

Abstract
N-Heterocyclic carbene (NHC)-mediated ring-opening polymerization of N-decylN-carboxylanhydride monomer (De-NCA) has been shown to occur in a controlled manner, yielding cyclic poly(N-decyl-glycine)s (c-PNDGs) with polymer molecular weights (MW) between 4.8 and 31 kg·mol−1 and narrow molecular weight distributions (PDI < 1.15). The reaction exhibits pseudo-first order kinetics with respect to monomer concentration. The polymer MW increases linearly with conversion, consistent with a living polymerization. ESI MS and SEC analysesconfirm the cyclic architectures of the forming polymers. DSC and WAXS studies reveal that the c-PNDG homopolymers are highly crystalline with two prominent first order transitions at 72–79°C (Tm,1) and 166–177°C (Tm,2), which have been attributed to the side chain and main chain melting respectively. A series of amphiphilic cyclic diblock copolypeptoids [i.e.,poly(N-methyl-glycine)-b-poly(N-decyl-glycine) (c-PNMG-b-PNDG)] with variable molecular weight and composition was synthesized by sequential NHC-mediated polymerization of the corresponding N-methyl N-carboxyanhydride (Me-NCA) and De-NCA monomers. 1H NMR analysis reveals that adjusting the initial monomer to NHC molar ratio can readily control the block copolymer chain length and composition. Time-lapsed light scattering and cryogenic transmission electron microscopy (cryo-TEM) analysis of c-PNDG-b-PNMG samples revealed that the amphiphilic cyclic block copolypeptoids self-assemble into spherical micelles that reorganize into micron-long cylindrical micelles with uniform diameter in room temperature methanol over the course of several days. An identical morphological transition has also been noted for the linear analogs, which occurs more rapidly than for the cyclic copolypeptoids. We tentatively attribute this difference to the different crystallization kinetics of the solvophobic block (i.e., PNDG) in the cyclic and linear block copolypeptoids.
Introduction
Cyclic polymersexhibit physical properties that are distinctly different from their linear analogs both in the solid state and in solution.1, 2 Without chain ends, cyclic polymers are physically more compact, as manifested by a smaller radius of gyration (Rg) in good and theta solvents, as well as a reduced intrinsic viscosityrelative to their linear counterparts.3–5They also have reduced conformational freedom as compared to their linear analogs, and this feature contributes to their diminished dynamics in porous media,6, 7 and promotes polymer crystallization8 and novel micellar morphologies9–12and stability.13
Recent advancements in synthetic methodologies have led to a renaissance in cyclic polymers, allowing their fundamental properties and potential utilities to be explored. Cyclic polymers are most commonly synthesized by (1) end-to-end coupling of the linear precursors, (2) ring-chain equilibrium or (3) ring expansion polymerization.1The first approach requires high dilution conditions, thereby limiting the synthetic efficiency. The second approach is unsuitable for accessing high molecular weight cyclic polymers because the ring-chain equilibrium is unfavorably biased towards the linear species as the degree of polymerization increases.14 The third approach has successfully produced high molecular weight cyclic polymers in high efficiency and purity without requiring dilution. The cyclic architecture is enforced by the molecular design of the cyclic catalytic species where the entropic penalty for cyclization is paid in advance15–19 or by a zwitterionic polymerization pathway where the propagating chain-ends are kept in close proximity throughout the polymerization reaction by ionic interactions.20–25
While the above mentioned approaches can afford cyclic homopolymers, their utility for thesynthesis of well-defined cyclic multiblock copolymers has not been extensively explored. Some of the ring expansion polymerization methods are unsuited for block copolymer synthesis due to either rapid chain termination or inter-chain transfer.18,22While high dilution conditions limit the synthetic efficiency of end-to-end coupling of linear block copolymer precursors, the approach is versatile with respect to providing block copolymers with diverse structures.26,27 A recent report showed that one could bypass the need for high dilution by forcing specially designed linear triblock precursors to self-assemble into micelles, thereby allowing end-to-end coupling to occur at the micelle interface.28
Polypeptoids, featuring an N-substituted polyglycine backbone, are structural mimics of polypeptides. While polypeptides adopt secondary structures (e.g., helix or sheet) that are stabilized by intra- or intermolecular hydrogen bonding, the folding of polypeptoids into well-defined secondary structures is dictated by side chain stereoelectronic effects.24,29–33 Studies conducted on oligomericpeptoids (n < 20) have demonstrated their biocompatibility, enhanced enzymatic stability34 – 36 and cell permeability relative to polypeptides.37 They have been extensively investigated for biological and therapeutic applications.34,35,38–41Recent investigations into the self-assembly of diblock peptoid oligomers and alternating peptoid oligomers have demonstrated the formation of novel homochiralsuperhelices and two dimensional bimolecular sheets.42, 43 In contrast to peptoid oligomers, polypeptoids or peptoid-based polymers are emerging as a new class of polymers24,25, 44 with potential biomedical uses owing to their demonstrated biocompatibility45, 46 and possible backbone degradability, as well as their structural analogy to existing bioinspired polymers such as poly(2-oxazoline)s and polypeptides.47Unlike polypeptides, the lack of extensive hydrogen bonding makes polypeptoids amenable to thermal processing techniques that are commonly used for thermoplastics.48 In summary, the combination of these features makes polypeptoids attractive peptidomimeticmaterials that are potentially useful for various biomedical applications (e.g., drug delivery and tissue engineering).
Cyclic oligomericpeptoidshaving controlled sequences have also been synthesized by end-to-end coupling of their linear precursors through stepwise solid-state synthesis methods.49–51 These cyclic peptoids exhibit enhanced conformational homogeneity relative to their linear analogs.49–51However, the ring size is limited in this coupling method. We have recently discovered that N-heterocyclic carbenes (NHC) can initiate the zwitterionic polymerization of N-substituted N-carboxyanhydride in a quasi-living manner to yield cyclic polypeptoids with large ring size (DP=400) and narrow molecular weight distribution (PDI<1.1).24Further, cyclic polypeptoids with random coil24 or PPI helical conformations25 can be readily accessed, and this method has not been extensively explored for block copolypeptoid synthesis. In this regard, Luxenhofer and coworkers have recently reported the synthesis of linear amphiphilic diblock copolypeptoids by primary amine-initiated polymerization of N-substituted N-carboxyanhydrides.44
In this contribution, we report the NHC-mediated zwitterionic polymerization of N-decylN-carboxyl anhydride (De-NCA) and the synthesis of cyclic poly(N-methyl-glycine)-b-poly(N-decyl glycine)s, an amphiphilic cyclic diblock copolypeptoid, by sequential monomer addition. Time-lapsed light scattering and cryo-TEM analysis of selected cyclic diblock copolypeptoids reveals the initial formation of spherical micelles in room temperature methanol, followed by the reorganization of these micelles into micron-long cylindrical micelles of uniform diameter over the course of several days. An identical morphological transition has been noted for linear analogs independently prepared by the primary amine-initiated polymerization of Me-NCA and De-NCA; however, the morphological transition occurs more rapidly for the linear polymers than their cyclic counterparts.
Experimental
Materials
All the solvents used in this study were purchased from Sigma-Aldrich and purified by passing through alumina columns under argon. Other chemicals were purchased from Sigma-Aldrich, and used as received. 1Hand 13C{1H}NMR spectra were recorded on a Bruker AV-400 spectrometer, and the chemical shifts in parts per million (ppm) were referenced relative to protio impurities or the 13C isotopes of CDCl3, CD3OD, C6D5CD3, or CF3COOD respectively. N-decylN-carboxyanhydride (De-NCA, M1),24N-Methyl N-carboxyanhydride (Me-NCA, M2)24 and 2,6-diisopropylphenylimidazol-2-ylidene (NHC)52 were synthesized by adapting literature procedures.
Instrumentations
ESI MS spectra were collected usinga Waters Synapt HDMS quadrupole / time-of-flight (Q/TOF) mass spectrometer (Waters, Milford, MA) under the following experimental conditions: ESI capillary voltage, 3.5 kV; sample cone voltage, 35 V; extraction cone voltage, 3.2 V; desolvation gas flow, 500 L·h−1 (N2); trap collision energy (CE), 6 eV; transfer CE, 4 eV; trap gas flow, 1.5 mL·min−1 (Ar); IM gas flow, 22.7 mL·min−1 (N2); sample flow rate, 5 μL·min−1; source temperature, 40 or 100 °C; desolvation temperature, 60 or 150 °C. The sprayed solutions were prepared by dissolving 0.3 mg of sample in 1 mL of CHCl3/MeOH (v/v, 50/50). Data analysis was conducted usingWaters MassLynx 4.1.
