repairs εC DNA lesions. (A) Denaturing gels of DNA products from double-stranded DNA substrates reacted with the indicated samples. (B) Quantification of ALKBH2-catalyzed repair for each lesion. The percent repaired substrate represents the amount of repaired product to unrepaired substrate normalized against the signal in the buffer control. Input represents the starting labeled oligonucleotide. (C) Kinetic analysis of ALKBH2-catalyzed repair of 3-meC or εC in double-stranded DNA substrates. The percent repaired substrate at select time points was quantified as in (B) and plotted with time. Quantification in (B) and (C) represent the average of three independent experiments.
In addition, we show that ALKBH2 displayed robust repair activity on εC, representing a novel DNA repair substrate for a human AlkB protein (Fig. 3A). ALKBH2 repaired εC in either ds- or ssDNA with the extent of εC repair reaching a plateau of >75% of the total substrate after 1 hour of incubation, which was comparable to that of 1-meA or 3-meC and greater than that of εA repair (Fig. 3B, Supplemental Fig. 1). Since the majority of ethenobase lesions can be detected in dsDNA, we investigated the kinetics of εC repair by ALKBH2 in double-stranded oligonucleotides for comparison to the known ALKBH2 substrate, 3-meC (Fig. 3C). Repair assays were performed under single turnover conditions using a saturating concentration of ALKBH2 (see Materials and Methods). Based upon this analysis, we found that ALKBH2 repaired εC in double-stranded DNA substrates with a relatively fast activity rate constant (kobs= 0.108 min−1 ± 0.02) that was slightly slower than 3-meC (kobs= 0.314 ± 0.05) but similar to that seen for ALKBH2 on εA substrates [24]. These results reveal εC as a substrate for direct reversal by the human ALKBH2 dioxygenase.
3.3 AAG inhibits repair of εC lesions by ALKBH2
Prior studies have shown that AAG can recognize and bind to a variety of AlkB substrates including the aforementioned 1-meA and 3-meC lesions as well as εA and εC [11]. Based upon the crystal structure of AAG bound to an εA substrate, AAG flips out the εA base from the DNA double helix to position it into the active site pocket for subsequent bond cleavage [32]. AAG uses a similar base-flipping mechanism for εC to bury the lesion in the same active site but because AAG is unable to protonate the εC base, there is no subsequent base excision [13]. The recognition of particular DNA base lesions by AAG without subsequent bond cleavage seems likely to influence the processing and repair of such lesions.
To test whether AAG substrate recognition influences ALKBH2-catalyzed repair, we analyzed the effect of adding AAG to the ALKBH2 repair reactions. For the majority of studies below, we used an extensively characterized AAG variant (AAGΔ79) that is truncated for the first 79 amino acids to facilitate purification; AAGΔ79 exhibits glycosylase activity similar to full-length AAG [11, 33]. The addition of AAGΔ79 greatly inhibited ALKBH2-mediated repair of εC lesions in a concentration-dependent manner (Fig. 4A, B). We observed AAG-dependent inhibition of ALKBH2 repair of εC starting at an equimolar protein concentration with complete repair inhibition at 20-fold excess of AAGΔ79 (Fig. 4B). To determine if this inhibition was due to non-specific effects caused by protein addition, we found that an equivalent 20-fold excess of either bacterial single strand binding protein (SSB) or bovine serum albumin (BSA) had no effect on εC repair by ALKBH2 (Fig. 4C). Notably, AAGΔ79 addition had little to no effect on ALKBH2 repair of either 1-meA or 3-meC even at the highest concentration of AAGΔ79 (Fig. 4A, B). As further confirmation, full-length AAG also displayed a specific inhibition of ALKBH2 repair of εC but not 1-meA or 3-meC (Supplemental Fig. 2). These results indicate that AAG inhibition of ALKBH2 repair is selective for εC lesions and that AAG's recognition of specific substrates inhibits ALKBH2 repair of these particular substrates rather than AAG having a general inhibitory effect on ALKBH2 enzymatic activity.
Figure 4.
