

Abstract
Infections with variant Creutzfeldt-Jakob disease (vCJD) have almost exclusively occurred in young patients, but the reasons for this age distribution are uncertain. Our data suggest that the pathogenesis of many peripherally acquired transmissible spongiform encephalopathy (TSE) agents is less efficient in aged individuals. Four vCJD cases linked to transfusion of vCJD-contaminated blood or blood products have been described. Three cases occurred in elderly patients, implying that intravenous exposure is more efficient in aged individuals than other peripheral routes. To test this hypothesis, young (6 to 8 weeks old) and aged (600 days old) mice were injected intravenously with a TSE agent. In aged and young mice, the intravenous route was more efficient than other peripheral routes of TSE agent exposure. However, in aged mice, disease pathogenesis was significantly reduced. Although most aged mice failed to develop clinical disease during their life spans, many showed histopathological signs of TSE disease in their brains. Thus, the effects of age on intravenous TSE pathogenesis may lead to significant levels of subclinical disease in the population. After peripheral exposure, many TSE agents accumulate upon follicular dendritic cells (FDCs) in lymphoid tissues before they infect the brain. In aged spleens, PrPC expression and TSE agent accumulation upon FDCs were reduced. Furthermore, the splenic marginal zone microarchitecture was substantially disturbed, adversely affecting the delivery of immune complexes to FDCs. This study is the first to suggest that the effects of aging on the microarchitecture and the function of the splenic marginal zone significantly influence the pathogenesis of an important pathogen.
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs) (prion diseases) are subacute neurodegenerative diseases affecting both humans and animals. These diseases are defined by a number of characteristic pathological changes in the central nervous system (CNS), including vacuolation of the neuropil, gliosis, and aggregations of PrPSc, an abnormally folded isoform of the cellular prion protein (PrPc). The precise nature of the TSE agent is uncertain, but PrPSc is considered to constitute the major, if not sole, component of the infectious agent (27). Infectious, sporadic, and familial forms of human TSE disease have been identified. Following peripheral exposure to many natural and experimental TSEs, including natural scrapie in sheep (1), chronic wasting disease (CWD) in cervids (45), and variant Creutzfeldt-Jakob disease (vCJD) in humans (19), early agent accumulation occurs upon follicular dendritic cells (FDCs) within the secondary lymphoid tissues prior to the spread of infection to the CNS (termed “neuroinvasion”). The accumulation of TSE agents upon FDCs is a critical stage in the neuroinvasion process as disease susceptibility is reduced in their absence (32, 34, 39).
Dietary exposure to bovine spongiform encephalopathy (BSE)-contaminated meat products is considered the most likely source of vCJD in humans (5). In the United Kingdom, most clinical cases of vCJD have occurred almost exclusively in young adults (median age at onset of disease, 26 years; median age at death, 28 years) (40). These data starkly contrast those for sporadic CJD cases, which have predominantly occurred in the elderly (median age at onset of disease, 67 years) (40). The factors responsible for the age-related incidence of vCJD are not known as the suggestion that this is simply due to exposure to greater levels of BSE through dietary preference is unproven (2). Data from experimental mouse models show that the underdeveloped or reduced functional status of FDCs in either very young (neonatal) or aged mice significantly impairs TSE neuroinvasion following intraperitoneal (i.p.) or oral exposure (4, 21, 42). This implies that the pathogenesis of many acquired TSE infections which accumulate in lymphoid tissues prior to neuroinvasion, including natural sheep scrapie, CWD, and vCJD, may likewise be much less efficient in the aged.
In the United Kingdom, four cases of vCJD have been reported in recipients of blood or blood products derived from vCJD-infected donors (18, 28, 43, 48). In contrast to the overwhelming majority of the clinical vCJD cases considered to be acquired through dietary exposure to BSE, three of the four blood transfusion-associated vCJD cases were reported in elderly patients. Two cases were preclinical and confirmed postmortem by detection of PrPSc within secondary lymphoid tissues (18, 43). However, a third case was reported as clinical vCJD in an elderly patient 6.5 years after receiving a transfusion of red blood cells from a vCJD-infected donor (28). Since the age of this patient was substantially beyond that of the majority of the cases considered to be acquired through dietary exposure (62 years at time of transfusion with vCJD-contaminated red cells), this raises the possibility that intravenous (i.v.) exposure may be more efficient in the elderly than other peripheral routes of exposure. To test this hypothesis, groups of young mice (6 to 8 weeks old) and aged mice (∼600 days old) were injected i.v. with a TSE agent. We show that, in aged mice as in young adult mice, the i.v. route is more efficient than other peripheral routes of TSE agent exposure (24). However, disease pathogenesis in i.v. exposed aged mice was significantly impaired compared to that in young mice, with most failing to develop clinical disease during their life spans. Cells within the marginal zone (MZ) of the spleen play an important role in the capture of blood-borne antigens and immune complexes and their efficient transport to FDCs (7, 38). We show that the microarchitecture of the MZ is disturbed in aged mice, significantly impairing the delivery of immune complexes to FDCs. TSE agents are likewise considered to be acquired by FDCs as complement-bound complexes (26, 30, 31, 50). Thus, these data imply that the impaired TSE pathogenesis in aged mice after i.v. exposure is likewise a consequence of the reduced transport of TSE agent-containing complement-bound complexes from the MZ to FDCs.
MATERIALS AND METHODS
Mice.
C57BL/Dk mice were aged to approximately 600 days under specific-pathogen-free (SPF) conditions prior to exposure to the scrapie agent. Six- to 8-week-old C57BL/Dk mice were used as young adults. tga20 mice overexpressing PrPc (15) were maintained on a C57BL/6 background. Prnp−/− mice were bred and maintained on a 129/Ola background (35). All experimental procedures were approved by The Roslin Institute's Ethical Review Committee and conducted according to the strict regulations of the UK Home Office Animals (Scientific Procedures) Act 1986.
Scrapie agent exposure and disease monitoring.
For intracerebral (i.c.) or intraperitoneal (i.p.) exposure, mice were injected with 20 μl of a 1% (wt/vol) scrapie brain homogenate prepared from mice terminally affected with the ME7 scrapie agent strain (containing approximately 1 × 104 i.c. 50% infectious dose [ID50] units). For i.v. exposure, mice were injected into the tail vein with 20 μl of a 0.1% dilution of scrapie brain homogenate. Following scrapie agent exposure, mice were coded, assessed weekly for signs of clinical disease, and culled at a standard clinical endpoint. The clinical endpoint of disease was determined by rating the severity of clinical signs of TSE disease exhibited by the mice. Following clinical assessment, mice were scored as “unaffected,” “possibly affected,” and “definitely affected” using standard criteria which typically present in mice clinically affected with ME7 scrapie. Clinical signs for the ME7 scrapie agent may include the following: weight loss, starry coat, hunched, jumpy behavior (at early onset) progressing to limited movement, upright tail, wet genitals, decreased awareness, discharge from eyes and/or blinking eyes, and ataxia of hind legs. The clinical endpoint of disease was defined in one of the following ways: (i) the day on which a mouse received a second consecutive “definite” rating, (ii) the day on which a mouse received a third “definite” rating within four consecutive weeks, or (iii) the day on which a mouse was culled in extremis.
The following criteria were used to help distinguish between the clinical signs of aging (senility) in mice from those of TSE disease. The fur of aged mice may lose color and appear less sleek. Body shape may gradually change. Senile mice may have a “vacant stare,” whereby the face looks thinner and the eyes not as bright. Mice beginning to display clinical signs of TSE disease move quicker and become more conspicuous, whereas those displaying definite positive signs of scrapie are immobile and less interactive with their cage mates. In contrast, senile mice still move around their cages and interact with their cage mates.