SEC analyses for thepoly(N-methyl-glycine)s (PNMGs) were conductedusing an Agilent 1200 system equipped with three Phenomenex 5 μm, 300×7.8 mm columns, Wyatt DAWN EOS multi-angle light scattering (MALS) detector (GaAs 30 mW laser at λ=690 nm) and Wyatt OptilabrEX differential refractive index (DRI) detector with a 690 nm light source. DMF containing 0.1M LiBr was used as the eluent at a flow rate of 1.0 mL·min−1. The column temperature was 50 °C, and the detector temperature was 25 °C. All data analyses were conducted using Wyatt Astra V 5.3 software. Polymer molecular weight (MW) and molecular weight distribution (PDI) were obtained by the Zimm model fit of the MALS-DRI data. The absolute molecular weights of the cyclic and linear PNMGs were determined using the reported dn/dc values [i.e., 0.0991 (8) mL·g−1 and 0.0987 (17) mL·g−1in DMF/0.1 M LiBr, respectively].24
SEC analyses for the poly(N-decyl-glycine)s (PNDGs) were conducted using an Agilent 1100 system (a Gastorr 704 degasser, isocratic pump, and Agilent 1100 auto sampler) equipped with two Phenomenex 10 μm, 300×7.8 mm columns [105 Å, linear MXM, and guard column], Wyatt DAWN DSP-F multi-angle light scattering (MALS) detector (a He-Ne 5 mW laser at λ = 632.8 nm), and Agilent 1200 differential refractive index (DRI) detector. THF containing 250 ppm BHT (2,6-di-tert-butyl-4-methylphenol) was used as the elution solvent at a flow rate of 1.0 mL·min−1. The columns were kept at room temperature. The temperatures of the MALS and DRI detector were room temperature and 35°C, respectively. All data analyses were performed using Wyatt Astra V 4.7 software. The MW and PDI of polymers were obtained by the Zimm model fit of MALS-DRI data. The absolute molecular weights of the cyclic and linear PNDGswere determined using the experimentally measureddn/dcvalues[i.e., 0.0876 (9) and 0.0863 (5) mL·g−1 in THF at 35°C,respectively].
The refractive index increment(dn/dc) of cyclic or linear PNDGs was measured using Wyatt's rEX DRI detector and Astra software dn/dc template. The polymers were purified by dissolving inTHF and twice precipitatedby the addition of methanol. Six THF solutions with different and precisely knownPNDG concentrations (0.25– 2.0mg·mL−1) were prepared and injected to the DRI detector. The dn/dc value was obtained from the linear fit to a plot of refractive index versus polymer concentration.
DSC studies of a series of cyclic PNDGs were conducted using a TA DSC 2920 calorimeter under nitrogen. Powder samples sealed into the aluminum pans were first heated from room temperature to 250°C at 10°C·min−1, cooled to 0°C at 5°C·min−1, and second heated to 250°C at 10°C·min−1.
WAXS experiments were conducted on the synchrotron beamline at the Center for Advanced Microstructures and Devices (CAMD, Baton Rouge, LA). Solid samples were annealed at 200°C under vacuum for 4 d. X-ray scattering patterns were collected under vacuum for 30 min using an image plate with a resolution of 200 μm per pixel (Molecular Dynamics storage phosphor screen with an active area of 20×25 cm). The wavelength of the X-ray beam was 1.55 Å. The sample-to-detector distance was 193.1 mm, giving aq range of 0.1 Å−1 to 2.5Å−1. WAXS data were reduced using FIT2D 12.077 developed by Dr. Andy Hammersley with the European Synchrotron Radiation Facility (ESRF).15 The domain spacing, dBragg, was calculated using Bragg's law: d = 2π/q, q = 4πsin(θ)/λ, where θ is the angle between the incident beam and the scattering planes.
Cryo-TEM experiments wereconducted on a Tecnai G2 12 Twin TEM operating at 120 keV, with a Gatan digital camera and analyzed using GatanDigitalMicrograph software. Samples were prepared on holey carbon coated 400 mesh copper grids, glow discharged in a plasma cleaner to render the carbon film hydrophilic. Grids were prepared using a FEI Vitrobot. The polymer solution (2–4 μL) was pipetted on to the carbon-coated side. The grids were then blotted to remove excess solution and plunged into liquid ethane near its freezing point, resulting in a vitrified thin film (~100–300 nm). Grids were then transferred to a Gatancryo-stage for analysis.
A representative procedure for cryo-TEM sample preparation is as follows. The block copolymer was mixed in methanol at 1 mg·mL−1. Then the mixture was heated to 70–80 °C and held at the temperature for 40 min in an oil bath; the opaque suspension became transparent upon heating. The solution was slowly cooled in an oil bathto room temperature. The copolymer solution then was passed through a 0.2 μm filter (Whatman, PTFE membrane), and centrifuged at 14,000 rpm for 30 min. Finally the solution was passed through a 0.02 μm filter (Anotop 25) and analyzed at varying time intervals over a period of 0–15 d (sample incubated at room temperature).
Light scattering measurements were taken using a Lexel Laser Inc. 488 nm laser operating at 100 mW coupled with a Brookhaven Instruments Corporation goniometer. Data were collected at 90°, and the scattered intensity (turbidity) was measured over the first 2 days after sample preparation.
Synthesis of 2-(n-decylamino)acetic acid hydrochloride(1)
Glyoxylic acid (14.8 g, 161 mmol) was stirred with n-decylamine (16 mL, 80.5 mmol) in CH2Cl2 (400 mL) for 24 h at room temperature. The solvent was removed under vacuum to yield a yellow viscous liquid to whichaqueous HCl (400 mL, 1M) was added. Thereaction mixture was heated at reflux for 20 h. The water was removed by rotary evaporation to afford a white solid which was further purified by recrystallization from methanol/diethyl ether at 0 °C (14.2 g, 70 % yield). 1H NMR (δ in DMSO, 400 MHz, ppm): 0.85 ppm (t, CH3(CH2)7CH2CH2-); 1.24 ppm (m, CH3(CH2)7CH2CH2-); 1.61 ppm (m, CH3(CH2)7CH2CH2-); 2.86 ppm (m, CH3(CH2)7CH2CH2-); 3.81 ppm (s, -COCH2-); 9.22 ppm (s, HNHCl). 13C{1H} NMR (δ in DMSO, 100 MHz, ppm): 14.0 (CH3(CH2)8CH2-); 22.1 (CH3CH2(CH2)7CH2-); 25.2 (CH3CH2CH2(CH2)6CH2-); 26.0 (CH3(CH2)2CH2(CH2)5CH2-); 28.5 (CH3(CH2)3CH2(CH2)4CH2-); 28.7 (CH3(CH2)4CH2(CH2)3CH2-); 28.8(CH3(CH2)5CH2(CH2)2CH2-), 28.9(CH3(CH2)6CH2CH2CH2-); 31.3 (CH3(CH2)7CH2CH2-); 46.7 (CH3(CH2)7CH2CH2-&-COCH2-); 168.0 (HOCO-).
