

Abstract
Osteogenesis imperfecta (OI) is caused by dominant mutations in the type I collagen genes. In principle, the skeletal abnormalities of OI could be treated by transplantation of patient-specific, bone-forming cells that no longer express the mutant gene. Here, we develop this approach by isolating mesenchymal cells from OI patients, inactivating their mutant collagen genes by adeno-associated virus (AAV)-mediated gene targeting, and deriving induced pluripotent stem cells (iPSCs) that were expanded and differentiated into mesenchymal stem cells (iMSCs). Gene-targeted iMSCs produced normal collagen and formed bone in vivo, but were less senescent and proliferated more than bone-derived MSCs. To generate iPSCs that would be more appropriate for clinical use, the reprogramming and selectable marker transgenes were removed by Cre recombinase. These results demonstrate that the combination of gene targeting and iPSC derivation can be used to produce potentially therapeutic cells from patients with genetic disease.
Introduction
Osteogenesis imperfecta (OI) or brittle bone disease is an excellent candidate disease for an induced pluripotent stem cells (iPSC)-based therapeutic approach. Severe, life-threatening forms of OI are incurable and typically caused by dominant mutations in COL1A1 or COL1A2 that disrupt the Gly-X-Y repeat and alter the collagen triple helix even when mutant and wild-type protein chains are mixed.1 Thus an effective treatment requires either the elimination or correction of the mutant collagen alleles in bone-forming cells, rather than the delivery of additional wild-type collagen genes. Because heterozygous null mutations in COL1A1 and COL1A2 result in minimal or no disease symptoms,2,3 conversion of the mutant allele to a null form would be therapeutic. We previously showed that the mutant collagen genes in mesenchymal stem cells (MSCs) from OI patients could be disrupted by gene targeting.4,5 However, this process requires significant expansion of cells with limited proliferative capacity, and the pool of senescing, gene-targeted MSCs produced cannot provide sufficient cell numbers for transplantation and engraftment. In clinical studies of OI, transplantation of MSCs resulted in limited engraftment that did not persist.6
iPSCs derived from somatic human cells7,8 may eventually be used in autologous cell transplantation therapies without the risk of graft rejection due to allogeneic histocompatibility factors. Because iPSCs can be expanded to large numbers before in vitro differentiation and transplantation, they have the potential to overcome the limitations seen with MSCs. In addition, human ESCs can be differentiated down the mesenchymal and osteogenic lineages in vitro,9,10 suggesting that iPSCs should also have osteogenic potential. For this paradigm to succeed, patient-specific OI iPSCs must first be engineered to provide appropriate expression of the wild-type collagen genes, and the pluripotent iPSCs must be differentiated down the MSC lineage to produce transplantable cells. Although experiments with human iPSCs have shown that aspects of other genetic diseases can be corrected by delivering the relevant gene with viral vectors in a gene addition strategy,11,12 this approach is not practical for OI due to the dominant negative effects of the mutant protein. Engineering the mutant OI cells by gene targeting would allow for inactivation of these dominant negative mutations and ensure that only wild-type collagen expression occurs.
Recently, mutant disease-causing genes have been targeted in human iPSCs by using electroporated human bacterial artificial chromosome constructs or helper-dependent adenoviral vectors.13,14 These approaches required the assembly of large targeting cassettes, multiple rounds of selection, and/or the screening of many independent clones. In the case of OI, gene targeting is even more problematic due to the lack of expression of COL1A1 and COL1A2 in pluripotent cells. Here, we develop an alternative strategy based on gene targeting with adeno-associated virus (AAV) vectors before iPSC derivation, which allows us to minimize the screening of iPSC clones (Figure 1). In this approach, mesenchymal cells are isolated from OI patients, an AAV gene-targeting vector is used to inactivate their mutant collagen genes, iPSCs are derived from these gene-targeted cells with a floxed, polycistronic reprogramming vector, all vector-encoded transgenes are deleted with Cre recombinase, the iPSCs are differentiated into mesenchymal and osteogenic cells in vitro, which produce bone in vivo after transplantation. Here, we show that each of these steps can be accomplished.
Figure 1.

Stem cell engineering strategy for osteogenesis imperfecta (OI). Schematic representation of proposed therapeutic approach for OI based on autologous, gene-targeted, transgene-free, pluripotent stem cells.