Survival times were recorded for mice that did not develop clinical signs of disease and were culled when they showed signs of intercurrent disease. Scrapie diagnosis was confirmed by histopathological assessment of vacuolation in the brain. For the construction of lesion profiles, vacuolar changes were scored in nine gray-matter areas of each brain, as described previously (16). Where indicated, some mice were culled 35 and 70 days postinjection (dpi) with scrapie, and spleens were taken for further analysis. For bioassay of scrapie agent infectivity, individual half-spleens were prepared as 10% (wt/vol) homogenates in physiological saline. Groups of four tga20 indicator mice were injected i.c. with 20 μl of each homogenate. The scrapie titer in each sample was determined from the mean incubation period in the indicator mice, by reference to a dose/incubation period response curve for ME7 scrapie-infected spleen tissue serially titrated in tga20 mice using the relationship: y = 9.4533 − 0.0595x (where y is log ID50 U/20 μl of homogenate, and x is the incubation period; R2 = 0.9562). As the expression level of cellular PrPc controls the TSE disease incubation period, tga20 mice overexpressing PrPc are extremely useful as indicator mice in scrapie agent infectivity bioassays as they succumb to disease with much shorter incubation times than conventional mouse strains (15).
IHC.
For the detection of disease-specific PrP (PrPd) in brains and spleens, tissues were fixed in periodate-lysine-paraformaldehyde fixative and embedded in paraffin wax. Sections (thickness of 6 μm) were deparaffinized and pretreated to enhance the detection of PrPd by hydrated autoclaving (15 min, 121°C, hydration) and subsequent immersion in formic acid (98%) for 5 min (37). Sections were then immunostained with 1B3 PrP-specific polyclonal antiserum (13). For the detection of astrocytes, brain sections were immunostained with anti-glial fibrillary acidic protein (anti-GFAP; Dako, Ely, United Kingdom). For the detection of microglia, deparaffinized brain sections were first pretreated with Dako target retrieval solution and subsequently immunostained with anti-ionized calcium-binding adaptor molecule 1 (Iba-1; Wako Chemicals GmbH, Neuss, Germany). Paraffin-embedded tissue (PET) immunoblot analysis was used to confirm that the PrPd detected by immunohistochemistry (IHC) was proteinase K (PK)-resistant PrPSc (44). Membranes were subsequently immunostained with 1B3 PrP-specific polyclonal antiserum.
To detect FDCs, frozen spleen sections (thickness, 10 μm) were fixed in acetone and visualized by staining with complement component C4-specific rat monoclonal antibody (MAb) FDC-M2 (AMS Biotechnology, Abingdon, United Kingdom) or MAb 8C12 to detect CR1 (CD35; BD Biosciences PharMingen, CA). Cellular PrPc was detected using 1B3 PrP-specific polyclonal antiserum. B cells were detected using monoclonal antibody B220 to detect CD45R (Caltag Laboratories, Buckingham, United Kingdom). Marginal zone (MZ) B cells were detected using MAb 1B1 to detect CD1d (BD Biosciences PharMingen). MZ sinus-lining cells were detected using MAb MECA-367 (BD Biosciences PharMingen) specific for mucosal vascular addressin cell-adhesion molecule 1 (MADCAM1). C-type lectin SIGNR1-expressing MZ macrophages were detected using MAb ER-TR9 (BMA Biomedicals, Augst, Switzerland), and sialic acid-binding immunoglobulin-like lectin 1 (SIGLEC1/CD169)-expressing MZ metallophilic macrophages were detected using MAb MOMA-1 (AdB Serotec, Kidlington, United Kingdom).
For light microscopy, immunolabeling was revealed using horseradish peroxidase (HRP) conjugated to the avidin-biotin complex (Novared kit; Vector Laboratories, Peterborough, United Kingdom). For fluorescence microscopy, following the addition of primary antibodies, species-specific secondary antibodies coupled to Alexa Fluor 488 (green), Alexa Fluor 594 (red), and Alexa Fluor 647 (blue) dyes (Invitrogen Life Technologies) were used.
Quantitative real-time PCR analysis of Prnp mRNA expression.
Total RNA was isolated from coded mouse brain (comprising the thalamus, hippocampus, and lateral cortex areas) samples using the RNeasy lipid tissue kit (Qiagen, Crawley, United Kingdom) followed by treatment with DNase I (Ambion, Warrington, United Kingdom). First-strand cDNA synthesis was performed using 1 μg of total RNA and Superscript III reverse transcriptase as described by the manufacturer (Invitrogen, Paisley, United Kingdom). Real-time quantitative PCR (qPCR) amplification was carried out in 10-μl reactions using FastStart Taq DNA polymerase (Roche, Burgess Hill, United Kingdom) and SYBR green I detection. The Prnp primers used were forward (5′-TCCAATTTAGGAGAGCCAAGC-3′) and reverse (5′-GCCGACATCAGTCCACATAG-3′); the reference gene primers were SSDHA, YWHAZ, GAPDH, RPL13A, and CALNX from the Quantace mouse normalization gene panel (Quantace, London, United Kingdom). After evaluation using the geNorm application (47), the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and SSDHA genes were chosen as the most stable reference genes for normalization. The PCR conditions used were 5 min at 94°C, followed by 40 cycles of 20 s at 94°C, 20 s at gene-specific annealing melting temperature (Tm), and 20 s at 72°C, followed by a dissociation curve analysis program. Data were analyzed using Rotor-Gene 3000 software (version 6.1.93; Corbett Life Science).
Quantitative real-time PCR analysis of C4b mRNA expression.
Total RNA was isolated from coded mouse liver samples using the RNeasy lipid tissue kit (Qiagen, Crawley, United Kingdom) followed by treatment with DNase I (Ambion, Warrington, United Kingdom). First-strand cDNA synthesis was performed using 1 μg of total RNA and Superscript III reverse transcriptase (SuperScript VILO cDNA synthesis kit) as described by the manufacturer (Invitrogen, Paisley, United Kingdom). PCR amplification reactions were carried out in 25-μl reaction mixtures using a Rotor-Gene SYBR green PCR kit (Qiagen, United Kingdom). All qPCR primers used were QuantiTect primer assays designed and purchased from Qiagen, for complement component 4B (QT01782011) and the reference gene, the primers used were eukaryotic translation initiation factor 2a (QT01050735), hypoxanthine guanine phosphoribosyl transferase (QT00166768), peptidylprolyl isomerase B (QT00169736), 18S rRNA (QT01036875), and ribosomal protein, large, P0 (QT00249375). After evaluation using the geNorm application (48), Hprt and Ppib were chosen as the most stable reference genes for normalization. The PCR conditions used were 5 min at 95°C, followed by 40 cycles of 5 s at 95°C and 15 s at 60°C, followed by a dissociation curve analysis program. Data were analyzed using Rotor-Gene 3000 software (version 6.1.93; Corbett Life Science), and statistical differences were assessed with a nonparametric Mann-Whitney U test (two-tailed probabilities) using GraphPad Prism version 5.04 (GraphPad Software).
Plasma C4 analysis.
Blood from mice from each group was collected into sterile EDTA-treated containers and immediately centrifuged at 1,000 × g for 20 min at 4°C. Plasma was collected and stored at −80°C prior to use. Plasma C4 concentrations were measured using a mouse complement component 4 enzyme-linked immunosorbent assay (ELISA) kit (Unsc Life Science, Inc., Wuhan, China) according to the manufacturer's instructions.
