

Abstract
To efficiently repair DNA, human alkyladenine DNA glycosylase (AAG) must search the million-fold excess of unmodified DNA bases to find a handful of DNA lesions. Such a search can be facilitated by the ability of glycosylases, like AAG, to interact with DNA using two affinities: a lower-affinity interaction in a searching process and a higher-affinity interaction for catalytic repair. Here, we present crystal structures of AAG trapped in two DNA-bound states. The lower-affinity depiction allows us to investigate, for the first time, the conformation of this protein in the absence of a tightly bound DNA adduct. We find that active site residues of AAG involved in binding lesion bases are in a disordered state. Furthermore, two loops that contribute significantly to the positive electrostatic surface of AAG are disordered. Additionally, a higher-affinity state of AAG captured here provides a fortuitous snapshot of how this enzyme interacts with a DNA adduct that resembles a one-base loop.
Genomic DNA is under constant attack from endogenous and exogenous DNA damaging agents, with most damage occurring in the form of DNA base lesions.1 While approximately 10000 of these lesions occur daily,1,2 most do not go on to harm the cell because they are repaired by endogenous pathways. One of the most prevalent DNA repair pathways is known as base excision repair (BER), which is initiated when a DNA glycosylase hydrolyzes the N-glycosidic bond of a lesion base. In humans, the abasic site produced by a monofunctional glycosylase is repaired by the subsequent action of AP endonuclease I, DNA polymerase β, and DNA ligase I or III.3,4
Human alkyladenine DNA glycosylase (AAG) is one of the monofunctional glycosylase enzymes responsible for initiating BER. AAG catalyzes the removal of a diverse group of purine lesions, including those caused by damage from alkylation (3-methyladenine, 3-methylguanine, and 7-methylguanine) and reactive oxygen and nitrogen species (hypoxanthine, 1,N6-ethenoadenine (εA), and 1,N2-ethenoguanine).5,6 Removal of these lesions is paramount as they can cause cytotoxicity and mutagenesis.7 To access lesion bases, AAG, like most glycosylases, uses the canonical nucleotide flipping mechanism wherein the nucleoside with the damaged base is flipped out of the double helix and into the active site while a protein residue intercalates the DNA, effectively substituting for the flipped base. This nucleotide flipping has been observed in the crystal structure of a catalytically active N-terminal truncation mutant of AAG (denoted Δ79AAG) in which the protein is bound to DNA containing substrate εA.8 This structure shows that Tyr162 of AAG intercalates DNA while the lesion fits snugly into the binding pocket. This tight interaction observed structurally is supported by the nanomolar affinity of AAG for its substrates in vitro.9−11 Interestingly, AAG also binds with high affinity to DNA containing lesions that it cannot excise, such as inhibitor 3,N4-ethenocytosine (εC).5,9,12 Structural studies show that εC is also flipped out of the DNA into the active site of AAG and that an extra hydrogen bond between AAG and εC accounts for the 2-fold higher affinity for the inhibitor versus the substrate DNA.9 Finally, AAG can also bind with high affinity to DNA with a base loop structure, shielding it from repair and leading to frameshift mutations.13 These highly specific interactions (outlined in Scheme 1) between AAG and DNA are even more intriguing when one considers the massive search that must be undertaken by DNA glycosylases to find damaged DNA bases in the human genome.
Scheme 1. DNA Adducts to Which AAG Binds with High Affinity, Lesions (A) εC and (B) εA and (C) One-Base Loop Structures.

Given the ∼1010 nucleotides in the human genome and the ∼104 lesions per cell per day,1,2 there are approximately 1 million normal bases for every lesion present in DNA. Even for an abundant protein like AAG (∼2 × 105 molecules per nucleus14), each enzyme would have to inspect tens of thousands of normal bases before finding one lesion to excise. Such a task would be seemingly impossible if it involved a strict three-dimensional search, where proteins float through the cell in a stochastic hunt for a scarce number of lesions. To limit the search space, Berg et al.15 and Schurr16 have proposed that DNA binding proteins could nonspecifically bind and track along DNA in a one-dimensional search. Recently, protein “sliding” on DNA was observed directly in single-molecule fluorescence studies for a number of enzymes, including several glycosylases.17−19 Such a nonspecific search has been indirectly observed for AAG using kinetic assays in which the ability of the enzyme to excise two lesions contained in one piece of duplex DNA was examined.20,21 Kinetic data are also available that indicate AAG is able to search both strands of substrate DNA and avoid obstacles using a “hopping” mechanism.20 While the ability to slide or hop along DNA requires a lower-affinity and nonspecific complex between protein and DNA, base excision requires high-affinity and specific interactions. Thus, one would expect AAG, and related enzymes, to have differential modes of DNA binding. Evidence in support of this idea is available for other glycosylase systems, including crystal structures of a functional homologue of AAG from Escherichia coli (AlkA),22,23 as well as a crystal structure24 and single-molecule data17 for human 8-oxoguanine DNA glycosylase (reviewed in ref (25)).
