

Abstract
Hypoxia and hypoxia-inducible factors (HIFs) play an important role in the Kaposi's sarcoma-associated herpesvirus (KSHV) life cycle. In particular, hypoxia can activate lytic replication of KSHV and specific lytic genes, including the replication and transcription activator (RTA), while KSHV infection in turn can increase the levels and activity of HIFs. In the present study, we show that hypoxia increases the levels of mRNAs encoding KSHV latency-associated nuclear antigen (LANA) in primary effusion lymphoma (PEL) cell lines and also increases the levels of LANA protein. Luciferase reporter assays in Hep3B cells revealed a moderate activation of the LANA promoter region by hypoxia as well as by cotransfection with degradation-resistant HIF-1α or HIF-2α expression plasmids. Computer analysis of a 1.2-kb sequence upstream of the LANA translational start site identified six potential hypoxia-responsive elements (HRE). Sequential deletion studies revealed that much of this activity was mediated by one of these HREs (HRE 4R) oriented in the 3′ to 5′ direction and located between the constitutive (LTc) and RTA-inducible (LTi) mRNA start sites. Site-directed mutation of this HRE substantially reduced the response to both HIF-1α and HIF-2α in a luciferase reporter assay. Electrophoretic mobility shift assays (EMSA) and chromatin immunoprecipitation (ChIP) assays demonstrated binding of both HIF-1α and HIF-2α to this region. Also, HIF-1α was found to associate with RTA, and HIFs enhanced the activation of LTi by RTA. These results provide evidence that hypoxia and HIFs upregulate both latent and lytic KSHV replication and play a central role in the life cycle of this virus.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also called human herpesvirus 8 (HHV-8), is the causative agent of Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD) (7, 8, 43). Like other herpesviruses, KSHV can establish persistent (latent) or lytic infection in target cells. During latent infection, a limited number of viral genes are expressed. These include the latency-associated nuclear antigen (LANA) encoded by ORF73, a viral cyclin (v-cyclin) encoded by ORF72, a viral FLICE inhibitory protein (vFLIP) encoded by ORF71, viral interferon regulatory factors encoded by K10, and kaposin encoded by K12 (10, 37). LANA in particular plays a key role in the maintenance of latency. LANA tethers the KSHV episome to cellular chromosomes and segregates the KSHV genome during host cell division (34, 40). In addition, LANA interacts with a variety of cellular proteins to help create a suitable environment for latent viral persistence (20, 35).
In the KSHV genome, three of the latent proteins, LANA (ORF73), v-cyclin (ORF72), and v-FLIP (ORF71), are located in a single cluster, and the mRNAs for these proteins have been found to originate from the same promoter (10, 37, 38, 45). Transcription of the multicistronic mRNAs encoding ORF71 to ORF73 is regulated by a cis-regulatory region that is primarily located between ORF73 and K14 (encoding v-OX2), a lytic gene oriented in the opposite direction (Fig. 1A) (10, 38, 45). During latency, these multicistronic RNAs are transcribed from a constitutively active promoter (LTc) initiating from nucleotide 127880 (also mapped nucleotide positions 127900 and 127948) (10, 31, 38, 44, 45). Interestingly, the KSHV replication and transcription activator (RTA), encoded by ORF50, has been found to activate transcription of the mRNAs for ORF71 to ORF73, but in this case using an alternate inducible promoter (LTi), with mRNA transcripts initiating ∼270 bp downstream of LTc (31). There is also evidence that RTA packaged with KSHV virions can assist in the establishment of latency through activation of LTi (25).
Fig 1.

Hypoxia increases ORF73 (LANA) promoter activity. (A) Schematic diagram of the genomic organization of the region spanning ORFK12 (Kaposin) through K14 (v-OX2) in the KSHV genome; this region includes ORF71 through ORF73 (LANA) as well as the KSHV miRNA cluster. The numbers above the closed arrows correspond to positions of initiation/termination codons of the ORFs. Two arrows between K14 (v-OX2) and LANA denote the LANA constitutive (LTc) and RTA-inducible (LTi) mRNA start sites. The location of the probes used in Northern blots for ORF73 and K12 are shown as lines above the respective genes. Shown below is an expanded diagram of LANA and the LANA promoter region surrounding LTc and LTi, with the nucleotide positions for the mRNA start sites indicated. The LTc mRNA start site has been alternatively mapped to nucleotide positions 127900 and 127948 (10, 38). The LANA promoter region contains six potential hypoxia response elements (HREs), shown as rectangular boxes labeled 1 through 6. R denotes HREs in the reverse orientation to the transcription of LANA. (B) Schematic diagram of luciferase reporter constructs of the LANA promoter region. pGL3-LANA(c&i)p-luc [LANA(c&i)p] contains a 1,201-bp DNA segment that includes LTc and LTi and the six potential HREs. pGL3-LANA(c)p-luc [LANA(c)p] contains a 795-bp DNA segment that includes the LTc start site and four potential HREs (1, 2, 3R, and 4R). pGL3-LANA(i)p-luc [LANA(i)p] contains a 570-bp DNA segment that includes the LTi promoter start site but not the LTc start site and only three of the potential HREs (4R, 5R, and 6). (C) Hypoxia and CoCl2 treatments induce ORF73 (LANA) promoter activity in Hep3B cells. A fixed amount (700 ng) of reporter plasmid pGL3-LANA(c&i)p-luc [LANA(c&i)p] or a pGL3-basic plasmid (control) was transfected into Hep3B cells cultured in triplicate wells of 12-well culture plates. At 24 h of posttransfection, cells were exposed to hypoxia or treated with CoCl2 for 16 h at 37°C. Cells were harvested, and the cell lysate was analyzed for luciferase activity. Mean light units were normalized to the total cellular protein estimated by the Bradford method. The fold activation of promoter activity was determined based on the ratio between the normalized mean light units and that of the pGL3-basic control construct cultured under normoxia. All values represent the means of results from three experiments, each done in triplicate. Error bars represent the standard deviations. Similar results were obtained in initial experiments when luciferase activity was normalized using a Renilla control (results not shown).
Cells latently infected with KSHV can be induced to undergo lytic replication by treatment with chemical agents such as 12-O-tetradecanoyl 13-acetate (TPA) or sodium butyrate (1, 32, 54). Activation of RTA, the lytic switch gene product encoded by ORF50, is central to this process, as RTA can by itself initiate lytic activation through activation of key downstream lytic genes by binding to RTA response elements (RRE) or interacting with other regulatory factors (27, 28, 30, 52, 55). Our lab has previously reported that KSHV could be induced to lytic replication by hypoxia (9). In addition, we found that the promoter region of RTA could be activated to a limited extent by hypoxia or by hypoxia-inducible factor (HIF) and that certain other lytic genes, ORF34 through ORF37, could also be directly activated by hypoxia or HIFs (16, 17). HIFs are heterodimeric transcription factors, comprised of HIFα and HIFβ, whose levels increase rapidly in the nucleus in hypoxic cells (12, 46, 48). HIFs activate hypoxia-responsive genes through binding to hypoxia response elements (HREs) in the promoter regions (12, 41, 46, 47, 51). The core HRE consensus sequence is 5′-RCGTG-3′ (51). Two principal isoforms of HIFα, HIF-1α and HIF-2α, have been identified. HIF-2α has a high degree of similarity and function with HIF-1α, although it preferentially activates certain genes and plays a more important role in certain cell types, such as endothelial cells (12, 46, 48).