Synthesis of 2-(N,N-tert-butoxycarbonyl-n-decylamino)acetic acid (2)
A mixture of 1(13 g, 52 mmol), di-tert-butyl dicarbonate (28.2 g, 129 mmol) and triethylamine (36 mL, 258 mmol) in distilled water (200 mL)was stirred at room temperaturefor 20 h. The reaction mixture was extracted with hexane (2×200 mL) to remove any unreacted di-tert-butyl dicarbonate. The aqueous phase was separated, acidified with aqueous HCl (50 mL, 4 M), and extracted with ethyl acetate (3×100 mL). The organic phase was then separated, washed with brine, and dried overanhydrous MgSO4. Subsequent filtration and solvent removal afforded paleyellow oil (11.7 g, 72 % yield). 1H NMR (δ in CDCl3, 400 MHz, ppm): 0.75 (t, CH3(CH2)7CH2CH2-); 1.15 (m, CH3(CH2)7CH2CH2-); 1.29 & 1.34 (s, -(CH3)3); 1.38 (m, CH3(CH2)7CH2CH2-); 3.13 (m, CH3(CH2)7CH2CH2-); 3.86 & 3.76 (s, -COCH2-). 13C{1H} NMR (δ in CDCl3, 100 MHz, ppm): 14.1 (CH3(CH2)8CH2-); 22.7 (CH3CH2(CH2)7CH2-); 26.8 (CH3CH2CH2(CH2)6CH2-); 28.1 (CH3(CH2)2CH2(CH2)5CH2-); 28.3 & 28.4 (-(CH3)3); 29.3 (CH3(CH2)3CH2(CH2)4CH2-); 29.4 (CH3(CH2)4CH2(CH2)3CH2-); 29.6(CH3(CH2)5CH2(CH2)2CH2-), 29.7(CH3(CH2)6CH2CH2CH2-); 31.9 (CH3(CH2)7CH2CH2-); 48.4 & 49.0 (CH3(CH2)7CH2CH2-&-COCH2-); 80.5 (-OC(CH3)3); 155.4 & 156.3 (-NCOO-); 175.2 & 175.5 (HOCO-).
Synthesis of N-decylN-carboxyanhydride (M1)
Compound2(10.3g, 32.8 mmol) was dissolved in anhydrous CH2Cl2(250 mL) under a nitrogen atmosphere. The solution was cooled to 0 °C and phosphorous trichloride (2.5 mL, 26.2 mmol) was added dropwise.The reaction mixture was stirred at 0 °C for 2 h, and then the volatileswere removed under vacuum.Inside the glovebox, the solid residue was extracted with anhydrous CH2Cl2 (15 mL) and filtered. The filtrate was concentrated and layered with hexane to recrystallize at −20°C overnight. White crystals were isolated by filtration and further purified by sublimation (4.5 g, 57 % yield). 1H NMR (δ in CDCl3, ppm): 0.87 (t, CH3(CH2)7CH2CH2-); 1.25 (m, CH3(CH2)7CH2CH2-); 1.58 (m, CH3(CH2)7CH2CH2-); 3.38 (t, CH3(CH2)7CH2CH2-); 4.09 (s, -COCH2-). 13C{1H} NMR (δ in CDCl3, 100 MHz, ppm): 14.2 (CH3(CH2)8CH2-); 22.8 (CH3CH2(CH2)7CH2-); 26.6 (CH3CH2CH2(CH2)6CH2-); 27.4 (CH3(CH2)2CH2(CH2)5CH2-); 29.2 (CH3(CH2)3CH2(CH2)4CH2-); 29.4 (CH3(CH2)4CH2(CH2)3CH2-); 29.6 [CH3(CH2)5CH2(CH2)2CH2-, CH3(CH2)6CH2CH2CH2-]; 32.0 (CH3(CH2)7CH2CH2-); 43.8 (CH3(CH2)8CH2-); 49.0 (-COCH2-); 152.2 (-NCOOCO-); 165.9 (-NCOOCO-).
Representative synthetic procedure for the cyclic poly(N-decyl-glycine)s (c-PNDGs)
In theglovebox, M1 (270 mg, 1.1 mmol, [M1]0=0.4 M) was dissolved in anhydrous THF (2.5 mL). A known volume of NHC/THF stock solution (246 μL, 11μmol, 45.5 mM, [M1]:[NHC]0 = 100:1) was added to the monomer solution. Polymerization was allowed to proceed at 70 °C for 24 h under nitrogen andthen quenched by the addition of cold methanol. The white precipitate was collected and washed with ample methanol and hexane to remove any unreacted monomer or initiator. Drying under vacuum yielded a white solid (180 mg, 82% yield). 1H NMR (δ in CDCl3/CF3COOD,400 MHz, ppm): 0.87 (CH3(CH2)7CH2CH2-); 1.25 (CH3(CH2)7CH2CH2-); 1.60 (CH3(CH2)7CH2CH2-); 2.95–3.29 (CH3N-); 3.28 (CH3(CH2)7CH2CH2-); 4.00 (-COCH2-). 13C{1H} NMR (δ in CDCl3/CF3COOD, 100MHz, ppm): 14.2 (CH3(CH2)8CH2-); 22.8 (CH3CH2(CH2)7CH2-); 26.8 (CH3CH2CH2(CH2)6CH2-); 28.1 (CH3(CH2)2CH2(CH2)5CH2-); 29.4 & 29.7 [CH3(CH2)3CH2(CH2)4CH2-, CH3(CH2)4CH2(CH2)3CH2-)]; 32.1 (CH3(CH2)7CH2CH2-); 48.2 (CH3(CH2)8CH2-); 49.7 (-NCOCH2-); 170.2 (-NCOCH2-).
Representative synthetic procedure for the cyclic poly(N-methyl-glycine)-b-poly(N-decyl-glycine) block copolymers(c-PNMG-b-PNDGs)
In the glovebox, Me-NCA (M2)(118 mg, 1.02 mmol, [M2]0=0.4 M) was dissolved in anhydrous acetonitrile (2.5 mL). A known volume of NHC/THF stock solution (450 μL, 20μmol, 45.5 mM, [M2]0:[NHC]0=50:1) was added using a syringe. Polymerization was allowed to proceed for 24 h at room temperature under a nitrogen atmosphere. Aliquots of the reaction mixture were taken and analyzed for conversion by 1H NMR and polymer MW by SEC.An acetonitrile solution (2.5 mL) containing De-NCA (M1) (247 mg, 1.02 mmol, [M1]0=0.4 M, [M1]0:[NHC]0=50:1) was then added into the above reaction mixture, which was heated at 70 °C for additional 48 h to reach complete conversion. The polymer product was then precipitated by the addition of methanol and collected by filtration. The polymer was further washed with ample methanol and followed by THF to remove any unreacted monomers or homopolymers, and dried under vacuum (218 mg, 79% yield). For the copolymers that contain high molar fractions of PNMGs (e.g., [M2]0:[M1]0:[NHC]0=100:10:1), the polymer products were obtained by precipitation in hexane and washed with ample THF. 1H NMR (δ in CDCl3/CF3COOD, 400 MHz, ppm): 0.87 (CH3(CH2)7CH2CH2-); 1.25 (CH3(CH2)7CH2CH2-); 1.60 (CH3(CH2)7CH2CH2-); 2.9–3.2 (CH3N-); 3.28 (CH3(CH2)7CH2CH2-); 4.09–4.45 (-COCH2-). 13C{1H} NMR (δ in CDCl3/CF3COOD, 100 MHz, ppm): 14.0 (CH3(CH2)8CH2-); 22.6 (CH3CH2(CH2)7CH2-); 26.6 (CH3CH2CH2(CH2)6CH2-); 27.9 (CH3(CH2)2CH2(CH2)5CH2-); 29.2 [CH3(CH2)3CH2(CH2)4CH2-,CH3(CH2)4CH2(CH2)3CH2-]; 29.4 [CH3(CH2)5CH2(CH2)2CH2-,CH3(CH2)6CH2CH2CH2-]; 31.8 (CH3(CH2)7CH2CH2-); 36.1 (CH3N-); 47.8 (CH3(CH2)8CH2-); 49.3–50.4 (-NCOCH2-); 170.0 (-NCOCH2-).
Synthetic procedure for the linearpoly(N-methyl-glycine)112-b-poly(N-decyl-glycine)16 block copolymer (l-PNMG112-b-PNDG16)
The butylamine initiator used in this synthesis was stirred over CaH2 for 24 h and distilled under vacuum. In the glovebox, Me-NCA (M2) (156 mg, 1.36mmol, [M2]0=0.4 M) was dissolved in anhydrous acetonitrile (3.3 mL). A known volume of BuNH2/THF stock solution (134 μL, 14μmol, 101.2mM, [M2]0:[NHC]0=100:1) was added using a syringe. Polymerization was allowed to proceed for 24 h at room temperature under a nitrogen atmosphere. Aliquots of the reaction mixture were taken and analyzed for conversion by 1H NMR and polymer MW by SEC. An acetonitrile solution (0.5 mL) containing De-NCA (M1) (49 mg, 0.2mmol, [M1]0=0.4 M, [M1]0:[NHC]0=15:1) was then added into the above reaction mixture, which was heated at 70 °C for additional 48 h to reach complete conversion. The polymer product was then precipitated by the addition of hexane and collected by filtration. The polymer was further washed with ample THF and MeOH to remove any unreacted monomers or homopolymers, and dried under vacuum (105 mg, 77% yield). The 1H and 13C{1H} NMR spectra of linear PNMG-b-PNDGs are identical with those of cyclic counterparts.
