

SUMMARY
There is an urgent need for the discovery and development of new antitubercular agents that target new biochemical pathways and treat drug resistant forms of the disease. One approach to addressing this need is through high throughput screening of medicinally relevant libraries against the whole bacterium in order to discover a variety of new, active scaffolds that will stimulate new biological research and drug discovery. Through the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (www.taacf.org), a large, medicinally relevant chemical library was screened against M. tuberculosis strain H37Rv. The screening methods and a medicinal chemistry analysis of the results are reported herein.
Keywords: TAACF, antitubercular, high throughput screening methods, medicinal chemistry analysis, medicinally relevant chemical library
The Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) Program was established through the National Institute of Allergy and Infectious Diseases in 1994 to allow researchers access to high quality screening services in order to encourage tuberculosis drug discovery research. Unique to that program was the required involvement of trained medicinal chemists from the outset in order to recruit high quality, medicinally relevant samples into the screening program. Furthermore, the medicinal expertise is available to researchers, biologists, and synthetic chemists that supply compounds to the program. More recently, the purchase of libraries such as the ChemBridge library described herein has been a TAACF function with the goal of making a large volume of new screening data against tuberculosis publicly accessible to the tuberculosis research community. This library and the functions of the TAACF program are described in a recent publication.1
TAACF medicinal chemists have begun the process of reviewing the screening data from the ChemBridge library. Compounds are prioritized for further analysis and pursuit. The initial analysis of the data from the screening of the ChemBridge library of compounds against Mycobacterium tuberculosis is described on the following pages, and the full analysis will be made publicly available at www.taacf.org and http://pubchem.ncbi.nlm.nih.gov/.
Tuberculosis (TB) represents one of the top public health concerns worldwide. One-third of the world’s population is infected with Mycobacterium tuberculosis, the etiological agent of TB causing 9.2 million new cases and 1.7 million deaths in 2006.2 The multi-drug regimen for treating TB established since the 1970s and still recommended today by the WHO has not been sufficient to eliminate TB due to the advent of HIV/AIDS, failure of treatment programs and enhanced transmission in hospitals and prisons.3 The recommended combination therapy for TB is lengthy and cumbersome since it can involve up to four drugs and requires daily medication for 6 to 12 months. Limited health care system resources often lead to treatment interruption or failure, exacerbating drug resistance problems.4
Multidrug resistant TB (MDR-TB), defined as resistance to at least the first-line drugs isoniazid and rifampicin, and extensively drug–resistant TB (XDR-TB), defined as resistance to rifampin, isoniazid, fluoroquinolones and to at least one of the injectable second-line drugs, have contributed to the resurgence of TB.5-6 Estimates for the global burden of drug resistant TB (including XDR-TB estimated at 40,000 XDR-TB cases per year) are likely a lower limit of the real case burden.7 Forty-nine countries have now reported XDR-TB infections as of June 2008 according to the WHO Stop TB Department (http://www.who.int/entity/tb/challenges/xdr/xdr_map_june08.pdf). Mathematical modeling reveals that if TB treatment does not improve globally, XDR-TB could represent 70% of the MDR-TB cases within 60-70 years.8 At present we know very little about how to reproducibly cure XDR-TB. Several reports attempted to validate treatment outcomes for XDR-TB, but in many cases the exact clinical history and specific use of antitubercular drugs is not fully known.9-19 Equally inadequate is the treatment available for the latent TB infection present in at least two billion individuals, which represents an enormous human reservoir of the infecting organism.20
The need for novel, more effective drugs to improve TB control is evident. Treatment of active disease needs to be shortened, simplified, and should not interfere with the administration of antiretroviral agents. It is especially desirable to identify new types of TB drugs acting on novel drug targets with no cross-resistance to existing drugs. Modern high-throughput screening (HTS) systems provide an immensely powerful strategy to identify new lead compounds in a relatively short amount of time. Many of the current in vitro assays for drug testing against M. tuberculosis are not well adapted for HTS.21 In this study we have adapted the microdilution Alamar blue assay22 to 384-well plate format and used it to screen a 100,997 compound library for antitubercular activity on a BSL3-contained HTS platform.
MATERIALS AND METHODS
Bacterial strain, growth conditions and media
M. tuberculosis H37Rv (ATCC 27294) was obtained from the American Type Culture Collection (Manassas, VA). To prepare permanent frozen stocks, H37Rv was grown as five mL subcultures (50 mL conical tubes, 36-37°C) in Middlebrook 7H9 broth (Becton Dickinson) supplemented with 0.2% glycerol (Becton Dickinson), 0.05% Tween 80 (Becton Dickinson), and 10% ADC enrichment (albumin, dextrose, catalase; Becton Dickinson). The subculture was mixed periodically and used to inoculate (5% inoculum) a second subculture (30 mL in 250 mL screw cap flask) when the turbidity reached a density similar to a #1 McFarland turbidity standard (A600 nm ~0.2). The subcultures were incubated with periodic mixing for 18-21 days until the turbidity reached a #3-#4 McFarland turbidity standard (A600nm ~0.6-0.8, 4-8 × 107 CFU/mL). The caps on both the conical tubes and flasks were loosened and wrapped in parafilm to allow for adequate gas exchange and to prevent evaporation during incubation. Prior to harvest, samples from all cultures were spotted onto Trypticase Soy Agar (TSA) plates and incubated for 3-4 days to check for contamination. M. tuberculosis grows poorly on TSA which supports the growth of most potential contaminating microorganisms. Each culture was then transferred to a 50 mL tube and allowed to settle at ambient temperature for one hour. The upper half of each culture was aspirated and pooled in a flask. Aliquots of one mL were then transferred to two mL cryovials and frozen at −80 °C. At least three frozen stocks were thawed and used to determine the viable count by plating dilutions, prepared in supplemented 7H9 broth, onto Middlebrook 7H11 Agar followed by incubation for up to 21 days. A contamination check on the thawed cultures was also performed as described above.
Control Drugs and Compound Libraries
A chemical diversity library containing 100,997 compounds was purchased from ChemBridge Corporation (http://www.chembridge.com/). This library compounds was selected for diversity and drug-likeness using the Lipinski23 criteria for drug-like compounds. A majority of this library of compounds, for example, had molecular weights ranging from 250–450 (>80%), CLogP value ~ 3.5, number of rotatable bonds ~ 4, topological polar surface area (tPSA)24 ~ 60 Å2, hydrogen bond donors < 3, and hydrogen bond acceptors < 5. TAACF medicinal chemists worked further with ChemBridge to make sure that this library of compounds did not include any compounds containing reactive functional groups such as oxiranes, aziridines, thiiranes, aliphatic aldehydes, isonitriles, diazenes, crown ethers, sulfamates and thiosulfates. The selected library contained a variety of heterocyclic compounds such as: pyrroles, furans, thiophenes, indoles and their benzo analogs, isoindolines, imidazoles, pyrazoles, triazoles, isoxazoles, thiazoles, oxadiazoles, thiadiazoles, pyridines, quinolines, pyridazines, pyrimidines, pyrazines, quinazolines, quinoxalines, pyrrolidines, piperazines and morpholines. The compounds were used as supplied and no structure/purity characterization was done beyond that carried out by ChemBridge. In general, we recommend that chemical structure and purity be confirmed before embarking on any development program. From this set of 100,997 compounds, a diversity subset of 13,440 compounds was selected using the dissimilarity selection (dbdiss) method as implemented in the Selector module of Sybyl. A smaller diverse and representative subset of 3,200 compounds was selected from the 13,440 compounds set using the OptiSim (dbdiverse) algorithm. This 3,200 compound set as well as the Prestwick Chemical Library (http://www.prestwickchemical.fr/index.php?pa=26) consisting of 1,120 compounds were used for initial assay validation experiments. Amikacin sulfate (Sigma) and ethambutol (Sigma), used in the assay as positive control drugs, were solubilized at 10.24 mg/mL in sterile water and DMSO respectively, aliquoted and frozen at −80 °C. Aliquots were made from one common drug stock, and a previously unthawed aliquot was used for each experiment and discarded afterwards.
The ChemBridge library was screened initially in a single dose of 10 μg/mL and a final DMSO concentration of 1%. Identified hits were picked and screened in a dose-response assay using a stacked-plate method wherein each compound dilution is interplate rather than intraplate. In this method, all of the compounds on a plate are at a single concentration. This design is driven by the efficiency of liquid handling, making it quick to generate simultaneous concentration curves for up to 1,280 compounds. Compounds screened in dose-response were tested in 10 two-fold dilutions from 50 μg/mL to 0.098 μg/mL.
M. tuberculosis Assay
The M. tuberculosis HTS assay was modified from that described by Collins and Franzblau22 using black, clear-bottom, 384-well microtiter plates and 7H12 broth. Compounds stocks of 1.0 mg/mL in 100% DMSO were diluted in assay media and 25 μL of these diluted compounds were transferred to 384-well plates. Amikacin was included in the positive control wells in every assay plate in two concentrations, 0.13 and 2.5 μg/mL. The low concentration is the approximate MIC and is an indicator of proper assay performance of each plate. The high concentration completely inhibits growth and is used in lieu of uninoculated medium (background) to calculate percent inhibition by the test compounds for each plate. Plates containing test compounds (320 compounds/plate) and positive control compounds were transferred into the BSL3 facility for bacteria addition and incubation. The bacterial stock was diluted to 1-2 × 105 CFU/mL in the assay medium, Middlebrook 7H12 broth (7H9 broth supplemented with 0.1% casitone, 5.6 μg/mL palmitate, 0.5% bovine serum albumin and 4 μg/mL catalase) and 25 μL was plated over the compounds. Positive and negative control wells were included in each plate. Amikacin was included in one of the compound wells as an internal control in dose-response runs. Plates were placed in stacks of two inside double low density polyethylene bags and incubated for 7 days at 37°C with approximately 90% humidity. After 7 days of incubation, end point reagent (two parts Alamar blue (Trek Diagnostics) + 1.5 parts 18.2% Tween 80 (Difco) diluted in milli Q water) was added to all wells in a volume of 9 μL per well. The plates were returned to the incubator for an additional 18-20 hours. Plates were sealed and bottom read for fluorescence using a Perkin Elmer Envision plate reader at 535 nm excitation and 590 nm emission.
Each assay run contained one plate of uninoculated medium (sterility control), another plate containing inoculated medium (growth control), and a 96-well plate with ethambutol at the approximate MIC (0.5 μg/mL) and 20 times the MIC (10 μg/mL). In addition to fluorometric reads, these plates were read at an absorbance of 615 nm (the approximate peak wavelength for oxidized dye) and were used to monitor the quality of the Alamar blue as well as adequate growth of the organism. Expected absorbance readings were about 0.8 and 0.2 for a good dye reagent (medium only) and growth control wells, respectively. The ethambutol plate was used to help confirm that a contaminating organism was not present after incubation since mycobacteria are generally susceptible while other genera are resistant.
Cell Cytotoxicity Assay
The Vero cell line (ATCC CCL-81) was obtained from the ATCC (Manassas, VA) to assess compound toxicity. The cell line was cultured in ATCC-formulated Eagle’s Minimum Essential Medium supplemented with 10% fetal bovine serum, 100 IU/mL penicillin and 100 μg/mL Streptomycin and incubated at 37°C, 5% CO2 and high humidity. Vero cell stocks were frozen in culture media supplemented with 5% DMSO at a density of 6×106 cells/mL. For freezing, 2 mL aliquots were dispensed to Nunc vials. For use in the assay, cells were thawed in a 37 °C water bath, mixed in the vial by light vortexing, and diluted with an equal volume of culture media. Cells were then centrifuged, media removed, resuspended in culture media, counted, and diluted to 125,000 cells per mL. Twenty (20) μL of cells were added to all wells of the assay plate containing 5 μL of compounds or controls, which had been preplated. Plates were then incubated for 72 h at 37 °C, with 5% CO2 in a humidified incubator. Cell viability was assessed using CellTiter-Glo reagent (Promega) according to the manufacturer’s protocol. Luminescence was recorded using an integration time of 0.1 sec per well. Columns one and two in the assay plates contained media + 0.3% DMSO for a negative control, and columns 23 and 24 contained media in 0.3% DMSO and 100 μM hyamine as a positive control. A stacked-plate dose-response method was used and the final test concentrations for the compounds ranged from 15 μg/mL to 0.029 μg/mL in two-fold dilutions with a final DMSO concentration of 0.3%.
Raw Data Analysis
Data were analyzed using IDBS Activity Base. Results of the single-dose screen were expressed as percent inhibition (% Inhibition) which was calculated as: 100*((Median Cell Ctrl – High Dose Ctrl Drug) – (Test well-High Dose Ctrl Drug))/(Median Cell Ctrl – High Dose Ctrl Drug). Any compound with an inhibition of ≥90% was designated to be active (hit) in the single-dose primary screen. The dose-response data was analyzed using a four parameter logistic fit (Excel Fit equation 205) with the maximum and minimum locked at 100 and 0. From these curves, TB IC90 and TB IC50 values were calculated for M. tuberculosis and CC50 values calculated for Vero cell cytotoxicity. The Selectivity Index is defined as the CC50/TB IC90.
RESULTS
Assay Validation
The assay for H37Rv22 was miniaturized to 384-well format. Initially, experiments were run in 96- and 384-well formats to ensure that the conversion to 384-well plates yielded similar results. CV% for media only, M. tuberculosis, and M. tuberculosis plus either 10 μg/mL ethambutol or 2.5 μg/mL amikacin were <10%. Z-values calculated for either M. tuberculosis alone or M. tuberculosis plus either 10 μg/mL ethambutol or 2.5 μg/mL amikacin were usually >0.7 and were equivalent in either 96- or 384-well format.
HTS validation of the 384-well format single-dose assay was accomplished by running a 3,000-compound diversity set on two different days and comparing the results of the two runs. For the two 3,000-compound runs the Pearson correlation, a statistical measure of the agreement of the two sets of data, was 0.768.
The first proof-of-concept for the stacked-plate dose-response assay was an experiment in which the compound source plate contained amikacin in each compound well. This experiment resulted in 320 10-point dose-response curves for amikacin, i.e. equal to the number of wells reserved for compounds in the 384-well plate format. The IC50 value for these 320 dose-response curves was 0.072 ± 0.009 μg/mL. A second HTS validation of the stacked-plate dose-response procedure was performed using the Prestwick Chemical Library of 1,120 compounds on two different days and comparing the results of the two runs. The distribution and reproducibility of the hits for this validation experiment is shown in Fig. 1. In this validation assay, the Prestwick library was run in ten-point dose-response format with concentrations ranging from 20 – 0.04 μg/mL in duplicate. The Minimum Significant Ratio (MSR) for the assay was calculated and is useful in determining if one compound is really more active than another. 166 compounds had IC50 values in both runs; from these data the MSR for the assay was calculated as 2.05. This value means that compounds with IC50 values between 0.5 and 2.0 are actually equivalent in potency in this assay (a two-fold range centered on an IC50 of 1.0). As an internal control, amikacin was added to an empty well for each dose-response experiment during the screening campaign. The MSR for amikacin during the campaign was <2. Both of these MSRs indicate that the H37Rv HTS assay is extremely reproducible. Further validation of the stability of the assay was obtained when amikacin was run over the course of a five month period, giving IC50 values of 0.09 ± 0.02 μg/mL.
Figure 1.