Additional studies of KSHV showed that hypoxia and HIFs play several important roles in the viral life cycle. Cai et al. showed that robust activation of the RTA promoter was obtained with HIFs and KSHV LANA through a physical interaction between these two factors (2). In addition, a constitutively active G protein-coupled receptor encoded by KSHV ORF74 was found to enhance the activity of HIF-1α (42). Finally, KSHV latent infection was found to synergize with hypoxia in endothelial cells to induce increased levels of HIF-1α and HIF-2α, and this effect was caused at least in part by LANA (3, 5, 14). Thus, KSHV infection increases levels of HIFs, which in turn appears to play an important role in increasing KSHV lytic gene activation. Given this central role of hypoxia, we hypothesized that hypoxia and HIFs may also play a role in regulating the production of KSHV latent genes, including LANA. In this paper, we show that hypoxia and HIFs can induce transcription of LANA and that this effect is mediated at least in part through interactions with one or more HRE in the LANA promoter region. Moreover, we show that HIFs can enhance the activation of LANA by RTA through the LTi.
MATERIALS AND METHODS
Cell lines and culture conditions.
The PEL cell lines JSC-1 (gift from Richard Ambinder, Johns Hopkins University, Baltimore, MD), dually infected with Epstein-Barr virus (EBV) and HHV-8 (4), and BC-3 (ATCC, Rockville, MD) and BCBL-1 (National Institutes of Health AIDS Research and Reagent Program, Rockville, MD), harboring KSHV only (1, 36), were grown in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Scientific, Rockford, IL) at 37°C under normoxic conditions with 5% CO2. Where indicated, these cells were exposed to hypoxia by culturing in an incubator with 1% O2 and 5% CO2 (9) or were treated with 20 ng/ml of TPA (Sigma, St. Louis, MO) to induce KSHV lytic replication. Hep3B, a human hepatoma cell line (ATCC, Rockville, MD), was maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS.
RNA isolation and Northern and Western blot analysis.
Total cellular RNA was isolated from cells using TRIzol reagent (Invitrogen). Northern blot hybridization was performed using a nonisotopic digoxigenin-labeled probe as described previously (17). The location of the probes used for ORF73 and K12 are shown in Fig. 1A. For Western blot analysis, nuclear extracts were prepared using the NE-PER nuclear extraction kit (Pierce, Rockford, IL). Nuclear protein (20 μg) was electrophoresed on 4 to 12% NuPAGE gels (Invitrogen). The proteins were transferred to a nitrocellulose membrane and blocked with 5% skim milk prepared in washing buffer (1× TBST [Tris-buffered saline containing 0.05% Tween 20]). The membrane was incubated with antibodies to LANA (Advanced Biotechnologies Inc., Columbia, MD), HIF-1α (BD Biosciences, San Jose, CA), viral interferon regulatory factor (vIRF-3, also called LANA-2), and HIF-2α (both from Novus Biologicals, Littleton, CO) with appropriate dilutions as indicated. After being washed with washing buffer, the membrane was incubated with appropriate anti-rabbit, anti-mouse, or anti-rat secondary antibody conjugated to peroxidase (Sigma), and bands were visualized using chemiluminescent substrate (Pierce). Films from these and other gels were scanned using the Epson Perfection 4990 Pro Scanner with Epson Silverfast Software, version 6.4 (Seiko Epson Corp., Long Beach, CA). Contrast was adjusted minimally and equally on some scans using PowerPoint 2004 version 11.5.1 for the Macintosh (Microsoft Co., Redmond, WA). Band intensities were assessed using ImageJ Software (National Institutes of Health, Bethesda, MD).
Reporter plasmids.
The full-length LANA promoter construct [pGL3-LANA(c&i)p-luc] contains 1,201 nucleotides (nt) spanning positions 127297 to 128497 upstream of the ATG initiation codon of LANA (nt position 127297) (Fig. 1A and B) (31, 33, 37). This region includes the LTc and LTi mRNA start sites (31) and six potential HREs (MacVector, Cary, NC). The promoter region was amplified by PCR with specific primers containing KpnI and Bgl11 sites and cloned into the pGL3 basic vector (Promega, Madison, WI). A LANA promoter region that includes LTc, but not LTi, was cloned into the pGL3 basic vector and denoted pGL3-LANA(c)p-luc. This promoter region, which included HRE 1, 2, 3R, and 4R, contains 795 nt (spanning positions 127703 to 128497). A LANA promoter region that includes LTi [LANA(i)p] but not LTc was cloned into the pGL3 basic vector and denoted as pGL3-LANA(i)p-luc. This promoter, which excludes potential HRE 1, 2, and 3R but contains HRE 4R, 5R, and 6, contains 570 nt (spanning positions 127297 to 127866) upstream of the ATG initiation codon of LANA. Additional constructs were made through sequential deletion from the 5′ end of LANA(i)p that excludes HRE4R [pGL3-LANA(i)p(D1)] (nt position 127297 to 127758) and HRE5 [pGL3-LANA(i)p-luc(D2)] (nt position 127297 to 127646) to identify the potential HREs in this promoter. Using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), HRE4R was mutated from CTGCAC to GATTAC, and the mutated construct was designated LANA(i)p-luc (HRE4Rm).
Expression plasmids.
Expression plasmids encoding degradation-resistant forms of HIF-1α (pcDNA-HIF-1αm) (P402A, P564A), as well as HIF-2α (pcDNA-HIF-2αm) (P405A, P531A), which replace two prolines with alanines in the oxygen-dependent degradation domain of HIF-1α and HIF-2α, have been described previously (17, 49). An expression plasmid encoding KSHV RTA (pcDNA-RTA) (a kind gift from Keiji Ueda, Osaka University Graduate School of Medicine, Osaka, Japan) and pcDNA3.1 empty vector were used in cotransfection experiments.
Transfection and reporter assays.
Reporter experiments were performed in Hep3B cells using Genjet transfection reagent (SignaGen Laboratory, Gaithersburg, MD) according to the supplier's protocol. The day before transfection, 0.5× 106 cells per well were seeded in a 12-well tissue culture plate. On the following day, cells were transfected with LANA promoter luciferase constructs (700 ng) or cotransfected (300 ng) with expression plasmids (400 ng) and cultured at 37°C in normoxic conditions for 24 h or 48 h. At 24 h posttransfection, cells were exposed to hypoxia for 16 h or treated with CoCl2 (Sigma) (100 μM), an oxygen mimic, for 16 h in normoxia. At the end of this incubation period, cells were lysed with 1× reporter lysis buffer (Promega) (250 μl/well), frozen, and thawed. The cell lysate was centrifuged at 13,000 × g for 15 min, and supernatant protein extract was collected. Protein extract (20 μl) was used for the luciferase assay (Promega). The concentration of total protein was determined using Bradford reagent (Bio-Rad, Hercules, CA). In initial experiments, cells were also cotransfected with an internal control plasmid producing Renilla luciferase to normalize transfection efficiency. However, we found that the activity of Renilla was generally increased when cells were cotransfected with HIF-2α, and therefore, subsequent experiments were done normalizing mean light units to total cellular protein. The results normalizing to cellular protein paralleled those with the Renilla control, except to a variable degree with HIF-2α transfection, and in key experiments, we also assessed transfection by measuring the cellular levels of HIF-1α and HIF-2α protein. The fold activation of promoter activity was determined based on the ratio between the normalized mean light units and that of the pGL3-basic construct or pcDNA control.
HIF-1α binding and competition assay.
HIF-1α binding and competition assays were performed using a TransAM HIF-1 kit (Active Motif, Carlsbad, CA) as described previously (16). This kit measures the binding to a 26-bp HRE oligonucleotide from the erythropoietin (EPO) gene promoter attached to a 96-well plate. Nuclear extracts were prepared from Hep3B cells by using the NE-PER nuclear extraction kit (Pierce) from untreated cells or cells treated with 100 μM CoCl2 for 16 h. Five micrograms of nuclear extract was used per well for binding and competition assays. The following 30-bp wild-type (WT) and mutant (Mut) synthetic double-stranded oligonucleotide probes from the LANA(i)p HRE4R region were used for HIF-1α binding and competition experiments; only the sense strand is shown: WT HRE4R, 5′-ACGAAGCAGTCACGTCCCCAA GAGCAGCAG-3′; Mut HRE4R, 5′-ACGAAGCAGTCATTTCCCCAAGAGCAGCAG-3′ (the core HRE sequence is shown in bold and the mutated region is underlined). Epo WT and mutant oligonucleotides were provided with the kit; the manufacturer suggests that competition can be obtained with 20 pmol WT oligonucleotide, and this amount is denoted as 1×. To demonstrate the specificity of binding, competition experiments were carried out in which 20 pmol (designated 1×) to 200 pmol (10×) of either wild-type or mutated oligonucleotide of the EPO HRE or the LANA(i)p HRE was first added to the appropriate well and incubated for 10 min. Nuclear extracts were then added and incubated for 1 h. After three washes with wash buffer, HIF-1α binding was determined by incubation for 1 h each with anti-HIF-1α antibody and then with horseradish peroxidase-conjugated secondary antibody. After incubating with substrate for 10 min, the reaction was stopped using the stop solution, and the absorbance was read at 450 nm.