Kinetic studies of NHC-mediated ring-opening polymerization of M2
Polymerization was conducted in toluene-d8 at 70 °C inside a sealed J-Young NMR tube. 1H NMR spectrawerecollected at a constant time interval for more than four half-lives. Conversions were determined by integrating the four methylene proton resonances of the monomer at 2.7–3.0 ppm (k and j in Figure S6) against those of the polymer at 3.4–3.7 and 4.0–4.2 ppm (k′ and j′ in Figure S1). Kinetic experiments were repeated twice for each polymerization condition.
Results and Discussions
Synthesis and Characterization of Monomer and Cyclic Poly(N-decyl-glycine)s(c-PNDGs)
De-NCA (M2)has beensynthesized in good overall yield (50–60%) by adapting a previously reported procedure (Scheme 1)24 and purified by sublimation prior to polymerization. The desired monomer structure has been unambiguously verified by 1H and 13C{1H} NMR spectroscopy (Figure 1).
Scheme 1.

Figure 1.

1H and 13C{1H} NMR spectra of De-NCA (M1) in CDCl3.
We have previously demonstrated the N-heterocyclic carbene (NHC) can mediate the zwitterionic polymerization of N-butyl N-carboxyanhydride (Bu-NCA) in a quasi-living manner to yield cyclic poly(N-butyl-glycine)s.24 To ascertain that the polymerization of De-NCA (M1) occurs in a controlled manner, polymerization of M1 with various initial [M1]0:[NHC]0 ratio have been conducted (Table 1). The reactions were allowed to proceed in THF at 70 °C under a nitrogen atmosphere until of all the monomer was consumed. Reaction aliquots were taken and analyzed by 1H NMR spectroscopy to verify the conversion. The polymers were isolated by precipitation in cold methanol and dried under vacuum prior to further analysis.
Table 1.
NHC-mediated zwitterionic polymerizations of M1.a
| Entry# | [M1]0/[I]0 | Reaction time (h) | Conv. (%) | Mn(theor.)b (kg·mol−1) | Mn(SEC)c (kg·mol−1) | Mn(NMR)d (kg·mol−1) | PDIc |
|---|---|---|---|---|---|---|---|
| 1 | 25 | 8 | 89 | 4.8 | 4.8 | 5.6 | 1.14 |
| 2 | 50 | 16 | 93 | 9.6 | 10.2 | 11.1 | 1.03 |
| 3 | 100 | 16 | 91 | 18.3 | 20.4 | 20.0 | 1.15 |
| 4 | 200 | 21 | 83 | 33.1 | 31.0 | - | 1.09 |
Polymerizations were conducted in THF at 70 °C with [M1]0 = 0.4 M. SEC analyses were conducted by directly injecting the polymerization solutions into SEC columns after the designated reaction time
theoretical molecular weights were calculated based on conversion and the [M1]0:[NHC]0 ratio
determined by a tandem SEC-MALS-DRI system using the dn/dc [0.0876(9) mL·g−1] in THF at room temperature
determined by 1H NMR analysis. Entry 4: The NHC content of the high MW PNDG is too low to be accurately integrated and hence its MW cannot be reliably determined by 1H NMR.
The polymer structure has been characterized by 1H and 13C{1H} NMRspectroscopy (Figure 2 and S3) and ESI MS spectrometry (Figure 3). The solubility of the dry polymer in CDCl3 is limited and can be substantially improved by a few drops of CF3COOD. All proton resonances appear broad and their relative integrations are consistent with the desired polymer backbone structure (Scheme 2). ESI MS analysis of a low MW sample reveals one major (e, Figure 3) and four minor envelopes of doubly charged ions (a–d, Figure 3). The mass of the major set of ion corresponds to the sum of an integer number of the PNDG repeating unit mass (197.18), one NHC mass (388.288) and two proton ion masses (2.02), consistent with the zwitterionic or spirocyclic PNDG with a NHC affixed to the chain end (Scheme 2).24,53 One minor set of ions (c, Figure 3) corresponds to the same c-PNDG structure that is ionized by a potassium ion. Mass analysis of the other three minor sets of ions revealsseveral PNDG species with different end-groups (a, b, d, Figure 3). The PNDGs bearing carboxyl and amino chain ends (a, Figure 3) are likely to be formed by M1 polymerization that is initiated by residual water, whereas species b (Figure 3) is presumably formed by end-to-end cyclization of the zwitterionic propagating species prior to CO2 liberation (Scheme 2). While it has been demonstrated that NHC moieties can be readily eliminated from the NHC-terminated poly(ethylene oxide) zwitterions by reaction with nucleophiles (e.g., water or alcohol) in the NHC-mediated polymerization of ethylene oxide,54 we have found that treatment of NHC-polypeptoid adduct (c-PNDG, Scheme 2) with excess water did not result in increased formation of linear PNDGs (a, Figure 3).
Figure 2.

1H and 13C{1H} NMRspectra of a high MW cyclic PNDG in CDCl3/CF3COOD.
Figure 3.

Representative ESI MS spectra of a low MW cyclic PNDGsample synthesized from NHC-mediated ring-opening polymerization of M2.
Scheme 2.

While the ESI MS results suggest that the cyclic PNDGs with one NHC affixed to the chain end are the dominant species from the NHC-mediated polymerization of M2, high MW polymers cannot be analyzed by this method due to their poor ionization efficiency. To verify the architecture of high MW PNDG samples, SEC analysis of a high MW c-PNDG (Mn= 20.4 kg·mol−1, PDI=1.20) and its linear analog (Mn= 20.8 kg·mol−1, PDI=1.09) has been conducted (Figure S2). Both samples exhibit mono-modal SEC chromatograms with the c-PNDG sample eluting with a longer retention time than its linear analog, consistent with the former having a cyclic architecture and thus a smaller hydrodynamic volume than the latter.
The PNDGs prepared from the NHC-mediated polymerization of M1 with different [M1]0:[NHC]0 ratio have been analyzed by SEC-MALS-DRI. We note that it is difficult to completely redissolve the dry polymer in THF, even with heating, and the strong tendency of the polymer to aggregate causes erroneous and irreproducible SEC results. However, the aggregation is greatly diminished when the polymerization solution (in THF) is directly injected into SEC columns, which leads to reproducible SEC chromatograms (Figure 4). SEC-MALS-DRI analysis of the polymers reveals a systematic increase of polymer MW as the [M1]0:[NHC]0 ratio is raised (Table 1 and Figure 4). Furthermore, the experimental MWs determined by SEC are in good agreement with the theoretical MWs based on a NHC-initiated living polymerization. The SEC chromatograms are mono-modal for all polymer MWs (4.8–31 kg·mol−1), and the molecular weight distribution remains reasonably narrow (PDI = 1.03–1.15). The polymer MWs also have been determined by 1H NMR analysis. Specifically, integration of the two methylene proton resonances of the PNDG repeating unit (j in Figure S3) relative to the four methine proton resonances of the NHC (m in Figure S3) yields the number average degree of polymerization, assuming each polymer chain having one affixed NHC. The MWs obtained by 1H NMR analysis also agree reasonably well with the theoretical MWs based on single-site initiation by NHCs.
Figure 4.