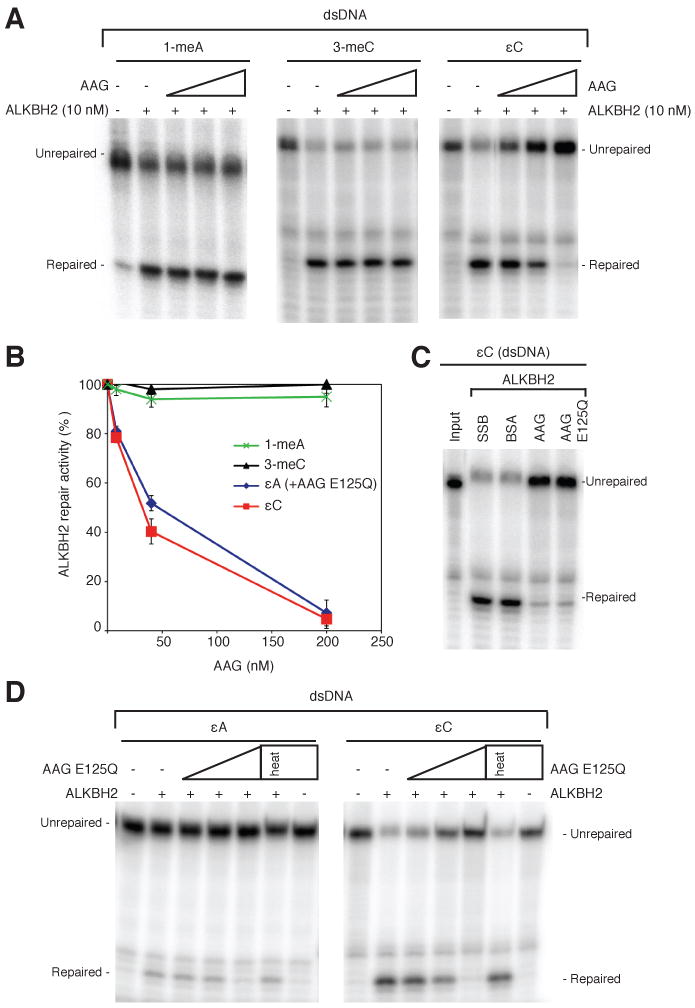
AAG specifically inhibits ALKBH2 repair of etheno lesions. (A) Increasing amounts of purified AAGΔ79 (8, 40 and 200 nM) was added together with ALKBH2 (10 nM) to the indicated DNA substrates followed by analysis. (B) Quantification of ALKBH2-catalyzed repair of the indicated lesions in the presence of AAG. The level of ALKBH2 repair activity represents the average of three to four independent experiments. (C) Neither BSA nor SSB protein inhibits ALKBH2 repair of εA or εC by ALKBH2. (D) Increasing amounts of catalytic-dead AAG E125Q were added to together with ALKBH2 to the indicated substrates as in (A) with analysis plotted in (C).
Since εA is a repair substrate for both AAG and ALKBH2, we also tested whether AAGΔ79 could affect ALKBH2 repair of εA adducts. To prevent excision of the εA base by AAG, we utilized an inactive AAGΔ79 variant (E125Q) that contains a mutation in the active site that abolishes glycosyl bond cleavage but retains virtually identical DNA binding and structural characteristics to wildtype AAGΔ79 [32, 34]. Similar to inhibition of ALKBH2-mediated εC repair by wildtype AAGΔ79, we find that addition of AAGΔ79 E125Q greatly inhibited ALKBH2 repair of εA as well as εC (Fig. 4D). While glycosylase activity is dispensable for the inhibition of εC repair, heat inactivation of AAG destroyed its ability to inhibit ALKBH2 repair of εC, indicating that substrate binding by AAG is necessary for blocking ALKBH2 repair (Fig. 4D). Together, these results reveal AAG as a potent inhibitor and competitor of ALKBH2 repair activity for etheno base lesions highlighting the potential consequences of substrate overlap between different DNA damage repair pathways.
4. Discussion
Ethenobase DNA lesions are highly prevalent byproducts of lipid peroxidation resulting from exposure to exogenous or endogenous sources of reactive oxygen and nitrogen species. These etheno lesions are highly mutagenic as well as toxic, and have been clearly linked to carcinogenesis [1, 3, 35]. Here, we find that ALKBH2 purified from human cells can repair etheno base lesions, including the novel ALKBH2 substrate, εC. Moreover, we find that the AAG base excision repair enzyme can specifically inhibit ALKBH2-catalyzed repair of εC DNA lesions without any effect on other ALKBH2 substrates.
Based upon our results, the kinetics of εC lesion recognition and repair would presumably depend upon the relative concentrations of AAG and ALKBH2 in each cell type. Prior studies have shown that AAG expression and activity can vary greatly across several different tissue types in mouse [9, 36]. Furthermore, transcriptome and protein expression profiling have revealed different levels of AAG and ALKBH2 in certain human tissue types [37]. Based upon antibody staining, AAG is highly expressed in the central nervous system and lymphoid cells while absent from bone marrow cells. In contrast, ALKBH2 is highly expressed in bone marrow but generally absent from lymphoid cells. Thus, it will be of great interest to analyze the levels of εC in the tissues of mice lacking AAG, ALKBH2 or both repair enzymes to uncover the relative contributions of these different repair pathways in the repair of εC lesions in vivo.