Electrophoretic mobility shift assay.
Hep3B cells were cultured in 10-cm dishes in normoxia, in hypoxia for 16 h, or after transfection with 5 μg degradation-resistant HIF-1α or HIF-2α for 48 h. Cells were lysed, and the nuclear extracts were prepared using the NE-PER nuclear extraction kit (Pierce). Electrophoretic mobility shift assays were performed using the digoxigenin (DIG) gel shift kit (Roche Applied Science, Indianapolis, IN). Synthetic oligonucleotides were annealed with their complementary strand and labeled with a DIG tag at their 3′ end using terminal transferase provided with the kit. Labeled DNA probes were incubated with 5 μg of nuclear extract for 15 min at room temperature. For competition assays, labeled probes were incubated with increasing amounts of cold unlabeled wild-type or mutant probes (100×, 150×) followed by nuclear extract. Protein-DNA complexes were separated on a 6% DNA retardation gel (Invitrogen) and transferred onto nylon membrane. After UV cross-linking, the membrane was blocked with 1× blocking buffer and then probed with antidigoxigenin antibody conjugated to alkaline phosphatase (1:10,000 dilution) for 30 min at room temperature. Following two washes in washing buffer, the membrane was developed using the chemiluminescent detection method.
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed using chromatin immunoprecipitation kits (Upstate, Hayward, CA) with some modifications as indicated below. BC-3 cells were treated with CoCl2 (100 μM) for 16 h at 37°C and then cross-linked with 37% formaldehyde at a final concentration of 1% at room temperature for 10 min. The unreacted formaldehyde was quenched using 2 M glycine for 5 min at room temperature before harvest. Cells were lysed, and nuclear extracts were prepared using the NE-PER nuclear extraction kit (Pierce). Nuclear extracts were sonicated using Misonix XL-3000 (Osonica, Newtown, CT) (20-s pulses for 6 min at 4°C) to obtain DNA with an average length of 200 to 1,000 bp. Sonicated lysate was centrifuged at 13,000 × g for 10 min at 4°C. Supernatant containing DNA-protein complexes were collected, and protein concentration was determined using the bicinchoninic acid reagent (Pierce). Equal amounts of DNA-protein complexes were distributed for each immunoprecipitation. The DNA-protein complexes in a 55-μl volume (300 μg) were diluted to 550 μl using ChIP dilution buffer (Upstate), and 20 μl (approximately 10 μg) was set aside for use as an input control. The remaining DNA-protein complexes were immunoprecipitated with 8 μg of mouse monoclonal antibody to HIF-1α or rabbit polyclonal antibody to HIF-2α (Novus Biologicals) and respective control rabbit (Rb) or mouse (Ms) IgG (Santa Cruz Biotechnology, Santa Cruz, CA) together with protein A magnetic beads overnight at 4°C on a rotating platform. Immune complexes were washed sequentially with high-salt immune complex buffer, low-salt immune complex buffer, lithium chloride immune complex buffer, and Tris-EDTA (TE) buffer, provided with the kit (Upstate), each for 4 min at room temperature on a rotating platform. The immune complexes and input samples were each mixed with elution buffer provided with the kit, and the DNA-protein cross-linking was reversed by heating at 62°C for 2 h with constant shaking. The DNA was purified using DNA purification columns (Zymo Research Corp, Irvine, CA) and analyzed by PCR using primers flanking the LANA-inducible promoter region and actin as a control. PCR products were electrophoresed with a 2% agarose gel stained with ethidium bromide.
Immunoprecipitation and Western blotting.
BCBL-1 cells were exposed to hypoxia for 48 h at 37°C. At the end of incubation period, cells were lysed and nuclear extracts were prepared using the NE-PER nuclear extraction kit (Pierce). Immunoprecipitation was performed using Dynabeads protein A or protein G (Invitrogen) with modifications. In other experiments, cells were cotransfected with degradation-resistant HIF-1α or HIF-2α and RTA expression plasmids. At 48 h posttransfection, cells were lysed and nuclear extracts were prepared in the same manner. Nuclear extracts were immunoprecipitated using anti-HIF-1α monoclonal antibody (4.0 μg) (Novus Biologicals) bound to protein G magnetic beads (Invitrogen) overnight at 4°C with constant rotation. Beads were washed for 10 min three times with washing buffer (Invitrogen) and resuspended in 20 μl of elution buffer (Invitrogen). The sample was subjected to SDS-PAGE (4 to 12% NuPAGE Tris-Bis). Western blot analyses were performed using anti-RTA rabbit serum (custom synthesized from Alpha Diagnostic International Inc., San Antonio, TX). The blot was developed using chemiluminescent substrate (Pierce).
RESULTS
Hypoxia increases LANA promoter activity.
The environs of the KSHV genome between ORF73 and K14 regulate the expression of the ORF71-ORF73 gene cluster in one direction and K14/ORF74 in the opposite direction (44) (Fig. 1A). Previous studies of this region have identified two mRNA start sites for LANA, a constitutive site (LTc) at nucleotide 127880, and an inducible site (LTi) at nucleotide 127611 or 127610 (10, 31). There is some variability in the LTc start site, and it has also been variously mapped to nucleotide positions 127900 and 127948 (10, 39, 45). Previous studies have shown that KSHV lytic replication, as well as certain lytic genes, are activated by hypoxia through HIFs, and we hypothesized that the expression of the LANA gene cluster may also be affected by hypoxia. To explore this hypothesis, we analyzed the region between the start codon of LANA (127297) and the terminus of v-OX2 (128930) using MacVector bioinformatics software (MacVector Inc., Cary, NC). Six potential HREs were identified, denoted here as 1, 2, 3R, 4R, 5R, and 6, where R denotes HREs in the reverse orientation to the transcription of LANA (Fig. 1A). Three of these potential HREs were located between the LANA coding start site and LTc, while the three remaining potential HREs were located upstream of LTc within the K14 coding region.
We next assessed whether the LANA promoter region was responsive to the effects of hypoxia or HIF. The LANA promoter region (127297 to 128497), including both LTi and LTc, was cloned into a luciferase reporter construct to make pGL3-LANA(c&i)p-luc (Fig. 1B), and the promoter activity was examined in Hep3B cells. We found that the basal activity of this construct was 11-fold greater than the pGL3 basic construct (control) when cultured under normoxic conditions (Fig. 1C). Moreover, the activity of the pGL3-LANA(c&i)p-luc construct was increased approximately 2-fold when it was exposed to hypoxia compared to normoxia, while the pGL3 basic promoter was not affected by hypoxia. Also, the activity of pGL3-LANA(c&i)p-luc was increased when it was exposed to CoCl2 (100 μM for 16 h), a hypoxia mimic (Fig. 1C), whereas the activity of the pGL3 basic promoter was not. These results provided evidence that the pGL3-LANA(c&i)p-luc construct had constitutive activity that was increased by exposure to hypoxia or CoCl2, a hypoxia mimic.
Hypoxia increases the production of LANA RNA and protein levels in PEL cell lines.