In vivo immune complex trapping.
To assess antigen trapping by FDCs in vivo, mice were passively immunized by i.p. injection with 100 μl preformed peroxidase-antiperoxidase (PAP) immune complexes (Sigma). Spleens were removed 48 h later, and the levels of FDC-associated immune complexes compared by IHC. Digital microscopy images were analyzed using ImageJ software (http://rsb.info.nih.gov/ij/), as described previously (22). Spleens from 6 mice from each group were analyzed. From each spleen, 4 sections were studied, and on each section, data from 3 randomly chosen 1,000- by 1,000-μm fields of view were collected.
Embryonic spleen transplantation.
Developing whole embryonic day 15 spleens from Prnp−/− or wild-type control mice were collected using a dissecting microscope and grafted under the kidney capsule of recipient wild-type mice (5 weeks old) as described previously (17). Recipient mice were culled 6 weeks later, and grafted spleens were collected and analyzed by IHC.
Statistical analysis.
Differences in the incidence of clinical disease and vacuolar pathology were compared between age groups within each exposure route using Fisher's exact test for 2-by-2 contingency tables (49). Brain lesion scores in animals with positive vacuolar pathology were compared between ages within each exposure route using multivariate analysis of variance (6).
RESULTS
Effect of host age on susceptibility to i.v. injected scrapie.
When young adult (6 to 8 weeks old) and aged (∼600 days old) C57BL/Dk mice were injected i.c. with the ME7 scrapie agent strain, no significant differences in disease incidence, the onset of clinical signs, or incubation period of disease were observed (P = 0.92, Fisher's exact test). The i.c. injected mice succumbed to clinical disease approximately 165 days after exposure (young, 163 ± 3 days, n = 5/6; aged, 167 ± 8 days, n = 6/7), clearly demonstrating that aging has no effect on TSE disease susceptibility when the infection is established directly in the CNS. In contrast, when mice were injected i.v. with the scrapie agent, a significant effect of host age on the incidence of clinical disease was observed (P < 0.001, Fisher's exact test). All young i.v. injected mice succumbed to clinical disease, with a mean incubation period of 222 ± 7 days (n = 9/9). In contrast, only 5 of 16 i.v. injected aged mice succumbed to clinical disease during their life spans, with individual incubation periods ranging from 216 to 324 days (Table 1). Due to the advanced ages of the aged mice used in this study, some were culled due to aging-related intercurrent disease, including senility, swollen abdomen, eye abscess, etc. Six of these mice were culled at times after exposure prior to the arrival and detection of the TSE agent within the brains of the i.v. injected young mice (individual survival times of 120, 120, 127, 134, 141, and 148 days). No vacuolar pathology or PrPd was detected in any of the brains from the mice culled ≤148 days postinjection. As a consequence, only the clinically negative aged mice that were culled after the first clinically positive, pathologically confirmed case in the young mouse group were included in our analysis (representing mice ranging from 190 to 317 days after scrapie exposure, which were 790 to 917 days old).
Table 1.
Effect of host age on susceptibility to i.v. scrapie infection
| Mouse model and incubation period/survival timea | Clinical disease | Vacuolar pathology in brainb | PrPd in: |
|
|---|---|---|---|---|
| Brainc | Spleen | |||
| Young | ||||
| Incubation period (days) | ||||
| 190, 197, 216, 216, 216, 225, 239, 239, 260 | Yes (9/9 mice) | Yes (9/9 mice) | Yes (9/9 mice) | Yes (9/9 mice) |
| Aged | ||||
| Incubation period (days) | ||||
| 216 | Yes | Yes | Yes | Yes |
| 255 | Yes | Yes | Yes | Yes |
| 274 | Yes | Yes | Yes | No |
| 317 | Yes | Yes | Yes | Yes |
| 324 | Yes | Yes | Yes | No |
| Survival time (days) | ||||
| 232 | No | Yes | Yes | Yes |
| 285 | No | Yes | Yes | Yes |
| 293 | No | Yes | Yes | Yes |
| 190 | No | No | Yes | Yes |
| 197 | No | No | Yes | No |
| 225 | No | No | Yes | No |
| 232 | No | No | Yes | No |
| 232 | No | No | No | No |
| 246 | No | No | Yes | NDe |
| 291 | No | No | No | No |
| 317 | No | No | Yes | No |
| Incidence (no. of mice affected/no. tested)d | 5/16 (P < 0.001) | 8/16 (P < 0.05) | 14/16 | 7/15 |
Young C57BL/Dk mice were 6 to 8 weeks old at the time of i.v. scrapie agent exposure. Aged C57BL/Dk mice were ∼600 days old at the time of i.v. scrapie agent exposure. Only aged mice that were culled after the first clinically positive case in the corresponding group of young mice (190 days postinjection) were included in the analysis. Six aged mice were culled <190 days postinjection (individual survival times of 120, 120, 127, 134, 141, and 148 days). No vacuolar pathology or PrPd was detected in any of the brains from the mice culled <190 days postinjection. The mean ± standard error of the mean (SEM) disease incubation period for young mice was 222 ± 7 days.
The magnitude of the TSE-specific vacuolation was scored using a scale of 0 to 5 in 9 distinct gray matter areas as described previously (16). A score of 1 (a small number of vacuoles) or greater in any region of the brain was considered positive.
The detection of PrPd in any region on the tissue section was considered positive.
Only aged mice that were culled after the first clinically positive case in the corresponding group of young mice were included in the analysis. Statistical differences in the incidence of clinical disease and vacuolar pathology in the brain were compared between age groups with Fisher's exact test for 2-by-2 contingency tables.
ND, not determined.
We next compared the susceptibility of aged mice to TSE infection via the i.v., i.p., or oral routes using data derived from two independent sets of experiments (Table 2). These data show that even though the i.p. injected aged mice were given a 10× higher dose of the TSE agent compared to the i.v. injected mice (i.p. route, 20 μl of 1.0% scrapie brain homogenate; i.v. route, 20 μl of 0.1% scrapie brain homogenate), only 1/21 i.p. injected mice succumbed to clinical disease during their life spans (Table 2). None of the aged mice developed clinical disease after oral exposure to the scrapie agent. Together, these data show that although as in young mice the i.v. route is more efficient than other peripheral routes of TSE agent exposure in aged mice (24), disease susceptibility is significantly reduced compared to that in young mice.
Table 2.
Effect of route of exposure on TSE disease susceptibility in aged mice
| Expta | Route of exposure | Disease incubation period or survival timeb | % inoculum dilution (wt/vol) | Clinical disease incidence (no. of mice affected/no. tested)b | No. of mice affected/no. tested |
|
|---|---|---|---|---|---|---|
| Vacuolar pathology in brain | PrPd in brain | |||||
| Expt 1 | i.v. | Incubation periods, 216, 255, 274, 317, and 324 days | 0.1 | 5/16 | 8/16 | 14/16 |
| Survival time, 190–317 days | ||||||
| i.p. | Incubation period, 321 days | 1.0 | 1/12 | 3/12 | 9/10c | |
| Survival time, 230–357 days | ||||||
| Expt 2 | i.p. | Survival time, 247–323 days | 1.0 | 0/9 | 3/9 | 6/9 |
| Oral | Survival time, 282–366 days | 1.0 | 0/12 | 5/12 | 12/12 | |
Experiment 1 represents the present study; experiment 2 represents data from Brown et al. (4). Young C57BL/Dk mice were 6 to 8 weeks old at the time of scrapie agent exposure. Aged C57BL/Dk mice were ∼600 days old at the time of scrapie agent exposure.