Results
Derivation of iPSCs from gene-targeted OI MSCs
For reprogramming we used either a previously described combination of four lentiviral (LV) vectors expressing OCT4, SOX2, LIN28, or NANOG,8 or floxed, polycistronic foamy virus (FV) vectors expressing OCT4, SOX2, KLF4, and MYC separated by peptide cleavage signals (Supplementary Figure S1a) as used in other polycistronic vectors.15 FV vectors are integrating retroviral vectors with a large packaging capacity that efficiently transduce human MSCs.16 A comparison of several FV vectors showed that an internal murine leukemia virus long-terminal repeat (LTR) promoter was most effective at reprogramming normal human fibroblasts (Supplementary Figure S1b), and the efficiency was further improved by including a short hairpin RNA cassette directed against the mRNA encoding the p53 protein as noted by others.17 This latter vector (ΔΦ53MOSKMETNW) was as efficient at producing iPSCs as the four LV vector combination. To generate OI iPSCs, we infected MSCs from several OI patients with these reprogramming vectors, cultured the cells under ESC conditions, then picked and expanded clonal iPSC lines. A total of 75 independent iPSC lines were derived from six individuals with different collagen mutations (Table 1). The different MSC cultures had a range of reprogramming frequencies of up to 10−4 that did not clearly correlate with patient age or the length of time in culture before reprogramming (Supplementary Table S1).
Table 1. I iPSC lines.
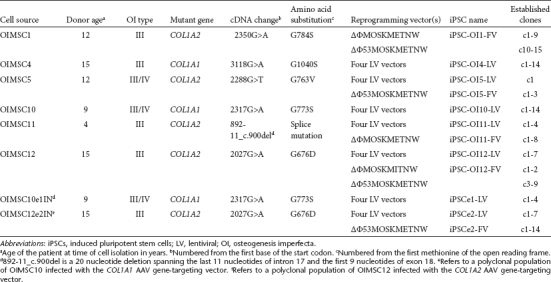
Gene-targeted iPSCs were derived from two OI MSC lines (OIMSC10 and OIMSC12 with COL1A1 and COL1A2 mutations respectively; Table 1). Each MSC line was infected with an AAV gene-targeting vector designed to knockout collagen expression by insertion of an IRES-neo-polyadenylation signal (pA) cassette in exon 1 of COL1A1 or exon 2 of COL1A2 (Figure 2a and Supplementary Figure S2). OI MSCs were infected with these AAV vectors and selected with G418 to produce polyclonal populations for subsequent iPSC derivation. iPSC lines were established after infection with either the four LV vectors or FV vector ΔΦ53MOSKMETNW (Table 1) and verified to be independent based on their provirus integration pattern (Supplementary Table S2 and data not shown). The reprogramming frequencies were two to tenfold lower than when parental, untargeted OI MSCs were infected with the same vectors (Table 1), which presumably reflects the additional culture period before iPSC derivation. Southern blot analysis showed that three of the four iPSC lines derived from OIMSC10 were targeted at one COL1A1 allele (Supplementary Figure S2), and 16 of 21 iPSC lines derived from OIMSC12 were targeted at one COL1A2 allele (Figure 2a and data not shown). Probing for neo sequences showed that none of these targeted clones contained random integrants, whereas the untargeted lines did (data not shown). Either the mutant or wild-type allele was targeted as determined by sequencing the mutation sites in complementary DNA (cDNA) prepared from iPSC-derived embryoid bodies (Figure 2b and Supplementary Table S2).
Figure 2.

Induced pluripotent stem cells (iPSCs) derived from gene-targeted mesenchymal stem cells (MSCs). (a) Southern blot identification of iPSC clones targeted at COL1A2. EV, EcoR V; neo, neomycin resistance gene; pA, polyadenylation site; S, stop codons; L, loxP site; OI12, parental OIMSC12 culture; Poly, polyclonal G418-resistant MSCs transduced with AAV vector. (b) Identification of targeted allele by complementary DNA (cDNA) sequencing in two gene-targeted clones, with controls of untransduced OI12 iPSCs (iPSC-OI12-FVc3) and a random integrant. TW, wild-type allele targeted; TM, mutant allele targeted. (c) Pluripotency gene expression as measured by microarray. (d) Teratoma assay of iPSCe2-FVc3. H&E, hematoxylin and eosin; DAPI, 4,6-diamidino-2-phenylindole; MAP-2, microtubule associated protein-2; AFP, α-fetoprotein; SMA, α-smooth muscle actin. Bar = 50 µm.