High Throughput Screening Campaign
The 100,997 compound library was screened in single-dose format against H37Rv at 10 μg/mL. The screen was successful with Z’-values > 0.7 for all plates (0.85 ± 0.04; Fig. 2). Compounds with ≥90% inhibition were classified as hits. Based on this criterion, 1,782 compounds were so classified from the screen. The 1,782 hits were cherry-picked from the library and screened in dose-response format. 1,593 compounds confirmed as actives in dose-response, giving a confirmation rate of 90%. A number of issues can contribute to compounds not confirming their activity in the dose response screen including liquid handling problems such as a blocked pipette tip. However, a 90% confirmation rate is quite good for a high throughput campaign. One of the compounds first identified as a hit was determined later to be autofluorescent and therefore excluded from the actives.
Figure 2.

A determination of compound cytotoxicity in a mammalian cell line is a useful parameter for determining which compounds to pursue as antitubercular leads, since compounds showing toxicity against mammalian cells would not generally be useful. Hits from the TB screen were tested for cytotoxicity using the Vero cell-based cytotoxicity assay described above. Forty-four (44) compounds had CC50 values <7.5 μg/mL and were considered toxic to mammalian cells. None of these compounds had a Selectivity Index (SI: CC50/TB IC90) greater than 9.
The 1,549 non-cytotoxic M. tuberculosis active compounds can be grouped into three categories based on their SI (Table 1). The highest activity group of 293 agents has an SI ≥10 and contains some extremely potent compounds that completely inhibited M. tuberculosis growth at all test concentrations. The medium activity group contains compounds that, despite moderate activity in the M. tuberculosis screen, possess an SI that may have been underestimated because the highest test concentration in the cell cytotoxicity assay was 15 μg/mL; if the actual Vero cell CC50 values could have been determined, the differential between the activity in the bacterial and mammalian cells might have been observed to be greater. The top test concentration was limited to 15 μg/mL by the DMSO sensitivity of the cells.
Table 1.
Distribution of 1,549 Non-Cytotoxic M. tuberculosis Actives
| Activity Category |
TB IC90 (μg/ml) H37Rv |
CC50 (μg/ml) Vero |
SI (CC50/IC90) |
Number of Compounds |
|---|---|---|---|---|
| High | <0.1 – 1.6 | 8 - >15 | 10 - >150 | 293 |
| Medium | 0.9 – 10.3 | 7.5 - >15 | 1.5 – 9.4 | 787 |
| Low | 6.3 - >50 | 7.8 - >15 | 0.2 – 1.4 | 469 |
DISCUSSION
In an effort to identify specific classes of compounds that show significant antitubercular activity, a structure based clustering analysis was performed on the set of compounds that was considered active with TB IC90 value of less than 10 μg/mL. The clustering analysis was performed using a hierarchical clustering method as implemented in LeadScope (LeadScope, Inc.) to identify common structural elements among this diverse set of structures. Clusters were separated using the ‘Complete Linkage (Furthest Neighbor)’ method with a cluster threshold distance of 0.7. Each identified cluster had a common core structure and a group of hits that share that common scaffold. Core structures were then prioritized through an enrichment analysis that compares the frequency of occurrence of a given scaffold in the active compounds set with its occurrence in the entire screening library. The enrichment was computed as the ratio of percentage of compounds containing a given core structure within the active set with percentage of compounds within the entire library of 100,997 compounds. In the following section, we have highlighted the activity profiles of selected high enrichment scaffolds as well as a few other scaffolds of interest, and we also discuss other relevant known biological activities of the chosen scaffolds. The identified scaffolds and clusters of active compounds appear to be novel classes of compounds that display significant antitubercular activity, and these may serve as leads for new antitubercular drug discovery and development.
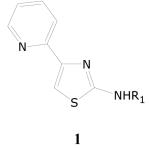
2-Aminothiazoles
The structure-activity relationship analysis revealed that compounds possessing the 2-amino-4-(2-pyridyl)thiazoles with an aryl or arylalkyl substituent on the amino group at the 2-position represent a particularly interesting scaffold. The entire database of screened compounds contained 18 compounds possessing the substructure 1. Of these 18 compounds, 15 compounds displayed >90% inhibition of growth of M. tuberculosis in the initial single-dose screen at 10 μg/mL (Table 2). In dose-response experiments, 13 of these 15 compounds displayed sub μg/mL TB IC90 values against M. tuberculosis and two compounds displayed TB IC90 values of 1.87 and 2.27 μg/mL, respectively. Most of these compounds (12 out of 15) also displayed selectivity ratios >10, indicating that compounds in this class are potent and selective. Most of these compounds, (13 of the 15) possess a substituted phenyl or pyridyl group on the 2-thiazolamine nitrogen. Two compounds, however, possess an arylalkyl substituent such as the 2-phenylethyl and a 3,4-dimethoxy-2-phenylethyl groups on the thiazolamine nitrogen. It appears that there are no literature reports on this class of compounds as antimicrobial agents and thus, the 4-(2-pyridyl)-2-thiazolamines represent a novel class of antituberculosis agents.
Table 2.
Antitubercular activities of representative thiazolamines
 |
|||
|---|---|---|---|
| Cpd.No. | R1 | TB IC90 | SI |
| 1a | 2-trifluoromethylphenyl | 0.10 | >150 |
| 1b | 6-methyl-2-pyridyl | 0.10 | >150 |
| 1c | 3-methyl-2-pyridyl | 0.10 | >150 |
| 1d | 5-chloro-2-pyridyl | 0.10 | >150 |
| 1e | 2-methylphenyl | 0.15 | >100 |
| 1f | 2,4-dimethoxyphenyl | 0.15 | >98 |
| 1g | 2-(3,4-dimethoxyphenyl)ethyl | 0.28 | >17 |
| 1h | 4-ethoxyphenyl | 0.32 | >47 |
| 1i | 4-bromophenyl | 0.32 | >46 |
| 1j | 3-methoxyphenyl | 0.34 | >43 |
| 1k | 4-fluorophenyl | 0.61 | >24 |
| 1l | 4-(dimethylamino)phenyl | 0.91 | 8.3 |
| 1m | 2-phenylethyl | 0.94 | 14 |
| 1n | 3-acetylphenyl | 1.87 | >8.0 |
| 1o | 2-methyl-4-hydroxyphenyl | 2.27 | >6.6 |
The pyridyl substituent at the 4-position of the thiazole ring system, particularly a 2-pyridyl substituent, appears to be a key structural requirement for potent antitubercular activity. Within the evaluated library of compounds, several equivalent pairs of 2-pyridyl and 4-pyridyl isomers are present. A comparison of the activity data on these pairs of compounds reveals that while the 2-pyridyl isomers (1b–e) are highly active the corresponding 4-pyridyl isomers are either inactive or weakly active. The 3-pyridyl isomer of 1a is also present as a member of the evaluated library of compounds. This 3-pyridyl isomer, however, was found to be inactive (0% inhibition) at 10 μg/mL. The deaza analog of 1a (i.e., 4-phenyl-[2-(6-methyl-2-pyridyl)amino]thiazole) was also found to be only weakly active (22% inhibition at 10 μg/mL).
Substituted Benzopyran-2-ones and Benzopyran-4-ones
Within the active compounds, three identified groups are all derived from a benzopyran core with several different substitution patterns. For two of these groups, the core is actually a 2H-1-benzopyran-2-one, commonly known as a coumarin, while for the other group, the core is a 4H-1-benzopyran-4-one. Below, each of these compound series is examined in terms of the structure activity relationships that exist, followed by summary comments. In examining the literature, there are very few reports on benzopyran-2-one and benzopyran-4-one moieties being evaluated for anti-tuberculosis activity. A few reports exist on coumarin derivatives that were evaluated against M. tuberculosis, including some natural products and analogs of natural products. The natural products calanolide A and B show some anti-TB activity,25,26 and in addition calanolide A shows activity against drug resistant strains of M. tuberculosis (H37Rv). A series of 4-methylumbelliferone derivatives was prepared that also show anti-TB activity against H37Rv.27 Finally, a series of coumarinyl thioureas was prepared with some activity against H37Rv.28
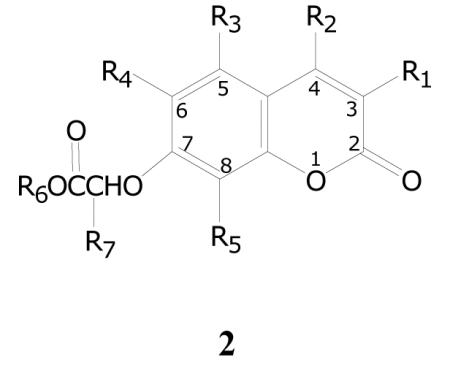
In the first of the two coumarin groups, the standard feature present in all of the compounds is a variously substituted acetic acid ester moiety attached through an oxygen to the 7-position of the coumarin core, as shown in the generic structure 2 in Table 3. The esters are variously methyl, ethyl, 2-propyl, cyclohexyl and benzyl groups. The series contains 87 compounds, of which 27 (31%) have a percent inhibition of greater than 90% in the initial assay. Additionally, a total of 50 compounds (57%) have an inhibition greater than 50%. As with the other series, compounds that were at 90% or greater percent inhibition in the initial evaluation were examined in the dose-response assay. The activity of 24 of these compounds was confirmed in the dose-response assay, though potencies varied and a few of them had only very weak activity. The most potent compounds all tended to have substitutions at C-4 along with the C-7 acetic acid moiety, and some in addition have substitutions at C-3, C-6 and/or C-8. Of the 24 compounds, 8 have TB IC90 values between 0.2 and 2.0 μg/mL, and another 12 have potencies between 2.0 and 10 μg/mL.
Table 3.
Antitubercular Activities of Substituted Benzopyran-2-ones
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd. No. |
R1 | R2 | R3 | R4 | R5 | R6 | R7 | TB IC90 |
SI |
| 2a | H | Ph | H | H | H | cC6H11 | H | 0.18 | >83 |
| 2b | H | 3-nitro phenyl |
H | H | H | i-Pr | H | 0.45 | >33 |
| 2c | H | n-butyl | H | Cl | H | Et | Me | 0.64 | >23 |
| 2d | H | n-butyl | H | Cl | H | CH2Ph | H | 0.66 | >23 |
| 2e | H | 3-nitro phenyl |
H | H | H | Me | H | 0.78 | >19 |
| 2f | CH2CO2Me | Me | H | H | Me | CH2Ph | H | 1.03 | >15 |
| 2g | H | 4- methoxy phenyl |
H | H | H | H | 1.7 | >9 | |
| 2h | Et | Me | H | H | Me | Me | H | 1.8 | >8 |
| 2i | 2- benzothiazolyl |
H | H | H | H | Et | H | 2.7 | >6 |
| 2j | -(CH2)5- | H | H | H | Et | H | 3.5 | >4 | |
| 2k | Cl | CH3 | OCH2CO2Me | H | H | Me | H | Inact. | - |
| 2l | -(CH2)3)- | H | H | H | Me | Ph | * | - | |
58% Inhibition in our initial single dose assay, not evaluated in the dose response assay.
Compounds with good activity within this group contain a variety of different esters, and those with significant activity in the dose response assay are presented in Table 3. It is possible that the esters are cleaved during the assay, and that the compounds are actually prodrugs of the corresponding carboxylic acids. A consideration in that regard, however, is the different cleavage rates of the variety of ester groups in the most active compounds. In some cases the acetic acid moiety has a group attached at the α-carbon. When that group is small, for example methyl, it has little effect on the activity. When the group is phenyl, however, it generally has the property of reducing the activity as compared to hydrogen or methyl as seen with 2l. Thirteen compounds have the α-phenyl substituent, and all of them have little or no activity.
Most compounds in this group have an alkyl or aryl group at either or both of the C-3 and C-4 positions. Additionally, other compounds have alkyl, aryl or chlorine groups at C-6 and C-8. Compounds vary from disubstituted up to tetrasubstituted, and activity can be seen in all of these variations. Some examples of various substitution patterns are provided in Table 3.
The compounds in this group have a rather narrow range of structures, but it is clear that there is a strong pattern of activity within the series. The opportunity for extensive SAR research exists using the core structure, and that research could move in many different directions.
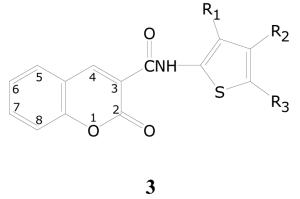
The other compound group with a coumarin core structure is embodied by the generic structure 3 (Table 4), which has a distinctive substitution pattern as compared to 2. This group has only one substitution on the coumarin ring, a carboxamide at C-3. The carboxamide is variously substituted at the nitrogen with a series of different groups, all of which contain a thiophen-2-yl ring substituted with an ester and/or an amide group. The group at R3 is generally either an ester or an amide, while substitutions at R4 and/or R5 are typically alkyl or aryl groups, with one example containing at ester group at R5.
Table 4.
Antitubercular Activity of Certain Benzopyran-2-one-3-carboxamides
 |
|||||
|---|---|---|---|---|---|
| Cpd.No. | R1 | R2 | R3 | TB IC90 |
SI |
| 3a | CO2Et | H | Et | 0.89 | >17 |
| 3b | CO2i-Pr | H | Me | 1.4 | >11 |
| 3c | CONH2 | -(CH2)6- | 2.9 | >5.2 | |
| 3d | CO2Me | -(CH2)3- | 7.7 | >1.9 | |
| 3c | CO2Et | H | 2-thienyl | 9.1 | >1.7 |
| 3f | CO2Me | H | n-Pr | 50 | - |
| 3g | CO2Et | Ph | Me | Inact. | - |
| 3h | CO2Et | 4-(CH3)Ph | H | * | - |
87% Inhibition in our initial single dose assay, not evaluated in the dose response assay.
The group is comprised of 25 compounds, of which 7 demonstrate 90% or greater inhibition and a total of 14 compounds show 50% or greater inhibition in the primary assay. In the dose-response assay, 5 of the 7 compounds demonstrated activity, and data on them are presented in Table 4. Of the other two compounds, one compound (3f) shows only very weak activity and the other one is inactive in the dose-response assay. Some compounds with no substituent at C-5 of the thiophene ring such as 3h have reasonable activity in the percent inhibition assay, but were not evaluated for dose response.
Though this group is again very narrowly defined, there is a clear pattern of activity. The potential for useful new SAR is quite high, given that all compounds have only thiophene rings attached to the amide, and that no compounds in this class that have substituents on the coumarin ring were in the library.
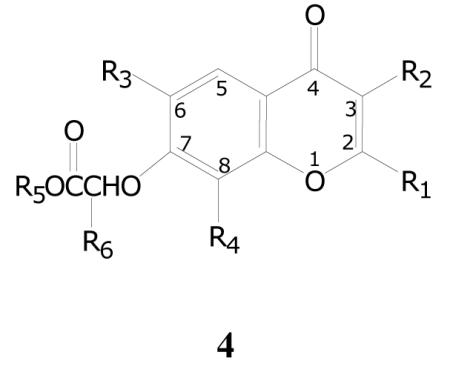
The other benzopyran group, comprised of a series of benzopyran-4-ones (4, Table 5), has two key features. First, it again has an acetic acid ester moiety attached through an oxygen to the 7-position of the benzopyran-4-one core. The esters again are comprised variously of methyl, ethyl, cyclohexyl, and benzyl, and all are present in compounds that have a >90% inhibition in the initial single dose assay. In some compounds the acetic acid moiety has a methyl group attached to the α carbon, and that attachment has little or no effect on the activity.
Table 5.