We next sought to determine the effects of hypoxia on LANA protein and LANA RNA in KSHV-infected PEL cells and also to determine which species of LANA RNA may be affected. Previous studies have shown that a latent polycistronic RNA transcript of approximately 5.8 kb is produced from LTc that encodes the ORF 73, 72, and 71 gene cluster (Fig. 2A) (10, 31, 33, 38, 44, 45). In addition, through alternative splicing, messages of approximately 5.4 kb and 1.7 kb are produced from the LTc. The longer (5.4-kb) transcript encodes ORFs 73, 72, and 71, while the shorter (1.7-kb) transcript encodes ORFs 72 and 71. It was subsequently found that a transcript of 5.5 kb that also encodes ORFs 73, 72, and 71 is produced from the LTi in response to RTA (Fig. 2A) (31).
Fig 2.

Hypoxia increases the production of LANA RNA and LANA protein levels. (A) Diagram of the mRNA transcripts produced from LTc and LTi as previously reported (10, 31, 33, 38, 44, 45). An unspliced transcript of 5.8 kb encoding ORFs 71 to 73 is produced from the LTc start site. Alternative splicing (as shown by dotted lines) generates mRNA transcripts of 5.4 kb and 1.7 kb for ORFs 71 to 73 and ORFs 71 to 72, respectively. A 5.5-kb transcript produced from the LTi start site also encodes ORFs 71 to 73 (31). (B and C) Northern blot analysis showing increased ORF73 (LANA) mRNA in BC-3 and JSC-1 cells exposed to hypoxia (1% O2). Cells were cultured in normoxia (N) or hypoxia (H) or in the presence of 20 ng/ml TPA (T). After 22 h, RNA was extracted and 5 μg of RNA was probed using probes specific for ORF73 (B) or K12 (C). LANA mRNAs are upregulated in hypoxia compared to normoxia but not by TPA treatment. K12 is upregulated substantially by exposure to TPA but only minimally or not at all by hypoxia. (D) Western blot showing protein levels of LANA in BC-3 and JSC-1 cells cultured in 1% O2 (H) for up to 72 h in hypoxia compared to those cultured in normoxia (N). After culture in hypoxia or normoxia, cells were lysed and 20 μg of protein was run on two gels. One gel (upper) was probed with antibody specific for LANA, HIF-1α, and actin, and the other (lower) was probed with antibody to HIF-2α and actin. The gels were stripped between successive probes. HIF-1α and HIF-2α levels were stabilized up to 48 h in hypoxia and then decreased in both BC-3 and JSC-1 cells. ImageJ analysis, shown in the right panel, showed LANA levels increased approximately 2- to 3-fold in BC-3 and 2.3-fold in JSC-1 cells. (E) vIRF3 levels in BC-3 and JSC-1 cells exposed to hypoxia for 24 h and through 72 h. Its levels remained essentially unchanged in hypoxia.
In the current study, BC-3 and JSC-1 cells were cultured in normoxia, in hypoxia (1% O2), or in normoxia with 20 ng/ml TPA. After 22 h, RNA was extracted and 5 μg of RNA was used for Northern blot analysis using digoxigenin-labeled probes specific for ORF73 and K12 as described previously (17). When probed for ORF73, mRNA bands at approximately 5.4 to 5.5 kb and approximately 5.8 kb were present in normoxia. In hypoxia, these bands increased by approximately 2-fold in JSC-1 cells and almost 1.5-fold in BC-3 cells, both normalized to actin (Fig. 2B). In contrast, exposure of the cells to TPA appeared to decrease the levels of these RNA species. It was relatively difficult to differentiate among the 5.4-kb, the 5.5-kb, and the 5.8-kb species because of their proximity on the gels. However, it appeared that both an upper band, corresponding to the 5.8-kb species initiating at the LTc start site, and a lower band that could represent the 5.4-kb and/or the 5.5-kb RNA species, were increased by exposure to hypoxia. Using a probe specific to K12, a latent gene that is reported to increase in response to lytic reactivation (26), a band of an expected 2-kb size was seen in BC-3 cells (but not in JSC-1 cells). This band was strongly upregulated in both cell lines after TPA treatment. However, there was little or no change in the intensity of this band when the BC-3 or JSC-1 cells were exposed to hypoxia (Fig. 2C). These results suggested that hypoxia enhanced production of LANA mRNA from the LTc start site and may also have enhanced production of the 5.5-kb message from the LTi site.
We next assessed whether hypoxia affected the protein levels of LANA in PEL cell lines. BC-3 and JSC-1 cells were cultured in normoxia or hypoxia. After 24 h, 48 h, and 72 h of incubation in hypoxia, cells were lysed, and 20 μg of nuclear lysate was probed using antibody specific to LANA, HIF-1α, HIF-2α, and actin. One blot was first probed for HIF-1α and subsequently stripped and probed for LANA and then actin; a second blot was first probed for HIF-2α and then for actin. As expected, levels of both HIF-1α and HIF-2α were upregulated through 48 h of hypoxia compared to those of normoxia (Fig. 2D) in both PEL cell lines. However, at 72 h of hypoxia, HIF levels were decreased in both cell lines. These changes in HIF levels are consistent with previous studies showing that HIF levels decreased after an initial increase in several cell lines tested (15, 18, 53). In BC-3 cells, LANA levels increased approximately 1.9-fold after 24 h of hypoxia and then increased further up to approximately 3-fold after 72 h of hypoxia. In JSC-1 cells, LANA levels were increased approximately 1.7-fold after 24 h of hypoxia and 2.3-fold after 48 h of hypoxia. However, at 72 h, levels of high-molecular-weight LANA decreased somewhat, while levels of lower-molecular-weight LANA isoforms (23) were elevated. Overall, these data suggested that total protein levels of LANA were increased with hypoxia from 24 h through at least 72 h in both PEL cell lines (Fig. 2D). In contrast, when a parallel blot made with another aliquot of the same cell lysate was probed using antibodies specific to the KSHV latent protein vIRF-3, which utilizes a different promoter, the protein levels remain unchanged in hypoxia (Fig. 2E). This provided evidence that hypoxia upregulated levels of LANA but not another latent gene that utilizes a different promoter. These results were consistent with the hypothesis that changes in HIFs played a key role in mediating effects of hypoxia on LANA.
The LANA promoter is induced by both HIF-1α and HIF-2α.
To evaluate the roles of HIF-1α and HIF-2α in mediating the response of LANA promoter to hypoxia, pGL3-LANA(c&i)p-luc, pGL3-LANA(c)p, or pGL3-LANA(i)p-luc was cotransfected into Hep3B cells along with the degradation-resistant mutant HIF-1α (HIF-1αm) or HIF-2α (HIF-2αm) expression plasmids or with a pcDNA control. Unlike wild-type HIFs, these mutants are not degraded under normoxic conditions. As shown in Fig. 3, top, activity of the LANA(c&i) promoter as well as the two segments was substantially induced by either HIF-1αm or HIF-2αm, with the LANA(i)p segment responding somewhat more than the LANA(c)p segment. Also, each of the constructs responded somewhat more to cells expressing HIF-2αm than HIF-1αm. In parallel experiments, vascular endothelial growth factor (VEGF) promoter activity was assessed following cotransfection with HIF-1αm or HIF-2αm. Consistent with previous reports (50, 53), VEGF promoter activity was upregulated both by HIF-1α and HIF-2α, with HIF-2α having a substantially greater effect (Fig. 3, bottom left).
Fig 3.