In the structural studies presented here, we have captured two novel states of AAG. One structure shows AAG making only nonspecific contacts with DNA, depicting a “lower-affinity” or “searching” protein–DNA complex. The other shows a higher-affinity complex in which AAG is bound to two pieces of single-stranded DNA each containing an εC lesion (ssεC) in an arrangement that resembles a single-base loop structure. By comparing these structures to each other and to previously determined structures of AAG bound with high affinity to double-stranded DNA (dsDNA),8,9,26 we can investigate the molecular basis for the differential affinities of this DNA repair protein for DNA and explore the recognition events involved in identifying DNA lesions.
Materials and Methods
AAG Plasmid Construction and Protein Preparation
The Δ79AAG plasmid was constructed as described previously.9 Briefly, 84 residues at the N-terminus of the protein were truncated in this construct, and four extra residues from a PreScission Protease cleavage site (GE Healthcare) (Gly80, Pro81, His82, and Met83) were left behind after histidine tag cleavage. Therefore, Thr84 begins the wild-type AAG sequence, but four residues precede Thr84 such that Gly80 is now the N-terminus. We refer to this truncated protein construct as Δ79AAG. It should be noted that the AAG protein from previous structural studies was also termed Δ79AAG.8,26 However, in those studies, all residues contained in the construct are of the wild-type sequence. The expression and purification of the Δ79AAG protein were performed as described previously.9
Crystallization of Δ79AAG with Single-Stranded εC DNA
An equimolar ratio of Δ79AAG and 13-mer single-stranded εC-containing DNA (ssεC) (5′-GAC ATG εCTT GCC T-3′) were mixed to yield a protein–DNA complex concentration of 0.3 mM in the complex buffer [20 mM HEPES-NaOH (pH 7.5), 100 mM NaCl, 0.1 mM EDTA, 5% (v/v) glycerol, and 1 mM DTT]. The complex was incubated on ice for 15 min and used for crystallization. Crystals were obtained by the hanging drop vapor diffusion method upon mixing 1 μL of the protein–DNA complex and 1 μL of reservoir solution [100 mM BIS-TRIS (pH 5.5), 200 mM cesium chloride, and 20% polyethylene glycol (PEG) 3350] over 0.5 mL of reservoir solution. Crystals appeared after incubation for 14 days at 22 °C. These crystals were cryoprotected with precipitation solution supplemented with 10% glycerol and flash-frozen in liquid nitrogen prior to the collection of data.
Data Collection and Structure Determination
X-ray diffraction data were collected at the Advanced Light Source (Berkeley, CA) on beamline 12.3.1 at 100 K to 2.0 Å resolution and processed using Denzo/Scalepack27 (Table S1 of the Supporting Information). The structure, with two molecules in the asymmetric unit, was determined by molecular replacement in PHASER28 using the coordinates from the Δ79AAG–pyr:T complex structure (PDB entry 1BNK(26)). Refinement was conducted in CNS29 and Refmac 5.4,30,31 using topology and parameter files for the εC lesion generated by XPLO2D.32 Additional rounds of refinement using TLS parameters and noncrystallographic symmetry restraints were very effective in improving the quality of the fit. Model building was performed using Coot,33 and figures were prepared using PyMOL.34 The final model converged to an R factor of 21.9 (Rfree = 26.5) (Table S1 of the Supporting Information) and was evaluated using PROCHECK35 and composite omit maps. As we observed in our previously determined structure using this protein construct,9 the positively charged N-termini of both molecules of Δ79AAG in the asymmetric unit occupy what was initially identified as a sodium ion site by Ellenberger and co-workers.8 Although this coordination of the N-terminus is common to our Δ79AAG structures, the packing of the molecules in this study allowed AAG to crystallize in a novel space group (P43). The following residues of the total sequence of residues 80–298 lack electron density and are therefore not included in the model: 201–208, 265, 266, and 294–298 in chain A (pseudoduplex structure) and 131–141, 199–206, 253, 254, 263–273, and 290–298 in chain B (lower-affinity AAG). Because of a lack of interpretable electron density for the side chains of some residues in the structure (L200, L209, and E250 in chain A and P144, I161, M164, and R212 in chain B), these residues were modeled as alanines. Nucleotides G1′ and C11′–T13′ of the strand containing the nucleotide-flipped εC lesion and T9–T13 of the pseudocomplement strand are disordered.