Only aged mice that were culled after the first clinically positive case in the corresponding group of young mice were included in the analysis.
Two brains were unavailable for PrPd analysis.
Effect of host age on the development of neuropathological changes.
Histopathological analysis of brain tissue from all of the clinically affected, i.c. injected, young and aged mice showed the characteristic spongiform pathology, astrogliosis, microgliosis, and disease-specific PrP (PrPd) accumulation associated with terminal TSE disease (Fig. 1A). The brains from all the clinically affected i.v. injected, young and aged mice likewise displayed the typical TSE-specific neuropathological changes and PrPd accumulation associated with infection with the ME7 scrapie agent strain (Fig. 1B, left-hand columns). Paraffin-embedded tissue (PET) immunoblot analysis can be used to demonstrate the presence of TSE agent-specific PrPSc on histological sections (44). During the tissue processing, sections are treated with PK to destroy the cellular PrPc, leaving only the PK-resistant PrPSc (if present). PET immunoblotting confirmed that the PrPd detected by immunohistochemistry within the brains of all of the clinically affected, i.v. injected, young and aged mice was TSE agent-specific PrPSc (Fig. 1B, panels q and r, respectively). In contrast, the majority (8/11) of the brains from the clinically negative aged mice did not show signs of spongiform pathology (Table 1) and had negligible levels of reactive astrocytes and microglia (Fig. 1B, right-hand columns). However, most (9/11) of the clinically negative aged mice displayed evidence of PrPSc accumulation in their brains (Table 1), which was typically restricted to a few small foci (Fig. 1B, panels o and s).
Fig 1.

Effect of host age on the development of neuropathology with the brain at the terminal stage of disease. (A) Histopathological analysis of brains from clinically scrapie-affected young and aged mice infected with the scrapie agent introduced directly into the brain by i.c. injection. (B) Histopathological analysis of brains from young and aged mice exposed to the scrapie agent by i.v. injection: panels a to d, spongiform pathology; panels e to h, reactive astrocytes expressing GFAP (brown); panels i to l, Iba-1-expressing microglia (brown); panels m to p, disease-specific PrP (PrPd; brown); panels q to t, PET immunoblot detection of PK-resistant PrPSc (blue or black). High levels of spongiform pathology, reactive astrocytes expressing GFAP, and active microglia expressing Iba-1 and large accumulations of PrPd/PrPSc were detected in the hippocampi of the brains of all clinically scrapie-affected young and aged mice (left-hand columns). In contrast, most brains from the i.v. injected, clinically negative aged mice did not show signs of spongiform pathology, and the levels of astrocytosis (g and h) and microgliosis (k and l) were likewise reduced in comparison to brains from mice with clinical disease (e and f and i and j, respectively). However, most of the brains from the clinically negative aged mice displayed evidence of PrPd/PrPSc deposition, which was typically restricted to a few small foci such as the thalamus (arrows, panel o). Scale bar in panels a to n and p, 20 μm; scale bar in panels q to t, 50 μm. Panel o shows the thalamus as no PrPSc accumulation was detected in the hippocamus. Scale bar, 20 μm. H&E, hematoxylin and eosin; Clin., presence of clinical signs of scrapie at the time of cull; Path., detection of spongiform pathology in the brain; dpi, days postinjection with the scrapie agent.
Spongiform vacuolation of the neuropil is a neuropathological characteristic of TSE infection. Although both young and aged mice succumbed to clinical TSE disease with similar incubation periods after i.c. exposure, the magnitude of the spongiform pathology was significantly lower in aged mice than that in young mice (Fig. 2A) (P < 0.05, multivariate analysis of variance). Similarly, the average severity of the spongiform pathology within the brains of the clinically affected, i.v. injected aged mice also was significantly lower than that observed in young mice (Fig. 2B) (P < 0.01, multivariate analysis of variance). The reasons for the reduced spongiform pathology in the brains of the clinically affected aged mice are not known. To determine whether this was due to the influence of host age on PrPc expression in the brain, we compared Prnp transcript levels in tissues from young and aged mice by quantitative real-time PCR analysis. No significant differences in Prnp transcript levels were observed between brains from young or aged mice (Fig. 2C) (P < 0.776, Mann-Whitney test), clearly demonstrating that the effects of host age on the reduced spongiform pathology and reduced susceptibility to i.v. injected scrapie were not due to effects on Prnp expression levels in the brain.
Fig 2.
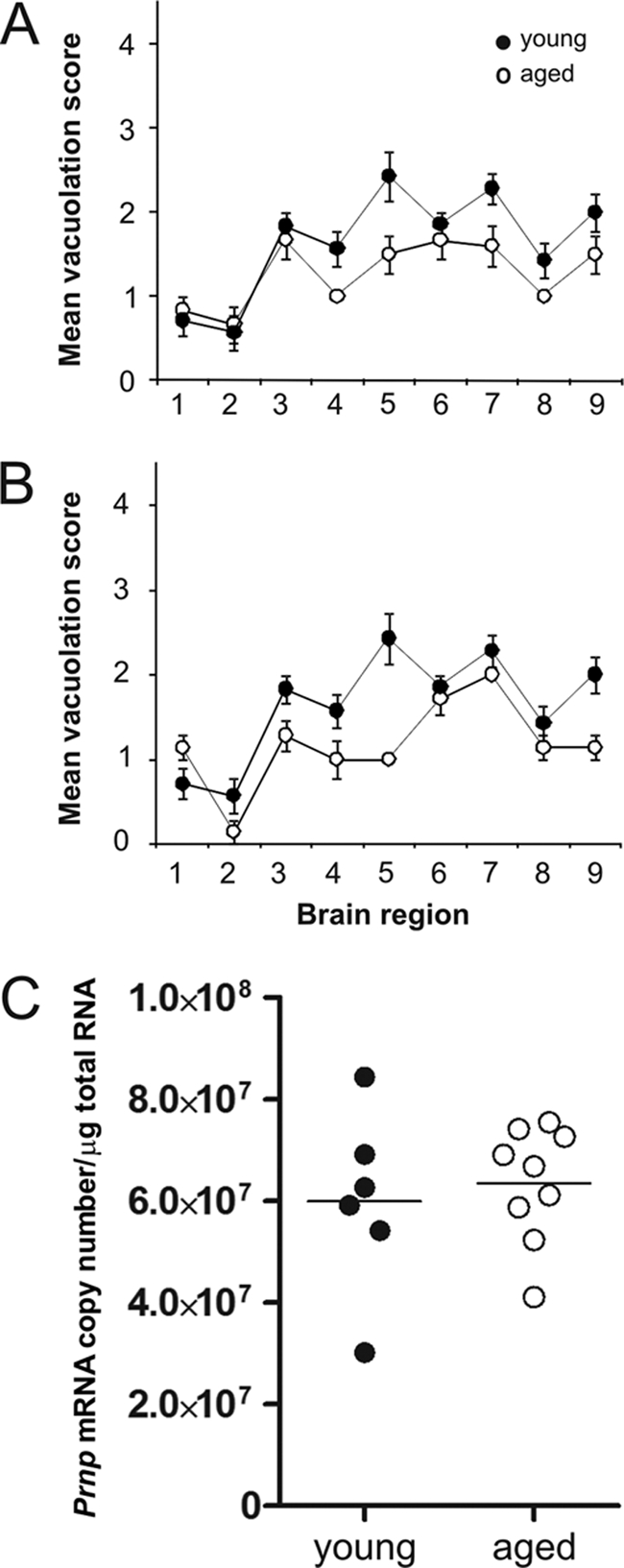
Effect of host age on TSE-specific neuropathology and Prnp expression in the brain. (A and B) Pathological assessment of the spongiform change (vacuolation) in the brains of young (closed circles) and aged (open circles) mice with positive vacuolar pathology after i.c. (A) or i.v. (B) injection with the scrapie agent. Vacuolation was scored on a scale of 1 to 5 in nine gray matter areas: 1, dorsal medulla; 2, cerebellar cortex; 3, superior colliculus; 4, hypothalamus; 5, thalamus; 6, hippocampus; 7, septum; 8, retrosplenial and adjacent motor cortex; 9, cingulate and adjacent motor cortex. Each point represents the mean vacuolation score ± standard error of the mean (SEM) for groups of 5 to 9 mice. The severity of the vacuolar pathology was significantly lower in the brains of aged mice (A, P < 0.05, n = 5 young, n = 6 aged; B, P < 0.01, n = 9 young, n = 5 aged; multivariate analysis of variance). (C) Prnp transcript levels in the brains of young and old mice. Real-time RT-qPCR analyses were performed on samples from brains of young (n = 6) and aged (n = 8) mice. No significant difference in Prnp transcript levels were observed between the two age groups of mice (P = 0.776, Mann-Whitney test).