The COL1A2-targeted iPSCs were chosen for further analysis. Transcriptional profiling of four iPSC lines showed they had a gene expression pattern similar to that of human ESCs (Supplementary Figure S3), and had activated pluripotency genes silent in the parental MSCs (Figure 2c). Two lines were tested for teratoma formation and shown to have trilineage developmental potential (Figure 2d and Supplementary Figure S4). We were concerned that the anti-p53 cassette in the FV reprogramming vector might lead to genomic instability, but two of the four iPSCe2-FV lines analyzed had cells with normal karyotypes (Supplementary Figure S5). Three of the four iPSCe2-FV lines had silenced the FV vector reprogramming cassette, whereas both iPSCe2-LV clones tested expressed the LV vector-encoded OCT4, SOX2, and NANOG transgenes (Supplementary Figure S6).
Mesenchymal differentiation of gene-targeted iPSCs
Mesenchymal cells were generated from four independent iPSC lines by embryoid body formation, culturing on gelatin-coated dishes, then passaging with trypsin and culturing in the presence of fetal bovine serum as described for human ESCs.9 These iPSC-derived MSCs (iMSCs) expressed type I collagen in a cellular pattern similar to OI MSCs (Figure 3a). The collagen proteins synthesized in iMSCs targeted at the mutant COL1A2 allele were not overmodified by hydroxylases as typically observed in OI cells.18 This overmodification slows the migration of both α1(I) and α2(I) peptides (encoded by COL1A1 and COL1A2 genes, respectively) during gel electrophoresis, as seen when comparing collagen from iMSCs targeted at the mutant allele to that from wild-type MSCs, OI MSCs, and iMSCs targeted at the wild-type allele or containing a random AAV vector integrant (Figure 3b). Similarly, the collagen produced by iMSCs targeted at the mutant allele had normal structural integrity as determined by its thermostability when treated with proteases (Figure 3c and Supplementary Figure S7). These findings demonstrate that disruption of the mutant allele by gene targeting results in normal collagen production by iMSCs.
Figure 3.

Mesenchymal differentiation of induced pluripotent stem cells (iPSCs). (a) Collagen expression detected by immunohistochemistry with anti-human α2 type I procollagen antibody in mesenchymal stem cells (MSCs) and iMSCs. Bar = 100 µm. (b) Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) separation of [3H]-proline-labeled collagen peptides from MSCs and iMSCs targeted at the wild-type allele (TW), mutant allele (TM) or containing a random AAV vector integrant (Ran). (c) Thermostability curves of collagen produced by MSCs and iMSCs. (d) Flow cytometry analysis of MSC surface markers with isotype controls filled in black. (e) Hierarchical clustering dendrogram of global gene expression profiles. Height is Euclidean distance. (f) Percent of cells expressing senescence-associated β-galactosidase (senescence assay) or incorporating bromodeoxyuridine (proliferation assay) measured in parental MSCs (OIMSC12) at passage 4 (P4) or 7 (P7), three gene-targeted OIMSC12 clones, three gene-targeted iMSCe2-FV clones, and three gene-targeted iMSCe2-FV clones that had undergone transgene excision with Cre. (g) Mean terminal restriction fragment (TRF) length of cell lines from panel f (gene-targeted OIMSC12 clone #3 could not be expanded enough for analysis). Comparisons using Student's t-test are shown. kb, kilobase.
Gene-targeted iMSCs were similar to parental OI MSCs in their expression of mesenchymal cell surface markers (Figure 3d and Supplementary Table S3) with the notable exception of CD106, which was not expressed in iMSCs. Global transcriptional profiling and hierarchical clustering showed that all four iMSC lines were closely related, but could be distinguished from three parental MSC lines (Figure 3e). Genes upregulated in iMSCs as compared to MSCs included those involved in cell growth and replication, whereas downregulated genes were involved in extracellular protein production and cell–cell interactions (Supplementary Figure S8 and Supplementary Table S4). There were also differences in the expression of genes related to cell aging (Supplementary Figure S9). These findings suggested that iMSCs were less senescent than MSCs, which was confirmed by the decreased expression of senescence-associated β-galactosidase and increased proliferation rate of gene-targeted iMSCs in comparison to gene-targeted MSC clones (Figure 3f and Supplementary Figure S10). Importantly, gene-targeted iMSCs could be further subcloned and expanded after transgene removal (see below), without a significant change in senescence-associated β-galactosidase expression or proliferation rate (Figure 3f). In contrast, gene-targeted MSCs could not be cloned and expanded after transgene removal (data not shown). MSCs that undergo replicative senescence have progressive shortening of their telomeres,19 so we measured mean terminal restriction fragment lengths and found that the telomeres of gene-targeted MSCs were significantly shorter than those of both gene-targeted and transgene-free iMSCs (Figure 3g). Finally, the lack of senescence in iMSCs was supported by the expression levels of the 14 genes previously shown to be greater than fourfold upregulated with extended passage of MSCs,20 13 of which were downregulated in iMSCs (Supplementary Table S5).