Results
The structure of Δ79AAG in the presence of stoichiometric amounts of single-stranded εC-containing DNA (ssεC) was determined to 2.0 Å resolution by molecular replacement using the previously determined structure of Δ79AAG bound to pyrrolidine-containing DNA (abbreviated Δ79AAG–pyr:T) (PDB entry 1BNK(26)) as a search model. The final structure, with two molecules of Δ79AAG in the asymmetric unit, has been refined to an R factor of 21.9 (Rfree = 26.5) (Table S1 of the Supporting Information). Instead of observing two Δ79AAG molecules each bound to one ssεC DNA, we obtained two different and unique structures of this protein. During crystallization, the ssεC oligonucleotides formed a self-complementary pseudoduplex, which is specifically recognized by a single molecule of Δ79AAG in the asymmetric unit (Figure 1A,B, orange). We will refer to this interaction as the pseudoduplex structure. Although the other molecule of Δ79AAG in the asymmetric unit is also interacting with an εC-containing DNA strand, it makes only nonspecific contacts with the phosphodiester backbone and leaves the εC lesion untouched (Figure 1B and Figure S1 of the Supporting Information). This nonspecific protein–DNA interaction will be termed the lower-affinity structure (Figure 1A,B, green). The 13-mer pseudoduplex piece of DNA that we observe crystallographically is highly unlikely to persist in solution, which precludes traditional binding measurements. We have studied the binding of Δ79AAG to 13-mer ssεC oligonucleotides by gel shift assays as previously described9 and found no measurable affinity (Figure S2 of the Supporting Information). These same assays have shown high-affinity Δ79AAG binding (Kd = 21 ± 3 nM) for preannealed 13-mer doubled-stranded εC oligonucleotides, and this highly specific interaction is depicted by a crystal structure with the same dsDNA.9 With these data in mind, the molecules of Δ79AAG shown in Figure 1 must have affinities for their 13-mer oligonucleotides that fall in the range from immeasurably weak, as observed for true single-stranded DNA, to high (Kd = 10–23 nM), as measured for preannealed dsDNA.9 Considering the green molecule (Figure 1) has only a few nonspecific contacts with the DNA whereas the orange molecule has many specific contacts and closely resembles the high-affinity structures determined previously with dsDNA,8,9,26 these structures appear to represent lower- and higher-affinity states, respectively, and will be referred to as such.
Figure 1.

Structures of Δ79AAG bound to εC DNA. (A) Δ79AAG crystallized in the presence of ssεC DNA has two Δ79AAG molecules in the asymmetric unit: one that makes few contacts with DNA and represents a lower-affinity complex (green) and one that makes multiple contacts with DNA and represents a higher-affinity complex (orange). The two strands of ssεC DNA, which form a pseudoduplex, are shown as sticks with cyan carbons. Panel I displays Tyr162 (orange sticks) intercalating DNA while the εC lesion is flipped into the active site. Panel II depicts the lower-affinity interaction between Δ79AAG and DNA where Tyr162 (green sticks) stacks with nucleotide A2′. Atoms are colored as follows: red for oxygen, blue for nitrogen, and orange for phosphorus. A blue star denotes the location of the empty active site of lower-affinity AAG. (B) Schematic illustration of the interactions between the two strands of ssεC DNA and amino acid side chains (three-letter code) and main chains (mc) of the Δ79AAG molecules. Amino acid labels from the lower- and higher-affinity (pseudoduplex-bound) Δ79AAG molecules are colored green and orange, respectively. Hydrogen bonds are indicated by solid lines and van der Waals interactions by dashed lines. DNA bases are shown as rectangles containing one-letter codes and numbers that signify their respective positions in the oligonucleotide (5′ to 3′). All DNA bases contained in the nucleotide-flipped εC lesion strand are denoted with a prime. Disordered nucleotides are shown in dashed lines. (C) Nucleotide interactions near the lesion in Δ79AAG−εC:G dsDNA (PDB entry 3QI5) (yellow carbons). Relevant distances shown by dashed lines are given in angstroms. (D) Nucleotide interactions near the lesion in the pseudoduplex Δ79AAG structure (cyan carbons). (E) van der Waals interactions with G19 in the Δ79AAG−εC:G structure. (F) van der Waals interactions with A2 in the pseudoduplex Δ79AAG structure.
Δ79AAG Pseudoduplex Structure
In the pseudoduplex structure, the canonical nucleotide flipping mechanism of DNA glycosylases can be observed with Tyr162 inserted into the DNA duplex while the lesion nucleoside (εC7′) from one ssεC strand is flipped into the enzyme active site (Figure 1A, panel I). Interestingly, the interactions of the active site with the εC lesion for this pseudoduplex structure are identical to those of Δ79AAG with dsDNA (abbreviated Δ79AAG−εC:G) (PDB entry 3QI5(9)), and both structures share a high degree of overall similarity with a root-mean-square deviation (rmsd) between α-carbons of 0.43 Å (Figure S3 of the Supporting Information). This pseudoduplex structure is also very similar to the structure of Δ79AAG with substrate lesion εA in dsDNA (abbreviated Δ79AAG−εA:T) (PDB entry 1F4R(8)), with an rmsd for α-carbons of 0.93 Å.