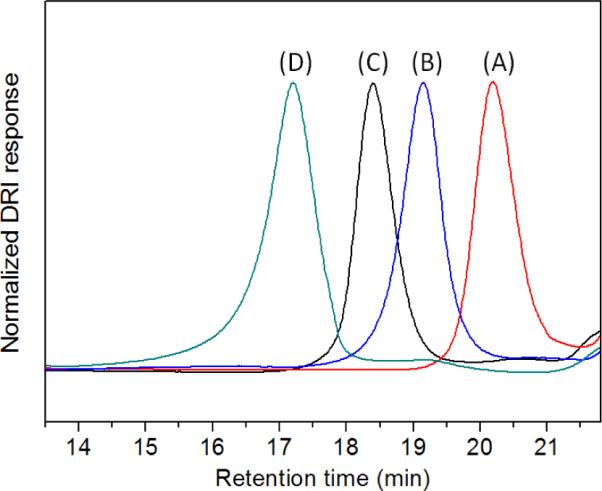
SEC chromatograms of the cyclic PNDGs prepared from NHC-mediated polymerization of M1 ([M1]0:[NHC]0 = 25:1 (A), 50:1 (B), 100:1 (C), and 200:1 (D), Table 1).
Polymerization kinetics were investigated at three different initial NHC concentrations (i.e., [NHC]0 = 3, 8 and 16 mM) and a constant initial monomer to initiator ratio ([M1]0:[NHC]0 = 50:1). All reactions exhibit a first-order dependence on the monomer concentration (i.e., d[M1]/ dt=kobs[M1]), consistent with a living polymerization. As the initial NHC concentration ([NHC]0) increases from 3 to 8 to 16 mM, the observed rate constant (kobs) increases from 0.62 (±5) to 1.74 (±10) and 2.77 (±6) h−1(Figure 5B). The plot of kobs against the initial NHC concentration reveals a linear relationship [i.e., kobs=kp[NHC]0, propagation rate constant kp= 178(9) M−1·h−1], indicative of single-site initiation by the NHC. Additionally, the plot of polymer MW as a function of conversion reveals a linear relationship (Figure S4), suggesting a constant concentration of propagating species, consistent with a living polymerization. The molecular weight distribution (PDI=1.03–1.22) also remains narrow throughout the reaction.
Figure 5.

(A) Plots of ln([M1]0/[M1]) versus the reaction time and their linearly fitted curves for the NHC-mediated polymerizations of M2 at 70 °C in toluene-d8 at three different initial NHC concentration (i.e., [NHC]0 = 3 (●), 8(▲), 16 mM (■)) and a constant initial monomer to NHC concentration (i.e., [M1]0:[NHC]0 = 50:1). (B) Plot of the observed rate constant (kobs) versus the initial NHC concentration and the linearly fitted curves.
The thermal properties of the different molecular weight c-PNDGs have been investigated by DSC. DSC thermograms reveal two prominent first-order exothermic transitions in the first cooling cycle (Tc,1= 47–51 °C, Tc,2= 135–141 °C) (Figure S5 and Table S1) and two first-order endothermic transitions in the second heating cycle (Tm,1= 72–79°C, Tm,2= 166–176 °C) (Figure 6A). No notable secondary transition was found in either cycle under the experimental conditions. Based on the magnitude of the enthalpic change (ΔHm,1 = 35–39 J·g−1 and ΔHm,2= 42–55 J·g−1) associated with the transitions, these are attributed to polymer crystallization and melting. In contrast, a previous crystallization study of oligomericpeptoidswith shorter alkyl side chains (e.g., n-butyl, n-hexyl, and n-octyl) revealed only one melting transition (Tm=155–220 °C).48 As a result, we tentatively attribute the two melting transitions (Tm,1= 72–79 °C, Tm,2= 166–176 °C) to crystalline packing of the c-PNDG side chains and the main chains, respectively. Alkyl side chains longer than ten carbons are presumably required for crystalline packing of the side chain in peptoid materials. Furthermore, thetwo melting transitions exhibit a clear molecular weight dependence, with the melting transition due to side chain packing (Tm,1) moving to lower temperature and the melting transition due to main chain packing (Tm,2) shifting to higher temperature as the polymer MW increases (Figure 6B). This suggests that increasing the polymer chain length promotes the packing of the main chains and inhibits the packing of the side chains.
Figure 6.

(A) DSC thermograms ofc-PNDGs having different molecular weight from the second heating cycle. (B) Plots of two melting points (Tm,1, Tm,2) as a function of polymer molecular weights.
The highly crystalline nature of the c-PNDGs also has been confirmed by variable-temperature WAXS analysis. At room temperature, a sharp principle scattering peak (d=24 Å) together with second and third order reflections are found for the thermally annealed c-PNDG (Mn = 7.7 kg·mol−1, PDI=1.26) (Figure 7A), attesting to a lamellar packing of the crystalline domain. The principle domain spacing is approximately twice the length of fully extended decyl group (i.e., 12.4 Å), consistent with the lamellar axis being parallel with the carbon chain of decyl group (Figure 7B). The peaks at q = 1.40 Å−1and 1.75 Å−1(d = 4.5 and 3.6 Å) have been assigned to the in-plane diffractions offthe polypeptoid main chains, where the inter-chain distance is 4.5 Å and the distance between adjacent repeating units is 3.6 Å, in agreement with those previously reported for oligomeric peptoids.42,43 The peak at q = 2.2 Å−1 (d = 2.7 Å) is thereby assigned to the (110) diffraction of an orthorhombic cell (a=4.5 Å, b=3.6 Å, c=24 Å).43In comparison, WAXS analysis of the as-prepared c-PNDG (Mn = 7.7 kg·mol−1, PDI=1.26) without thermal annealing at room temperature (Figure S6) reveals a diffraction patternanalogous to that of the annealed sample (Figure 7A), except that the second and third order reflections associated the principle scattering peaks (d=24 Å) are significantly weakened, suggesting that thermal annealing promotes the long range lamellar packing. At 100 °C, a temperature that is between the first and second melting transitions, no appreciable change was observed for the WAXS pattern of the as-prepared c-PNDG (Figure S6), suggesting that the diffractions are all related to the polymer backbone crystallinity and the melting of side chains do not significantly affect the lamellar packing. As a result, the diffraction peaks at q = 1.25 Å−1 (d = 5.0 Å) and q = 1.51 Å−1 (d = 4.2 Å) are tentatively assigned to the diffraction by polypeptoid main chains that crystallize in different polymorphs,where the d-spacing corresponds to the inter-chain distance in these polymorphs. At 250 °C which is above the second melting transition temperature, the WAXS diffractogram (Figure S6) exhibits two broad and featureless peaks, consistent with a lack of crystalline order due to complete polymer melting at this temperature.
Figure 7.

(A) WAXS diffractogram of a thermally annealed c-PNDG in the solid state (Mn = 7.7 kg·mol−1, PDI=1.26) at room temperature and (B) the proposed crystalline packing of c-PNDG based on WAXS data.
Synthesis and Characterization of Cyclic Poly(N-methyl-glycine)-b-poly(N-decyl-glycine) Diblock Copolymers (c-PNMG-b-PNDG)
Sequential NHC-mediated polymerizations of Me-NCA (M2) and De-NCA (M1) were conducted to synthesize cyclic poly(N-methyl-glycine)-b-poly(N-decyl-glycine) diblock copolymers (c-PNMG-b-PNDG) (Scheme 3). The PNMG homopolymers have poor solubility in THF or toluene, the solvents commonly used in NHC-mediated polymerizations of N-substituted NCAs. While acetonitrile appears to be a good solvent for PNMGs, the solubility of the polymer decreases as the MW increases. To ensure that polymerization occurs in a homogenous fashion, we targeted at a series of c-PNMG-b-PNDG block copolymers where the PNMG block length is kept constant [Mn(SEC-MALS-DRI)=10.6kg·mol−1, DPn(1H NMR)=105, PDI=1.04] and the PNDG block length is systematically increased. Under anaerobic conditions in room temperature acetonitrile, the NHC-mediated polymerization of M2 ([M2]0:[NHC]0=100:1, [M2]0= 0.4M) required 24 h for quantitative conversion. Aliquots of the reaction mixture that were taken to verify conversion and the number degree of polymerization by 1H NMR spectroscopy were also analyzed by SEC-MALS-DRI for the polymer MW and PDI. Variable amounts of M1 were subsequently introduced to the above reaction mixture, and the reaction was allowed tocontinue at 70 °C for another 48 h to reach full conversion. The diblock copolymers were isolated by precipitation with methanol and purified by washing with a mixture of methanol and THF. We have noted that c-PNMG-b-PNDGs with high PNDG content become sparingly soluble in common organic solvents once isolated and dried. This enables the removal of any homopolymer impurities by washing with selected solvents, i.e., methanol for PNMG and THF for PNDG. SEC-DRI analysis of a c-PNMG-b-PNDG sample with low PNDG content reveals an increase in the hydrodynamic size in comparison to the c-PNMG precursor, confirming successful enchainment by sequential monomer addition (Figure S7).