Bone production by gene-targeted iPSCs
In order to produce bone, iPSCs must first differentiate into osteoblasts. This was achieved by differentiation into iMSCs as described above, followed by further culture in the presence dexamethasone, β-glycerol phosphate, and ascorbic acid. Gene-targeted iPSCs that underwent this osteogenic differentiation protocol expressed the osteogencic genes ALPL and RUNX2 as well as the osteoblast-specific gene BGLAP (Figure 4a),21 and they deposited minerals in vitro as shown by alizarin red staining for calcium and von Kossa staining for phosphate (Figure 4b and Supplementary Figure S11). When hydroxyapatite/tricalcium phosphate matrices were seeded with different lines of osteogenic iMSCs and implanted into immmunodeficient mice, all four cell lines tested produced bone based on staining with Van Gieson's picric acid fuchsin, and two of these lines were further analyzed and shown to express type I human collagen and the osteoblast marker sialoprotein in areas of bone containing osteocytes (Figure 4c and data not shown). Teratomas were not observed in these implants. Thus iPSCs can progress through the complete developmental program of embryoid body to MSC to osteoblast to terminally differentiated osteocyte. Bone was also found in teratomas produced by injecting undifferentiated gene-targeted iPSCs into the subrenal capsule of immunodeficient mice (Figure 4d and Supplementary Figure S12), demonstrating that the entire osteogenic differentiation process can also occur in vivo.
Figure 4.

iMSCs form bone. (a) RT-PCR of ALPL, RUNX2, and BGLAP genes in the indicated cell lines after osteogenic differentiation, with GAPDH control. (b) Alizarin red and Von Kossa staining of wild-type mesenchymal stem cells (MSCs) and iMSCs (iMSCe2-FVc2) grown with and without additional osteogenic differentiation. (c) Serial histological sections of hydroxyapatite/tricalcium phosphate matrices seeded with osteogenic iMSCe2-FVc4s and stained with Van Gieson's picric acid fuchsin, Masson's trichrome and antibodies against human bone sialoprotein and human type I collagen. Bar = 500 µm. Two high power views of the Masson's trichrome stain are included with arrow indicating osteocytes. Bar = 30 µm. (d) Serial sections of a teratoma derived from iPSCe2-FVc3 and stained as indicated showing bone formation. Bar = 100 µm. iPSCs, induced pluripotent stem cells.
We seeded implants with undifferentiated iPSCs or iPSCs cultured directly under osteogenic conditions without prior differentiation into iMSCs to see if a more simplified protocol could still produce bone as observed in some teratomas. These implants contained little or no bone, but produced teratomas instead, even when iPSCs were grown for 3 weeks in the presence of fetal bovine serum (Supplementary Figure S13). Therefore an extended period of in vitro differentiation and/or passage through an embryoid body and iMSC intermediate is critical for preventing teratoma formation.
Transgene removal from iPSCs
The gene-targeted iPSCs contain reprogramming vector transgenes as well as a neo cassette at the targeted collagen locus. These transgenes should be removed before clinical use to eliminate foreign antigen expression and prevent unwanted side effects of reprogramming factors on differentiation or tumor formation. The polycistronic FV reprogramming vectors contain loxP sites in each LTR (Figure 5a) to allow for Cre-mediated excision of all provirus genes, leaving behind a single deleted LTR with no promoter/enhancer activity.22 We infected iPSCe2-FV clones targeted at the mutant COL1A2 allele with a nonintegrating FV vector (NIFV) that transiently expresses Cre, isolated subclones without selection and screened for Cre-mediated excision events by Southern blots (Figure 5b). Each intact provirus produces two LTR-hybridizing fragments as compared to one fragment after excision. For example, iPSCe2-FV c6 has two proviruses and four LTR bands in Figure 5b (P1a, P1b, P2a, and P2b) that were converted to two excised proviruses with two LTR bands (E1 and E2). The floxed neo cassette in the targeted COL1A2 allele was also excised in the Cre-treated subclones that had excised their reprogramming vectors (Figure 5c,d) to generate transgene-free, gene-targeted iPSCs. Three stop codons and a pA signal remain in exon 2 of COL1A2 after neo cassette excision to prevent expression of the targeted collagen gene.