Although nucleotide flipping is observed in the pseudoduplex structure, the interactions surrounding the intercalated residue (Tyr162) are not identical to those previously observed in the structure of AAG with εC-containing dsDNA. In the Δ79AAG−εC:G structure, a potential steric clash of Tyr162 with G19 (base opposite εC) is prevented by a shifting of G19 out of the minor groove, leaving it without a base pairing partner (Figure 1C,E).9 In contrast, A2 of the pseudoduplex structure avoids a steric clash with a sideways motion that allows for hydrogen bonding to T8′ of the opposite strand (Figure 1D,F). This sideways motion also changes the orientation of the neighboring base G1, such that it now hydrogen bonds to T9′ (Figure 1D). Although Met164 contacts the “opposite bases” (G19 and A2) in both structures, the orientation of the interaction is also different (Figure 1E,F).
Δ79AAG Lower-Affinity Structure
The lower-affinity structure of Δ79AAG shows only nonspecific interactions (Figure 1A,B, green), with hydrogen bonding contacts to the phosphodiester backbone by the side chains of Arg182, Arg197, and Arg207 and the main chain amides of Ser219 and Lys220 (Figures 1B and 2A). Interestingly, the protein residue that normally intercalates substrate DNA, Tyr162, is contacting the pseudoduplex by stacking with nucleotide A2′ (Figure 1A, panel II, and Figure 2B). While this Tyr162 adopts a similar orientation as found for intercalated Tyr162 residues from the higher-affinity complex structures (e.g., panel A vs panel B of Figure 3), it has higher B factors, indicating increased conformational flexibility (see Figure S4B of the Supporting Information). In addition to stacking with Tyr162, A2′ hydrogen bonds with T8, leaving εC7 orphaned in terms of base pairing (Figure 2C). This εC7 lesion also has no interaction with protein residues (Figure S1 of the Supporting Information).
Figure 2.

Tyr162 contacts in lower-affinity Δ79AAG. (A) Hydrogen bonding contacts (dashed lines, distances in angstroms) for lower-affinity Δ79AAG (green) with pseudoduplex DNA (cyan carbons), and non-carbon atoms colored as in Figure 1. (B) van der Waals radii for the protein and DNA are shown in gray spheres with all other representations and colors as in panel A. (C) Same depiction as panel B with the orientation changed slightly to show relevant distances (in angstroms) as depicted by dashed lines and to draw attention to the rotation of εC7 out of the pseudoduplex.
Figure 3.

Comparison of the lower-affinity Δ79AAG structure with the high-affinity Δ79AAG−εA:T structure. (A) Δ79AAG (purple cartoon) bound to an εA lesion (stick form with cyan carbons) with active site residue His136 and intercalating Tyr162 represented in stick form with purple and yellow carbons, respectively (PDB entry 1F4R(8)). Oxygen atoms are colored red and nitrogen atoms blue. Regions that become disordered in the absence of a bound DNA adduct are circled and labeled 1–3. (B) Lower-affinity AAG (green) with Tyr162 (yellow). Loops 1–3 and other atoms are colored as in panel A. (C) Binding pocket for the εA lesion shown with van der Waals surfaces for protein residues (gray spheres) and εA lesion (cyan sphere). (D) Disrupted binding pocket in lower-affinity AAG with van der Waals surfaces colored as in panel B. (E) Electrostatic representation of Δ79AAG−εA:T calculated in the absence of DNA where blue surfaces are more positive, red surfaces more negative, and white surfaces near neutral. The position of Tyr162 is denoted with a yellow star. (F) Electrostatic representation of lower-affinity AAG with colors and symbols as in panel E. (G) Same depiction as panel E but with substrate DNA modeled (orange cartoon). (H) Same electrostatic depiction as panel F aligned and superimposed with a cartoon model (purple) of Δ79AAG−εA:T. Disordered regions that affect electrostatic potential are circled and represent the same loops as in panels A and B.
Overall, the structure of lower-affinity Δ79AAG is similar to other structures of this protein, including the pseudoduplex structure described above (rmsd for α-carbons of 1.27 Å), the structure of the Δ79AAG−εC:G complex (rmsd for α-carbons of 1.26 Å),9 and the structure of Δ79AAG−εA:T (rmsd for α-carbons of 1.18 Å).8 While these rmsds are low, the lower-affinity structure has three distinct disordered regions compared to the Δ79AAG−εA:T structure (Figure 3A,B). Because there are no lattice contacts in this area (Figure S1 of the Supporting Information), we can attribute the disorder to the absence of a bound nucleotide in the active site of lower-affinity AAG. The residues that lack electron density in the low-affinity complex, and are thus considered disordered, include Glu131–Arg141 (loop 1), Gly263–Lys273 (loop 2), and C-terminal residues after Asp289 (loop 3) (Figure 3A,B). Loop 1 contains crucial active site residues, including Ala134–His136, which form a snug pocket for lesion bases (Figure 3C). This binding pocket is only partially formed in the absence of a bound nucleotide (Figure 3D). Disordered loops 2 and 3 are not involved in forming the active site but contribute to the electrostatic potential of the protein (Figure 3E–H).