The LANA promoter is activated by HIF-2α and HIF-1α. The upper panel shows activation of the LANA promoter containing both LTc and LTi mRNA start sites [LANA(c&i)p], only the LTc mRNA start site [LANA(c)p], or only the LTi mRNA start site [LANA(i)p] by degradation-resistant HIF-1α and HIF-2α (HIF-1αm or HIF-2αm). Hep3B cells were cotransfected with pGL3-LANA(c&i)p-luc, pGL3-LANA (c)p-luc, or pGL3-LANA(i)p-luc (300 ng) and plasmids encoding degradation-resistant mutants of HIF-1α or HIF-2α or a pcDNA control (400 ng). At 48 h of posttransfection, cells were lysed and promoter activity was determined as described in the legend to Fig. 1. Fold activation for all three promoters was calculated by normalizing to the pcDNA (400 ng) control for LANA(i)p, and results are shown as normalized to the respective pcDNA control. In the lower left panel, VEGF promoter activity was assessed in a similar manner. All values represent means of triplicate determinations in one representative experiment out of two. Error bars represent the standard deviations. In the lower right panel are shown Western blots of the cells transfected with pGL3-LANA(c&i)p-luc and plasmids encoding degradation-resistant mutants of HIF-1α or HIF-2α or a pcDNA control (400 ng). The top half of each blot was probed with either anti-HIF-1α or anti-HIF2α, while the lower half was probed with anti-β actin as described in Materials and Methods.
HRE4R contributes to the activation of the LTi LANA promoter by HIFs.
Previous studies have shown that LANA(i)p is activated by RTA (31), which is expressed in KSHV-infected cells exposed to hypoxia (2). Additionally, as shown in Fig. 3, activation of LANA(i)p by HIFs was greater than that of LANA(c)p, and in subsequent experiments we focused on the response of this inducible region to HIF, as well as RTA. This DNA segment contains a 570-bp region upstream from the LANA start codon; this region includes LTi and three potential HREs (6, 5R, and 4R) but excludes LTc (Fig. 1A and B). We first investigated the LTi promoter region for the contribution of the three potential HREs on the activation by HIFs. Promoter reporter constructs were made with deleted 4R [pGL3-LANA(i)p(D1)] or deleted 4R and 5R [pGL3-LANA(i)p(D2)] from the 5′ end of the LTi promoter region (Fig. 4A). Each promoter reporter construct was cotransfected with HIF-1αm or HIF-2αm expression plasmids. As shown in Fig. 4B, the activation of the promoter with all the three HREs was significantly higher in the HIF-transfected cells than in the pcDNA control, and the promoter activity was 2-fold greater in the presence of HIF-2αm than in the presence of HIF-1αm, consistent with previous experiments. Deletion of HRE4R resulted in a 3-fold decrease in HIF-1αm (from 4.3 to 1.4) and a 1.6-fold decrease in HIF-2αm (from 9.2- to 5.6-fold)-induced promoter activation compared to the 570-bp LTi promoter that contained all the HREs. However, deletion of HRE5R to form LANA(i)p(D2) did not result in a substantial further decrease. The above-described results suggest that HRE4R was a functional HRE in the RTA-inducible LANA promoter. Also, the residual activity remaining in LANA(i)p(D2) suggested that HRE6 may also contribute to the HIF-2α-induced activation.
Fig 4.

HRE4R can mediate much of the activation of the LTi LANA promoter by HIFs. (A) Schematic diagram of the LTi promoter luciferase reporter constructs with progressive deletions of HRE4R [pGL3-LANA(i)p-luc(D1)] and 5R [pGL3-LANA(i)p-luc(D2)]. The wild-type LTi promoter luciferase reporter construct is shown in Fig. 1B. (B) Deletion of HRE4R but not HRE5R substantially decreases LTi LANA promoter induced by HIF-1α and HIF-2α. Hep3B cells were cotransfected with 300 ng each of wild-type pGL3-LANA(i)p-luc, pGL3-LANA(i)p-luc(D1), and pGL3-LANA(i)p-luc(D2) constructs, respectively, and degradation-resistant mutants of HIF-1α or HIF-2α or pcDNA control plasmids (400 ng). At 48 h of posttransfection, cells were lysed and promoter activity was assessed as described in the legend to Fig. 1. Fold activation was calculated by normalizing to the pcDNA (400 ng) control results of LANA(i)p. All values represent means of triplicate determinations of a representative experiment out of two. Error bars represent the standard deviations. (C) Schematic diagram of the pGL3-LANA(i)p-luc(HRE4Rm) construct. This construct was created by mutating the HRE4R sequence in the 569-bp pGL3-LANA(i)p-luc construct by site-directed mutagenesis. The sequence shown is for the DNA strand encoding LANA; the HRE is in the reverse orientation with sequence GACGTG. Mutated nucleotides are represented in bold. (D) Mutation of HRE4R attenuates LTi LANA promoter activity induced by HIF-2α and HIF-1α. Hep3B cells were cotransfected with 300 ng each of wild-type pGL3-LANA(i)p-luc or the pGL3-LANA(i)p-luc(HRE4Rm) constructs and degradation-resistant mutants of HIF-1α or HIF-2α or pcDNA control plasmids (400 ng). At 48 h of posttransfection, cells were lysed and promoter activity was assessed as described in the legend to Fig. 1. Fold activation was calculated by normalizing to LANA(i)p with pcDNA (400 ng) control. All values represent means of triplicate determinations of a representative experiment out of two. Error bars represent the standard deviations.
To further assess whether HRE4R could act as a functional HRE in the LTi LANA promoter, we introduced a 2-bp mutation in the putative binding sequence in the HRE4R by site-directed mutagenesis (Fig. 4C). The reporter constructs having a wild-type LTi LANA promoter region [pGL3-LANA(i)p-luc] or mutated HRE4R [pGL3-LANA(i)p-luc (HRE4Rm)] were cotransfected with HIF-1αm or HIF-2αm expression plasmids or with a pcDNA control plasmid into Hep3B cells. This mutation decreased the response of the pGL3-LANA(i)p-luc promoter to HIF-2αm by almost half, from about 8-fold activation to about 4.2-fold activation (Fig. 4D). Also, this mutation decreased the response of the promoter to HIF-1αm cotransfection by about 44%, from approximately 3.6-fold activation to 2-fold activation. These data confirmed that HRE4R was a substantial contributor to activation of the LANA LTi promoter region by HIFs.
HIFs bind to the inducible LANA promoter.
To explore whether HIFs bind to the HRE4R in the LTi LANA promoter region, electrophoretic mobility shift assays were performed using nuclear extracts from Hep3B cells cultured in normoxia or hypoxia or transfected with HIF-1αm or HIF-2αm in normoxia. The DIG-labeled oligonucleotide probe containing HRE4R formed a complex with nuclear extracts from hypoxic, HIF-1αm-transfected, and HIF-2αm-transfected Hep3B cells, compared to a minimal amount of binding with extracts from normoxic Hep3B cells (Fig. 5A). HRE4R complex formation with HIF-1αm and with HIF-2αm was inhibited by competition with unlabeled wild-type HRE4R oligonucleotide at 150 times the concentration of labeled probe but not by identical concentrations of mutant HRE4R oligonucleotides, suggesting that binding was specific to the HRE4R sequence (Fig. 5A). For reasons that are not clear, slightly greater inhibition was seen with a 100-fold excess of labeled probe. These data demonstrated that both HIF-1αm and HIF-2αm can bind to HRE4R of the LTi LANA promoter.
Fig 5.