Figure 5.

Transgene removal from induced pluripotent stem cells (iPSCs). (a) Diagram of an integrated floxed reprogramming vector treated with a nonintegrating FV vector (NIFV-ECreW) expressing codon-optimized Cre to produce a single deleted long-terminal repeat (LTR) remnant. A, Avr II sites; E, EF1α promoter; W, WPRE. (b) Southern blot analysis of Avr II-digested genomic DNAs showing the removal of both integrated proviruses in iPSCe2-FVc6 subclones 4 and 5. P1a/b and P2a/b, nonexcised provirus LTR fragments; E1 and E2, excised provirus bands. (c) COL1A2 locus schematic showing untargeted, targeted and Cre-excised alleles with numbered exons. N, Nde I sites; ***, 3 stop codons. (d) Southern blot analysis demonstrating removal of the IRES-neo cassette in the genomic DNAs of Cre-treated iPSCe2-FVc5 and iPSCe2-FVc6 subclones.
Based on several experiments, ~30% of Cre-treated subclones underwent provirus excision (data not shown) and we isolated 11 fully excised subclones from three iPSCe2-FV clones, each of which contained two proviruses (Supplementary Table S2). These iPSC lines should have minimal genotoxicity, because all the mapped integrants were either outside genes or within introns (one provirus could not be mapped). In total, we determined the chromosomal locations of 27 reprogramming proviruses by inverse PCR in both OI and normal iPSC lines (Supplementary Table S2 and data not shown), and found that 11 integrants were outside genes, 15 were within introns, and 1 was in an exon. Two transgene-free clones were also differentiated into mesenchymal and osteogenic lineages and shown to form bone in vitro (Supplementary Figure S11).
Discussion
Here, we have shown that the deleterious mutations present in iPSCs derived from individuals with genetic diseases can be eliminated by gene targeting. We produced 75 different iPSC clones from the mesenchymal cells of six OI patients by using both LV and FV reprogramming vectors, thereby establishing an iPSC bank with different collagen mutations that will be useful for OI research. We also derived 25 iPSC lines from mesenchymal cells that had been transduced by AAV gene-targeting vectors, 19 of which were targeted at COL1A1 or COL1A2. In three iPSC subclones targeted at the mutant COL1A2 allele, we also removed all vector-encoded genes by Cre-mediated recombination.
Several techniques have been used for gene targeting in human ESCs or iPSCs, including plasmid electroporation,13,23,24 viral vectors,14,25,26,27 and zinc finger nucleases to stimulate recombination with site-specific double-strand breaks.28,29 Since COL1A1 and COL1A2 are expressed at low levels (if at all) in human iPSCs, we chose instead to target these genes in mesenchymal cells where they are highly expressed. This allowed us to use AAV promoter trap vectors, which efficiently target collagen genes in human mesenchymal cells without inducing potentially genotoxic double-strand breaks.4,5 By using this approach we generated polyclonal populations containing many independent targeted clones that could still be reprogrammed. AAV vectors can also target genes in human ESCs or iPSCs,25,26 but targeting before reprogramming may be especially useful for genes that are not expressed in pluripotent cells, which is the case for many disease-causing genes. A major advantage of the knockout approach we employed is that the same targeting vector can be used in multiple patients, because the homology arms do not need to overlap the causative mutations, making it an effective strategy for dominant diseases such as OI. Engineering the mutant OI cells by gene targeting would allow for inactivation of these dominant negative mutations and ensure that only wild-type collagen expression occurs. A similar strategy has been used to inactivate dominant mutations that cause epidermolysis bullosa in human keratinocytes.30
Our use of polycistronic, floxed reprogramming FV vectors was critical for efficiently generating iPSCs, even in cells that had been previously transduced and selected for gene targeting events. Eleven out of fourteen iPSCs derived with these vectors contained just one or two proviruses (Supplementary Table S2), simplifying subsequent Cre-mediated transgene excision. These FV vectors may have a less genotoxic integration site profile than analogous polycistronic LV vectors that have been used to reprogram human cells,12,31 because they do not preferentially integrate within genes,32 and the deleted LTR remnant produced by Cre-mediated excision has no promoter or enhancer activity.33 Ultimately, improvements in reprogramming methods based on nonintegrating vectors,34,35 RNA transfection,36,37 protein transduction,38 and/or chemical factors may eliminate the need for integrating vectors. An additional concern is the potential for iPSCs to acquire copy number variants and other mutations during reprogramming and extended culture.13,39 If iPSCs are ever to be used clinically, these types of abnormalities will need to be identified and their clinical significance determined.