Antitubercular Activities of Certain Benzopyran-4-ones
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd. No. |
R1 | R2 | R3 | R4 | R5 | R6 | TB IC90 |
SI |
| 4a | H | 2-methylphenyl | H | H | Et | H | 0.43 | >35 |
| 4b | H | 2-ethylphenyl | H | H | cC6H11 | H | 0.47 | >32 |
| 4c | CH3 | 2-chlorophenyl | H | H | Me | H | 0.82 | >18 |
| 4d | H | 2-chlorophenyl | H | H | Me | H | 0.86 | >17 |
| 4e | H |
|
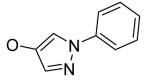
H | H | Et | H | 1.2 | >13 |
| 4f | H | 2-methoxyphenyl | H | H | CH2Ph | H | 1.2 | >12 |
| 4g | CH3 | 2-methoxyphenyl | H | H | CH2Ph | H | 1.4 | >11 |
| 4h | CH3 | 2-chlorophenyl | H | Me | Et | H | 1.8 | >9 |
| 4i | CH3 | 4-methoxyphenyl | H | H | Et | H | 2.0 | >7 |
| 4j | H | 4-chlorophenyl | H | H | Me | H | 2.1 | >7 |
| 4k | CH3 | 2,3-dimethoxyphenyl | H | H | i-Pr | H | 2.2 | >7 |
| 4l | H |
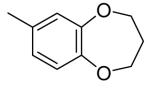
|
n-Pr | H | Et | H | 2.3 | >6 |
| 4m | H | 4-methoxyphenyl | Et | H | Me | H | 3.3 | >5 |
| 4n | H | 2-bromophenyl | H | H | Me | Me | 3.4 | >4 |
| 4o | H | 4-ethylphenyl | H | H | Me | Me | 3.5 | >4 |
| 4p | CH3 | 4-ethoxyphenyl | H | H | n-Bu | H | 4.0 | >4 |
| 4q | CH3 | 4-bromophenyl | H | H | Me | Me | 4.9 | >3 |
| 4r | CH3 | 4-methoxyphenoxy | H | H | Me | H | 5.1 | >3 |
| 4s | CH3 | 4-fluorophenoxy | H | H | Et | H | 5.2 | >3 |
| 4t | H | 2-chlorophenoxy | H | H | Et | H | 6.2 | >2 |
| 4u | H | 4-chlorophenyl | H | H | Me | Me | 7.17 | 2 |
| 4v | H |
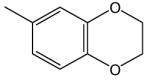
|
Et | H | i-Pr | H | 8.4 | >2 |
The second key feature is an aromatic group attached to C-3 of the benzopyranone ring in one of three ways. The attachment is either directly to C-3, through an oxygen atom to C-3, or through a pyrazole ring and an oxygen atom to C-3. Variously substituted benzenes have been examined and many different substitution patterns have been found to have high inhibition percentages.
Additionally, some of the compounds in this series have small alkyl groups, including methyl, trifluoromethyl, ethyl and butyl, variously at C-2, C-6 and C-8. Compounds are most commonly disubstituted, but are also tri- and tetra-substituted on the benzopyran-4-one ring, and again activity can be seen in all these variations.
The group is comprised of 68 compounds, of which 35 (51%) showed 90% or greater inhibition in the primary assay, and 53 (78%) showed greater than 50% inhibition. Of the compounds examined in the dose-response assay, 6 have TB IC90 value between 0.3-2.0 μg/mL, and another 12 have values between 2.0-10 μg/mL (Table 5). Compounds with the highest potency have of course the C-3 and C-7 substitutions noted above, and could also have almost any of the other substituents. In general, though, an aromatic moiety directly attached to C-3 is more potent than an aryloxy moiety at C-3. Compounds with a 2-CF3 group have either modest activity or no activity at all, whereas compounds with a 2-CH3 group are among the more potent compounds in the group. It should be noted that only two compounds out of the 68 total have C-5 substitutions, one a methyl and one a hydroxyl group, and both exhibit some percent inhibition. The hydroxyl-substituted compound, which produced 94% inhibition, when examined in dose-response demonstrated only weak activity. The structure-activity interpretations in this group need to be examined with care because the data for closely related compounds in some cases shows significant differences in TB IC90 values.
This series of compounds is again very narrowly defined, but has a very high percentage of active compounds within it. The potential for extended SAR exists, and could again proceed in many directions.
Oxadiazolylthio, Thiadiazolylthio and Triazolylthio Derivatives
Compounds possessing the thiazole, oxadiazole, and triazole are ring systems, due to their ready synthetic accessiblity, are frequently explored in the search for new antibacterial, antiparasitic, and antifungal agents. Recent literature also contains numerous examples of thiazoles, oxadiazoles, and triazoles being investigated as antimycobacterial agents.29-33 The library of compounds that we evaluated also contained a large number of thioether derivatives of 2-mercapto-1,3,4-oxadiazoles, 2-mercapto-1,3,4-thiadiazoles and 3-mercapto-1,2,4-triazoles. However, only 25 compounds possessing a thioacetic ester or amide function displayed significant activity against M. tuberculosis. Of these 25 compounds, 17 compounds that displayed TB IC90 values < 5.0 μg/mL are listed in Table 6. These results show that compounds belonging to this heterocyclic group are indeed endowed with promising antimycobacterial activity.
Table 6.
Antitubercular activities of Oxadiazolylthio, Thiadiazolylthio and Triazolylthio Derivatives
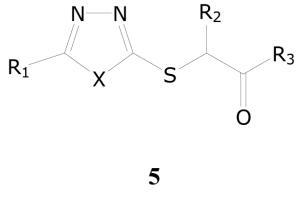 |
||||||
|---|---|---|---|---|---|---|
| Cpd. No. |
X | R1 | R2 | R3 | TB IC90 |
SI |
| 5a | O | 4-methoxyphenyl | H | NHCH2CH2Ph | 0.14 | >110 |
| 5b | O | 4-methoxybenzyl | H | NHCH2CH2Ph | 0.29 | >50 |
| 5c | O | 4-methoxyphenyl | H | NHC6H4CO2Et-p | ||
| 5d | O | 4-methoxyphenyl | H | NHC6H4-NO2-p | 0.87 | >17 |
| 5e | O | (3-methylphenoxy)methyl | H | NHCH2CH2Ph | 1.0 | >15 |
| 5f | O | 4-chlorophenyl | H | OMe | 1.5 | >10 |
| 5g | O | 4-bromophenyl | Me | OMe | 2.2 | >6 |
| 5h | O | (2,6-dimethylphenoxy)methyl | H | NHCH2CH2Ph | 3.3 | >5 |
| 5i | O | 4-bromophenyl | H | NHC6H4OMe-m | 3.5 | >4 |
| 5j | O | 4-dimethylaminophenyl | H | CH(CH3)2 | 4.0 | >3 |
| 5k | O | 2-chlorophenyl | H | NHC6H4Et-p | 4.2 | >3 |
| 5l | S | 4-tert-butylphenyl | H | OMe | 0.20 | >76 |
| 5m | S | 4-chlorobenzoylamino | H | OMe | 0.29 | >52 |
| 5n | S | amino | Me | NH-4,5,6,7- tetrahydro-2- benzothiazolyl |
0.85 | >17 |
| 5o | S | 1-naphthylthio | H | NHC6H4F-o | 1.6 | >9 |
| 5p | NH | 2-propoxyphenyl | H | OEt | 3.2 | >4 |
| 5q | NH | 4-chlorophenyl | H | NHC6H3ClF-m,p | 3.5 | >4 |
Phenoxyalkylimidazoles and Related Compounds
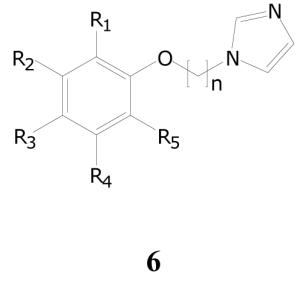
The screening set contains many examples of 1-(ω-phenoxyalkyl)-1-H-imidazoles (6, Table 7), a number of which possess very good (1-2 μg/mL) antitubercular activity. Though there is at present no evidence regarding mechanism of action, one can speculate from the simple, linear structures of these compounds, along with their potential amphiphilic nature, that membrane disruption is a possibility. Nonetheless, many of them show sufficient selectivity to warrant further investigation. Such compounds have been previously reported to inhibit nitric oxide synthase34 and thromboxane synthetase,35 as well as to possess anticonvulsant36 and metamorphosis-inducing37 activities; we have not found any reports of antimicrobial activities of this class.
Table 7.
Representative Antitubercular Activities of Phenoxyalkylimidazoles
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd. No. | R1 | R2 | R3 | R4 | R5 | n | TB IC90 |
SI |
| 6a | Cl | H | Br | H | H | 4 | 1.3 | >8.3 |
| 6b | Cl | H | Br | H | H | 3 | 24 | >0.6 |
| 6c | Cl | H | Br | H | H | 2 | Inact. | --- |
| 6d | H | H | s-Bu | H | H | 4 | 1.4 | >11 |
| 6e | H | H | s-Bu | H | H | 2 | 3.0 | >5 |
| 6f | H | Cl | Cl | H | H | 4 | 2.5 | 5.9 |
| 6g | H | Cl | Cl | H | H | 3 | 4.9 | >3.1 |
| 6h | H | Cl | Cl | H | H | 2 | 10 | --- |
| 6i | t-Bu | H | Cl | H | H | 4 | 2.2 | >6.9 |
| 6j | t- Bu | H | Cl | H | H | 2 | 2. 8 | >5.4 |
| 6k | t- Bu | H | H | H | Me | 2 | 11 | >1.4 |
| 6l | H | Me | i-Pr | H | H | 4 | 2.9 | >5.2 |
| 6m | H | Me | i-Pr | H | H | 3 | 5.9 | >2.5 |
| 6n | H | Me | H | i-Pr | H | 4 | 2.6 | >5.8 |
| 6o | Me | H | H | i-Pr | H | 4 | 2.5 | >6.1 |
| 6p | i-Pr | H | H | Me | H | 4 | 3.1 | >4.9 |
| 6q | Br | H | Cl | H | H | 4 | 1.7 | >9.0 |
| 6r | Cl | H | Cl | H | H | 5 | 4.4 | >3.4 |
There were 88 compounds containing this motif in the library, of which 27 compounds (30.7%) showed ≥ 90% inhibition during the single-dose screen, and 40 (45.5%) showing ≥ 50% inhibition. Table 7 illustrates the biological data of several examples from the class. As frequently happens during HTS, the SAR analysis is fragmentary because the structural clusters within the screening library were incomplete with respect to substitution pattern. Several trends do emerge from the data, however. For example, without exception among identically substituted aryl groups, potency increases with increasing alkyl chain length (cf. 6a, 6b, 6c; 6f, 6g, 6h). Whether this pattern continues beyond a four-carbon chain cannot be definitively determined, since the only example of a five-carbon analog in the screening set (6r) is not an exact analog of any shorter chain compound, and there are no examples of a six-carbon chain analog. It is noteworthy that the activity of 6r drops relative to that of its nearest four-carbon counterparts (cf. 6a, 6q). The trend of poorer activity with decreasing chain length, though discernable from analysis of exact congeners, is not reflected in the overall statistics: as the carbon chain length decreases, the overall fraction of those class members with single-dose inhibition ≥ 50% is essentially constant (n=4, 10 out of 22 compounds; n=3, 15 of 35 ; n=2, 14 of 30).
Also noteworthy is the size and variety of permissible aryl substituents. As one example, the ortho-tert-butyl substituted compounds form an interesting subclass, containing 15 active agents. Other active compounds contain as their aryloxy component substituted and unsubstituted α- and β-naphthyl groups, and a para-benzyloxyphenyl moiety (not depicted). Additional structural modifications that can retain activity include replacement of the oxygen by sulfur (7, TB IC90 3.16; Chart 1), substitution of the alkyl chain (8a (TB IC90 1.69) vs. 8b (TB IC90 5.96)), and substitution of the imidazole (e.g., 9, TB IC90 0.96, the most active member of the series). More generally, activity can be retained when the imidazole is replaced with alternative basic moieties (10a, TB IC90 7.09; 10b, TB IC90 4.16), though activity trends in this case are less well developed.
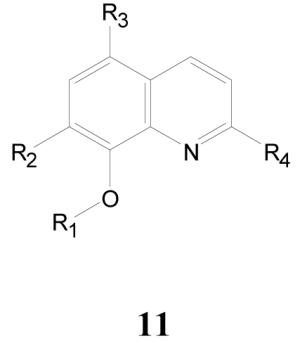
8-Hydroxyquinolines
There are over two hundred examples of this class in the screening set with an activity range (TB IC90) of less than 0.1 up to greater than 50 μg/mL. A small number of the highly active samples show selectivity for growth inhibition against M. tuberculosis versus eukaryotic Vero cells of greater than one hundred. Table 8 gives a representative set of ten highly active (TB IC90 < 1.0 μg/mL) and selective examples in this class. This cluster of actives contains 8-OH and 8-OR (R = acyloxy and alkoxy) substituted quinolines; the acyloxy, ester blocked 8-OH compounds are likely to be labile in vitro and in vivo and should generate the 8-OH forms over time depending on the lability of the ester linkage and ability to act as substrates for non-specific esterases. The alkoxy linkages should be relatively biologically stable. Interestingly, the majority of active examples from the screen are either the free 8-OH or a labile, ester blocked 8-OH that may speak to the issue of metal chelation discussed later.
Table 8.
Antitubercular Activities of 8-Hydroxyquinolines
 |
||||||
|---|---|---|---|---|---|---|
| Cpd. No. |
R1 | R2 | R3 | R4 | TB IC90 |
SI |
| 11a | COMe | H | H | trans-HC=CHPh(3-Br) | <0.10 | >150 |
| 11b | H | H | Br | H | <0.10 | >150 |
| 11c | COEt | H | H | trans-HC=CHPh(2-F) | 0.11 | >135 |
| 11d | COMe | H | H | trans-HC=CHPh(4-Cl) | 0.19 | >79 |
| 11e | H | H | H | trans-HC=CHPh(3-F) | 0.20 | >75 |
| 11f | H | H | H | trans-HC=CHPh(4-OMe) | 0.20 | >75 |
| 11g | COEt | H | H | trans-HC=CHPh(3,4-OCH2O-) | 0.29 | >51 |
| 11h | COPh(2-NO2) | Cl | Cl | H | 0.32 | >47 |
| 11i | COMe | H | H | trans-HC=CHPh(4-F) | 0.33 | >45 |
| 11j | CO-2-thiophene | H | Cl | H | 0.50 | >30 |
| 11k | H | H | H | trans-HC=CHPh(3-OMe, 4-OH) | 0.54 | >28 |
| 11l | COMe | H | H | trans-HC=CHPh(2-Cl, 6-F) | 0.94 | >16 |
The 8-hydroxyquinolines are known for their antimicrobial properties, among other activities.38-40 The activity of this class is broadly attributed to the ability of 8-hydroxyquinolines to act as bidentate metal chelators.41-43 This ability can be used to deliver toxic metals to target cells, scavenge essential trace metals within cells, or nonselectively bind within active sites of metal requiring enzymes.41 These activities are all considered to be nonselective, and the 8-hydroxyquinolones are primarily used topically for antimicrobial, anticaries activity.44 Other known drugs that potently bind metals include ethambutol, isonicotinic acid hydrazide, phenytoin, tetracycline, disulfuram, and D-penicillamine,41 suggesting that non-selective metal binding compounds can be appropriately modified to develop better, more selective drugs. While most activities of the class are attributed to metal chelation, substituted 8-hydroxyquinolines are known that potently inhibit brain catechol O-methyl transferase, and this inhibition did not appear to relate to metal chelating ability of the analogs.45 Others have attempted to synthesize selective inhibitors of HIF-1α-prolyl hydroxylase using the chelating function as a scaffold and building block for structure-based selectivity enhancement of enzyme binding.46 This approach has also been used to develop selective inhibitors of the HIV-1 integrase.47 A series of 8-hydroxyquinolines was investigated for its ability to affect cell wall synthesis in E. coli via inhibition of MurF, and a pharmacophore model of the scaffold was developed in order to search for analogs lacking the metal chelation properties of the starting scaffold.48 Although the perception exists that these agents are non-selective antibacterial agents for topical use only, the fact that the chelating core has been utilized as a building block to prepare selective enzyme inhibitors and the fact that several compounds (11a-e, Table 8) show potent and reasonably selective antitubercular activity suggest that certain of these active compounds may be worth further pursuit in terms of their selective activity and potential mechanism of action.