Scheme 3.

1H and 13C{1H} NMR spectra of the purified products are consistent with the desired backbone structure of the c-PNMG-b-PNDG block copolymers (Figure 8 and S9). The NHC moieties remain attached to the block copolymers, as evidenced by the1H NMR analysis of a low MW c-PNMG-b-PNDG sample (Figure S8). Polymer MWs of c-PNMG-b-PNDG diblock copolypeptoids with high PNDG content cannot be directly determined by SEC-MALS-DRI alone due to limited solubility and the strong tendency to aggregate in many common organic solvents (e.g., THF, CHCl3, methanol, DMF). As a result, 1H NMR spectroscopy has been used in conjunction with SEC-MALS-DRI to determine the molecular weight (Mn) of the block copolymers. Specifically, the molar ratios of the PNMG and PNDG repeating units were determined by integrating the methyl proton resonance of PNMGs (k, Figure 8) against the methyl proton resonance of PNDGs (j, Figure 8). As the absolute MW (and PDI) of the PNMG block can be determined by SEC-MALS-DRI and the 1H NMR analysis,24 the number average molecular weight of the block copolymers (Mn) can be deduced. All 1H NMR spectra of the block copolymers were collected either in CDCl3/CF3COOD or CD3OD/CF3COOD to ensure their complete solubility.
Figure 8.

1H NMR spectra of a series of cyclic diblock copolymers with various compositions in CDCl3/CF3COOD: (1) PNMG105-b-PNDG10, (2) PNMG105-b-PNDG25, (3) PNMG105-b-PNDG50 and (4) PNMG105-b-PNDG125.
Table 3 summarizes the experimentally determined c-PNMG-b-PNDGblock copolymer compositions and MWs. It is evident that c-PNMG-b-PNDGs with variable PNDG block length can be readily prepared by adjusting the ratio of [M2]0:[M1]0:[NHC]0. The copolymer MW and the relative weight fraction of the two blocks agree reasonably well with the theoretical prediction based on the [M2]0:[M1]0:[NHC]0 ratio and conversion (Table 3). The deviation of MW from the theoretical value is more pronounced for high MW polymers, with the copolymer having a longer PNDG block length than theoretically predicted (entry 4, Table 3). This increased deviation suggests the possibility of incomplete chain extension from the first PNMG block, presumably due to its limited solubility in the reaction medium.
Table 3.
Compositions and the polymer MWs of cyclic poly(N-methyl-glycine)-b-poly(N-decyl-glycine) [c-PNMG-b-PNDG] block copolymersa
| Entry # | [M2]0:[M1]0:[NHC]0 | Experimental compositionb | Mn (theor.) (kg·mol−1)c | Mn (expt.) (kg·mol−1)b | WPNMG(theor.)d | WPNMG(expt.)e | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 100:10:1 | PNMG105-b-PNDG10 | 9.5 | 9.8 | 0.78 | 0.79 | 85 |
| 2 | 100:25:1 | PNMG105-b-PNDG25 | 12.4 | 12.8 | 0.59 | 0.60 | 84 |
| 3 | 100:50:1 | PNMG105-b-PNDG50 | 17.4 | 17.7 | 0.42 | 0.43 | 85 |
| 4 | 100:100:1 | PNMG105-b-PNDG125 | 27.2 | 32.5 | 0.26 | 0.23 | 82 |
Polymerization of M2 and M1 were both allowed to reach full conversion
the number average degree of polymerization (DPn) of the PNMG block was determined by 1H NMR analysis, from which the PNMG molecular weights were deduced; the block copolymer compositions were determined by 1H NMR analysis of c-PNMG-b-PNDGs, from which the copolymer molecular weights were deduced
the theoretical molecular weights were determined from the [M2]0:[M1]0:[NHC]0 ratio
the theoretical weight fractions of the PNMG block were determined from the [M2]0:[M1]0:[NHC]0 ratio
the experimental weight fractions of the PNMG block were determined from the experimental compositions of the copolymers.
Solution Self-Assembly of Cyclic Poly(N-methyl-glycine)-b-poly(N-decyl-glycine) Diblock Copolypeptoid (c-PNMG-b-PNDG)
The PNMG-b-PNDG block copolypeptoids exhibit strong tendencies for aggregation in common solvents. For example, we noted that a turbid methanol or aqueous suspension of c-PNMG105-b-PDMG10 becomes clear upon gentle heating and remains so for an extended period of time (> 6 months) after cooling. To investigate the aggregate structures of the block copolypeptoids in methanol, a dilute methanol solution of c-PNMG105-b-PDMG10 (1 mg·mL−1) was characterized by both light scattering and cryogenic transmission electron microscopy (cryo-TEM) over a period of 15 d.
Light scattering analysis of the copolymer solutions in methanol (Figure S10) revealed an increase in turbidity over the first 2 days, suggesting an increase in size of the self-assembled structures. The linear copolypeptoid solution showed a more rapid increase in turbidity compared to the cyclic copolypeptoid solution. A kinetic cryo-TEM study of c-PNMG105-b-PDMG10 revealed the initial formation of spherical micelles in room temperature methanol solution (Figure 9A) within 30 min of sample preparation. After 1 h the spherical micelles remained; however, after 2 h the appearance of cylindrical micelles were evident (Figure 9B), which appeared to increase in length over time (Figure 9C). Over the course of the first 24 h the solution remained a mix of spherical and cylindrical micelles as the spherical micelles continued to aggregate and form longer cylindrical micelles. After 7 d cylindrical micelles were very much the dominant species, which continued to increase in length over the course of the next 15 d, after which, mostly micron length cylindrical micelles were evident in solution with a very small fraction of spherical micelles remaining (Figure 9C). The darker areas in the cryo-TEM images correspond to the PNDG-rich regions as a result of their higher electron density relative to that of the solvated PNMG blocks. Being solvophobic and highly crystalline, the PNDG blocks constitute the core of the micelles in methanol solution, whereas the solvophilic and amorphous PNMG blocks make up the shell of the micelles (Figure 9). The average core diameter of the spherical micelles is 10.3 (±2.9) nm (Figure 9A). Over the course of 15 d, the spherical micelles slowly reorganize into micron-long cylindrical micelles with a uniform core diameter of 12.2 (±1.8) nm (Figure 9C). The evolution of spherical micelles into cylindrical micelles was also observed for the linear poly(N-methyl-glycine)-b-poly(N-decyl-glycine)block copolypeptoid (l-PNMG112-b-PNDG16) that was prepared from sequential BuNH2-initiated polymerization of Me-NCA and De-NCA. Cryo-TEM analysis of l-PNMG112-b-PNDG16 reveals the morphological transition from spherical micelles [d=10.0 (±1.6) nm] (Figure 9D) into cylindrical micelles [d=12.5 (±1.7) nm] (Figures 9E and 9F) over a faster time scale than the cyclic copolypeptoid. The analysis of l-PNMG112-b-PNDG16 revealed the cylindrical micelles were the dominant species after only 8 days, with no spherical micelles remaining in solution after the full 15 d. The different rates of transition in the cyclic and linear copolypeptoid solutions revealed in the cryo-TEM data (Figure 9) are consistent with the light scattering data (Figure S8) where a faster increase in turbidity was measured for the linear copolypeptoid relative to the cyclic counterpart.
Figure 9.

Representative cryo-TEM images obtained from dilute methanol solutions of the cyclic c-PNMG105-b-PNDG10 block copolypeptoid after 1 h (A), 2 h (B), and 15 d (C), and the linear PNMG112-b-PNDG16 block copolypeptoid after 1 h (D), 2 h (E), and 7 d (F) in methanol.