Although human MSCs are able to proliferate significantly in vitro, their colony-forming and adipogenic potential decreases over time and they eventually senesce.40 In our experience, gene-targeted MSCs that underwent Cre-mediated transgene removal had little remaining proliferative capacity and could not be cloned or expanded, limiting the clinical utility of this approach. Because iMSCs are derived directly from immortal and pluripotent stem cells, they can overcome this constraint. The Cre recombination step is performed on undifferentiated, gene-targeted iPSCs, allowing for unlimited expansion before iMSC differentiation. In addition, the iMSCs derived from gene-targeted iPSCs are distinct from parental MSCs by several criteria, with important implications. First, iMSCs did not express the CD106 cell surface marker, which is also absent on MSCs isolated from adipose tissue,41 fetal lung,42 and amniotic fluid,43 consistent with the pluripotent origin of iMSCs as compared to bone-derived MSCs. Second, the global transcriptional profile of iMSCs was distinct from that of MSCs, and the most notable changes in gene expression levels were the opposite of those observed when comparing late-passage MSCs to early passage MSCs,20 suggesting that iMSCs may represent a type of MSC found earlier during development. Third, iMSCs were less senescent than MSCs based on senescence-associated β-galactosidase expression, proliferation rates, and telomere length. All these differences suggest that iMSCs will be more clinically useful than MSCs, with a greater capacity for clonal expansion, increased longevity, and possibly improved long-term engraftment with replacement of host MSC populations, which currently limits clinical MSC transplantation.6 Although it is concerning that iMSCs produced less extracellular matrix than bone-derived MSCs, the fact that iMSCs were able to differentiate into osteoblasts and produce bone in vivo demonstrates their clinical potential.
In summary, we have shown that the combination of gene targeting and iPSC derivation can be used to produce a population of patient-specific, expandable, bone-forming cells with normal collagen expression that may ultimately be useful for treating OI. We also removed all the reprogramming transgenes and selectable markers from these gene-targeted iPSCs, which will be necessary to avoid their unwanted effects on differentiation and oncogenicity, and to eliminate foreign antigens. While our use of OI MSCs to derive iPSCs could have provided residual epigenetic memory that favored subsequent mesenchymal differentiation,44 other cell types such as fibroblasts might produce similar results and be more easily accessible on a routine basis. Further research will be required to demonstrate the safety and efficacy of this approach, which has broad implications for regenerative medicine, since iPSC-derived cells may offer the same advantages when treating other genetic diseases where the appropriate adult stem cells cannot be isolated, genetically manipulated, and/or expanded adequately for transplantation.
Materials and Methods
Cell culture. Normal human fibroblasts (Coriell Institute for Medical Research, Camden, NJ repository number GM05387) were cultured at 37 °C in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) with 10% heat-inactivated fetal bovine serum (HyClone Laboratories, Logan, UT), 100 U/ml penicillin, and 100 µg/ml streptomycin. OI MSC lines were established from discarded bone fragments of affected individuals undergoing corrective surgery with institutional review board approval, and the mutations in these OI MSC lines were identified as described previously.5 OI MSCs and normal MSCs (Biowhittaker, Walkersville, MD) were grown at 37 °C in MSC medium consisting of Dulbecco's modified Eagle's medium with low glucose containing 10% characterized fetal bovine serum (HyClone Laboratories), 100 U/ml penicillin, 100 µg/ml streptomycin, and supplemented with 2 mmol/l -glutamine. Human iPSCs and ESCs were either cultured on irradiated mouse embryonic fibroblasts (MEFs) derived from the progeny of DR-4 mice45 crossed with CF-1 mice (Charles River Laboratories, Wilmington, MA) in Dulbecco's modified Eagle's medium/F12 medium supplemented with 20% knockout serum replacement (Invitrogen), 1% nonessential amino acid solution, 1% sodium pyruvate, 0.1 mmol/l β-mercaptoethanol, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 ng/ml basic fibroblast growth factor (ESC medium) as described8 or on ESC-qualified matrigel (BD Biosciences, Bedford, MA) in TeSR 2 medium (StemCell Technologies, Vancouver, BC).