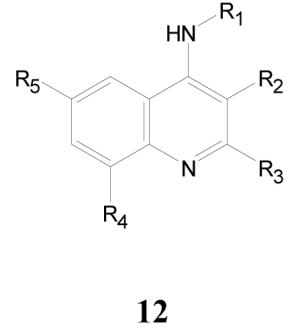
4-Aminoquinolines
The activity data from the library screen are slightly enriched in moderately active 4-aminoquinolines; ten active compounds resulted from a total set of seventy six analogs. The complete list of active compounds is shown in Table 9 and Figure 3.
Table 9.
Antitubercular Activities of 4-Aminoquinolines
 |
|||||||
|---|---|---|---|---|---|---|---|
| Cpd.No. | R1 | R2 | R3 | R4 | R5 | TB IC90 |
SI |
| 12a | 4-n-propoxyphenyl | H | Me | H | Cl | 0.83 | 18 |
| 12b | 4-benzyloxyphenyl | H | Me | H | COOMe | 1.9 | 7.7 |
| 12c | 4-N-piperidinylphenyl | H | Me | H | COOEt | 3.0 | 5.0 |
| 12d | 3,4-ethylenedioxyphenyl | COOEt | H | H | OMe | 3.4 | 4.5 |
| 12e | 4-ethoxyphenyl | H | Me | H | COOMe | 5.6 | 2.7 |
| 12f | 4-N,N-dimethylaminophenyl | H | Me | Me | H | 6.2 | 2.4 |
| 12g | 2-hydroxyphenyl | H | Me | H | OMe | 10 | --- |
Figure 3.

A variety of 4-substituted quinolines show antimycobacterial activity. These include mefloquine, 4-quinolone carboxylic acids, and 4-alkylquinolines.49-58 Mefloquine was first reported to show activity against M. avium, and genetic approaches were used to identify two potential targets.49 Screening through the TAACF suggested that mefloquine also was active against M. tuberculosis, and certain stereoisomers of the parent drug had superior activity.50,51 Chemical modification of the parent drug led to the identification of 4-hydrazone analogs of mefloquine with significant activity.51 Independently, a supplier to the TAACF program has pursued analogs similar to those identified through these screens; several examples showed good antitubercular activity.52-58 Overall, the analogs identified through the current screen are comparable in activity to previously reported antimycobacterial quinolines. It is notable that chloroquine, an antimalarial quinoline that is similar in structure to these and reported active quinolines, is a known inhibitor of autophagy.59 Autophagy appears to play an important role in macrophage processing of intracellular tubercle bacilli as part of their response to clear organisms that evade lysosomal processing.60 As such, it may be important to determine the effect of any similar quinolone on autophagy as part of assessing the role that these quinolines might play in antitubercular treatment.
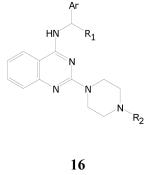
2,4-Diaminoquinazolines
The activity data from the library screen are slightly enriched in moderately active 2,4-diaminoquinazolines of two basic, and similar, types – see Tables 10 and 11 for active samples.
Table 10.
Antitubercular Activities of 2,4-Diaminoquinazolines
 |
|||||
|---|---|---|---|---|---|
| Cpd.No. | R1 | Ar | R2 | TB IC90 |
SI |
| 16a | H | Ph | CH2CH2OPh(4-Me) | 1.5 | 9.8 |
| 16b | Me | Ph | COPh(4-Cl) | 1.6 | 3.0 |
| 16c | H | Ph | CH2Ph | 1.7 | 8.8 |
| 16d | H | 2-Furyl | CH2C=CH2(CH3) | 6.1 | 2.5 |
| 16e | H | 2-Furyl | CH2CH2OPh(4-Me) | 9.6 | 1.6 |
Table 11.
Antitubercular Activities of 2,4-Diaminoquinazolines
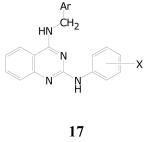 |
||||
|---|---|---|---|---|
| Cpd.No. | Ar | X | TB IC90 | SI |
| 17a | Ph | 3-OH | 1.3 | 3.9 |
| 17b | 2-Furyl | 3-Cl | 1.4 | 11 |
| 17c | 2-Furyl | 4-Me | 2.3 | 6.7 |
| 17d | 2-Furyl | 4-OEt | 2.3 | 2.8 |
| 17e | 2-Furyl | 3-Me | 2.5 | 2.3 |
| 17f | 2-Furyl | 2-Me | 3.2 | 4.6 |
| 17g | Ph | 2-OMe | 5.3 | 5.3 |
| 17h | 2-Furyl | 2-OH | 6.1 | >2.5 |
Substituted quinazolines are reported to show a variety of antibacterial activities including antimycobacterial activity. The general scaffold includes quinazoline analogs of the fluoroquinolone DNA gyrase inhibitors.61,62 Substituted 3-hydroxyquinazoline-2,4-diones also show good antibacterial activity.63 Derivatives of 2-aryl-3-aminoquinazoline-4(3H)-ones show good antibacterial and antitubercular activity.64-67 There are reports of quinazolines that are similar to the hits found in our screen (see Figure 4, structure 1868 19,69 and 2070).
Figure 4.

There is no indication from the literature reports as to a potential target for these compounds, and the small number of actives in each set impacts the ability to define a structure –activity profile. The good activity, however, of these drug-like small molecule scaffolds suggests that further work to identify a mode of action and prepare analogs to develop a clear SAR would be worthwhile.
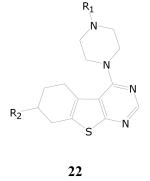
Thieno[2,3-d]pyrimidin-4-amines
In the library of compounds evaluated, there were 303 compounds possessing the thieno[2,3-d]pyrimidin-4-amine core structure 21. Of these 303 compounds, only 15 displayed >90% inhibition of growth of M. tuberculosis in the single-dose assay at 10 μg/mL. In dose-response experiments, only one compound (22a) displayed activity with TB IC90 value of 4.6 μg/mL and two other compounds (22b and 22c) were weakly active with TB IC90 values of 24 μg/mL and 28 μg/mL, respectively (Table 12).
Table 12.
Antitubercular activities of representative thienopyrimidinamines
 |
||||
|---|---|---|---|---|
| Cpd. No. | R1 | R2 | TB IC90 | SI |
| 22a | 6-methyl-2-pyridyl | H | 4.6 | >3.3 |
| 22b | Me | 1,1-dimethylpropyl | 24 | >0.6 |
| 22c | 2-phenylethyl | H | 28 | >0.5 |
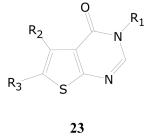
Thieno[2,3-d]pyrimidin-4-ones
In the library of compounds evaluated, there were a large number of compounds possessing the 4-oxothienopyrimidine core structure 23. Of the 1,043 compounds possessing this core structure, only a small set of eight compounds emerged as active or moderately active against M. tuberculosis. Structurally, all of these contained an α-methylacetic acid ester as the alkyl substituent on the pyrimidinone nitrogen at the 3-position. There were a total of 46 compounds possessing this substructure in the library. Of these, nine compound displayed >90% inhibition of growth of M. tuberculosis in the single-dose assay at 10 μg/mL. Of the nine, eight compounds displayed confirmatory activity in the dose-response experiments. Six of these eight compounds displayed TB IC90 values in the range of 1.0–6.6 μg/mL while the two other analogs displayed weak activity with TB IC90 values of 12.4 μg/mL and 12.7 μg/mL, respectively (Table 13). From the available data set it is difficult to arrive at any firm SAR conclusions regarding the effect of substituents (alkyl or aryl) on the thiophene ring or of the ester substituents (alky or benzyl) on the activity of this group of compounds.
Table 13.
Antitubercular activities of representative thienopyrimidinones
 |
|||||
|---|---|---|---|---|---|
| Cpd.No. | R1 | R2 | R3 | TB IC90 |
SI |
| 23a | CH(Me)CO2C6H4-Cl-o | C6H4-Me-p | H | 1.0 | >15 |
| 23b | CH(Me)CO2-cC6H11 | C6H3-(Me)2-m,p | H | 1.7 | >8.6 |
| 23c | CH(Me)CO2-cC6H11 | CH3 | Me | 1.8 | >8.4 |
| 23d | CH(Me)CO2CH2C6H4 | C6H4-Br-p | H | 2.2 | >6.8 |
| 23e | CH(Me)CO2CH2C6H4 | CH2CH2CH(Me)CH2 | 3.1 | >4.8 | |
| 23f | CH(Me)CO2CH2CH3 | C6H4-Et-p | H | 6.6 | >2.2 |
| 23g | CH(Me)CO2CH(CH3)2 | C6H4-Br-p | H | 12 | >1.2 |
| 23h | CH(Me)CO2-cC6H11 | C6H4-OMe-p | H | 12 | >1.2 |
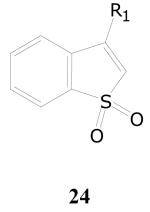
Benzothiophene 1,1-dioxides, Dihydrothiophene 1,1-dioxides and Related compounds
In the library of compounds evaluated, there was a relatively small group of compounds possessing the benzo(b)thiophene 1,1-dioxide structure. Of the 14 compounds, one compound contained a pyrrolidine as the substituent at the 3-position, four compounds contained a chlorine or bromine, two compounds contained an aryl ether function and the remaining seven compounds contained a thioether function. Of these 14 compounds, only five compounds, all possessing a heteroarylthio group at the 3-position displayed potent (>90%) inhibition of growth of M. tuberculosis in the single-dose assay at 10 μg/mL. Three of these compounds (24a–c) emerged as potent inhibitors in the dose-response experiment (Table 14). The 5-(4-methylphenyl) analog 24d, however, was found to display only weak activity with an IC50 value of 16.4 μg/mL.
Table 14.
Antitubercular activities of representative benzothiophene 1,1-dioxides
 |
|||
|---|---|---|---|
| Cpd.No. | R1 | TB IC90 | SI |
| 24a | 2-benzothiazolylthio | 0.10 | >150 |
| 24b | 5-phenyl-1,3,4-oxadiazol-2-ylthio | 0.10 | >33 |
| 24c | 5-(4-pyridyl)-1,3,4-oxadiazol-2-ylthio | 2.8 | >5.3 |
| 24d | 5-(4-methylphenyl)-1,3,4-oxadiazol-2-ylthio | 16 | >0.91 |
| 24e | [2-(4-morpholinyl)ethyl]thio | Inact. | ND |
| 24f | 4,6-diphenyl-2-pyrimidinylthio | Inact. | ND |
| 24g | pyrrolidino | Inact. | ND |
| 24h | 4-fluorophenoxy | Inact. | ND |
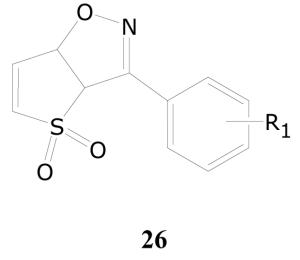
In the library of compounds evaluated, there were forty 3-substituted-2,3-dihydrothiophene-1,1-dioxides possessing the generic structure 25. None of these 40 compounds, however, displayed any significant inhibitory activity in the single-dose experiment at 10 μg/mL. Interestingly, there were three bicyclic dihydrothienoisoxazole 4,4-dioxides 26a–c in the library all of which displayed potent (>99%) inhibition of the growth of M. tuberculosis in the single-dose assay at 10 μg/mL. In dose-response experiments, these three compounds displayed activity with IC50 values of 1.4, 3.4, and 12.0 μg/mL, respectively (Table 15).
Table 15.
Antitubercular activities of representative thienoisoxazole 4,4-dioxides
 |
|||
|---|---|---|---|
| Cpd.No. | R1 | TB IC90 | SI |
| 26a | 4-chloro | 1.4 | >10 |
| 26b | 4-trifluoromethyl | 3.4 | 3.6 |
| 26c | 2,6-dichloro | 12 | >1.2 |
Pyrazolopyrimidinones
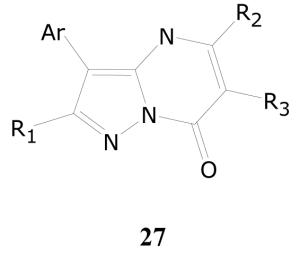
Of the 24 compounds in this cluster, 6 had percent inhibitions of greater than 90% in the single dose assay at 10 μg/mL. As shown in Table 16, there were two single dose active compounds (27a and 27b) with percent inhibitions of 99. The percent inhibitions of the other single dose assay actives ranged from 98 to 92. When five of these single dose assay actives were further tested in the dose response assay, compound 27b was found to be the most active, with TB IC90 of 0.4 μg/mL and an SI of 37. Compounds 27f, 27d, and 27c were marginally active, while 27a was essentially inactive.
Table 16.
Antitubercular activities of 3-Phenylpyrazolo[1,5-α]pyrimidin-7(4H)-ones
 |
||||||
|---|---|---|---|---|---|---|
| Cpd.No. | Ar | R1 | R2 | R3 | TB IC90 |
SI |
| 27a | 1-napthyl | benzyl | Ph | H | 50 | ND* |
| 27b | Ph | H | benzyl | H | 0.4 | 37 |
| 27c | 4-chlorophenyl | H | Me | CH2COOEt | 6.0 | 2.5 |
| 27d | 4-methoxyphenyl | Me | Me | benzyl | 4.2 | 3.6 |
| 27e | Ph | H | Me | benzyl | ND | ND |
| 27f | 3-bromophenyl | H | Me | CH2COOEt | 3.4 | 4.4 |
ND = not determined.
A search of the literature for biological activity for 3-phenylpyrazolo[1,5-a]pyrimidin-7(4H)-ones showed that related compounds inhibit xanthine oxidase and that similar compounds act as antischistosomals. No reported antibacterial or antitubercular activity was found.
1,4-Naphthoquinones
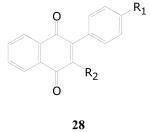
A small cluster of 1,4-naphthoquinones that was highly enriched in moderately active samples resulted from the library screen. The complete set is represented in Table 17. Such a small set does not allow development of a clear SAR, but some substitution is tolerated at both R groups. A large biphenyl (28d), however, does not appear to be tolerated. Other samples in this class are available commercially, and they should be investigated in order to further probe SAR.
Table 17.