The effect of host age on scrapie agent accumulation in the spleen.
Following peripheral exposure, the early accumulation of TSE agents upon FDCs in the secondary lymphoid tissues is a crucial stage in the neuroinvasion process (32, 34, 39). Heavy PrPd accumulations, consistent with localization upon FDCs, were detected immunohistochemically in the spleens of all young adult mice at 5 and 10 weeks after i.v. injection with the scrapie agent (Fig. 3A and B, upper panels) and were maintained until the terminal stages of disease (Table 1). PET immunoblotting confirmed the presence of TSE agent-specific PrPSc upon the surfaces of the FDCs in spleens from young mice (Fig. 3A and B, lower panels). In contrast, in aged spleens PrPSc was only detected in 1 of 2 spleens at 5 weeks postinfection (Fig. 3A). Similarly, the same spleens from the young adult mice collected 5 weeks postinfection contained high levels of scrapie agent infectivity (5.6 and 6.3 log ID50 U/g, respectively). However, only one of the aged spleens contained high levels of scrapie agent infectivity (5.4 log ID50 U/g); the other contained at least 100× less agent infectivity than that measured in the young spleens (3.4 logID50 U/g). By 10 wk after i.v. exposure, PrPSc was detected in both aged spleens, although in one of the samples, the PrPSc was only observed in association with a single FDC network (Fig. 3B).
Fig 3.

Effect of host age on TSE agent accumulation in the spleen. High levels of PrPd (red, upper rows) were detected in association with FDCs in spleens from young mice at 5 weeks (A) and 10 weeks (B) after i.v. injection with the scrapie agent. Analysis of adjacent sections by PET immunoblot analysis confirmed the presence of PK-resistant PrPSc (blue or black, lower rows). In contrast, PrPSc was only detected in 1 of 2 aged spleens at 5 weeks postinfection (B). At 10 weeks after i.v. injection, PrPSc was detected in both aged spleens, although in one, the PrPSc was only detected in association with a single FDC network (B, far-right column). Arrows indicate sites of PrPSc accumulation upon FDCs. Upper rows, scale bar, 20 μm; lower rows, scale bar, 50 μm.
Following peripheral exposure of young mice to the ME7 scrapie agent, strong levels of PrPSc accumulations are maintained upon the surfaces of FDCs for the duration of disease (33 and 34). However, when we analyzed spleens from aged mice taken at the end of the experiment (from both clinically scrapie-affected and clinically negative mice), considerable variation in the detection of PrPSc was observed (Table 1). Despite the presence of PrPSc in the brains of most mice, in many spleens, PrPSc was undetectable, and in two mice, PrPSc was observable only upon occasional FDC networks.
The retention of complement component C4 and expression of PrPc upon FDCs is impaired in the spleens of aged mice.
FDCs characteristically trap and retain native antigen on their surfaces in the form of immune complexes, consisting of antigen-antibody and/or opsonizing complement components. Whereas high levels of complement component C4 and cellular PrPc were expressed on the surfaces of FDCs from young mice, this was severely reduced on most FDC networks in the spleens of aged mice (Fig. 4A). Morphometric analysis confirmed that the magnitude of complement component C4 retained upon FDCs in the spleens of aged mice was significantly reduced compared to that in young mice (Fig. 4B) (P = 0.0025; n = 36 fields of view/group). FDCs in mice characteristically express high levels of complement receptor 1 (CR1/CD35) (34, 50). However, when we compared the levels of expression of CR1/CD35 by FDCs, similar levels were detected in the spleens of both young and aged mice (Fig. 4C). Since the expression of CD35 by aged FDCs appeared to be unaffected, we next compared complement component C4 levels between each mouse group to determine whether expression was reduced in aged mice. The comparison of C4b mRNA expression (which encodes C4 in the mouse) across 95 individual microarray data sets representing a wide range of mouse tissues and cell-lineages clearly shows that the liver is the major source of complement component C4 (see Fig. S1A in the supplemental material; http://biogps.gnf.org). The levels of C4b expressed by FDCs themselves are negligible in comparison (see Fig. S1B). We therefore compared C4b transcript levels in livers from young and aged mice by quantitative real-time PCR analysis. No significant difference in C4b transcript levels were observed between livers from young or aged mice (Fig. 4D) (P < 0.286, Mann-Whitney test). Similarly, the synthesis of C4 was not adversely affected as no significant difference was observed in plasma C4 levels between young and aged mice (Fig. 4E) (P < 0.191, Student's t test). We also compared the transcriptional profiles of other complement component-encoding genes (C1qa, C1qb, C1qc, C1r, C1s, C1qbp, C3, Cfh, and Cfi) across a large microarray data set of livers from aging C57BL/6 mice (age range, 29 to 722 days old; n = 40) (51). Consistent with our data for C4b expression (Fig. 4D), which was not represented on the microarrays used (51), no significant effect of aging on the expression of any of these genes in the liver was observed (see Tables S1 and S2 in the supplemental material).
Fig 4.

Effect of host age on FDC status. (A) Triple immunofluorescent analysis of FDC status. High levels of complement component C4 (blue) and cellular PrPc (red) were expressed on the surfaces of FDCs within the B-cell follicles (B cells, CD45R, green) of young mice (left-hand panels). In contrast, on the FDCs in the spleens of aged mice, the detection of complement component C4 and cellular PrPc was dramatically reduced (right-hand panels). Scale bar, 20 μm. (B) Morphometric analysis confirmed that the level of complement component C4 colocalized upon the surfaces of FDCs in spleens of aged mice was significantly lower than that observed in spleens from young mice (P < 0.0025, n = 36 fields of view/group). (C) Comparison of complement receptor 1 (CR1/CD35; brown) expression upon young and aged FDCs. Scale bar, 20 μm. (D) Real-time RT-qPCR analysis of C4b transcript levels in livers of young and old mice (n = 5 and 4/group, respectively). No significant differences in transcript levels were observed between the two age groups of mice (P < 0.286, Mann-Whitney test). (E) ELISA analysis revealed no significant difference in the complement component C4 concentration in plasma from young and aged mice (Fig. 4D) (P < 0.191, Student's t test). (F) FDCs do not passively acquire significant levels of PrPC from other cell types. Whole embryonic day 15 spleen tissue from Prnp−/− mice or wild-type (WT) controls was transplanted under the kidney capsule of WT recipient mice. Grafted spleens were analyzed 6 weeks after transplant. FDCs expressing high levels of PrPC were detected in the B-cell follicles of the WT donor spleens grafted into WT recipients (WT→WT; left-hand panels). However, no PrPC was detected upon FDCs within the Prnp−/− spleens grafted into WT mice (Prnp−/−→WT; right-hand panels). Upper panels, scale bar, 200 μm; lower panels, scale bar, 50 μm.