Electrostatic surfaces for Δ79AAG−εA:T (after removing the DNA) and for lower-affinity Δ79AAG are considerably different (Figure 3E,F) [all electrostatic depictions were calculated using the Adaptive Poisson–Boltzmann Solver (APBS) software plug-in36 for PyMOL34]. The Δ79AAG−εA:T complex shows a continuous, and richly positive, DNA-binding surface as would be expected for a protein that contacts DNA with high affinity (Figure 3E,G). In contrast, the DNA-binding surface for the lower-affinity structure is more neutral with disordered loops 1–3 disrupting positive patches observed in the Δ79AAG−εA:T high-affinity complex (Figure 3E–H). Other regions of the protein show little difference in the ordered electrostatic surface, such as the area near the intercalating residue Tyr162 (marked with a star in Figures 3 and 4). Also, both structures display a positively charged electrostatic surface that circles the protein molecule from the top of Δ79AAG to the bottom (see the middle panels of Figure 4A,B), as well as a negative electrostatic region located opposite the DNA binding surface (Figure 4).
Figure 4.

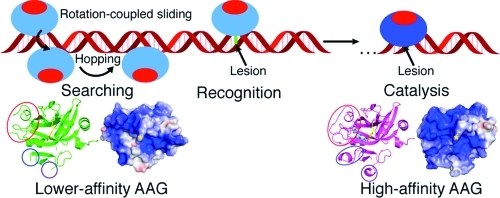
Proposal for how AAG can recognize DNA with two different affinities. The electrostatic representations from panels E and F of Figure 3 are displayed in panels A and B for Δ79AAG−εA:T and lower-affinity Δ79AAG, respectively, with the same coloring and symbols as in Figure 3. The orientations of the molecules start as in Figure 3 and are then rotated 120° counterclockwise (in two 60° steps) along the vertical axis such that the continuous positive surface can be visualized. (C) Cartoon depiction of the search on DNA by AAG, where blue and red represent positively and negatively charged surfaces, respectively. Relevant steps are labeled, and a lesion base is denoted as a green line. The AAG catalysis complex is a darker shade of blue to represent the more ordered positive surface visualized crystallographically (panel A vs panel B above).
Discussion
DNA glycosylases are charged with the formidable task of locating and repairing potentially harmful DNA lesions while avoiding the million-fold excess of normal, healthy DNA bases. The difficulty of this searching process can be partially overcome by the formation of a weak complex between protein and DNA, effectively creating a nonspecific, one-dimensional search. However, to maintain fidelity and genomic integrity, the enzyme must also be able to form a stronger, highly specific complex for lesion recognition and excision. Therefore, the ability to adopt both low- and high-affinity conformations appears to be advantageous. Here, we have trapped a human DNA glycosylase, AAG, in both lower- and higher-affinity complexes with DNA (Figure 1), providing two snapshots of this enzyme that relate to this search process.
Interestingly, AAG has been shown to bind with high affinity to DNA damage that it can repair (such as εA lesions) as well as to damage that it cannot repair (such as εC lesions and one-base loops).9−11,13 Crystallographic studies have provided molecular insight into how AAG recognizes both εA and εC lesions within dsDNA,8,9 but no structure of AAG bound to a one-base loop or in a low-affinity complex with DNA has been determined. The pseudoduplex structure that we present here appears to be the best representation available of how AAG could bind to such a DNA loop structure. In a one-base loop, one nucleoside is “looped out” of the DNA, and the base opposite the looped out base hydrogen bonds with an adjacent base instead (Scheme 1C). This arrangement of bases resembles what we observe in the pseudoduplex structure and represents a major departure from the hydrogen bonding pattern of nucleotides observed in other AAG–DNA complexes (e.g., Figure 1C). The looped out base is nicely accommodated in the AAG active site with interactions identical to those observed previously for an εC lesion (Figure S3B of the Supporting Information).9 In addition, the close resemblance of the pseudoduplex structure to previously determined structures of Δ79AAG bound to dsDNA8,9,26 is consistent with the idea that this structure represents a high-affinity complex between AAG and DNA. This observation is in agreement with the high-affinity binding observed between AAG and a one-base loop structure in vitro.13 A physiological rationale for why AAG binds to DNA damage that it cannot repair remains to be determined: while tight binding of AAG to lesions it can repair such as εA can be beneficial to the cell,37 tight binding of AAG to base loop structures shields them from repair, increasing mutation rates.13 As the physiological significance of this behavior of AAG is elucidated, our work suggests a molecular basis for the recognition of base loops by this human DNA repair protein.