Antitubercular Activities of 1,4-Naphthoquinones
 |
||||
|---|---|---|---|---|
| Cpd. No. | R1 | R2 | TB IC90 | SI |
| 28a | H | H | 2.9 | 5.1 |
| 28b | Me | COOEt | 3.3 | 4.5 |
| 28c | Et | H | 6.9 | 2.2 |
| 28d | Ph | COOEt | Inact. | --- |
| 28e | OMe | H | 4.1 | 3.7 |
| 28f | OMe | COOEt | 8.5 | 0.3 |
The 1,4-naphthoquinone scaffold is represented in numerous natural products that are known for a variety of activities including antiparasitic, anticancer, antibacterial and antifungal.71 The natural product, 7-methyljugalone, a substituted 1,4-naphthoquinone, shows activity against M. tuberculosis, and it may act as a subversive substrate for mycothiol disulfide reductase.72 The combination of 7-methyljugalone with other antitubercular agents suggests synergistic activity, and thus active naphthoquinones may fit well into current antitubercular regimens.73
Numerous synthetic analogs have been prepared to investigate the various medicinal activities of 1,4-naphthoquinones.74 Among the synthetic agents, atovaquone is a clinically used drug for the treatment of Plasmodium falciparum.75 Atovaquone (see Figure 5), a substituted 2-hydroxy naphthoquinone, is considered an analog of ubiquinone (Figure 5, coenzyme Q – CoQ), a critical component of the respiratory chain and synthesis of ATP.76 The cidal activity of this drug is thought to involve the competitive and irreversible inhibition of cytochrome b in the bc1 complex within the inner mitochondrial membrane.77 CoQ moves electrons from complex I and complex II to complex III, and inhibition of this process disrupts electron transport in the mitochondrial membrane, thereby altering the transmembrane proton gradient, oxidative phophorylation, and ATP synthesis. As such, atovaquone alters fundamental energy production in the malaria parasite. It is used to treat other eukaryotic pathogens as well. Many of these functions are recapitulated in bacteria including M. tuberculosis, distant relatives of eukaryotic mitochondria, although menaquinone (Figure 5) is the primary electron carrier.78 ATP synthesis in M. tuberculosis is considered a new target that is receiving considerable attention in the research community.79 Although speculative at this point, the structural similarity of these active naphthoquinones with menaquinone and atovaquone, as well as the known role of atovaquone in inhibiting the parasitic respiratory chain, suggest a hypothetical target in M. tuberculosis that might be explored further. Atovaquone had modest activity against M. tuberculosis in support of this hypothesis.80 It is notable that structure 28d in Table 17 is comparable to atovaquone (biphenyl versus trans-cyclohexylphenyl) suggesting that, while large steric groups may be tolerated at this position of the naphthoquinone, a specific orientation of the moiety appears to be favored.
Figure 5.

Piperidinamines
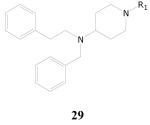
Of the 14 compounds in this group, 4 had percent inhibitions of greater than 90%, ranging from 92% to 100% in our single dose assay at 10 μg/mL (Table 18). Only one other had a percent inhibition of greater than 50%; R = 2-pyridylmethyl, percent inhibition = 82%. All of these actives were 1-N-arylmethyl substituted compounds, and the most active compound of these in the single dose assay was compound 29a, which showed a percent inhibition of 100%. When the four single dose assay actives were further tested in the dose-response assay, the most active was found to be 29d with TB IC90 of 1.6 μg/mL and a selectivity index of 9.6. There do not appear to be any literature reports relating to antibacterial activity of this general structure.
Table 18.
Antitubercular activities of 1-N-Aryl-4-N-(2-phenylethyl)-N-(phenylmethyl)-piperidinamines
 |
|||
|---|---|---|---|
| Cpd. No. | R1 | TB IC90 | SI |
| 29a | 1H-indol-3-yl-methyl- | 4.0 | 3.8 |
| 29b | 2,4-dimethoxyphenylmethyl- | 3.3 | 3.9 |
| 29c | 2,3-dimethoxyphenylmethyl- | 9.1 | 1.6 |
| 29d | 5-norbornenyl-2-methyl- | 1.6 | 9.6 |
Thioamides, Ureas and Thioureas
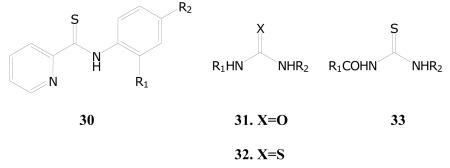
The discussion of these classes is being combined due to similarities in structures and the likely required activation of thione-containing compounds for antitubercular activity. It must be clearly stated, however, that the unified discussion does not imply similar targets or modes of action. The first set of three samples in Table 19 (30a-c) are closely related to the known, second line antitubercular drug, ethionamide (2-ethylthioisonicotinamide).81-83 Interestingly, ethionamide is relatively inactive (TB IC90 > 100 μg/mL) in the TAACF screens, and the increased activity of two of these analogs may be due to higher lipophilicity and/or an alternative mechanism of action. It is highly likely that these analogs are prodrugs of an activated form of the compounds as is the case with ethionamide.84-86 Thioamide-containing tuberculosis drugs are known to be activated within M. tuberculosis, and drug resistant strains are available that lack these activation mechanisms.87-89 The activity of these samples (30a-b) may warrant investigation as alternative ethionamide drugs, although they should be screened versus ethionamide-resistant M. tuberculosis strains.
Table 19.
Antitubercular Activities of Various Thioamides, Ureas and Thioureas
 |
||||
|---|---|---|---|---|
| Cmpd. No. |
R1 | R2 | TB IC90 |
SI |
| 30a | H | Me | 1.0 | >14 |
| 30b | H | Et | 2.7 | >5.0 |
| 30c | Me | Me | >50 | --- |
| 31a | CH2CH(Me)-2-exo-norbornanyl | 3-trifluoromethylphenyl | <0.1 | >150 |
| 31b | 3,5-Bis-carboethoxyphenyl | 3-chlorophenyl | 0.20 | >72 |
| 31c | 4-trifluoromethoxyphenyl | 3-nitrophenyl | 0.20 | >72 |
| 31d | 4-chloro-3- trifluoromethylphenyl |
3-nitrophenyl | 0.21 | >70 |
| 31e | 4-trifluoromethylphenyl | 3-chlorophenyl | 0.30 | >50 |
| 31f | 5-methyl-2-nitrophenyl | 3-chlorophenyl | 0.31 | >48 |
| 32a | 3,4-dichlorophenyl | 2-furyl(3-COOMe, 5-Et) | 0.23 | 37 |
| 32b | 1-methyl-2-indoyl | Ph | 0.38 | >39 |
| 32c | 3,4-dichlorophenyl | 3-nitrophenyl | 0.48 | >31 |
| 32d | 3-chloro-4-fluorophenyl | 4-carboethoxyphenyl | 0.53 | >29 |
| 32e | N-benzyl-4-piperidinyl | 2-thiophene(3-COOMe, 5-Ph) |
0.85 | >18 |
| 32f | 4-phenoxyphenyl | 4-carboethoxyphenyl | 0.85 | >18 |
| 33a | 1-naphthyl | 5-t-butyl-3-chloro-2- hydroxyphenyl |
<0.10 | >150 |
| 33b | 3-pyridyl | 5-s-butyl-2- hydroxyphenyl |
0.10 | >150 |
| 33c | 5-bromopyridyl | 5-s-butyl-2- hydroxyphenyl |
0.22 | >69 |
| 33d | Phenylethyl | 4-N-piperidinylphenyl | 0.33 | >46 |
| 33e | 1-naphthyl | 5-ethyl-2-hydroxyphenyl | 0.34 | >44 |
| 33f | 4-ethoxy-3-hydroxyphenyl | 4-n-butylphenyl | 0.37 | >41 |
There are approximately fifty examples of ureas that were screened in the overall library and activity (TB IC90) ranged from less than 0.10 to greater than 50 μg/mL; 35 of these samples gave TB IC90 of less than 10 μg/mL. Overall, this set of compounds is highly enriched in active analogs, and several of the compounds appear to be relatively nontoxic as indicated by the selectivity index. Table 19 gives a representative set of six highly active (TB IC90 < 0.5 μg/mL) and selective examples in this class (31a-f). As a broad class, the substituted ureas were reported to have antibacterial,87-89 and, specifically, antimycobacterial activity.90 Targets have been suggested, but the exact mechanism of action is currently unclear. Among these mechanisms, the potential to target the bacterial enzyme inosine monophosphate dehydrogenase (IMPDH) is an area that is actively being pursued.91 Although a purely speculative connection, substituted ureas are known to compete into ATP binding sites of human kinases,92-94 and bacterial signaling kinases and ATP binding proteins are receiving increased attention as new drug targets against tuberculosis.95-98
As a class, the thioureas have been known for some time to have antitubercular activity.99 For example, isoxyl (4,4′-diisoamyloxydiphenylthiourea), is an approved antitubercular drug in Europe. A variety of analogs were reported, and a mechanism of action that includes proactivation of the thione group has been discussed.100-102 It is notable that thiourea analogs act as histamine receptor antagonists,103 and antihistamines have antitubercular activity (see Prestwick Library screening data at www.taacf.org).104 The current screening set includes fifty seven thiourea analogs ranging in activity (TB IC90) of less than 0.4 to greater than 50 μg/mL – see Table 19 for examples (32a-f). Approximately 35 have TB IC90 of less than or equal to 10 μg/mL. Several compounds from this set show comparable activity relative to isoxyl and reported analogs.
The current screening data contains a large number of acylthioureas (seventy eight samples), and fifty of these samples show activity in the range of greater than 0.10 to 10 μg/mL. Twenty-three of these relatively active samples give selectivity indices of less than or equal to ten, suggesting that the class may be a rich source for new active drugs. Table 19 gives a representative set of active acylthioureas (33a-f). On the other hand, it is highly likely that the thione group may undergo further modification to an active intermediate within the tubercle bacillus as do other thione containing antitubercular drugs, and a profile of these active compounds against drug resistant strains (isoniazid, ethionamide, isoxyl, and thiacetazone) should be assessed. As a class, the acylthioureas show a variety of activities,105-107 but there are no reports regarding antibacterial, especially antitubercular, activity.
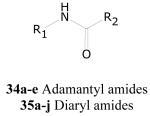
Adamantyl Amides and Amines
A small group of 1-adamantyl amines and amides, 1-exo-norbornyl amides, and corresponding urea (see earlier section) analogs show good to potent antitubercular activity. Table 20 lists a representative set of these analogs (34a-e). Over 200 examples of this class are in the screening set with an activity range (TB IC90) of less than 0.1 up to greater than 50 μg/mL. A small number of the highly active samples show selectivity for growth inhibition against M. tuberculosis versus eukaryotic Vero cells of greater than 100. Table 20 gives a representative set of ten highly active (TB IC90 < 1.0 μg/mL) and selective examples in this class.
Table 20.
Antitubercular Activities of Various 1-Adamantyl Amides
 |
||||
|---|---|---|---|---|
| Cmpd. No. |
R1 | R2 | TB IC90 |
SI |
| 34a | 1-Ad | 3-chloro-4-fluorophenyl | <0.10 | >150 |
| 34b | 1-AdCH2 | 1-naphthyl | <0.10 | >150 |
| 34c | 1-Ad | 4-methylphenyl | 0.20 | >72 |
| 34d | Ph(4-Me) | 1-AdCH2CH2 | 0.31 | >49 |
| 34e | Ph(4-Br) | 1-AdCH2CH2 | 0.44 | >34 |
| 35a | Ph(4-Cl, 3-[2-(5-Me- benzoxazolyl]) |
benzyl | <0.10 | >150 |
| 35b | 2-imidazolyl | 4-t-butylphenyl | <0.10 | >150 |
| 35c | Ph(2-Me, 4-NO2) | 3-iodo-4-methylphenyl | <0.10 | >150 |
| 35d | Ph(4-O-CH2COOMe) | 2,4-dimethylphenyl | <0.10 | >150 |
| 35e | Ph(4-O-CH2COOMe) | 4-phenylphenyl | <0.10 | >150 |
| 35f | (4-OMe)PhCH2CH2 | 5-t-butylethynyl-2-thiazolyl | <0.10 | >150 |
| 35g | Ph(2-Me, 4-Cl) | α-methyl- carboxyclohexyloxymethoxy |
<0.10 | >150 |
| 35h | Ph(4-CF3) | 5-(2-methylthio)pyrimidinyl | <0.10 | >150 |
| 35i | Ph(2-Me, 4-Cl) | 4-carboethoxymethoxyphenyl | 0.15 | >99 |
| 35j | Ph(2-OH, 5-Cl) | 2-methoxy-4-nitrophenyl | 0.16 | >94 |
1-Ad = 1-adamantyl
Other related actives are exemplified by 36 and 37 shown in Chart 2.
Adamantane-containing antiviral drugs have been known for some time.108,109 More recently, the adamantyl moiety has been used as a large, spherically symmetrical and hydrophobic group to occupy appropriate lipid-requiring drug binding sites. These include multidrug resistant transporters,110 11-ß-hydroxysteroid dehydrogenase,111 human isoprenylcysteine carboxyl methyltransferase,112 epoxide hydrolase,113 and lysosomal glucocerbrosidase.114 Adamantyl analogs have been shown to bind in substrate binding sites in ATPases and kinases such as sphingosine kinase.115-116 Furthermore, adamantyl-substituted aglycones of vancomycin and quinoline carboxamides show antibacterial and antimycobacterial activity.117-118 It is not clear at this time, however, just how the adamantyl-containing actives reported herein are acting. The similarity of the actives in Table 20 with reported adamantyl-containing sphingosine kinase inhibitors is intriguing as lipid kinases may play an important role in the life cycle and pathogenesis of M. tuberculosis.119
The diaryl amides (35a-j) are in many respects similar to the adamantyl amides and diaryl ureas previously discussed. These are relatively simple to more complex structures, and examples of the class are presented in Table 20. These types of compounds show various activities that include antiviral, anticancer and analgesic activity.109,120,121 As well, similar structures have been reported to inhibit the enoyl acyl carrier protein InhA in M. tuberculosis.122,123 As with many of the other actives seen in these phenotypic, whole cell assays, it is not clear by what mechanism they are acting, but the potent antitubercular activity in combination with the relatively low toxicity would suggest that these analogs may be worthy of further pursuit as leads for antitubercular drug discovery.
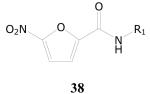
5-Nitrofuran-2-carboxamides
Lee first reported on a series of 5-nitrofuran-2-carboxamides as antitubercular agents.124 A recent review125 of the data showed that aromatic amides were more potent than aliphatic amides, a phenyl ring was favored over a heteroaromatic ring, the nitro functionality was required, and secondary amides were favored over tertiary amides. Secondary amine-substituted amides were also examined by Lee to allow for salt formation to increase solubility and absorption.
Of a total 52 nitrofuranyl compounds in the library 30 were nitrofuranylamides and among these 16 were found active (90% or greater inhibition in the primary screen). The MIC for these 16 active compounds ranged from < 0.1 to 12.4 μg/mL and SI values ranged from 0.37 to > 150. The active compounds are listed in Table 21.
Table 21.
5-Nitrofuran-2-carboxamides
 |
|||
|---|---|---|---|
| Cmpd. No. | R1 | TB IC90 |
SI |
| 38a |
|
< 0.1 | > 150 |
| 38b |
|
0.34 | ≥ 22.2 |
| 38c |
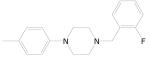
|
0.38 | ≥ 39.5 |
| 38d |
|
0.51 | 2.57 |
| 38e |
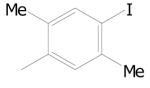
|
0.73 | ≥ 20.5 |
| 38f |
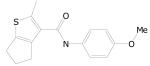
|
0.85 | ≥ 17.6 |
| 38g |
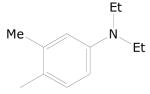
|
1.00 | ≥ 15 |
Data on additional nitrofurans will be submitted to PubChem, however, two other potent compounds of interest with SI values of 10 or greater are as 39 and 40.
Miscellaneous compounds
One group of structurally interesting leads from the non-clustered active compounds is the 2-benzamido-3,4-diphenyloxazoles such as 41 and 42. Excellent activities were seen for these compounds (TB IC90 values of <0.1 and 0.21 μg/mL and SIs of 150 and 72, respectively). Interestingly, the structurally similar 2-thio-substituted 43 was essentially inactive (TB IC90 of 13.8 μg/mL), suggesting that the benzamide (and especially one with an electron withdrawing substituent) is important for the activity of this group.