Vectors. AAV gene-targeting vectors designed to disrupt the COL1A1 and COL1A2 genes were prepared as described previously.4,5 FV reprogramming vectors plasmids pΔΦEOSKMITNW, pΔΦCOSKMITNW, pΔΦMOSKMITNW, pΔΦMOSKMETNW, and pΔΦ53MOSKMETNW (corresponding vectors shown in Supplementary Figure S1) were derived from pΔΦ and vector stocks were prepared as described.16 The NIFV plasmid pNIFV-ECreW was constructed by replacing the MSCV promoter of pNIFV-MscvCre22 with an EF1α promoter and inserting a WPRE element downstream of Cre. NIFV vector stocks were prepared with an integrase mutant helper plasmid as described.22 LV vectors plasmids 16,579, 16,577, 16,578, and 16,580 expressing reprogramming factors were obtained from Addgene (Cambridge, MA), and stocks were prepared as described.46 Plasmid sequences are available on request.
Gene targeting. AAV-mediated gene targeting was performed by infecting 5 × 104 OI MSCs with an AAV targeting vector at an multiplicity of infection of 2,000 genome-containing particles/cell in one well of a 24-well plate, passaging the cells to two 10-cm dishes the next day, then selecting in 0.45 mg/ml of active G418 for 10 days to produce a pooled, polyclonal population of G418-resistant MSCs.
Generation of iPSCs. LV reprogramming experiments were conducted as previously described.25 For FV reprogramming experiments, 1 × 105 fibroblasts or MSCs were seeded per well of a 6-well plate in their somatic cell culture medium on day 0, and infected with FV reprogramming vector stocks on day 1 at a multiplicity of infection of 5 G418 transducing units/cell. On day 2, the cells were treated with trypsin and replated in 10-cm dishes preseeded with MEFs in their somatic cell culture medium. On day 4, the medium was changed to ESC culture medium and the cells were cultured for 10 additional days followed by culture in MEF-conditioned ESC medium. Colonies with ESC morphology were observed 20–30 days after transduction, isolated, and expanded for further characterization.
Karyotyping. Standard chromosome G-banding analysis was carried out in the Cytogenetics Laboratory of the Department of Pathology, University of Washington (Seattle, WA).
RNA isolation and analysis. Total RNA was extracted using RNeasy mini kit (Qiagen, Valencia, CA) with on-column DNAse digestion. ESCs and iPSCs were passaged three times on matrigel in TeSR II medium (StemCell Technologies) before RNA isolation and microarray analysis to eliminate MEF feeder cells. cDNA synthesis was performed from total RNA using the Superscript III First-Strand Synthesis System, as per the manufacturer's instructions (Invitrogen). PCR was performed with GoTaq Flexi DNA polymerase (Promega, Madison, WI) and 1 µg of cDNA per reaction. Targeted COL1A1 and COL1A2 iPSC clones were identified by sequencing PCR products amplified using cDNA primers that flank the known collagen mutations. Transcripts of marker genes RUNX2, ALPL, and BGLAP were detected with gene specific primers. LV vector transgene expression was determined using specific forward primers located in the OCT4, SOX2, NANOG, or LIN28 transgenes and a reverse primer located in the IRES element. FV vector transgene expression was detected using the Foamy-f and Foamy-r primers. All primer sequences are shown in Supplementary Table S6.
DNA isolation and analysis. Genomic DNA was isolated using Puregene DNA purification system (Gentra Systems, Minneapolis, MN). Southern blot analysis, plasmid preparation, and restriction digests were performed according to standard protocols. Radiolabeled probes were synthesized by random priming using Rediprime II (GE Healthcare, Piscataway, NJ). Telomere lengths were determined by the TeloTAGGG Telomere Length Assay kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions.