The SEC-DRI chromatogram of a cyclic copolypeptoid (i.e., c-PNMG105-b-PDMG15) obtained after 17 days in room temperature methanol is identical to that of the original sample (Figure S11), suggesting the architecture of the cyclic copolypeptoids remain intact under these conditions. We tentatively attribute the crystallization of the PNDG hydrophobic core as the driving force for the morphological transition, which we believe is retarded in the cyclic copolypeptoid aggregates, leading to the slower structural transitions. Previous studies on the self-assembly of amorphous-crystalline diblock copolymers [i.e. polyisoprene-b-polyferrocenylsilane,55–58 poly(3-hexylthiophene)-b-poly(dimethylsiloxane),59 poly(lactide)-b-poly(acrylic acid)60] have shown that the formation of cylindrical micelles is the result of maximizing the packing of the short crystalline block. The cylindrical micelles undergo epitaxial growth in a living manner with the addition of the block copolymer unimers.55–58 In contrast, cryo-TEM images of the block copolypeptoid samples taken between 1 h and 15 d reveal that spherical micelles are initially formed before merging into one another to generate the cylindrical micelles (Figures 9B and 9E) that continue to grow in length (Figures 9C and 9F), suggesting an aggregation induced morphological transition.
While topological constraints have been shown to induce completely different micellar morphologies in the self-assembly of cyclic and linear polystyrene-b-polyisoprene (PS-b-PI) in n-heptane or n-decane, the two constituent blocks are both amorphous.9–12 To verify that the morphological transition observed in the c/l-PNMG-b-PNDG is due to crystallization of the solvophobic block (PNDG), the synthesis of amorphous-amorphous cyclic and linear diblock copolypeptoid analogs and investigation of their solution self-assembly behavior is currently in progress.
Conclusions
We have demonstrated that NHC mediates the zwitterionic polymerization of N-decylN-carboxyanhydride (De-NCA) to afford cyclic poly(N-decyl-glycine) (c-PNDG) having controlled polypeptoid MW and narrow PDI. The reaction exhibits characteristics of a living polymerization and has been successfully extended towards the synthesis of amphiphilic cyclic block copolypeptoids [i.e. cyclic poly(N-methyl-glycine)-b-poly(N-decyl-glycine) (c-PNMG-b-PNDG)] by sequential monomer addition. While the exact composition of the block copolypeptoids appear to deviate somewhat from the theoretical values, the relative PNMG and PNDG content in the amphiphilic block copolypeptoids can be adjusted by controlling the initial monomer to NHC ratio and the conversion. This new class of cyclic amphiphilic block copolymers exhibit a strong tendency towards aggregation, forming discrete supramolecular assemblies (e.g., spherical or cylindrical micelles). The synthesis of these block copolypeptoids sets the foundation for systematic investigations of their self-assembly properties in solution and in the solid state. Their self-assembly also provides a means to access novel peptidomimetic materials of variable length scale and dynamics which are potentially useful for various biomedical applications such as drug delivery carriers or bioactive coatings.
Supplementary Material
Table 2.
Melting temperatures and heat of fusionof c-PNDGs having different MWs.
| Entry # | Mn(kg·mol−1) | Tm,1 (°C) | Tm,2 (°C) | ΔHm,1 (J·g−1) | ΔHm,2 (J·g−1) |
|---|---|---|---|---|---|
| 1 | 7.7 | 78 | 167 | 39 | 49 |
| 2 | 13.5 | 77 | 174 | 39 | 55 |
| 3 | 24.3 | 73 | 176 | 35 | 42 |
| 4 | 63.2 | 72 | 175 | 37 | 42 |
Acknowledgements
The authors thank Dr. Xiaopeng Li for conducting ESI MS experiments (Department of Chemistry, University of Akron). The authors also thank Dr. Rafael Cueto for assisting the SEC studies. This work was supported byLouisiana State University, National Science Foundation (CHE-0955820) and Louisiana State Board of Regents [LEQSF(2008-11)-RD-A-11]. T.P.S and T.H.E thank the NIH-NCRR COBRE (#P20RR017716) and NIST, U.S. Department of Commerce (#70NANB7H6178) for financial support. The statements herein do not reflect the views of NIH or NIST.We acknowledge the University of Delaware Keck Microscopy Facility for use of their TEM and Vitrobot, and the University of Delaware Center for Molecular and Engineering Thermodynamics for the use of their light scattering instrument.
Footnotes
Supporting Information Available Representative 1H NMR spectrum of a NHC-mediated polymerization of M1 showing the formation of c-PNDG and unreacted M1, SEC chromatograms of a high MW c-PNDG sample and its linear analog (l-PNDG), 1H NMR spectrum of a low MW c-PNDG in CDCl3/CF3COOD, plot of polymer molecular weight determined by SEC-MALS-DRI or 1H NMR analysis, and PDI of c-PNDG versus conversion for the NHC-mediated polymerization of M2, DSC thermograms of c-PNDGs having different molecular weight from the first cooling cycle, crystallization temperatures and heat of fusionsof c-PNDGs having different MWs, WAXS diffractogram of c-PNDGs in the solid state at different temperature, SEC-DRI chromatograms of c-PNMG105 and c-PNMG105-b-PNDG15 with low PNDG content, 13C{1H} NMR spectra of c-PNMG105-b-PNDG50in CDCl3/CF3COOD, 1H NMR spectrum of a low MW cyclic poly(N-Me-glycine)72-b-poly(N-De-glycine)8 diblock copolymers in CD3OD,turbidity measurements of linear and cyclic block copolypeptoid solutions, SEC-DRI chromatograms of c-PNMG105-b-PNDG15 obtained after 17 days in room temperature methanol and the original sample, 1H and 13C{1H} NMR spectra of 2-(n-decylamino)acetic acid hydrochloride(1) in DMSO-d6, and 1H and 13 C{1H} NMR spectra of 2-(N,N-tert-butoxycarbonyl-n-decylamino)acetic acid (2) in CDCl3. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Kricheldorf HR. J. Polym. Sci., Part A: Polym. Chem. 2010;48:251–284. [Google Scholar]
- 2.Semlyen JA. Cyclic Polymers. 2nd Ed. Kluwer Academic Publishers; Dordrecht, Netherland: 2000. [Google Scholar]
- 3.Douglas JF, Freed KF. Macromolecules. 1984;17:2344–2354. [Google Scholar]
- 4.Ohta Y, Masuoka K, Takano A, Matsushita Y. Physica B: Condensed Matter. 2006;385:532–534. [Google Scholar]
- 5.Zimm BH, Stockmayer WH. J. Chem. Phys. 1949;17:1301–1314. [Google Scholar]
- 6.Fox ME, Szoka FC, Fréchet JM. J. Acc. Chem. Res. 2009;42:1141–1151. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasongkla N, Chen B, Macaraeg N, Fox ME, Fréchet JMJ, Szoka FC. J. Am. Chem. Soc. 2009;131:3842–3843. doi: 10.1021/ja900062u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin EJ, Jeong W, Brown HA, Koo BJ, Hedrick JL, Waymouth RM. Macromolecules. 2011;44:2773–2779. [Google Scholar]
- 9.Minatti E, Borsali R, Schappacher M, Deffieux A, Soldi V, Narayanan T, Putaux JL. Macromol.Rapid Commun. 2002;23:978–982. [Google Scholar]
- 10.Minatti E, Viville P, Borsali R, Schappacher M, Deffieux A, Lazzaroni R. Macromolecules. 2003;36:4125–4133. [Google Scholar]