HIFs bind to the LANA promoter. (A) HIFs bind to the HRE4R in vitro as detected by electrophoretic mobility shift assay. DIG-labeled synthetic oligonucleotide containing wild-type (WT) HRE4R was incubated with nuclear extracts (NE) from Hep3B cells cultured in 10-cm dishes in normoxia (N) or in hypoxia (H) for 16 h or transfected with 5 μg degradation-resistant mutants of HIF-1α or HIF-2α for 48 h. The mixture was then analyzed on a 6% DNA retardation gel. Where indicated, unlabeled wild-type (WT) or mutant (Mut) oligonucleotides (at concentrations 100× and 150× that of the labeled probe) were added to the reaction to assess binding competition (Comp.). Protein-DNA complexes were separated, blotted onto a nylon membrane, and probed with antidigoxigenin antibody conjugated to alkaline phosphatase. The sequence of the labeled WT probe and the WT and Mut competing oligonucleotides are shown at the bottom. The WT HRE4R sequence is underlined, and nucleotide changes in the Mut sequences are shown in bold. The positions of the DIG-labeled HIF complexes and free probe are indicated with arrows. (B) Diagrammatic representation of the LANA promoter region showing the location of forward and reverse primers used in the chromatin immunoprecipitation (ChIP) assay in the region surrounding LTi. Amplification of DNA flanking the primers produces an expected band of 450 bp. Sequences of forward (F) and reverse (R) primers are shown in the lower panel. (C) HIFs bind to HRE in the region of the LANA promoter surrounding LTi in vivo as detected by ChIP assay. Duplicate samples of 107 BC-3 cells were treated with CoCl2 (100 μM) for 16 h at 37°C. Cells were cross-linked with 37% formaldehyde (1% final concentration), and unreacted formaldehyde was quenched using 2 M glycine. The nuclear extracts were sonicated (Misonix 3000; 20 s pulses for 6 min at 4°C) to obtain DNA with an average length of 200 to 1,000 bp. Sonicated lysate was centrifuged at 13,000 × g for 10 min at 4°C. Supernatant containing DNA-protein complexes was collected, and protein estimation was done by the BCA method. DNA-protein complexes in a 55-μl volume (300 μg) were diluted to 10× using ChIP dilution buffer (Upstate, California), and 20 μl (approximately 10 μg) was set aside for use as an input. The remaining DNA-protein complexes were immunoprecipitated with 8 μg of monoclonal mouse antibody to HIF-1α (lanes 3 and 4) or polyclonal rabbit antibody to HIF-2α (lanes 5 and 6) and respective control rabbit (Rb) antibody (lane 7) or mouse (Ms) (lane 8) IgG. After de-cross-linking, the DNA was purified and amplified using primers flanking the LTi LANA promoter region as diagrammed in Fig. 5B. The more-intense 450-bp bands of HIF-1α and HIF-2α immunoprecipitated samples compared to that of their IgG controls suggest the binding of HIFs to the LTi LANA promoter region. (D) HIF-1α binding to the HRE4R as detected by in vitro binding and competition assay. Nuclear extracts were prepared from Hep3B cells cultured in normoxia or treated with CoCl2 (100 μM) for 18 h at 37°C. A total of 5 μg of nuclear extract was incubated with 20 pmol (1×), 100 pmol (5×), or 200 pmol (10×) of a wild-type (WT) 26-bp probe from the EPO gene promoter encompassing an HRE or a mutant (Mut) probe with a mutated HRE sequence. At the same time, parallel extracts were incubated with equivalent amounts of a 30-bp LANA(i)p HRE 4R synthetic oligonucleotide probe from the LANA promoter region encompassing wild-type (WT) HRE4R or a similar probe with a mutated HRE4 (Mut). The binding of HIF-1α in the Hep3B nuclear extracts to EPO HRE bound to a well in the microtiter plate was then assessed as described in Materials and Methods; results shown are the absorbance at 450 nm. All values represent means of duplicate determinations in a representative experiment out of two.
To further assess whether HIFs might activate LANA by binding to the LTi LANA promoter region, we performed ChIP assays using BC-3 cells treated with 100 μM CoCl2 for 16 h in normoxia. Primers were designed that covered 450 bp in the inducible LTi promoter region as shown in Fig. 5B. Formaldehyde-cross-linked nuclear lysate from BC-3 cells was sonicated and immunoprecipitated with mouse antibody specific to HIF-1α or rabbit antibody specific to HIF-2α (Fig. 5C). The complexes were then de-cross-linked and used as a template for PCR using the above-described primers. As expected, the PCR product of 450 bp was observed as having greater intensity with HIF-1α or HIF-2α immunoprecipitated samples than their respective IgG controls. Bands observed with antibody to HIF-2α were darker; however, this could have been due to the greater affinity of this antibody compared to that of the antibody to HIF-1α. Beta-actin amplification using the DNA template from respective input samples served as an internal control for the equal amounts of DNA-protein complex used for immunoprecipitation (data not shown) (Fig. 5C). These results confirmed that HIFs bind to the LANA-inducible promoter in vivo in PEL cells.
To assess HIF-1α binding to HRE4R using a different technique, we utilized an HIF-1α-specific enzyme-linked immunosorbent assay (ELISA)-based competition assay using nuclear lysate from CoCl2-treated Hep3B cells (Fig. 5D). For this assay, both wild-type and mutant probes specific for HRE4R were designed, and their abilities to inhibit binding of HIF-1α to an HRE in the enhancer region of the erythropoietin (EPO) gene were used. As seen in Fig. 5D, nuclear extracts from CoCl2-treated Hep3B cells efficiently bound to the EPO HRE bound to the plates, and this binding was inhibited by wild-type EPO HRE but not by mutant EPO HRE. In a similar manner, 1× LANA WT HRE4R inhibited binding of HIF-1α to the EPO HRE, while up to 10× Mut HRE 4R had a substantially lesser effect. This provided further evidence that HIF-1α could bind to LANA HRE 4R.
Hypoxia or CoCl2 treatment enhances the RTA-mediated induction of the LANA promoter.
Previous studies have found that the LANA-inducible promoter has RTA response elements (Fig. 6A) and that the promoter was responsive to RTA (31). Given our results described above that the LANA promoter was also responsive to hypoxia, we wondered if hypoxia (or HIFs) might cooperate with RTA in inducing the LANA(i)p. Hep3B cells were cotransfected with an RTA expression plasmid or with a pcDNA control together with the LTi LANA promoter reporter. At 24 h of posttransfection, cells were exposed to hypoxia (1% O2) for 16 h at 37°C. We observed that under normoxic conditions, RTA transfection induced an approximately 75-fold activation of the LTi promoter region compared to that of the control. However, after treatment with CoCl2 (100 μM) for 16 h in normoxia, the activation of LTi promoter was increased to approximately 118-fold over that of the control in normoxia, an increase of approximately 1.6-fold over the RTA transfected cells in normoxia. Similarly, the activation of LTi promoter by RTA was increased to approximately 123-fold in hypoxia, an increase of approximately 1.6-fold compared to that of RTA transfected cells in normoxia (Fig. 6 B).
Fig 6.

Hypoxia or CoCl2 treatment enhances the RTA-mediated inducible LANA promoter activity (A) Schematic representation of the RTA-inducible LANA promoter region showing the presence of RTA response elements (RRE) in relation to the putative HRE sequences. RRE are shown as rectangular boxes filled with black color. (B) Hypoxia or CoCl2 enhance RTA-inducible LANA promoter activity. Hep3B cells were transfected with 400 ng empty vector or an RTA expression plasmid along with a fixed amount (300 ng) of pGL3-LANA(i)p-luc. At 24 h posttransfection, cells were exposed to hypoxia or treated with CoCl2 (100 μM) for 16 h. At the end of incubation period, cells were lysed and promoter activity was determined as described in the legend to Fig. 1. All results are normalized to the empty vector control in normoxia. CoCl2 or hypoxia alone induced the inducible LANA promoter by approximately 1.6-fold and 2-fold, respectively. All values represent means of triplicate determinations of a representative experiment out of two. Error bars represent the standard deviation. (C) HIFs cooperate with RTA to increase the LTi LANA promoter activity. Hep3B cells were cotransfected with 300 ng of pGL3-LANA(i)p-luc along with 200 ng of degradation-resistant HIF-1α or HIF-2α and/or 200 ng RTA expression plasmid as shown. The total amount of DNA was normalized where appropriate using the pcDNA control (200 or 400 ng). At 48 h posttransfection in normoxia, cells were lysed and promoter assay was assessed as described in the legend to Fig. 1. Fold activation was calculated by normalization to the pcDNA control. All values represent means of triplicate determinations. Error bars represent the standard deviations.