Purines that are substituted by various groups at the 2, 6, and 9-positions have been identified through past TAACF screening.1,126-128 Notably, a single, similar purine analog (44) was present in the ChemBridge library and showed significant activity (MIC < 0.1 μg/mL) and selectivity (SI > 150) against M. tuberculosis H37Rv. Currently, a mechanism of action for these types of inhibitors is unknown, although potential targets have been proposed.126,127
Conclusions
The discovery of new lead compounds active against M. tuberculosis is only the initial step in the development of a new antitubercular drug. Analysis of cytotoxicity is a rudimentary measure of potential toxicity, but can serve to prioritize compounds for further study. Additional in vitro evaluations that would provide useful information are 1) the evaluation of activity against other non-mycobacterial bacterial pathogens for identification of TB-specific drugs; 2) evaluation of bactericidal activity; 3) testing on strains with target-specific reporter such as the iniBAC promoter fusion that responds to cell wall inhibitors;123 4) evaluation of activity against non-replicating bacteria adapted to low oxygen;129 5) classification based on gene expression studies using microarrays; 6) testing against purified enzyme targets, for example see 130-137 (also see the www.taacf.org for additional enzyme targets amenable to screening); and 7) bioinformatic analysis for target identification and development of predictive algorithms for drug engineering. Additional in vivo evaluations that could be used to rapidly evaluate compounds include: 1) efficacy in the γ-interferon knockout mice;138 and 2) rapid pharmacological analysis in mice.21 Hopefully medicinal chemistry programs centered on one or more of the scaffolds identified will lead to the development of new antitubercular drugs.
Over the last decade several new TB-focused research entities and product development partnerships have evolved with the goal of contributing new candidate tuberculosis drugs to the antimicrobial armamentarium. Among these are the Global Alliance for TB Drug Development (TB Alliance), the Novartis Institute for Tropical Diseases, New Medicines for Tuberculosis, Institute for Tuberculosis Research, Astrazeneca India, Sequella, Inc., the Lilly TB Drug Discovery Initiative, GSK’s Diseases of the Developing World Drug Discovery Center, the Seattle Biomedical Research Institute, the Centers for Disease Control and Prevention’s Tuberculosis Trials Consortium, and the Vertex Global TB Research Network. For the first time in over 40 years, new chemical entities for TB are in human clinical trials cosponsored by large pharmaceutical companies or partnerships involving Bayer, Otsuka, Lupin, Sanofi-Aventis, and Tibotec. However, the existing pipeline is not sufficient to ensure the success and approval of 1-3 new drugs for TB. Investigators in these enterprises and at universities and research foundations engaged in TB drug discovery research throughout the world are likely to find the data described here and in the complete dataset of positive and negative results useful to identify new leads or metabolic probes for chemical biology initiatives. Phenotypic screening campaigns using commercially available compounds may contribute to new research avenues for medicinal chemistry exploration. It is the authors’ hope that screening centers throughout the world may join forces in sharing experiences and making more results publicly available to further accelerate development of new medicines to shorten therapy for TB and to treat resistant forms of this deadly disease.
Figure 6.

Figure 7.

ACKNOWLEDGMENTS
This work was supported by contracts N01-A- 95364 (JAS) and N01-AI-15449 (ELW). The complete data sets for the HTS campaign will be deposited in PubChem. The authors thank Dr. Scott Franzblau who generously provided us with his protocol for the Alamar Blue M. tuberculosis assay at the beginning of this program. The authors are appreciative of the work by Sara McKellip, Nicole Kushner and Kristen Southworth for their assistance in running the H37Rv screen, Anna Manouvakhova for assisting in the data analysis and Shuang Feng for the statistical analyses.
Footnotes
CONFLICT OF INTEREST STATEMENT Competing interests: Drs. Laughon and Goldman are NIAID staff members who either in the past or currently provide oversight for the project that generated the data used as the basis for this work.
REFERENCES
- 1.Goldman RC, Laughon BE, Reynolds RC, Secrist JA, III, Maddry JA, Guie M-A, Poffenberger AC, Kwong CA, Ananthan S. Programs to facilitate tuberculosis drug discovery: The Tuberculosis Antimicrobial Acquisition and Coordinating Facility. Infect Disord – Drug Targets. 2007;7:92–104. doi: 10.2174/187152607781001790. [DOI] [PubMed] [Google Scholar]
- 2.WHO: World Health Organization Global Tuberculosis Control 2008. Surveillance, Planning, Financing. 2008.
- 3.Schneider E, Castro KG. Tuberculosis trends in the United States, 1992-2001. Tuberculosis (Edinb) 2003;83:21–29. doi: 10.1016/s1472-9792(02)00075-6. [DOI] [PubMed] [Google Scholar]
- 4.Duncan K. Progress in TB drug development and what is still needed. Tuberculosis (Edinb) 2003;83:201–207. doi: 10.1016/s1472-9792(02)00076-8. [DOI] [PubMed] [Google Scholar]
- 5.Espinal MA. The global situation of MDR-TB. Tuberculosis (Edinb) 2003;83:44–51. doi: 10.1016/s1472-9792(02)00058-6. [DOI] [PubMed] [Google Scholar]
- 6.Mondal R, Jain A. Extensively drug-resistant Mycobacterium tuberculosis, India. Emerg Infect Dis. 2007;13:1429–1431. doi: 10.3201/eid1309.070443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen T, Murray M. Modeling epidemics of multidrug-resistant M. tuberculosis of heterogeneous fitness. Nat Med. 2004;10:1117–1121. doi: 10.1038/nm1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blower S, Supervie V. Predicting the future of XDR tuberculosis. Lancet Infect Dis. 2007;7:443. doi: 10.1016/S1473-3099(07)70143-3. [DOI] [PubMed] [Google Scholar]
- 9.Banerjee R, Allen J, Westenhouse J, Oh P, Elms W, Desmond E, Nitta A, Royce S, Flood J. Extensively drug-resistant tuberculosis in California, 1993-2006. Clin Infect Dis. 2008;47:450–457. doi: 10.1086/590009. [DOI] [PubMed] [Google Scholar]
- 10.Blaas SH, Mutterlein R, Weig J, Neher A, Salzberger B, Lehn N, Naumann L. Extensively drug resistant tuberculosis in a high income country: A report of four unrelated cases. BMC Infect Dis. 2008;8:60. doi: 10.1186/1471-2334-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan ED, Strand MJ, Iseman MD. Treatment outcomes in extensively resistant tuberculosis. N Engl J Med. 2008;359:657–659. doi: 10.1056/NEJMc0706556. [DOI] [PubMed] [Google Scholar]
- 12.Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368:1575–1580. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]
- 13.Kim H-R, Hwang SS, Kim HJ, Lee SM, Yoo C-G, Kim YW, Han SK, Shim Y-S, Yim J-J. Impact of extensive drug resistance on treatment outcomes in non-HIV-infected patients with multidrug-resistant tuberculosis. Clin Infect Dis. 2007;45:1290–1295. doi: 10.1086/522537. [DOI] [PubMed] [Google Scholar]
- 14.Migliori GB, Besozzi G, Girardi E, Kliiman K, Lange C, Toungoussova OS, Ferrara G, Cirillo DM, Gori A, Matteelli A, Spanevello A, Codecasa LR, Raviglione MC, SMIRA/TBNET Study Group Clinical and operational value of the extensively drug-resistant tuberculosis definition. Eur Respir J. 2007;30:623–626. doi: 10.1183/09031936.00077307. [DOI] [PubMed] [Google Scholar]
- 15.Migliori GB, Lange C, Centis R, Sotgiu G, Mutterlein R, Hoffmann H, Kliiman K, De Iaco G, Lauria FN, Richardson MD, Spanevello A, Cirillo DM, TBNET Study Group Resistance to second-line injectables and treatment outcomes in multidrug-resistant and extensively drug-resistant tuberculosis cases. Eur Respir J. 2008;31:1155–1159. doi: 10.1183/09031936.00028708. [DOI] [PubMed] [Google Scholar]
- 16.Migliori GB, Lange C, Girardi E, Centis R, Besozzi G, Kliiman K, Ortmann J, Matteelli A, Spanevello A, Cirillo DM, SMIRA/TBNET Study Group Extensively drug-resistant tuberculosis is worse than multidrug-resistant tuberculosis: Different methodology and settings, same results. Clin Infect Dis. 2008;46:958–959. doi: 10.1086/528875. [DOI] [PubMed] [Google Scholar]
- 17.Migliori GB, Ortmann J, Girardi E, Besozzi G, Lange C, Cirillo DM, Ferrarese M, De Iaco G, Gori A, Raviglione MC, SMIRA/TBNET Study Group Extensively drug-resistant tuberculosis, Italy and Germany. Emerg Infect Dis. 2007;13:780–782. doi: 10.3201/eid1305.060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitnick CD, Shin SS, Seung KJ, Rich ML, Atwood SS, Furin JJ, Fitzmaurice GM, Viru FA Alcantara, Appleton SC, Bayona JN, Bonilla CA, Chalco K, Choi S, Franke MF, Fraser HSF, Guerra D, Hurtado RM, Jazayeri D, Joseph K, Llaro K, Mestanza L, Mukherjee JS, Munoz M, Palacios E, Sanchez E, Sloutsky A, Becerra MC. Comprehensive treatment of extensively drug-resistant tuberculosis. N Engl J Med. 2008;359:563–574. doi: 10.1056/NEJMoa0800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raviglione MC. Facing extensively drug-resistant tuberculosis - A hope and a challenge. N Engl J Med. 2008;359:636–638. doi: 10.1056/NEJMe0804906. [DOI] [PubMed] [Google Scholar]
- 20.Spigelman MK. New tuberculosis therapeutics: A growing pipeline. J Infect Dis. 2007;196(Suppl 1):S28–34. doi: 10.1086/518663. [DOI] [PubMed] [Google Scholar]
- 21.Gruppo V, Johnson CM, Marietta KS, Scherman H, Zink EE, Crick DC, Adams LB, Orme IM, Lenaerts AJ. Rapid microbiologic and pharmacologic evaluation of experimental compounds against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2006;50:1245–1250. doi: 10.1128/AAC.50.4.1245-1250.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 24.Ertl P, Rhode B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 25.Xu Z-Q, Barrow WW, Suling WJ, Westbrook L, Barrow E, Lin Y-M, Flavin MT. Anti-HIV natural product (+)-calanolide A is active against both drug-susceptible and drug-resistant strains of Mycobacterium tuberculosis. Bioorg Med Chem. 2004;12:1199–1207. doi: 10.1016/j.bmc.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 26.Nayyar A, Jain R. Recent advances in new structural classes of anti-tuberculosis agents. Curr Med Chem. 2005;12:1873–1886. doi: 10.2174/0929867054546654. [DOI] [PubMed] [Google Scholar]
- 27.Yee SW, Shah B, Simons C. Synthesis and antimycobacterial activity of 7-O-substituted-4-methyl-2H-2-chromenone derivatives vs Mycobacterium tuberculosis. J Enzyme Inhib and Med Chem. 2005;20:109–113. doi: 10.1080/14756360400002015. [DOI] [PubMed] [Google Scholar]
- 28.Gupta AS, Merchant JR. Synthesis of new coumarylthioureas with antitubercular activity. Curr Sci. 1977;46:813–814. [Google Scholar]
- 29.Mamolo MG, Zampieri D, Vio L, Fermeglia M, Ferrone M, Pricl S, Scialino G, Banfi E. Antimycobacterial activity of new 3-substituted 5-(pyridin-4-yl)-3H-1,3,4-oxadiazol-2-one and 2-thione derivatives. Preliminary molecular modeling investigations. Bioorg Med Chem. 2005;13:3797–3809. doi: 10.1016/j.bmc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Hadizadeh F, Vosooghi R. Synthesis of alpha-[5-(5-amino-1,3,4-thiadiazol-2-yl)-2-imidazolylthio]acetic acids. J Heterocycl Chem. 2008;45:1477–1479. [Google Scholar]
- 31.Foroumadi A, Kargar Z, Sakhteman A, Sharifzadeh Z, Feyzmohammadi R, Kazemi M, Shafiee A. Synthesis and antimycobacterial activity of some alkyl [5-(nitroaryl)-1,3,4-thiadiazol-2-ylthio]propionates. Bioorg Med Chem Lett. 2006;16:1164–7. doi: 10.1016/j.bmcl.2005.11.087. [DOI] [PubMed] [Google Scholar]
- 32.Wujec M, Swatko-Ossor M, Mazur L, Rzaczynska Z, Siwek A. Synthesis, structure and investigations of tuberculosis inhibition activities of new 4-methyl-1-substituted-1H-1,2,4-triazole-5(4H)-thione. J Heterocycl Chem. 2008;45:1893–1896. [Google Scholar]
- 33.Kücükgüzel I, Tatar E, Kücükgüzel SG, Rollas S, De Clercq E. Synthesis of some novel thiourea derivatives obtained from 5-[(4-aminophenoxy)methyl]-4-alkyl/aryl-2,4-dihydro-3H-1,2,4-triazole-3-thiones and evaluation as antiviral/anti-HIV and anti-tuberculosis agents. Eur J Med Chem. 2008;43:381–392. doi: 10.1016/j.ejmech.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Salerno L, Sorrenti V, Guerrera F, Sarva MC, Siracusa MA, Di Giacomo C, Vanella A. 1-[aryloxyalkyl]-1H-imidazoles as inhibitors of neuronal nitric oxide synthase. Pharm Pharmacol Comm. 1999;5:491–494. [Google Scholar]