These data clearly demonstrate that the effects of host age on C4 retention by FDCs were not due to effects on the synthesis of opsonizing complement components. Instead, these data imply that the ability of aged FDCs to trap and retain C4-containing immune complexes was adversely affected.
Host cells must express cellular PrPC to replicate TSE agents (35), and FDCs appear to express high levels of PrPC on the cell membrane in uninfected mice (3, 25). Many cell types secrete small membrane vesicles termed exosomes that are enriched in cell-specific protein (11, 14). These microvesicles enable FDCs to passively acquire and display proteins on their surfaces that they do not express at the mRNA level (12). To determine whether FDCs likewise passively acquire significant levels of PrPC from other cell lineages, whole embryonic day 15 spleen tissue from PrPC-deficient (Prnp−/−) mice or wild-type (WT) controls was transplanted under the kidney capsule of WT recipient mice (17). Six weeks after transplant, recipient kidneys were collected and the grafted spleen tissue was analyzed by IHC. Within the grafted spleens, the stromally-derived FDCs are of the donor Prnp genotype, whereas the majority of the hematopoietically derived cells (lymphocytes, macrophages, etc.) within the graft and all cell lineages within the rest of the mouse are of the host Prnp genotype. If FDCs do acquire PrPC from other cell lineages, one would expect to detect PrPC upon the surfaces of the FDCs within the PrPC-deficient spleens grafted into WT mice (Prnp−/−→WT). As anticipated, FDCs expressing high levels of PrPC were detected in the B-cell follicles of the WT donor spleens grafted into WT recipients (WT→WT; Fig. 4F). However, no PrPC was detected upon FDCs within the Prnp−/− spleens grafted into WT mice (Prnp−/−→WT; Fig. 4F). These data therefore confirm that FDCs express high levels of PrPC and do not simply acquire it from other cell populations. Taken together, these data suggest that the expression of PrPC by FDCs themselves is considerably reduced in the spleens of aged mice.
The abilities of FDCs to retain complement-bound complexes and express high levels of PrPc are both considered important for the trapping and amplification of TSE agents upon their surfaces (3, 25, 26, 30, 31, 50). Data in the present study clearly show that both of these properties are dramatically impaired in the spleens of aged mice.
The effects of host age on the splenic marginal zone impair immune complex trapping by FDCs.
Our data show that the ability of FDCs to retain complement-bound immune complexes was impaired in the spleens of aged mice despite the expression of high levels of CR1 on their surfaces. Therefore, we next investigated whether the transport of immune complexes to FDCs in the spleens of aged mice was adversely affected. Blood-borne antigens enter the spleen via the sinus of the marginal zone (MZ), where they are rapidly cleared by macrophages. Within the MZ, a specialized subset of B cells also plays an important role in the capture of immune complexes and facilitates their delivery to FDCs (7). We determined whether aging adversely affected the microarchitecture of the MZ and, as a consequence, systemic antigen capture and delivery to FDCs. In the spleens of young mice, a distinct channel of sinus-lining cells expressing mucosal vascular addressin cell-adhesion molecule 1 (MADCAM1) forms a barrier between the MZ and the white pulp (Fig. 5A). Two distinct populations of macrophages also reside in the MZ. Within an outer layer are a ring of C-type lectin SIGNR1-expressing MZ macrophages, and in a continuous inner ring close to the white pulp are the sialic-acid-binding immunoglobulin-like lectin 1 (SIGLEC1/CD169)-expressing MZ metallophilic macrophages (38) (Fig. 5A). IHC analysis of the MZ microarchitecture within the spleens of aged mice revealed that the distribution and density of the MAdCAM1-expressing sinus-lining cells, MZ macrophages, and MZ metallophilic macrophages were all severely disrupted compared to those of young mice (Fig. 5A, right-hand panels).
Fig 5.

Effect of host age on the microarchitecture of the splenic MZ. (A) In the spleens of young mice, MADCAM1-expressing sinus-lining cells form a barrier between the MZ and the white pulp (WP) (brown, arrows). RP, red pulp. Two distinct populations of macrophages also reside in the MZ: SIGNR1-expressing MZ macrophages and SIGLEC1/CD169-expressing MZ metallophilic macrophages (middle and lower panels, respectively; brown). In the spleens of aged mice, the distribution and density of the MAdCAM1-expressing sinus-lining cells, MZ macrophages, and MZ metallophilic macrophages were all severely disrupted compared to those in young mice (right-hand panels). Scale bar, 100 μm. (B) In the spleens of young mice, abundant CD1d-expressing MZ B cells (upper panels, green, arrow; lower panels, brown, arrow) were present within the MZ and B-cell follicles (Fol). In the spleens of aged mice, the distribution of MZ B cells appeared disrupted. Upper panel, scale bar, 50 μm; lower panel, scale bar, 100 μm. (C) Mice were passively immunized with preformed PAP immune complexes, and 24 h later, the presence of immune complexes (green) upon FDCs (CR1/CD35+ cells, red) was assessed by IHC. Scale bar, 50 μm (n = 6 spleens/group). (D) Morphometric analysis confirmed that the level of immune complexes colocalized upon the surfaces of FDCs in spleens of aged mice was significantly lower than that observed in spleens from young mice (P < 0.014).
In mice, the MZ B cells can be readily discriminated by their expression of high levels of the nonclassical major histocompatibility complex molecule CD1d (7, 36). In the spleens of young mice, abundant CD1d-expressing MZ B cells were present within the MZ and B-cell follicles, whereas in the spleens of aged mice, their distribution appeared disrupted (Fig. 5B). Since the shuttling of MZ B cells between the MZ and B-cell follicles provides an efficient mechanism for the delivery of systemic antigen to FDCs (7), we reasoned that the disrupted MZ microarchitecture in aged mice might impair immune complex trapping by FDCs. To test this hypothesis, mice were passively immunized with preformed PAP immune complexes, and 48 h later, the presence of FDC-associated immune complexes was identified by IHC. Consistent with our observation of reduced complement component C4 accumulation upon aged FDCs (Fig. 4A and B), the magnitude of immune complex trapping detected upon FDCs in the spleens of aged mice was also significantly reduced compared to that of young mice (Fig. 5C and D) (P = 0.014; n = 40/group). These data clearly show that the effects of host age on the microarchitecture of the splenic MZ significantly impair the delivery and retention of immune complexes upon FDCs. TSE agents are likewise considered to be initially acquired by FDCs as complement-bound complexes (26, 30, 31, 50). Thus, these data imply that the impaired scrapie pathogenesis in aged mice after intravenous exposure is likewise a consequence of the reduced transport of TSE agent containing complement-bound complexes from the MZ to FDCs.