To determine whether HIFs could be responsible for the cooperative activation by hypoxia, Hep3B cells were cotransfected with RTA and HIF-1αm or HIF-2αm or pcDNA plasmids. At 48 h posttransfection, cells were lysed and promoter assays were performed. As expected, LTi LANA promoter was induced by HIF-1α (10.7-fold), HIF-2α (26.5-fold), and RTA (95.3-fold) individually compared to the pcDNA control (Fig. 6C). Moreover, activation of the LTi promoter was increased to 156-fold in HIF-1α and RTA cotransfected cells and to 190-fold in RTA and HIF-2α cotransfected cells. To exclude the possibility that RTA protein levels could have been stabilized following cotransfection with HIF and by this mechanism contributing to the activation of the LTi promoter, we examined RTA protein levels in cells cotransfected with RTA and HIF-1αm. We observed that while HIF-1α levels were substantially increased after transfection, RTA protein levels remained unchanged (data not shown). Taken together, these results suggest the possible cooperation between RTA and HIFs in activating the LTi LANA promoter and suggest that a stronger effect is observed with HIF-2α.
RTA interacts with HIF-1α and HIF-2α in vivo in BCBL-1 cells.
Because HIF and RTA appeared to cooperate in the activation of the LTi LANA promoter region, we hypothesized that the two may physically interact with each other. In initial experiments, we observed coimmunoprecipitation of RTA and either HIF-1α or HIF-2α in Hep3B or 293T cells cotransfected with degradation-resistant HIF-1α or HIF-2α and with RTA expression plasmids (results not shown). To study this in KSHV-infected cells, BCBL-1 cells were exposed to hypoxia for 48 h at 37°C. At the end of incubation period, nuclear extracts were prepared and immunoprecipitated (IP) using anti-HIF-1α monoclonal antibody and subsequently immunoblotted using anti-RTA rabbit serum. As shown in Fig. 7, RTA was immunoprecipitated with HIF-1α, suggesting that during hypoxia, RTA interacts physically with HIF-1α in vivo in BCBL-1 cells. These results suggest that the cooperative effects of hypoxia or HIFs and RTA in activating LANA may result from their physically interacting with each other and binding to adjacent sites on the promoter region.
Fig 7.
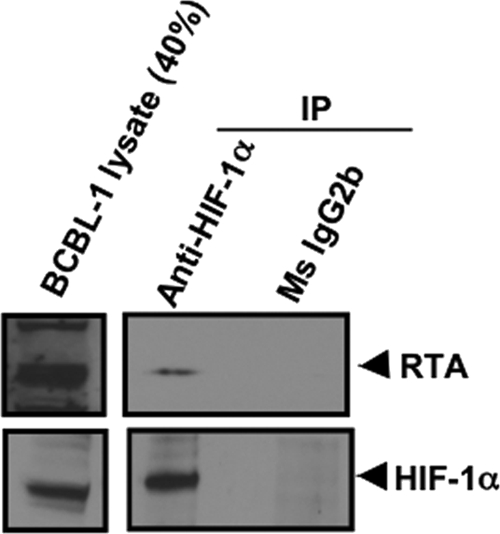
RTA interacts with HIF-1α as detected by immunoprecipitation assay. BCBL-1 cells were exposed to hypoxia for 48 h at 37°C. At the end of the incubation period, cells were lysed and nuclear extracts were prepared. Fifty micrograms of nuclear extracts were immunoprecipitated using anti-HIF-1α mouse monoclonal antibody and respective control mouse monoclonal antibody (Ms). The immunoprecipitated material was then subjected to SDS-PAGE and Western blot analysis performed using anti-RTA rabbit serum. Total lysate (40%) was also probed with anti-RTA rabbit serum as a control. A parallel blot was also probed using anti-HIF-1α mouse monoclonal antibody. Results suggest that RTA associates with HIF-1α in vivo in BCBL-1 cells in hypoxia.
DISCUSSION
Here, we show that hypoxia can increase the levels of LANA and that this can occur through activation of the LANA promoter by HIFs. Moreover, we show that this effect can be mediated by either the LTi or the LTc promoter region, although the effect is more pronounced for the LTi region. This promoter region has several potential HREs, and we show that the activity is mediated at least in part by one of these, HRE 4R, which can be activated by either HIF-1α or HIF-2α. Finally, we show that HIFs can physically interact with and cooperate with RTA in activating the LTi.
Our group has previously shown that hypoxia and HIFs can activate lytic KSHV replication and in addition specifically activate certain lytic genes (e.g., the ORF34 to ORF37 gene cluster) (9, 16, 17). Subsequent studies have shown that HIFs can cooperate with LANA to induce transcription of RTA, the lytic switch protein (2). In this report, we extend these results to show that hypoxia and HIFs also play a major role in activating transcription of the latency transcript cluster encoding ORF71 to ORF73 and can thus promote both the latent and lytic modes of replication. This effect was mediated at least in part by the binding of HIFs to HRE4R in the LTi. Evidence in support of this included the effect of deletion mutations on reporter constructs, the effects of site-directed mutagenesis of the HRE4R, and evidence for binding of HIFs to HRE4R by EMSA, ChIP assay, and an HIF-1α-specific competition ELISA. In cotransfection experiments, both HIF-1α and HIF-2α were found to activate the LTi, and both of these factors were also found to bind to the LTi HRE in the EMSA and the ChIP assay. There was some evidence that HIF-2α had a greater effect in transfection assays and bound better to the HRE in these assays; however, such differences can be affected by the level of HIF in transfected cells and the relative avidity of the antibodies used in the binding assays, making quantitative comparisons difficult. However, the results of these experiments at a minimum do indicate that the LTi can be induced by both HIF-1α and HIF-2α.
The results of this study provide evidence that HIFs can activate both the LTi and the LTc start sites. Analysis of the LANA mRNA species produced in PEL cell lines exposed to hypoxia (Fig. 2B) show a major transcript or transcripts of about 5.4 kb to 5.5 kb, which is consistent with previously described species starting from LTc and LTi (10, 31, 33, 38, 44, 45). In addition, a slightly longer mRNA species of about 5.8 kb is observed, suggesting that hypoxia can also enhance this species of mRNA production from LTc. Consistent with these results, transfection experiments suggest that both the LTi and the LTc start site can be activated by hypoxia in the absence of RTA. In addition to these direct effects on the LANA promoter, we found that hypoxia, HIF-1α, and HIF-2α enhance the activation of the LTi by RTA. Moreover, we found that HIF-1α physically interacted with RTA (Fig. 7), suggesting possible mechanisms for this effect. RRE on the LTi are located between putative HRE 5R and 4R (31, 44), and binding of RTA to HIFs could enhance binding of these factors to their respective response elements. Alternatively, tethering of the complex to HRE could assist in the indirect binding of RRE to the LTi through factors such as RBP-Jκ (25, 44).
Hypoxia can induce RTA production and lytic KSHV activation, primarily through the effects of HIFs interacting with LANA and the RTA promoter (2, 9, 16). The current paper provides evidence that hypoxia and HIFs also activate the mRNAs of the latency transcript cluster, which encodes for LANA, v-cyclin, and vFLIP. Moreover, HIFs cooperate with RTA in the activation of this cluster; this may assist the effective production of KSHV progeny by inhibiting p53 and blocking apoptosis (11, 13). The effects of hypoxia on LANA production may also play a key role in the induction of latency during initial infection. There is evidence that RTA packaged with KSHV virions can activate LANA (and other RTA-responsive genes) during the initial stages of infection and that this can help in the establishment of latent infection (22, 25). The LANA produced during the initial stages of infection then suppresses production of RTA, helping to maintain the latent state (24, 25, 29). If KSHV infects a cell in a hypoxic environment, the effects of HIFs on the LANA promoter may help produce LANA even without RTA and would also serve to boost the production of LANA (and other genes of the latency transcript cluster) in combination with RTA. This may serve to enhance early LANA production under hypoxic conditions through effects such as the prevention of apoptosis and thus help establish latent infection. Also, the effects of LANA and other KSHV genes to increase cellular levels and activity of HIF (3, 5, 14, 42) may provide a positive feedback loop that helps sustain LANA production and latent KSHV infection. The effects of LANA and HIFs in inducing RTA in this environment (2, 16) may in turn help a percentage of the cells establish lytic infection and infect other cells.