- 11.Lecommandoux S, Borsali R, Schappacher M, Deffieux A, Narayaman T, Rochas C. Macromolecules. 2004;37:1843–1848. [Google Scholar]
- 12.Putaux JL, Minatti E, Lefebvre C, Borsali R, Schappacher M, Deffieux A. Faraday Discuss. 2005;128:163–178. doi: 10.1039/b403157a. [DOI] [PubMed] [Google Scholar]
- 13.Honda S, Yamamoto T, Tezuka Y. J. Am. Chem. Soc. 2010:14759–14769. doi: 10.1021/ja104691j. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson H, Stockmayer WH. J. Chem. Phys. 1950;18:1600–1606. [Google Scholar]
- 15.Bielawski CW, Benitez D, Grubbs RH. J. Am. Chem. Soc. 2003;125:8424–8425. doi: 10.1021/ja034524l. [DOI] [PubMed] [Google Scholar]
- 16.Boydston AJ, Holcombe TW, Unruh DA, Frechet JMJ, Grubbs RH. J. Am. Chem. Soc. 2009;131:5388–5389. doi: 10.1021/ja901658c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boydston AJ, Xia Y, Kornfield JA, Gorodetskaya IA, Grubbs RH. J. Am. Chem. Soc. 2008;130:12775–12782. doi: 10.1021/ja8037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia Y, Boydston AJ, Yao Y, Kornfield JA, Gorodetskaya IA, Spiess HW, Grubbs RH. J. Am. Chem. Soc. 2009;131:2670–2677. doi: 10.1021/ja808296a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Jérôme R, Lecomte P. Macromolecules. 2008;41:650–654. [Google Scholar]
- 20.Culkin DA, Jeong W, Csihony S, Gomez ED, Balsara NP, Hedrick JL, Waymouth RM. Angew. Chem., Int. Ed. 2007;119:2681–2684. doi: 10.1002/anie.200604740. [DOI] [PubMed] [Google Scholar]
- 21.Jeong W, Hedrick JL, Waymouth RM. J. Am. Chem. Soc. 2007;129:8414–8415. doi: 10.1021/ja072037q. [DOI] [PubMed] [Google Scholar]
- 22.Jeong W, Shin EJ, Culkin DA, Hedrick JL, Waymouth RM. J. Am. Chem. Soc. 2009;131:4884–4891. doi: 10.1021/ja809617v. [DOI] [PubMed] [Google Scholar]
- 23.Shin EJ, Brown HA, Gonzalez S, Jeong W, Hedrick JL, Waymouth RM. Angew.Chem. Int. Ed. 2011;50:6388–6391. doi: 10.1002/anie.201101853. [DOI] [PubMed] [Google Scholar]
- 24.Guo L, Zhang D. J. Am. Chem. Soc. 2009;131:18072–18074. doi: 10.1021/ja907380d. [DOI] [PubMed] [Google Scholar]
- 25.Guo L, Li J, Brown Z, Ghale K, Zhang D. Biopolym.: Peptide Sci. 2011;96:596–603. doi: 10.1002/bip.21597. [DOI] [PubMed] [Google Scholar]
- 26.Eugene DM, Grayson SM. Macromolecules. 2008;41:5082–5084. [Google Scholar]
- 27.Zhang Y, Wang G, Huang J. J. Polym. Sci., Part A: Polym. Chem. 2011;49:4766–4770. [Google Scholar]
- 28.Hu J, Zheng R, Wang J, Hong L, Liu G. Macromolecules. 2009;42:4638–4645. [Google Scholar]
- 29.Baldauf C, Günther R, Hofmann HJ. Phys.Biol. 2006;3:S1–S9. doi: 10.1088/1478-3975/3/1/S01. [DOI] [PubMed] [Google Scholar]
- 30.Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. J. Am. Chem. Soc. 2008;130:16622–16632. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]
- 31.Paul B, Butterfoss GL, Boswell MG, Renfrew PD, Yeung FG, Shah NH, Wolf C, Bonneau R, Kirshenbaum K. J. Am. Chem. Soc. 2011;133:10910–10919. doi: 10.1021/ja2028684. [DOI] [PubMed] [Google Scholar]
- 32.Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KTV, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc. Natl. Acad. Sci. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armand P, Kirshenbaum K, Falicov A, Dunbrack JRL, Dill KA, Zuckermann RN, Cohen FE. Folding and Design. 1997;2:369–375. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 34.Patch JA, Barron AE. Curr.Opin. Chem. Biol. 2002;6:872–877. doi: 10.1016/s1367-5931(02)00385-x. [DOI] [PubMed] [Google Scholar]
- 35.Barron AE, Zuckermann RN. Curr.Opin. Chem. Biol. 1999;3:681–687. doi: 10.1016/s1367-5931(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 36.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg. Med. Chem. Lett. 1994;4:2657–2662. [Google Scholar]
- 37.Kwon Y, Kodadek T. J. Am. Chem. Soc. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc. Natl. Acad. Sci. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fowler SA, Blackwell HE. Org. Biomol. Chem. 2009;7:1508–1524. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodson B, Ehrhardt A, Ng S, Nuss J, Johnson K, Giedlin M, Yamamoto R, Moos WH, Krebber A, Ladner M. Antimicrob. Agents Chemother. 1999;43:1429–1434. doi: 10.1128/aac.43.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuckermann RN, Kodadek T. Curr. Opin. Mol. Ther. 2009;11:299–307. [PubMed] [Google Scholar]
- 42.Murnen HK, Rosales AM, Jaworski JN, Segalman RA, Zuckermann RN. J. Am. Chem. Soc. 2010;132:16112–16119. doi: 10.1021/ja106340f. [DOI] [PubMed] [Google Scholar]
- 43.Nam KT, Shelby SA, Choi PH, Marciel AB, Chen R, Tan L, Chu TK, Mesch RA, Lee B-C, Connolly MD, Kisielowski C, Zuckermann RN. Nat. Mater. 2010;9:454–460. doi: 10.1038/nmat2742. [DOI] [PubMed] [Google Scholar]
- 44.Fetsch C, Grossmann A, Holz L, Nawroth JF, Luxenhofer R. Macromolecules. 2011;44:6746–6758. [Google Scholar]
- 45.Kanzaki T, Horikawa Y, Makino A, Sugiyama J, Kimura S. Macromol.Biosci. 2008;8:1026–1033. doi: 10.1002/mabi.200800071. [DOI] [PubMed] [Google Scholar]
- 46.Makino A, Kizaka-Kondoh S, Yamahara R, Hara I, Kanzaki T, Ozeki E, Hiraoka M, Kimura S. Biomaterials. 2009;30:5156–5160. doi: 10.1016/j.biomaterials.2009.05.046. [DOI] [PubMed] [Google Scholar]
- 47.Hoogenboom R, Schlaad H. Polymers. 2011;3:467–488. [Google Scholar]
- 48.Rosales A, Murnen H, Zuckermann R, Segalman R. Macromolecules. 2010;43:5627–5636. [Google Scholar]
- 49.Shin S, Yoo B, Todaro L, Kirshenbaum K. J. Am. Chem. Soc. 2007;129:3218–3225. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- 50.Holub J, Jang H, Kirshenbaum K. Org. Lett. 2007;9:3275–3278. doi: 10.1021/ol071169l. [DOI] [PubMed] [Google Scholar]
- 51.Yoo B, Shin S, Huang M, Kirshenbaum K. Chem. Eur. J. 2010;16:5528–5537. doi: 10.1002/chem.200903549. [DOI] [PubMed] [Google Scholar]
- 52.Arduengo AJ, Krafczyk R, Schmutzler R, Craig HA, Goerlich JR, Marshall WJ, Unverzagt M. Tetrahedron. 1999;55:14523–14534. [Google Scholar]
- 53.Li X, Guo L, Casiano-Maldonado M, Zhang D, Westdemiotis C. Macromolecules. 2011;44:4555–4564. [Google Scholar]
- 54.Raynaud J, Christelle A.; Gnanou Y, Taton D. J. Am. Chem. Soc. 2009;131:3201–3209. doi: 10.1021/ja809246f. [DOI] [PubMed] [Google Scholar]
- 55.Massey JA, Temple K, Cao L, Rharbi Y, Raez J, Winnik MA, Manners I. J. Am. Chem. Soc. 2000;122:11577–11584. [Google Scholar]
- 56.Cao L, Manners I, Winnik M. Macromolecules. 2002;35:8258–8260. [Google Scholar]
- 57.Wang X, Guerin G, Wang H, Wang Y, Manners I, Winnik M. Science. 2007;317:644–646. doi: 10.1126/science.1141382. [DOI] [PubMed] [Google Scholar]
- 58.Gilroy JB, Gädt T, Whittell GR, Chabanne L, Mitchels JM, Richardson RM, Winnik MA, Manners I. Nat. Chem. 2010;2:566–570. doi: 10.1038/nchem.664. [DOI] [PubMed] [Google Scholar]
- 59.Patra SK, Ahmed R, Whittell GR, Lunn DJ, Dunphy EL, Winnik MA, Manners I. J. Am. Chem. Soc. 2011;133:8842–8845. doi: 10.1021/ja202408w. [DOI] [PubMed] [Google Scholar]
- 60.Petzetakis N, Dove AP, O'Reilly RK. Chem. Sci. 2011;2:955–960. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.