Excitingly, our crystallization conditions have also yielded the first nonspecific or lower-affinity depiction of AAG, providing insight into a conformation of the protein likely responsible for inspecting DNA for damage. Although the top side of the active site, including the position of the putative catalytic water, agrees well with high-affinity lesion-bound structures (Figure S3B of the Supporting Information), the residues comprising the active site floor are disordered (Figure 3). This observation of a partially ordered active site suggests an order of events for the binding of AAG to DNA in which a lesion base is first identified by a more dynamic state of the protein and is later recognized with high affinity as the active site pocket closes around the nucleotide-flipped lesion. Our structural studies are consistent with fluorescence-based kinetic assays, which have provided evidence of a two-state lesion recognition process for AAG, where the active site experiences changes in environment prior to nucleotide flipping.10,38 This initial state observed kinetically has been likened to the initial recognition complexes suggested for other glycosylases.25,39
With the lesion flipped into the active site, an intercalating residue (Tyr162 for AAG) maintains the double-helical DNA structure. An interesting point of discussion in the DNA repair literature is whether intercalating residues play an active or passive role in lesion recognition, in other words, whether the intercalating residue directly interrogates base pairs (active) or the success of the search relies on the intercalating residue filling the gap left behind by a flipped lesion (passive). Two recent structural studies on the glycosylases MutM and the functional homologue of AAG from E. coli, AlkA, have provided conflicting answers to this question. In both studies, the glycosylases were linked to undamaged DNA in a stable complex using disulfide cross-links,22,39 and the position of the intercalating residue was evaluated. For MutM, the intercalating residue (Phe114) is fully inserted into the DNA duplex, buckling the bases with which it interacts, as the protein simultaneously bends the DNA, suggestive of an active interrogation mode.39 In contrast, the structures of AlkA with undamaged DNA show snapshots of a glycosylase in a more passive interrogation mode, with the intercalating residue (Leu125) situated completely outside of a double helix, which maintains all base stacking interactions and remains mostly linear.22 In our lower-affinity structure, the intercalating residue of AAG, Tyr162, has increased flexibility but still maintains the same average position for its side chain as is found in the higher-affinity structures (e.g., panels A vs panel B of Figure 3). Tyr162 is also still involved in a stacking interaction with a nucleotide (A2′) even when intercalation is not possible (Figure 2). This observation suggests that Tyr162 is capable of forming both lower- and higher-affinity interactions with DNA, possibly playing roles both in a lower-affinity searching process and in a higher-affinity “recognition” process. Consistent with an ability to form different types of interactions, the Tyr162 loop is flexible, displaying B factors approximately 2-fold higher than average for this crystal structure (Figure S4 of the Supporting Information).
Just as residues in the active site of AAG are disordered in the absence of a tightly bound DNA lesion, residues that contribute to the positive electrostatic surface are also disordered (loops 2 and 3 in Figure 3 and Figure S1 of the Supporting Information). The highly positively charged and complementary surface of AAG that binds DNA with high affinity (Figure 3E,G) is disrupted in the low-affinity structure (Figure 3F,H). Loops 2 and 3 are not preordered, ready to bind with high affinity to a DNA lesion. Instead, they are highly mobile, suggesting that they could play an active role in interrogating DNA.
In terms of interrogating DNA, there is strong evidence that the searching process of DNA binding proteins is not a strictly linear scan of DNA. A single-molecule study of eight different DNA binding proteins, including three glycosylases, found that the movements of these proteins along DNA were better described by a rotation-coupled sliding mechanism.17 Such movement would orient the enzyme so that its binding surface always faces the axis of the DNA double helix. In essence, these proteins circle the DNA while diffusing along it. The electrostatic potential surface calculated for AAG is consistent with this rotation-coupled search mechanism. In both higher- and lower-affinity AAG complexes, a positive electrostatic surface is found to wrap around the protein (Figure 4A,B). This surface could be used to “roll” or “rock” back and forth along the negatively charged DNA backbone, while the presence of a negatively charged electrostatic cap on the opposite face of AAG (red in Figure 4) would maintain the correct orientation for lesion recognition. Hopping, another DNA search method, has been established for AAG through the use of kinetic assays.20 Hopping, or short-range dissociation–association events, allows AAG to search both DNA strands simultaneously and avoid obstacles, such as a DNA-encasing endonuclease like EcoRI, that may be present along the search path.20 Rotation-coupled sliding and hopping are not mutually exclusive, and we consider both in the proposed search mechanism for AAG that is outlined in Figure 4C.