How might this process help KSHV maintain itself in its human host population? One possibility is that it may help establish infection after exposure. KSHV is shed in saliva (6, 21), and it is possible that a principal portal of entry is through small cuts in the skin or oropharynx. The edges of wounds are hypoxic, and the effects of hypoxia on LANA production described here may assist in the establishment of initial latent infection. Alternatively, it may help spread KSHV and establish latent infection in areas of hypoxia and endothelial cell growth in an infected individual.
KSHV appears to be unique among the viruses studied to date in its extensive interface with hypoxia and the HIFs in particular. Hypoxia and HIFs can activate RTA and lytic infection and in addition can directly activate certain lytic KSHV genes (2, 9, 16, 17). Moreover, KSHV LANA and ORF74 can enhance the levels and activity of HIFs in infected cells (3, 5, 42), and HIF-1 escapes suppression of cellular genes by KSHV shutoff exonuclease (OR37) (14). The current paper extends our understanding of this relationship to show that hypoxia and HIFs activate LANA and the latency transcript cluster and play a significant role in regulating both latent and lytic life cycles of KSHV (Fig. 8). Additional studies may help shed light on how these molecular interactions affect the interactions of KSHV with its human host.
Fig 8.

Schematic model showing some of the key interrelationships between HIF and LANA. A plus sign indicates upregulation, with greater activation indicated by a larger size. LANA mRNA is constitutively produced and also induced by RTA (19, 25, 31, 44). HIF is normally degraded under conditions of normoxia, but when cells are exposed to hypoxia or certain other conditions, this process is inhibited and levels of HIF increase. As shown in the present study, HIF can activate production of LANA and also enhances LANA production induced by RTA. Also, as previously shown, KSHV latent infection and LANA act to increase the levels of HIF (3, 5, 14, 31). This provides a positive feedback loop that can lead to sustained levels of HIF and LANA in KSHV-infected cells.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute.
We thank Joseph Ziegelbauer of the National Cancer Institute for helpful comments and his critical reading of the manuscript.
Footnotes
Published ahead of print 16 November 2011
REFERENCES
- 1. Arvanitakis L, et al. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88:2648–2654 [PubMed] [Google Scholar]
- 2. Cai Q, et al. 2006. Kaposi's sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: latency control under low oxygen conditions. J. Virol. 80:7965–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cai Q, Murakami M, Si H, Robertson ES. 2007. A potential alpha-helix motif in the amino terminus of LANA encoded by Kaposi's sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia. J. Virol. 81:10413–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cannon JS, et al. 2000. A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J. Virol. 74:10187–10193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carroll PA, Kenerson HL, Yeung RS, Lagunoff M. 2006. Latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 80:10802–10812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Casper C, et al. 2007. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J. Infect. Dis. 195:30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 8. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 9. Davis DA, et al. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250 [DOI] [PubMed] [Google Scholar]
- 10. Dittmer D, et al. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Djerbi M, et al. 1999. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 190:1025–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ema M, et al. 1997. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. U. S. A. 94:4273–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894 [DOI] [PubMed] [Google Scholar]
- 14. Glaunsinger B, Ganem D. 2004. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 200:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Graven KK, Bellur D, Klahn BD, Lowrey SL, Amberger E. 2003. HIF-2alpha regulates glyceraldehyde-3-phosphate dehydrogenase expression in endothelial cells. Biochim. Biophys. Acta 1626:10–18 [DOI] [PubMed] [Google Scholar]
- 16. Haque M, Davis DA, Wang V, Widmer I, Yarchoan R. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. J. Virol. 77:6761–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. 2006. Genetic organization and hypoxic activation of the Kaposi's sarcoma-associated herpesvirus ORF34-37 gene cluster. J. Virol. 80:7037–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hussein D, Estlin EJ, Dive C, Makin GW. 2006. Chronic hypoxia promotes hypoxia-inducible factor-1alpha-dependent resistance to etoposide and vincristine in neuroblastoma cells. Mol. Cancer Ther. 5:2241–2250 [DOI] [PubMed] [Google Scholar]
- 19. Jeong J, Papin J, Dittmer D. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 75:1798–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knight JS, Cotter MA, II, Robertson ES. 2001. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus transactivates the telomerase reverse transcriptase promoter. J. Biol. Chem. 276:22971–22978 [DOI] [PubMed] [Google Scholar]
- 21. Koelle DM, et al. 1997. Frequent detection of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in saliva of human immunodeficiency virus-infected men: clinical and immunologic correlates. J. Infect. Dis. 176:94–102 [DOI] [PubMed] [Google Scholar]
- 22. Krishnan HH, et al. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwun HJ, et al. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81:8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lan K, Kuppers DA, Robertson ES. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lan K, et al. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Komatsu T, Dezube BJ, Kaye KM. 2002. The Kaposi's sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J. Virol. 76:11880–11888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang Y, Ganem D. 2004. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818–6826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao W, et al. 2003. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77:9399–9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsumura S, Fujita Y, Gomez E, Tanese N, Wilson AC. 2005. Activation of the Kaposi's sarcoma-associated herpesvirus major latency locus by the lytic switch protein RTA (ORF50). J. Virol. 79:8493–8505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller G, et al. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pearce M, Matsumura S, Wilson AC. 2005. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 79:14457–14464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 75:3948–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121–1127 [DOI] [PubMed] [Google Scholar]
- 36. Renne R, et al. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346 [DOI] [PubMed] [Google Scholar]
- 37. Russo JJ, et al. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sarid R, Wiezorek JS, Moore PS, Chang Y. 1999. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 73:1438–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwam DR, Luciano RL, Mahajan SS, Wong L, Wilson AC. 2000. Carboxy terminus of human herpesvirus 8 latency-associated nuclear antigen mediates dimerization, transcriptional repression, and targeting to nuclear bodies. J. Virol. 74:8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Semenza GL, Roth PH, Fang HM, Wang GL. 1994. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269:23757–23763 [PubMed] [Google Scholar]
- 42. Sodhi A, et al. 2000. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60:4873–4880 [PubMed] [Google Scholar]
- 43. Soulier J, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 44. Staudt MR, Dittmer DP. 2006. Promoter switching allows simultaneous transcription of LANA and K14/vGPCR of Kaposi's sarcoma-associated herpesvirus. Virology 350:192–205 [DOI] [PubMed] [Google Scholar]
- 45. Talbot SJ, Weiss RA, Kellam P, Boshoff C. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257:84–94 [DOI] [PubMed] [Google Scholar]
- 46. Tian H, McKnight SL, Russell DW. 1997. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 11:72–82 [DOI] [PubMed] [Google Scholar]
- 47. Wang GL, Jiang BH, Rue EA, Semenza GL. 1995. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A. 92:5510–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. 2005. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 65:3299–3306 [DOI] [PubMed] [Google Scholar]
- 49. Wang V, Davis DA, Veeranna RP, Haque M, Yarchoan R. 2010. Characterization of the activation of protein tyrosine phosphatase, receptor-type, Z polypeptide 1 (PTPRZ1) by hypoxia inducible factor-2 alpha. PLoS One 5:e9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warnecke C, et al. 2004. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 18:1462–1464 [DOI] [PubMed] [Google Scholar]
- 51. Wenger RH, Stiehl DP, Camenisch G. 2005. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005:re12. [DOI] [PubMed] [Google Scholar]
- 52. West JT, Wood C. 2003. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22:5150–5163 [DOI] [PubMed] [Google Scholar]
- 53. Wiesener MS, et al. 1998. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 92:2260–2268 [PubMed] [Google Scholar]
- 54. Yu Y, et al. 1999. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J. Gen. Virol. 80(Part 1):83–90 [DOI] [PubMed] [Google Scholar]
- 55. Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]