While AAG blocks the repair of etheno lesions by ALKBH2 in vitro, it is important to note that the effects of AAG binding could be different in vivo. Indeed, the levels of both εA and εC lesions increase in the colon of Aag-null mice compared to wildtype mice upon chronic inflammation, even though AAG can only excise εA lesions [3]. This indicates that the presence of AAG is contributing to the efficient repair of εC in vivo, even though AAG itself cannot excise this base. AAG bound to εC lesions could serve as a signal to recruit other repair enzymes, including perhaps ALKBH2 itself. While AAG antagonizes ALKBH2 repair of εC in vitro, endogenous AAG could recruit ALKBH2 to εC lesions through unknown factors that facilitate repair. The recruitment of a DNA repair enzyme by an inactive DNA repair protein has been observed previously with the alkyltransferase-like proteins that bind O6-methylguanine lesions to shunt the alkylated DNA into the nucleotide excision repair pathway [38]. Thus, the dynamics of AAG, ALKBH2 and other repair proteins at endogenous εC lesions deserve further investigation.
Supplementary Material
Highlights.
Human ALKBH2 dioxygenase catalyzes direct reversal of ethenocytosine lesions in DNA.
ALKBH2 repairs ethenocytosine in either double- or single-stranded DNA.
Binding of human AAG glycosylase to ethenocytosine in DNA blocks repair by ALKBH2.
Acknowledgments
We thank Dr. Manjappa Lingaraju Gondichatnahalli for purification of the AAG enzyme used in this study. D.F was funded by an American Cancer Society Postdoctoral Fellowship [116155-PF-08-255-01-GMC]. L.D.S. is an American Cancer Society Research Professor. This work was supported by NIH grants CA055042 and ES002109.
Abbreviations
- AAG
alkyladenine DNA glycosylase
- ALKBH2
AlkB homolog 2
- εA
1,N6-ethenoadenine
- εC
3,N4-ethenocytosine
- 1-meA
1-methyladenine
- 3-meC
3-methylcytosine
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nair U, Bartsch H, Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: a review of published adduct types and levels in humans. Free Radic Biol Med. 2007;43:1109–1120. doi: 10.1016/j.freeradbiomed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Shrivastav N, Li D, Essigmann JM. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010;31:59–70. doi: 10.1093/carcin/bgp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, Schauer DB, Dedon PC, Fox JG, Samson LD. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Speina E, Zielinska M, Barbin A, Gackowski D, Kowalewski J, Graziewicz MA, Siedlecki JA, Olinski R, Tudek B. Decreased repair activities of 1,N(6)-ethenoadenine and 3,N(4)-ethenocytosine in lung adenocarcinoma patients. Cancer Res. 2003;63:4351–4357. [PubMed] [Google Scholar]
- 5.Nair J, Sinitsina O, Vasunina EA, Nevinsky GA, Laval J, Bartsch H. Age-dependent increase of etheno-DNA-adducts in liver and brain of ROS overproducing OXYS rats. Biochem Biophys Res Commun. 2005;336:478–482. doi: 10.1016/j.bbrc.2005.08.114. [DOI] [PubMed] [Google Scholar]
- 6.Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singer B, Antoccia A, Basu AK, Dosanjh MK, Fraenkel-Conrat H, Gallagher PE, Kusmierek JT, Qiu ZH, Rydberg B. Both purified human 1,N6-ethenoadenine-binding protein and purified human 3-methyladenine-DNA glycosylase act on 1,N6-ethenoadenine and 3-methyladenine. Proc Natl Acad Sci U S A. 1992;89:9386–9390. doi: 10.1073/pnas.89.20.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JH, Samson LD. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc Natl Acad Sci U S A. 1997;94:13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hang B, Singer B, Margison GP, Elder RH. Targeted deletion of alkylpurine-DNA-N-glycosylase in mice eliminates repair of 1,N6-ethenoadenine and hypoxanthine but not of 3,N4-ethenocytosine or 8-oxoguanine. Proc Natl Acad Sci U S A. 1997;94:12869–12874. doi: 10.1073/pnas.94.24.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CY, Delaney JC, Kartalou M, Lingaraju GM, Maor-Shoshani A, Essigmann JM, Samson LD. Recognition and processing of a new repertoire of DNA substrates by human 3-methyladenine DNA glycosylase (AAG) Biochemistry. 2009;48:1850–1861. doi: 10.1021/bi8018898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gros L, Maksimenko AV, Privezentzev CV, Laval J, Saparbaev MK. Hijacking of the human alkyl-N-purine-DNA glycosylase by 3,N4-ethenocytosine, a lipid peroxidation-induced DNA adduct. J Biol Chem. 2004;279:17723–17730. doi: 10.1074/jbc.M314010200. [DOI] [PubMed] [Google Scholar]