DISCUSSION
Here we show that in aged mice as in young adult mice (24), the i.v. route is more efficient than other peripheral routes of TSE agent exposure. However, disease pathogenesis in i.v. exposed aged mice is significantly impaired compared to that in young mice, with most failing to develop clinical disease during their life spans. While multiple factors may contribute to the effects of host age on TSE susceptibility, we consider the major influence is most likely due to disturbances to the microarchitecture of the splenic MZ which adversely affect the transport and deposition of immune complexes on FDCs. As a consequence, the ability of FDCs to trap and accumulate complement-bound TSE agents after i.v. exposure may also be dramatically reduced, impairing disease pathogenesis. This hypothesis is consistent with the demonstration that aging had no effect on Prnp expression in the brain or TSE disease incubation period and susceptibility when the infection was established directly in the CNS. Indeed, previous data show that the requirement for amplification upon FDCs prior to neuroinvasion is bypassed when the TSE agent is injected directly into the brain (3, 32–34).
However, some aged mice did develop clinical disease, and the majority showed evidence of PrPSc in their brains. We cannot exclude the possibility that if these mice had survived much longer they all would have eventually developed clinical disease, but it is important to note that the clinically negative i.v. injected mice were between 790 and 917 days old when they were culled. Although the MZs in the majority of the spleens of the aged mice were severely disrupted, and their FDCs lacked PrPc expression and displayed a reduced capacity to retain complement-opsonized immune complexes, small numbers of intact PrPc-expressing FDC networks were observed. When we measured TSE agent accumulation in the spleens of aged mice, the levels were variable. For example, one aged spleen contained at least 100× less agent infectivity than that observed in young mice. After peripheral exposure, many host factors will aid the elimination of the initial inoculum from the host. Following oral exposure, much of the inoculum will be excreted or digested by enzymes in the gastrointestinal tract. After i.v. exposure, it is plausible that a larger proportion of the initial inoculum is delivered intact to the spleen to establish infection upon FDCs. Thus, the few remaining PrPC-expressing FDCs in the spleens of the aged mice at the time of exposure were most likely sufficient to eventually amplify the TSE agent above the threshold required for neuroinvasion, causing clinical disease in a small number of mice but extending the disease incubation period in most instances beyond the life span of the mouse.
Despite the evidence of PrPSc accumulation in the brains of most i.v. injected aged mice at the end of the experiment, its presence in the spleens of the same mice was variable. Our data imply that in aged mice, the TSE agent initially accumulated upon the remaining PrPC-expressing FDCs, facilitating subsequent neuroinvasion (albeit at a much slower rate than in young mice), but was most likely later cleared from the spleen by macrophages as the status of the FDCs declined. The lack of direct correlation between the presence of PrPSc in the brain and spleen of the i.v. injected aged mice has important practical implications as there are currently no reliable TSE-specific preclinical diagnostics available. The detection of PrPSc in lymphoid tissue biopsy specimens such as tonsils and appendix has been proposed as a useful preclinical assay to help predict the incidence of vCJD infection in the human population (8, 10, 20, 23). However, as PrPd was absent in the spleens of many i.v. injected aged mice, despite its presence in the brains of the same animals, these data suggest that tests based on the detection of the TSE agent in lymphoid tissues may be much less sensitive when used on samples from aged individuals.
MZ B cells continuously shuttle between the MZ and B-cell follicles and play an important role in the capture and transport of blood-borne, complement-bound antigens to FDCs (7, 36). Our data show that aging adversely affected the MZ microarchitecture and as a consequence substantially reduced the delivery of immune complexes to FDCs. TSE agents are likewise considered to be initially acquired by FDCs as complement-bound complexes (26, 30, 31, 50). These data suggest the effects of aging on i.v. TSE pathogenesis are most likely due to the reduced transport of complement-opsonized TSE agents from the MZ to FDCs. The mechanisms by which aging affects the status of the splenic MZ are not known. B cells play an important role in the organization and maintenance of the MZ (9, 41, 46). Whether aging adversely affects the expression of chemokines and cytokines, such as lymphotoxins (46), which influence the expression of adhesion molecules by reticular fibroblasts and MZ sinus-lining cells remains to be determined.
Our data show that FDCs express high levels of PrPC and do not simply passively acquire it from other cell types—for example, via exosomes, which have been shown to permit intercellular transfer of both PrPC and PrPSc (14). In the spleens of aged mice, most FDCs lacked expression of PrPC. The aging-related factors responsible for the downregulation of PrPC expression by FDCs are uncertain. Data suggest that PrPC expression by FDCs is upregulated by immune complex trapping (29). In the absence of complement component C1q, the upregulation of PrPC could not be provoked. The reduced expression of PrPC by aged FDCs may likewise be a consequence of their dramatically reduced ability to trap complement-opsonized immune complexes. Of course, we cannot exclude the possibility that the reduced expression of PrPC and C4 on FDCs was in part due to death of a fraction of the aged FDCs. However, our analysis of CD35 (CR1) expression suggested no observable difference in the density of the FDCs between the two aged groups.
In conclusion, we show that in aged mice as in young adult mice, the i.v. route is more efficient than other peripheral routes of TSE agent exposure. However, disease pathogenesis in i.v. injected aged mice is significantly impaired compared to that in young mice, with most failing to develop clinical disease during their life spans. Together, these data suggest that the effects of age on the pathogenesis of i.v. TSE infection may lead to significant levels of subclinical TSE disease in the population. Our data suggest that the impaired TSE pathogenesis in the spleens of aged mice after i.v. exposure is most likely a consequence of aging-related disturbances in the MZ that dramatically impair the transport of complement-opsonized TSE agents from the MZ to FDCs. This study is the first to show that the effects of aging on the microarchitecture and function of the splenic MZ significantly influence the pathogenesis of an important pathogen.
Supplementary Material
ACKNOWLEDGMENTS
We thank Irene McConnell, Fraser Laing, Simon Cumming, Bob Fleming, and the Pathology Services Group [The Roslin Institute and R(D)SVS, University of Edinburgh, United Kingdom] for excellent technical support, Christine Farquhar [The Roslin Institute & R(D)SVS] for provision of pAb 1B3; Caroline McCorquodale [The Roslin Institute and R(D)SVS] for statistical advice, Graham Anderson and Andrea White (MRC Centre for Immune Regulation, University of Birmingham, United Kingdom) for embryonic spleen transplantation expertise, and Daniel Mitchell (University of Warwick, United Kingdom) for helpful advice.
This work was supported grant funding from the European Commission (FP7 project no. 222887: PRIORITY) and by project (BB/526429-1) and Institute Strategic Programme Grant funding from the Biotechnology and Biological Sciences Research Council.