- 35.Cross PE, Dickinson RP, Parry MJ, Randall MJ. Selective thromboxane synthetase inhibitors. 1. 1-[(Aryloxy)alkyl]-1H-imidazoles. J Med Chem. 1985;28:1427–1432. doi: 10.1021/jm00148a009. [DOI] [PubMed] [Google Scholar]
- 36.Robertson DW, Beedle EE, Lawson R, Leander JD. Imidazole anticonvulsants: Structure-activity relationships of [(biphenylyloxy)alkyl]imidazoles. J Med Chem. 1987;30:939–943. doi: 10.1021/jm00388a035. [DOI] [PubMed] [Google Scholar]
- 37.Shimazu K, Shimizu M, Suzuki K, Kuwano E. Precocious metamorphosis-inducing activity of 1-substituted imidazoles. Nippon Noyaku Gakkaishi. 1996;21:337–339. [Google Scholar]
- 38.Albert A. Selective Toxicity: The Physico-chemical Basis of Therapy. Chapman and Hall Ltd.; New York: 1985. pp. 469–714. [Google Scholar]
- 39.Rhode W, Mikelens P, Jackson J, Blackman J, Whitcher J, Levinson W. Hydroxyquinolines inhibit ribonucleic acid-dependent deoxyribonucleic acid polymerase and inactivate Rous sarcoma virus and herpes simplex virus. Antimicrob Agents Chemother. 1976;10:234–240. doi: 10.1128/aac.10.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scheibel LW, Adler A. Antimalarial activity of selected aromatic chelators III. 8-Hydroxyquinolines (oxines) substituted in positions 5 and 7, and oxines annelated in position 5,6 by an aromatic ring. Mol Pharmacol. 1982;22:140–144. [PubMed] [Google Scholar]
- 41.Weismann K. Chelating drugs and zinc. Dan Med Bull. 1986;33:208–211. [PubMed] [Google Scholar]
- 42.Levinson W, Rhode W, Mikelens P, Jackson J, Antony A, Ramakrishnan T. Inactivation and inhibition of Rous sarcoma virus by copper-binding ligands: Thiosemicarbazines, 8-hydroxyquinolines, and isonicotinic acid hydrazide. Ann NY Acad Sci. 1977;284:525–532. doi: 10.1111/j.1749-6632.1977.tb21985.x. [DOI] [PubMed] [Google Scholar]
- 43.Levinson W, Mikelens P, Jackson J. Anti-tumor virus activity of copper-binding drugs. Adv Exp Med Biol. 1977;91:161–178. doi: 10.1007/978-1-4684-0796-9_12. [DOI] [PubMed] [Google Scholar]
- 44.Mirth DB, Chite AF, Schuster GS. 8-Hydroxyquinolines with the potential for long-term anticaries activity: Design, synthesis, and in vitro evaluation. J Dent Res. 1978;57:65–71. doi: 10.1177/00220345780570012401. [DOI] [PubMed] [Google Scholar]
- 45.Borchardt RT. Catechol O-methyltransferase. 2. In vitro inhibition by substituted 8-hydroxyquinolines. J Med Chem. 1973;16:382–387. doi: 10.1021/jm00262a016. [DOI] [PubMed] [Google Scholar]
- 46.Warshakoon NC, Wu S, Boyer A, Kawamoto R, Sheville J, Renock S, Xu K, Pokross M, Zhou S, Winter C, Walter R, Mekel M, Evdokimov AG. Structure-based design, synthesis, and SAR evaluation of a new series of 8-hydroxyquinolines as HIF-1α prolyl hydroxylase inhibitors. Bioorg Med Chem Lett. 2006;16:5517–5522. doi: 10.1016/j.bmcl.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 47.Zhuang L, Wai JS, Embrey MW, Fisher TE, Egbertson MS, Payne LS, Guare JP, Jr, Vacca JP, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Witmer MV, Moyer G, Schleif WA, Gabryelski LJ, Leonard YM, Lynch JJ, Jr, Michelsoin SR, Young SD. Design and synthesis of 8-hydroxy-[1,6]naphthyridines as novel inhibitors of HIV-1 integrase in vitro and in infected cells. J Med Chem. 2003;46:453–456. doi: 10.1021/jm025553u. [DOI] [PubMed] [Google Scholar]
- 48.Baum EZ, Crespo-Carbone SM, Klinger A, Foleno BD, Turchi I, Macielag M, Bush K. A MurF inhibitor that disrupts cell wall biosynthesis in Escherichia coli. Antimicrob Agents Chemother. 2007;51:4420–4444. doi: 10.1128/AAC.00845-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danelishvili L, Wu M, Young LS, Bermudez LE. Genomic approach to identifying the putative target of and mechanisms of resistance to mefloquine in mycobacteria. Amtimicrob Agents Chemother. 2005;49:3707–3714. doi: 10.1128/AAC.49.9.3707-3714.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jayaprakash S, Iso Y, Wan B, Franzblau SG, Kozikowski AP. Design, synthesis, and SAR studies of mefloquine-based ligands as potential antituberculosis agents. Chem Med Chem. 2006;1:593–597. doi: 10.1002/cmdc.200600010. [DOI] [PubMed] [Google Scholar]; Chem Med Chem. 2006;1:667. Erratum in. [Google Scholar]
- 51.Mao J, Wang Y, Wan B, Kozikowski AP, Franzblau SG. Design, synthesis, and pharmacological evaluation of mefloquine-based ligands as novel antituberculosis agents. Chem Med Chem. 2007;2:1624–1630. doi: 10.1002/cmdc.200700112. [DOI] [PubMed] [Google Scholar]
- 52.Nayyar A, Jain R. Recent advances in new structural classes of anti-tuberculosis agents. Curr Med Chem. 2005;12:1873–1886. doi: 10.2174/0929867054546654. [DOI] [PubMed] [Google Scholar]
- 53.Jain R, Vaitilingam B, Nayyar A, Palde PB. Substituted 4-methylquinolines as a new class of anti-tuberculosis agents. Bioorg Med Chem Lett. 2003;13:1051–1054. doi: 10.1016/s0960-894x(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 54.Vangapandu S, Jain M, Jain R, Kaur S, Singh PP. Ring-substituted quinolines as potential anti-tuberculosis agents. Bioorg Med Chem. 2004;12:2501–2508. doi: 10.1016/j.bmc.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 55.Vaitilingam B, Nayyar A, Palde PB, Monga V, Jain R, Kaur S, Singh PP. Synthesis and antimycobacterial activities of ring-substituted quinolinecarboxylic acid/ester analogues. Part 1. Bioorg Med Chem. 2004;12:4179–4188. doi: 10.1016/j.bmc.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 56.Nayyar A, Malde A, Jain R, Coutinho E. 3D-QSAR study of ring-substituted quinoline class of anti-tuberculosis agents. Bioorg Med Chem. 2006;14:847–856. doi: 10.1016/j.bmc.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 57.Nayyar A, Malde A, Coutinho E, Jain R. Synthesis, anti-tuberculosis activity, and 3D-QSAR study of ring-substituted-2/4-quinolinecarbaldehyde derivatives. Bioorg Med Chem. 2006;14:7302–7310. doi: 10.1016/j.bmc.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 58.Nayyar A, Monga V, Malde A, Coutinho E, Jain R. Synthesis, anti-tuberculosis activity, and 3D-QSAR study of 4-(adamantan-1-yl)-2-subtituted quinolines. Bioorg & Med Chem. 2007;15:626–640. doi: 10.1016/j.bmc.2006.10.064. [DOI] [PubMed] [Google Scholar]
- 59.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Purdy GE, Russell DG. Ubiquitin trafficking to the lysosome: Keeping the house tidy and getting rid of unwanted guests. Autophagy. 2007;3(4):399–401. doi: 10.4161/auto.4272. [DOI] [PubMed] [Google Scholar]; Alonso S, Pethe K, Russell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci USA. 2007;104:6031–6036. doi: 10.1073/pnas.0700036104. Addendum to: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huband MD, Cohen MA, Zurack M, Hanna DL, Skerlos LA, Sulavik MC, Gibson GW, Gage JW, Ellsworth E, Stier MA, Gracheck SJ. In Vitro and in vivo activities of PD 0305970 and PD 0326448, new bacterial gyrase/topoisomerase inhibitors with potent antibacterial activities versus multidrug-resistant gram-positive and fastidious organism groups. Antimicrob Agents Chemother. 2007;51:1191–1201. doi: 10.1128/AAC.01321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tran TP, Ellsworth EL, Sanchez JP, Watson BM, Stier MA, Showalter HDH, Domagala JM, Shapiro MA, Joannides ET, Gracheck SJ, Nguyen DQ, Bird P, Yip J, Sharadendu A, Ha C, Ramesani S, Wu X, Singh R. Structure-activity relationships of 3-aminoquinazolinediones, a new class of bacterial type-2 topoisomerase (DNA gyrase and topo IV) inhibitors. Bioorg Med Chem Lett. 2007;17:1312–1320. doi: 10.1016/j.bmcl.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 63.Tran TP, Ellsworth EL, Stier MA, Domagala JM, Showalter HDH, Gracheck SJ, Shapiro MA, Joannides TE, Singh R. Synthesis and structural-activity relationships of 3-hydroxyquinazoline-2,4-dione antibacterial agents. Bioorg Med Chem Lett. 2004;14:4405–4409. doi: 10.1016/j.bmcl.2004.06.063. [DOI] [PubMed] [Google Scholar]
- 64.Raghavendra NM, Thampi P, Gurubasavarajaswamy PM, Sriram D. Synthesis, antitubercular and anticancer activities of substituted furyl-quinazolin-3(4H)-ones. Arch Pharm Chem Life Sci. 2007;340:635–641. doi: 10.1002/ardp.200700096. [DOI] [PubMed] [Google Scholar]
- 65.Nanda AK, Ganguli S, Chakraborty R. Antibacterial activity of some 3-(arylideneamino)-2-phenylquinazoline-4(3H)-ones: Synthesis and preliminary QSAR studies. Molecules. 2007;12:2413–2426. doi: 10.3390/12102413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raghavendra NM, Thampi P, Gurubasavarajaswamy PM, Sriram D. Synthesis and antimicrobial activities of some novel substituted 2-imidazolyl-N-(4-oxoquinazolin-3(4H)-yl)-acetamides. Chem Pharm Bull. 2007;55:1615–1619. doi: 10.1248/cpb.55.1615. [DOI] [PubMed] [Google Scholar]
- 67.Alagarsamy V, Rajasolomon V, Meena R, Ramseshu KV. Synthesis, analgesic, anti-inflammatory and antibacterial activities of some novel 2-butyl-3-substituted quinazolin-4-(3H)-ones. Biol Pharm Bull. 2005;28:1091–1094. doi: 10.1248/bpb.28.1091. [DOI] [PubMed] [Google Scholar]
- 68.Nasr MN, Gineinah MM, El-Bendary ER. Synthesis and in vitro antibacterial evaluation of novel imidazo[2′,1′:5,1]-1,2,4,-triazolo[4,3-c]-quinazoline derivatives of 5-thioxo-1,2,4-triazole, 4-oxothiazolidine, and their open-chain counterparts. Arch Pharm Pharm Med Chem. 2003;336:560–566. doi: 10.1002/ardp.200300809. [DOI] [PubMed] [Google Scholar]
- 69.Bedi PMS, Kumar V, Mahajan MP. Synthesis and biological activity of novel antibacterial quinazolines. Bioorg Med Chem Lett. 2004;14:5211–5213. doi: 10.1016/j.bmcl.2004.07.065. [DOI] [PubMed] [Google Scholar]
- 70.De La Fuente R, Sonawane ND, Arumainayagam D, Verkman AS. Small molecules with antimicrobial activity against E. coli and P. aeruginosa identified by high-throughput screening. Br J Pharmacol. 2006;149:551–559. doi: 10.1038/sj.bjp.0706873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tandon VK, Singh RV, Yadav DB. Synthesis and evaluation of novel 1,4-naphthoquinone derivatives as antiviral, antifungal and anticancer agents. Bioorg Med Chem Lett. 2007;14:2901–2904. doi: 10.1016/j.bmcl.2004.03.047. [DOI] [PubMed] [Google Scholar]
- 72.Mahapatra A, Mativandlela SPN, Binneman B, Fourie PB, Hamilton CJ, Meyer JJM, van der Kooy F, Houghton P, Lall N. Activity of 7-methyljuglone derivatives against Mycobacterium tuberculosis and as subversive substrates for mycothiol disulfide reductase. Bioorg Med Chem. 2007;15:7638–7646. doi: 10.1016/j.bmc.2007.08.064. [DOI] [PubMed] [Google Scholar]
- 73.Bapela NB, Lall N, Fourie PB, Franzblau SG, Van Rensburg CEJ. Activity of 7-methyljuglone in combination with antituberculous drugs against Mycobacterium tuberculosis. Phytomedicine. 2006;13:630–635. doi: 10.1016/j.phymed.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 74.Tandon VK, Yadav DB, Singh RV, Vaish M, Chaturvedi AK, Shukla PK. Synthesis and biological evaluation of novel 1,4-naphthoquinone derivatives as antibacterial and antiviral agents. Bioorg Med Chem Lett. 2005;15:3463–3466. doi: 10.1016/j.bmcl.2005.04.075. [DOI] [PubMed] [Google Scholar]
- 75.Tandon VK, Yadav DB, Singh M, Chaturvedi AK, Shukla PK. Synthesis and biological evaluation of novel (L)-α-amino acid methyl ester, heteroalkyl, and aryl substituted 1,4-naphthoquinone derivatives as antifungal and antibacterial agents. Bioorg Med Chem Lett. 2005;15:5324–5328. doi: 10.1016/j.bmcl.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 76.Kaneshiro ES, Basselin M, Merali S, Kayser O. Ubiquinone synthesis and its regulation in Pneumocystis carinii. J Eukaryot Microbiol. 2006;53:435–444. doi: 10.1111/j.1550-7408.2006.00127.x. [DOI] [PubMed] [Google Scholar]
- 77.Kessl JJ, Lange BB, Merbitz-Zahradnik T, Zwicker K, Hill P, Meunier B, Pálsdóttir H, Hunte C, Meshnick S, Trumpower BL. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem. 2003;278:31312–31318. doi: 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]
- 78.Teh JS, Yano T, Rubin H. Type II NADH:menaquinone oxidoreductase of Mycobacterium tuberculosis. Infect Disord – Drug Targets. 2007;7:169–181. doi: 10.2174/187152607781001781. [DOI] [PubMed] [Google Scholar]
- 79.Laurenzi M, Ginsberg A, Spigelman M. Challenges associated with current and future TB treatment. Infect Disord – Drug Targets. 2007;7:105–119. doi: 10.2174/187152607781001817. [DOI] [PubMed] [Google Scholar]
- 80.See Prestwick Library Data. http://www.taacf.org.