In the initial search, we propose that AAG closely resembles the lower-affinity structure, interacting with DNA nonspecifically through its positive electrostatic surface. Incorrect orientation of AAG would be avoided because of the negative electrostatic patch opposite the active site (Figure 4B). The positive surface that wraps around AAG would promote a rotation-coupled sliding search of the DNA, while still allowing for the hopping events described above. As a lesion is recognized, disordered regions of AAG, including the active site pocket, become more ordered (Figure 3). After nucleotide flipping, AAG adopts a higher-affinity conformation such as the pseudoduplex structure (Figure 1A, orange) or dsDNA structures published previously.8,9,26 Here, previously disordered loops are completely ordered to display the full potential of a continuous electrostatic surface for binding DNA; Tyr162 is fully inserted into the DNA, and a base lesion is bound tightly in the AAG active site. This lesion recognition complex would interact very strongly with the DNA, halting the search by AAG. In cases where the lesion is a substrate, base excision would follow. After the release of the base–lesion contact, the active site and other loops of AAG would become partially disordered, decreasing the extent of order of the DNA binding surface, ultimately leaving AAG in its lower-affinity, nonspecific searching state once again. In cases where the lesion cannot be repaired, AAG would remain fixed in its higher-affinity state, providing a rationale for the abortive AAG−εC complexes observed in vivo.12
The two novel structures of human AAG presented here help provide a molecular understanding of this intriguing DNA repair protein, both in terms of understanding how AAG can recognize different types of DNA damage, such as base lesions and one-base loops, and in terms of how it may search the genome for DNA damage. With recent literature describing an enhanced ability of AAG both to repair base lesions5 and to identify DNA damage that it cannot repair,5,9,13 this study provides important insight into the molecular basis of AAG interactions.
Glossary
Abbreviations
- AAG
human alkyladenine DNA glycosylase
- BER
base excision repair
- εA
1,N6-ethenoadenine
- Δ79AAG
N-terminal truncation mutant of AAG
- εC
3,N4-ethenocytosine
- AlkA
E. coli alkyladenine DNA glycosylase
- ssεC
single-stranded 3,N4-ethenocytosine-containing DNA
- dsDNA
double-stranded DNA
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- EDTA
ethylenediaminetetraacetic acid
- DTT
dithiothreitol
- BIS-TRIS
2,2-bis(hydroxymethyl)-2,2′,2″-nitrilotriethanol
- PEG
polyethylene glycol
- Δ79AAG–pyr:T
Δ79AAG bound to pyrrolidine-containing dsDNA
- PDB
Protein Data Bank
- Kd
dissociation constant
- Δ79AAG−εC:G
Δ79AAG bound to 3,N4-ethenocytosine-containing dsDNA
- rmsd
root-mean-square deviation
- Δ79AAG−εA:T
Δ79AAG bound to 1,N6-ethenoadenine-containing dsDNA
- APBS
Adaptive Poisson–Boltzmann Solver
- EcoRI
restriction endonuclease, type II from E. coli.
Supporting Information Available
Crystallographic data collection and refinement statistics (Table S1), additional figures of the crystal structure (Figures S1, S3, and S4), and raw gel mobility shift assay data (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.W.S. and G.M.L. are co-first authors.
Author Present Address
# Friedrich Miescher Institute for Biomedical Research, Maulbeerstrasse 66, CH-4058 Basel, Switzerland.
Author Present Address
∇ Bethune-Cookman University, 640 Dr. Mary McLeod Bethune Blvd., Daytona Beach, FL 32114.
This work was supported, in part, by National Institutes of Health Grants P30-ES002109 (to C.L.D. and L.D.S.), GM65337 (to C.L.D.), GM65337-03S2 (to C.A.D.), and CA055042 and CA092584 (to L.D.S.). C.L.D. is an investigator of The Howard Hughes Medical Institute. L.D.S. is an American Cancer Society Research Professor. J.W.S. is supported by a Repligen KIICR Graduate Fellowship. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.