Footnotes
Published ahead of print 26 October 2011
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Andreoletti O., et al. 2000. Early accumulation of PrP[sup]Sc in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 81:3115–3126 [DOI] [PubMed] [Google Scholar]
- 2. Boelle P-Y, Cesbron J-Y, Valleron A-J. 2004. Epidemiological evidence of higher susceptibility to vCJD in the young. BMC Infect. Dis. 4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown KL, et al. 1999. Scrapie replication in lymphoid tissues depends on PrP-expressing follicular dendritic cells. Nat. Med. 5:1308–1312 [DOI] [PubMed] [Google Scholar]
- 4. Brown KL, Wathne GJ, Sales J, Bruce ME, Mabbott NA. 2009. The effects of host age on follicular dendritic cell status dramatically impair scrapie agent neuroinvasion in aged mice. J. Immunol. 183:5199–5207 [DOI] [PubMed] [Google Scholar]
- 5. Bruce ME, et al. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389:498–501 [DOI] [PubMed] [Google Scholar]
- 6. Chatfield C, Collins AJ. 1986. Introduction to Multivariate analysis (revised edition). Chapman & Hall, London, United Kingdom [Google Scholar]
- 7. Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr., Cyster JG. 2008. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 9:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clewley JP, et al. 2009. Prevalence of disease related prion protein in anonymous tonsil specimens in Britain: cross sectional opportunistic survey. BMJ 338:b1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crowley MT, Reilly CR, Lo D. 1999. Influence of lymphocytes on the presence and organization of dendritic cell subsets in the spleen. J. Immunol. 163:4894–4900 [PubMed] [Google Scholar]
- 10. de Marco MF, Linehan J, Gill ON, Clewley JP, Brandner S. 2010. Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J. Pathol. 222:380–387 [DOI] [PubMed] [Google Scholar]
- 11. Denzer K, Kleijmeer MJ, Heijnen HFG, Stoorvogel W, Geuze HJ. 2000. Exosome: from internal vesicle of the multivesicular body to intercellular signalling device. J. Cell Sci. 113:3365–3374 [DOI] [PubMed] [Google Scholar]
- 12. Denzer K, et al. 2000. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 165:1259–1265 [DOI] [PubMed] [Google Scholar]
- 13. Farquhar CF, Somerville RA, Ritchie LA. 1989. Post-mortem immunodiagnosis of scrapie and bovine spongiform encephalopathy. J. Virol. Methods 24:215–222 [DOI] [PubMed] [Google Scholar]
- 14. Fevrier B, et al. 2004. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U. S. A. 101:9683–9688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer M, et al. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15:1255–1264 [PMC free article] [PubMed] [Google Scholar]
- 16. Fraser H, Dickinson AG. 1968. The sequential development of the brain lesions of scrapie in three strains of mice. J. Comp. Pathol. 78:301–311 [DOI] [PubMed] [Google Scholar]
- 17. Glanville SH, et al. 2009. Transplantation of embryonic spleen tissue reveals a role for adult non-lymphoid cells in initiating lymphoid tissue organization. Eur. J. Immunol. 39:280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Health Protection Agency 17 February 2009, posting date. vCJD abnormal protein found in a patient with haemophilia at post mortem. Health Protection Agency, London, United Kingdom: http://www.hpa.org.uk/cjd [Google Scholar]
- 19. Hilton D, Fathers E, Edwards P, Ironside J, Zajicek J. 1998. Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. Lancet 352:703–704 [DOI] [PubMed] [Google Scholar]
- 20. Hilton DA, et al. 2004. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J. Pathol. 203:733–739 [DOI] [PubMed] [Google Scholar]
- 21. Ierna MI, Farquhar CF, Outram GW, Bruce ME. 2006. Resistance of neonatal mice to scrapie is associated with inefficient infection of the immature spleen. J. Virol. 80:474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Inman CF, et al. 2005. Validation of computer-assisted, pixel-based analysis of multiple-colour immunofluorescence histology. J. Immunol. Methods 302:156–167 [DOI] [PubMed] [Google Scholar]
- 23. Ironside JW, et al. 2006. Variant Creutzfeldt-Jakob disease: prion protein genotype analysis of positive appendix tissue samples from a retrospective prevalence study. Br. Med. J. 332:1186–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kimberlin RH, Walker CA. 1978. Pathogenesis of mouse scrapie: effect of route of inoculation on infectivity titres and dose-response curves. J. Comp. Pathol. 88:39–47 [DOI] [PubMed] [Google Scholar]
- 25. Klein MA, et al. 1998. PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat. Med. 4:1429–1433 [DOI] [PubMed] [Google Scholar]
- 26. Klein MA, et al. 2001. Complement facilitates early prion pathogenesis. Nat. Med. 7:488–492 [DOI] [PubMed] [Google Scholar]
- 27. Legname G, et al. 2004. Synthetic mammalian prions. Science 305:673–676 [DOI] [PubMed] [Google Scholar]
- 28. Llewelyn CA, et al. 2004. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363:417–421 [DOI] [PubMed] [Google Scholar]
- 29. Lötscher M, Recher M, Hunzinker L, Klein MA. 2003. Immunologically induced. complement-dependent up-regulation of the prion protein in the mouse spleen: follicular dendritic cells versus capsule and trabeculae. J. Immunol. 170:6040–6047 [DOI] [PubMed] [Google Scholar]
- 30. Mabbott NA, Bruce ME. 2004. Complement component C5 is not involved in scrapie pathogenesis. Immunobiology 209:545–549 [DOI] [PubMed] [Google Scholar]
- 31. Mabbott NA, Bruce ME, Botto M, Walport MJ, Pepys MB. 2001. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat. Med. 7:485–487 [DOI] [PubMed] [Google Scholar]
- 32. Mabbott NA, Mackay F, Minns F, Bruce ME. 2000. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 6:719–720 [DOI] [PubMed] [Google Scholar]
- 33. Mabbott NA, et al. 2000. Tumor necrosis factor-alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J. Virol. 74:3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mabbott NA, Young J, McConnell I, Bruce ME. 2003. Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J. Virol. 77:6845–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manson JC, et al. 1994. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 8:121–127 [DOI] [PubMed] [Google Scholar]
- 36. Martin F, Kearney JF. 2002. Marginal-zone B cells. Nat. Rev. Immunol. 2:323–335 [DOI] [PubMed] [Google Scholar]
- 37. McBride P, Eikelenboom P, Kraal G, Fraser H, Bruce ME. 1992. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J. Pathol. 168:413–418 [DOI] [PubMed] [Google Scholar]
- 38. Mebius RE, Kraal G. 2005. Structure and function of the spleen. Nat. Rev. Immunol. 5:606–616 [DOI] [PubMed] [Google Scholar]
- 39. Montrasio F, et al. 2000. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 288:1257–1259 [DOI] [PubMed] [Google Scholar]
- 40. National CJD Surveillance Unit 2009. Creutzfeldt-Jakob disease surveillance in the UK. Seventeenth Annual Report. 2008. National CJD Surveillance Unit, Western General Hospital, Edinburgh, United Kingdom: www.cjd.ed.ac.uk [Google Scholar]
- 41. Nolte MA, et al. 2004. B cells are crucial for both development and maintenance of the splenic marginal zone. J. Immunol. 172:3620–3627 [DOI] [PubMed] [Google Scholar]
- 42. Outram GW, Dickinson AG, Fraser H. 1973. Developmental maturation of susceptibility to scrapie in mice. Nature 241:536–537 [DOI] [PubMed] [Google Scholar]
- 43. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. 2004. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364:527–529 [DOI] [PubMed] [Google Scholar]
- 44. Schulz-Schaeffer WJ, et al. 2000. The paraffin-embedded tissue blot detects PrP[sup]sc early in the incubation time in prion diseases. Am. J. Pathol. 156:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sigurdson CJ, et al. 1999. Oral transmission and early lymphoid tropism of chronic wasting disease PrP[sup]res in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 80:2757–2764 [DOI] [PubMed] [Google Scholar]
- 46. Tumanov AV, et al. 2002. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity 239:239–250 [DOI] [PubMed] [Google Scholar]
- 47. Vandesompele J, et al. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:research0034–research0034.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wroe SJ, et al. 2006. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 368:2061–2067 [DOI] [PubMed] [Google Scholar]
- 49. Yates F. 1984. Tests of significance for 2x2 contingency tables. J. R. Stat. Soc. Series A 147:426–463 [Google Scholar]
- 50. Zabel MD, et al. 2007. Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J. Immunol. 179:6144–6152 [DOI] [PubMed] [Google Scholar]
- 51. Zahn JM, et al. 2007. AGEMAP: a gene expression database for aging mice. PLoS Genet. 3:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.