- 81.Dover LG, Alahari A, Gratraud P, Gomes JM, Bhowruth V, Reynolds RC, Besra GS, Kremer L. EthA, a common activator of thiocarbamide-containing drugs acting on different mycobacterial targets. Antimicrobial Agents Chemother. 2007;51:1055–1063. doi: 10.1128/AAC.01063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeBarber AE, Mdluli K, Bosman M, Bekker L-G, Barry CE., III Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Nat Acad Sci USA. 2000;97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fraaije MW, Kamerbeek NM, Heidekamp AJ, Fortin R, Janssen DB. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J Biol Chem. 2004;279:3354–3360. doi: 10.1074/jbc.M307770200. [DOI] [PubMed] [Google Scholar]
- 84.Sousa EHS, Pontes DL, Diogenes ICN, Lopes LGF, Oliveira JS, Basso LA, Santos DS, Moreira IS. Electron transfer kinetics and mechanistic study of the thionicotinamide coordinated to the pentacyanoferrate (III)/(II) complexes: A model system for the in vitro activation of thioamides anti-tuberculosis drugs. J Inorg Biochem. 2005;99:368–375. doi: 10.1016/j.jinorgbio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 85.Qian L, de Montellano PR Ortiz. Oxidative activation of thiacetazone by the Mycobacterium tuberculosis flavin monooxygenase EtaA and human FMO1 and FMO3. Chem Res Toxicol. 2006;19:443–449. doi: 10.1021/tx050328b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barry CE, III, Slayden RA, Sampson AE, Lee RE. Use of genomics and combinatorial chemistry in the development of new antimycobacterial drugs. Biochem Pharmacol. 2000;59:221–231. doi: 10.1016/s0006-2952(99)00253-1. [DOI] [PubMed] [Google Scholar]
- 87.Reddy PVG, Reddy CS, Raju CN. Synthesis and antimicrobial activity of N-(substituted)-N’-(2,3-dihydro-2-oxido-5-benzoyl-1H-1,3,2-benzodiazaphosphol-2-yl) ureas. Chem Pharm Bull. 2003;51:860–863. doi: 10.1248/cpb.51.860. [DOI] [PubMed] [Google Scholar]
- 88.Kiran YB, Gunasekar D, Reddy CD, Reddy CS, Tran K, Le T, Berlin KD, Srinivasan S, Devi MC. Synthesis and bioactivity of some new N-aryl/alkyl/cyclohexyl-N’-(2,3-dihydro-2-oxo-4H-benz[e][1,3,2]oxazaphosphorin-2-yl) ureas. Pest Manag Sci. 2005;61:1016–1023. doi: 10.1002/ps.1067. [DOI] [PubMed] [Google Scholar]
- 89.Cetinkaya B, Cetinkaya E, Kucukbay H, Durmaz R. Synthesis and antimicrobial activity of electron rich olefin derived cyclic ureas. Arzneim-Forsch/Drug Res. 1996;46:154–1158. [PubMed] [Google Scholar]
- 90.Tewari N, Tiwari VK, Mishra RC, Tripathi RP, Srivastava AK, Ahmad R, Srivastava R, Srivastava BS. Synthesis and bioevaluation of glycosyl ureas as α-glucosidase inhibitors and their effect on mycobacterium. Bioorg Med Chem. 2003;11:2911–2922. doi: 10.1016/s0968-0896(03)00214-1. [DOI] [PubMed] [Google Scholar]
- 91.Shu Q, Nair V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med Res Rev. 2008;28:219–232. doi: 10.1002/med.20104. [DOI] [PubMed] [Google Scholar]
- 92.Ilovich O, Jacobson O, Aviv Y, Litchi A, Chisin R, Mishani E. Formation of fluorine-18 labeled diaryl ureas–labeled VEGFR-2/PDGFR dual inhibitors as molecular imaging agents for angiogenesis. Bioorg Med Chem. 2008;16:4242–4251. doi: 10.1016/j.bmc.2008.02.081. [DOI] [PubMed] [Google Scholar]
- 93.Dai Y, Hartandi K, Soni NB, Pease LJ, Reuter DR, Olson AM, Osterling DJ, Doktor SZ, Albert DH, Bouska JJ, Glaser KB, Marcotte PA, Stewart KD, Davidsen SK, Michaelides MR. Identification of aminopyrazolopyridine ureas as potent VEGFR/PDGFR multitargeted kinase inhibitors. Bioorg Med Chem Lett. 2008;18:386–390. doi: 10.1016/j.bmcl.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 94.Tao ZF, Chen Z, Bui M-H, Kovar P, Johnson E, Bouska J, Zhang H, Rosenberg S, Sowin T, Lin N-H. Macrocyclic ureas as potent and selective Chk1 inhibitors: An improved synthesis, kinome profiling, structure-activity relationships, and preliminary pharmacokinetics. Bioorg Med Chem Lett. 2007;17:6593–6601. doi: 10.1016/j.bmcl.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 95.Drews SJ, Hung F, Av-Gay Y. A protein kinase inhibitor as an antimycobacterial agent. FEMS Microbiol Lett. 2001;205:369–374. doi: 10.1111/j.1574-6968.2001.tb10974.x. [DOI] [PubMed] [Google Scholar]
- 96.Av-Gay Y, Everett M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000;8:238–244. doi: 10.1016/s0966-842x(00)01734-0. [DOI] [PubMed] [Google Scholar]
- 97.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, III, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: Structure-activity relationships of the nucleobase domain of 5′-O-[N-salicyl)sulfamoyl]adenosine. J Med Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bhave DP, Muse WB, 3rd, Carroll KS. Drug targets in mycobacterial sulfur metabolism. Infect Disord Drug Targets. 2007;7:149–158. doi: 10.2174/187152607781001772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Phetsuksiri B, Jackson M, Scherman H, McNeil M, Besra GS, Baulard AR, Slayden RA, DeBarber AE, Barry CE, III, Baird MS, Crick DC, Brennan PJ. Unique mechanism of action of the thiourea drug isoxyl on Mycobacterium tuberculosis. J Biol Chem. 2003;278:53123–53130. doi: 10.1074/jbc.M311209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kordulakova J, Janin YL, Liav A, Barilone N, Dos Vultos T, Rauzier J, Brennan PJ, Gicquel B, Jackson M. Isoxyl activation is required for bacteriostatic activity against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:3824–3829. doi: 10.1128/AAC.00433-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhowruth V, Brown AK, Reynolds RC, Coxon GD, Mackay SP, Minnikin DE, Besra GS. Symmetrical and unsymmetrical analogues of isoxyl; Active agents against Mycobacterium tuberculosis. Bioorg Med Chem Lett. 2006;16:4743–4747. doi: 10.1016/j.bmcl.2006.06.095. [DOI] [PubMed] [Google Scholar]
- 102.Liav A, Angala SK, Brennan PJ, Jackson M. N-d-Aldopentofuranosyl-N’-[p-(isoamyloxy)phenyl]-thiourea derivatives: Potential anti-TB therapeutic agents. Bioorg Med Chem Lett. 2008;18:2649–2651. doi: 10.1016/j.bmcl.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 103.Stark H, Purand K, Ligneau X, Rouleau A, Arrang J-M, Garbarg M, Schwartz J-C, Schunack W. Novel carbamates as potent histamine H3 receptor antagonists with high in vitro and oral in vivo activity. J Med Chem. 1996;39:1157–1163. doi: 10.1021/jm9507688. [DOI] [PubMed] [Google Scholar]
- 104.Meindl W, Friese-Kimmel A, Lachenmayr F, Buschauer A, Schunack W. Influence of agonists and antagonists of the histamine H1 and H2 receptor on the growth of Mycobacterium tuberculosis H37Ra. Arch Pharm (Weinheim) 1989;323:267–272. doi: 10.1002/ardp.19903230504. [DOI] [PubMed] [Google Scholar]
- 105.Ranise A, Spallaross A, Bruno O, Schenone S, Fossa P, Menozzi G, Bondavalli F, Mosti L, Capuano A, Mazzeo F, Falcone G, Filippelli W. Synthesis of N-substituted-N-acylthioureas of 4-substituted piperazines endowed with local anaesthetic, antihyperlipidemic, antiproliferative activities and antiarrythmic, analgesic, antiaggregating actions. II Farmaco. 2003;58:765–780. doi: 10.1016/S0014-827X(03)00132-0. [DOI] [PubMed] [Google Scholar]
- 106.Cesarini S, Spallarossa A, Ranise A, Schenone S, Rosano C, La Colla P, Sanna G, Busonera B, Loddo R. N-Acylated and N,N’-diacylated imidazolidine-2-thione derivatives and N,N’-diacylated tetrahydropyrimidine-2(1H)-thione analogues: Synthesis and antiproliferative activity. Eur J Med Chem. 2008:1–13. doi: 10.1016/j.ejmech.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 107.Katritzky AR, Cai X, Rogovoy BV. Solid phase synthesis and application of trisubstituted thioureas. J Comb Chem. 2003;5:392–399. doi: 10.1021/cc020109y. [DOI] [PubMed] [Google Scholar]
- 108.Aigami K, Inamoto Y, Takaishi N, Hattori K, Takatsuki A, Tamura G. Biologically active polycycloalkanes. 1. Antiviral adamantane derivatives. J Med Chem. 1975;18:713–721. doi: 10.1021/jm00241a015. [DOI] [PubMed] [Google Scholar]
- 109.Van Derpoorten K, Balzarini J, DeClercq E, Poupaert JH. Anti-HIV activity of N-1-Adamantyl-4-aminophthalimide. Biomed Pharmacother. 1975;51:464–468. doi: 10.1016/s0753-3322(97)82327-x. [DOI] [PubMed] [Google Scholar]
- 110.De Rosa MF, Ackerley C, Wang B, Ito S, Clarke DM, Lingwood C. Inhibition of multidrug resistance by adamantylGb3, a globotriaosylceramide analog. J Biol Chem. 2008;283:4501–4511. doi: 10.1074/jbc.M705473200. [DOI] [PubMed] [Google Scholar]
- 111.Webster SP, Ward P, Binnie M, Craigie E, McConnell KMM, Sooy K, Vinter A, Seckl JR, Walker BR. Discovery and biological evaluation of adamantyl amide 11β-HSD1 inhibitors. Bioorg Med Chem Lett. 2007;17:2838–2843. doi: 10.1016/j.bmcl.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 112.Donelson JL, Hodges HB, MacDougall DD, Henriksen BS, Hrycyna CA, Gibbs RA. Amide-substituted farnesylcysteine analogs as inhibitors of human isoprenylcysteine carboxyl methyltransferase. Bioorg Med Chem Lett. 2006;16:4420–4423. doi: 10.1016/j.bmcl.2006.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim I-H, Heirtzler FR, Morisseau C, Nishi K, Tsai H-J, Hammock BD. Optmization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J Med Chem. 2005;48:3621–3629. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu Z, Sawkar AR, Whalen LJ, Wong C-H, Kelly JW. Isofagomine- and 2,5-anhydro-2,5-imino-D-glucitol-based glucocerebrosidase pharmacological chaperones for Gaucher disease intervention. J Med Chem. 2007;50:94–100. doi: 10.1021/jm060677i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Whetstone H, Lingwood C. 3′ Sulfogalactolipid binding specifically inhibits Hsp70 ATPase activity in vitro. Biochemistry. 2003;42:1611–1617. doi: 10.1021/bi026735t. [DOI] [PubMed] [Google Scholar]
- 116.Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53:997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Printsevskaya SS, Solovieva SE, Olsufyeva EN, Mirchink EP, Isakova EB, De Clercq E, Balzarini J, Preobrazhenskaya MN. Structure-activity relationship studies of a series of antiviral and antibacterial aglycon derivatives of the glycopeptide antibiotics vancomycin, eremomycin, and dechloroeremomycin. J Med Chem. 2005;48:3885–3890. doi: 10.1021/jm0500774. [DOI] [PubMed] [Google Scholar]
- 118.Monga V, Nayyar A, Vaitilingam B, Palde PB, Jhamb SS, Kaur S, Singh PP, Jain R. Ring-substituted quinolines. Part 2: Synthesis and antimycobacterial activity of ring-substituted quinolinecarbohydrazide and ring-substituted quinolinecarboxamide analogues. Bioorg Med Chem. 2004;12:6465–6472. doi: 10.1016/j.bmc.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 119.Owens RM, Hsu FF, VanderVen BC, Purdy GE, Hesteande E, Giannakas P, Sacchettini JC, McKinney JD, Hill PJ, Belisle JT, Butcher BA, Pethe K, Russell DG. M. tuberculosis Rv2252 encodes a diacylglycerol kinase involved in the biosynthesis of phosphatidylinositol mannosides (PIMs) Mol Microbiol. 2006;60:1152–1163. doi: 10.1111/j.1365-2958.2006.05174.x. [DOI] [PubMed] [Google Scholar]
- 120.Bolós J. Structure-activity relationships of p38 mitogen-activated protein kinase inhibitors. Mini-Rev in Med Chem. 2005;5:857–868. doi: 10.2174/1389557054867048. [DOI] [PubMed] [Google Scholar]
- 121.Fletcher SR, McIver E, Lewis S, Burkamp F, Leech C, Mason G, Boyce S, Morrison D, Richards G, Sutton K, Jones AB. The search for novel TRPV1-antagonists: From carboxamides to benzimidazoles and indazolones. Bioorg Med Chem Lett. 2006;16:2872–2876. doi: 10.1016/j.bmcl.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 122.He X, Alian A, de Montellano PR Ortiz. Inhibition of the Mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides. Bioorg Med Chem. 2007;15:6649–6658. doi: 10.1016/j.bmc.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alland D, Steyn AJ, Weisbrod T, Aldrich K, Jacobs WR., Jr Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J Bacteriol. 2000;182:1802–1811. doi: 10.1128/jb.182.7.1802-1811.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tangallapally RP, Yendapally R, Daniels AJ, Lee REB, Lee RE. Nitrofurans as novel anti-tuberculosis agents: Identification, development and evaluation. Curr Top Med Chem. 2007;7:509–526. doi: 10.2174/156802607780059772. [DOI] [PubMed] [Google Scholar]
- 125.Tangallapally RP, Yendapally R, Lee RE, Hevener K, Jones VC, Lenaerts AJ, McNeil MR, Wang Y, Franzblau S, Lee RE. Synthesis and evaluation of nitrofuranylamides as novel antituberculosis agents. J Med Chem. 2004;47:5276–5283. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 126.Pathak AK, Pathak V, Seitz LE, Suling WJ, Reynolds RC. Antimycobacterial agents: Thio analogues of purine. J Med Chem. 2004;47:273–276. doi: 10.1021/jm030389b. [DOI] [PubMed] [Google Scholar]
- 127.Molle V, Reynolds RC, Alderwick LJ, Besra GS, Cozzone AJ, Fütterer K, Kremer L. EmbR2, a structural homologue of EmbR, inhibits the Mycobacterium tuberculosis kinase/substrate pair PknH/EmbR. Biochem J. 2008;410:309–317. doi: 10.1042/BJ20071384. [DOI] [PubMed] [Google Scholar]
- 128.Braendvang M, Gundersen LL. Synthesis, biological activity, and SAR of antimycobacterial 2- and 8-substituted 6-(2-furyl)-9-(p-methoxybenzyl)purines. Bioorg Med Chem. 2007;15:7144–7165. doi: 10.1016/j.bmc.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 129.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bhat J, Rane R, Solapure SM, Sarkar D, Sharma U, Harish MN, Lamb S, Plant D, Alcock P, Peters S, Barde S, Roy RK. High-throughput screening of RNA polymerase inhibitors using a fluorescent UTP analog. J Biomol Screening. 2006;11:968–976. doi: 10.1177/1087057106291978. [DOI] [PubMed] [Google Scholar]
- 131.Carlson EE, May JF, Kiessling LL. Chemical probes of UDP-galactopyranose mutase. Chem Biol. 2006;13:825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 132.Ma Y, Stern RJ, Scherman MS, Vissa VD, Yan W, Jones VC, Zhang F, Franzblau SG, Lewis WH, McNeil MR. Drug targeting Mycobacterium tuberculosis cell wall synthesis: Genetics of dTDP-rhamnose synthetic enzymes and development of a microtiter plate-based screen for inhibitors of conversion of dTDP-glucose to dTDP-rhamnose. Antimicrob Agents Chemother. 2001;45:1407–1416. doi: 10.1128/AAC.45.5.1407-1416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saini DK, Tyagi JS. High-throughput microplate phosphorylation assays based on DevR-DevS/Rv2027c 2-component signal transduction pathway to screen for novel antitubercular compounds. J Biomol Screening. 2005;10:215–224. doi: 10.1177/1087057104272090. [DOI] [PubMed] [Google Scholar]
- 134.Scherman MS, Winans KA, Stern RJ, Jones V, Bertozzi CR, McNeil MR. Drug targeting Mycobacterium tuberculosis cell wall synthesis: Development of a microtiter plate-based screen for UDP-galactopyranose mutase and identification of an inhibitor from a uridine-based library. Antimicrob Agents Chemother. 2003;47:378–382. doi: 10.1128/AAC.47.1.378-382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weinstein EA, Yano T, Li L-S, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. Inhibitors of type II NADH: menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci USA. 2005;102:4548–4553. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.White EL, Southworth K, Ross L, Cooley S, Gill RB, Sosa MI, Manouvakhova A, Rasmussen L, Goulding C, Eisenberg D, Fletcher TM., III A novel inhibitor of Mycobacterium tuberculosis pantothenate synthetase. J Biomol Screening. 2007;12:100–105. doi: 10.1177/1087057106296484. [DOI] [PubMed] [Google Scholar]
- 137.Zhang W, Jones VC, Scherman MS, Mahapatra S, Crick D, Bhamidi S, Xin Y, McNeil MR, Ma Y. Expression, essentiality, and a microtiter plate assay for mycobacterial GlmU, the bifunctional glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase. Int J Biochem Cell Biol. 2008;40:2560–2571. doi: 10.1016/j.biocel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lenaerts AJ, Gruppo V, Brooks JV, Orme IM. Rapid in vivo screening of experimental drugs for tuberculosis using gamma interferon gene-disrupted mice. Antimicrob Agents Chemother. 2003;47:783–785. doi: 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]