- 13.Lingaraju GM, Davis CA, Setser JW, Samson LD, Drennan CL. Structural basis for the inhibition of human alkyladenine DNA glycosylase (AAG) by 3,N4-ethenocytosine containing DNA. Journal of Biological Chemistry. 2011 doi: 10.1074/jbc.M110.192435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 15.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, Schar P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009;7:e91. doi: 10.1371/journal.pbio.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abu M, Waters TR. The main role of human thymine-DNA glycosylase is removal of thymine produced by deamination of 5-methylcytosine and not removal of ethenocytosine. J Biol Chem. 2003;278:8739–8744. doi: 10.1074/jbc.M211084200. [DOI] [PubMed] [Google Scholar]
- 18.Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, Matsumoto T, Fujiyama S, Shirode Y, Yamaoka I, Kitagawa H, Takeyama K, Shibuya H, Ohtake F, Kato S. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009;461:1007–1012. doi: 10.1038/nature08456. [DOI] [PubMed] [Google Scholar]
- 19.Delaney JC, Smeester L, Wong C, Frick LE, Taghizadeh K, Wishnok JS, Drennan CL, Samson LD, Essigmann JM. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat Struct Mol Biol. 2005;12:855–860. doi: 10.1038/nsmb996. [DOI] [PubMed] [Google Scholar]
- 20.Yi C, Jia G, Hou G, Dai Q, Zhang W, Zheng G, Jian X, Yang CG, Cui Q, He C. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature. 2010;468:330–333. doi: 10.1038/nature09497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishina Y, Yang CG, He C. Direct repair of the exocyclic DNA adduct 1,N6-ethenoadenine by the DNA repair AlkB proteins. J Am Chem Soc. 2005;127:14594–14595. doi: 10.1021/ja055957m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundheim O, Talstad VA, Vagbo CB, Slupphaug G, Krokan HE. AlkB demethylases flip out in different ways. DNA Repair (Amst) 2008;7:1916–1923. doi: 10.1016/j.dnarep.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 23.Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M, Sundheim O, Bjoras M, Slupphaug G, Seeberg E, Krokan HE. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 24.Ringvoll J, Moen MN, Nordstrand LM, Meira LB, Pang B, Bekkelund A, Dedon PC, Bjelland S, Samson LD, Falnes PO, Klungland A. AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA. Cancer Res. 2008;68:4142–4149. doi: 10.1158/0008-5472.CAN-08-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci U S A. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu D, Collins K. Human telomerase and Cajal body ribonucleoproteins share a unique specificity of Sm protein association. Genes Dev. 2006;20:531–536. doi: 10.1101/gad.1390306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu D, Brophy JA, Chan CT, Atmore KA, Begley U, Paules RS, Dedon PC, Begley TJ, Samson LD. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol. 2010;30:2449–2459. doi: 10.1128/MCB.01604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacCoss MJ, Wu CC, Yates JR., 3rd Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal Chem. 2002;74:5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 29.Ringvoll J, Nordstrand LM, Vagbo CB, Talstad V, Reite K, Aas PA, Lauritzen KH, Liabakk NB, Bjork A, Doughty RW, Falnes PO, Krokan HE, Klungland A. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. Embo J. 2006;25:2189–2198. doi: 10.1038/sj.emboj.7601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee DH, Jin SG, Cai S, Chen Y, Pfeifer GP, O'Connor TR. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J Biol Chem. 2005;280:39448–39459. doi: 10.1074/jbc.M509881200. [DOI] [PubMed] [Google Scholar]
- 31.Tsujikawa K, Koike K, Kitae K, Shinkawa A, Arima H, Suzuki T, Tsuchiya M, Makino Y, Furukawa T, Konishi N, Yamamoto H. Expression and sub-cellular localization of human ABH family molecules. J Cell Mol Med. 2007;11:1105–1116. doi: 10.1111/j.1582-4934.2007.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A. 2000;97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Brien PJ, Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem. 2004;279:9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 34.O'Brien PJ, Ellenberger T. Human alkyladenine DNA glycosylase uses acid-base catalysis for selective excision of damaged purines. Biochemistry. 2003;42:12418–12429. doi: 10.1021/bi035177v. [DOI] [PubMed] [Google Scholar]
- 35.Bolt HM. Vinyl chloride-a classical industrial toxicant of new interest. Crit Rev Toxicol. 2005;35:307–323. doi: 10.1080/10408440490915975. [DOI] [PubMed] [Google Scholar]
- 36.Roth RB, Samson LD. 3-Methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance. Cancer Res. 2002;62:656–660. [PubMed] [Google Scholar]
- 37.Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, Persson A, Ottosson J, Wernerus H, Nilsson P, Lundberg E, Sivertsson A, Navani S, Wester K, Kampf C, Hober S, Ponten F, Uhlen M. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.