Accession Codes
The atomic coordinates have been deposited as Protein Data Bank entry 3UBY.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Lindahl T.; Barnes D. (2000) Repair of endogenous DNA damage. Cold Spring Harbor Symp. Quant. Biol. 65, 127–134. [DOI] [PubMed] [Google Scholar]
- David S. S.; Williams S. D. (1998) Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair. Chem. Rev. 98, 1221–1262. [DOI] [PubMed] [Google Scholar]
- Kubota Y.; Nash R. A.; Klungland A.; Schär P.; Barnes D. E.; Lindahl T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Lee C.-Y. I.; Delaney J. C.; Kartalou M.; Lingaraju G. M.; Maor-Shoshani A.; Essigmann J. M.; Samson L. D. (2009) Recognition and Processing of a New Repertoire of DNA Substrates by Human 3-Methyladenine DNA Glycosylase (AAG). Biochemistry 48, 1850–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O′Brien P. J.; Ellenberger T. (2004) Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J. Biol. Chem. 279, 9750–9757. [DOI] [PubMed] [Google Scholar]
- Shrivastav N.; Li D.; Essigmann J. M. (2010) Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 31, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. Y.; Wyatt M. D.; Glassner B. J.; Samson L. D.; Ellenberger T. (2000) Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc. Natl. Acad. Sci. U.S.A. 97, 13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju G. M.; Davis C. A.; Setser J. W.; Samson L. D.; Drennan C. L. (2011) Structural basis for the inhibition of human alkyladenine DNA glycosylase (AAG) by 3,N4-ethenocytosine containing DNA. J. Biol. Chem. 286, 13205–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe A. E.; O'Brien P. J. (2009) Kinetic mechanism for the flipping and excision of 1,N6-ethenoadenine by human alkyladenine DNA glycosylase. Biochemistry 48, 11357–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O′Brien P. J.; Ellenberger T. (2003) Human alkyladenine DNA glycosylase uses acid-base catalysis for selective excision of damaged purines. Biochemistry 42, 12418–12429. [DOI] [PubMed] [Google Scholar]
- Gros L.; Maksimenko A.; Privezentzev C. (2004) Hijacking of the Human Alkyl-N-purine-DNA Glycosylase by 3,N4-Ethenocytosine, a Lipid Peroxidation-induced DNA Adduct. J. Biol. Chem. 279, 17723–17730. [DOI] [PubMed] [Google Scholar]
- Klapacz J.; Lingaraju G. M.; Guo H. H.; Shah D.; Moar-Shoshani A.; Loeb L. A.; Samson L. D. (2010) Frameshift mutagenesis and microsatellite instability induced by human alkyladenine DNA glycosylase. Mol. Cell 37, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye N.; Holmquist G. P.; O'Connor T. R. (1998) Heterogeneous repair of N-methylpurines at the nucleotide level in normal human cells. J. Mol. Biol. 284, 269–285. [DOI] [PubMed] [Google Scholar]
- Berg O. G.; Winter R. B.; von Hippel P. H. (1981) Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 20, 6929–6948. [DOI] [PubMed] [Google Scholar]
- Schurr J. M. (1979) The one-dimensional diffusion coefficient of proteins absorbed on DNA. Hydrodynamic considerations. Biophys. Chem. 9, 413–414. [PubMed] [Google Scholar]
- Blainey P. C.; Luo G.; Kou S. C.; Mangel W. F.; Verdine G. L.; Bagchi B.; Xie X. S. (2009) Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 16, 1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granéli A.; Yeykal C. C.; Robertson R. B.; Greene E. C. (2006) Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 103, 1221–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey P. C.; van Oijen A. M.; Banerjee A.; Verdine G. L.; Xie X. S. (2006) A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 103, 5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M.; O'Brien P. J. (2010) Hopping Enables a DNA Repair Glycosylase To Search Both Strands and Bypass a Bound Protein. ACS Chem. Biol. 5, 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M.; O'Brien P. J. (2008) Human Alkyladenine DNA Glycosylase Employs a Processive Search for DNA Damage. Biochemistry 47, 11434–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman B. R.; Lee S.; Wang S.; Verdine G. L. (2010) Structure of Escherichia coli AlkA in complex with undamaged DNA. J. Biol. Chem. 285, 35783–35791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis T.; Ichikawa Y.; Ellenberger T. (2000) DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 19, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner S. D.; Norman D. P.; Verdine G. L. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403, 859–866. [DOI] [PubMed] [Google Scholar]
- Friedman J. I.; Stivers J. T. (2010) Detection of Damaged DNA Bases by DNA Glycosylase Enzymes. Biochemistry 49, 4957–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A. Y.; Scharer D.; Samson L. D.; Verdine G. L.; Ellenberger T. (1998) Crystal Structure of a Human Alkylbase-DNA Repair Enzyme Complexed to DNA: Mechanisms for Nucleotide Flipping and Base Excision. Cell 95, 249–258. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Murshudov G.; Vagin A.; Dodson E. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D50, 760–763. [DOI] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A. (1998) Databases in protein crystallography. Acta Crystallogr. D54, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Delano W. L. (2002) The PyMOL Molecular Graphics System, Schrödinger, LLC, New York. [Google Scholar]
- Laskowski R.; MacArthur M.; Moss D. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meira L. B.; Bugni J. M.; Green S. L.; Lee C.-W.; Pang B.; Borenshtein D.; Rickman B. H.; Rogers A. B.; Moroski-Erkul C. A.; McFaline J. L.; Schauer D. B.; Dedon P. C.; Fox J. G.; Samson L. D. (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest. 118, 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot J. M.; Wolfe A. E.; O'Brien P. J. (2011) Substitution of Active Site Tyrosines with Tryptophan Alters the Free Energy for Nucleotide Flipping by Human Alkyladenine DNA Glycosylase. Biochemistry 50, 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.; Santos W. L.; Verdine G. L. (2006) Structure of a DNA glycosylase searching for lesions. Science 311, 1153–1157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.