iPSC differentiation and characterization. Embryoid bodies were prepared as described25 and used to determine which collagen allele was targeted by cDNA sequencing. iMSCs were derived from four independent iPSC clones as described for human ESCs.9 iMSCs and OI MSCs were stained with the anti-human α2 type I procollagen antibody Pro-COL1A2 (Y-18; Santa Cruz Biotechnology, Santa Cruz, CA). Surface marker expression was detected using the Human Multipotent Mesenchymal Stromal Cell Marker Antibody Panel (R&D Systems, Minneapolis, MN) per manufacturer's instructions. Collagen protein analysis and melting curves were performed on iMSC and MSC proteins as described.18,47 iMSCS and MSCs were induced to form bone using osteogenic induction medium consisting of MSC medium plus dexamethasone, -ascorbic acid, and β-glycerol phosphate as described.48 Alizarin red and Von Kossa staining were performed as previously described.48,49 For in vivo bone formation, cells were seeded in hydroxyapatite/tricalcium phosphate ceramic-type I bovine fibrillar collagen matrices (Zimmer, Warsaw, IN) after in vitro differentiation then grown in immunodeficient mice for 8 weeks as previously described.4 Teratomas were grown in NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ mice (The Jackson Laboratory, Bar Harbor, ME) as described.50 Histological analysis of teratomas and implants included staining with hematoxylin and eosin, hematoxylin and Van Gieson's picric acid fuchsin, Von Kossa stain Masson's trichrome stain or antibodies against-human type I collagen monoclonal antibody (Sigma, St Louis, MO) or human bone sialoprotein (Millipore, Billerica, MA) following the manufacturers' instructions.
Transcriptional profiling array analysis. RNA samples were hybridized to HumanHT-12 v3 Expression BeadChip arrays (Illumina, San Diego, CA). Arrays were processed by the Fred Hutchinson Cancer Research Center's Genomics Shared Resource (Seattle, WA). The gene expression levels of all RefSeq genes were quantile normalized. Hierarchical clustering was performed by means of the Euclidean distance with average linkage method using TM4 software (http://www.tm4.org/). The similarity metric for comparison between different cell lines is indicated on the height of cluster dendrogram. Cell line comparisons were conducted using the Bioconductor package “limma” (http://bioconductor.org/) by calculating the 75th percentile of negative controls for each array and using this as a minimum signal intensity threshold value to be applied to each array. For probe sets considered detectable, microarray data normalized to the median and log2 ratios were calculated for iMSCs versus MSCs. Genes that were more than twofold up- or downregulated in iMSCs were further classified by GeneOntology analysis using the David web server (http://david.abcc.ncifcrf.gov/).
Cellular senescence and proliferation assays. To determine cellular senescence, iMSCs or MSCs were plated at a density of 50,000 cells per well of a 12-well plate and allowed to attach overnight. The next day, cells were stained with bromo-chloro-indolyl-galactopyranoside at pH 6.0 using the Cellular Senescence Assay kit (Millipore) per the manufacturer's instructions. Staining for bromodeoxyuridine incorporation into proliferating cells was accomplished by plating iMSC or MSCs at a density of 20,000 cells per chamber of a Lab-Tek 4 chamber slide (Nalge Nunc International, Rochester, NY), allowing the cells to attach overnight, and incubating with bromodeoxyuridine at 10 µmol/l for 24 hours. The cells were then fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 minutes, washed once with PBS, permeabilized in 2 N hydrochloric acid for 20 minutes at 37 °C, and washed again in PBS. Cells were blocked in 10% rabbit serum plus 0.4% Triton X-100 for 60 minutes, and then stained with rat anti-bromodeoxyuridine antibody (1:100 dilution; Abcam, Cambridge, MA) overnight. The next day the cells were washed with PBS and stained with a rabbit anti-rat secondary antibody (Alexa 488; Millipore) for 60 minutes, washed with PBS, and then coverslips were mounted with VECTASHIELD mounting medium and DAPI (Vector Laboratories, Burlingame, CA).
Cre-mediated excision of transgenes. To generating transgene-excised iPSCs clones, 25,000 cells were plated per well of a 48-well plate preseeded with MEFs on day 1. On day 2, iPSCs were transduced with NIFV-ECreW at an multiplicity of infection of 5,000 vector genomes per cell. Medium was changed on day 3 and the cells were passaged to one well of a 12-well plate on day 6. The iPSC colonies were disaggregated into single cells using accutase (Stemgent, San Diego, CA) on day 8, counted and plated at densities of 1,000, 2,000, and 4,000 cells per well of a 6-well plate. Cells were grown in medium without selection and then individual colonies were picked 18–20 days later and expanded. Genomic DNA was extracted, digested with Avr II and analyzed by Southern blots.
SUPPLEMENTARY MATERIAL Figure S1 Reprogramming vectors. Figure S2 iPSCs derived from OI MSCs targeted at COL1A1. Figure S3 Global and pluripotency gene expression analysis of ESCs, iPSCs, MSCs, and iMSCs. Figure S4 Trilineage teratoma assay of iPSCe2-LVc4. Figure S5 Normal karyotypes of iPSCe2-FV cell lines. Figure S6 Expression of reprogramming transgenes in iPSCs. Figure S7 Collagen thermostability. Figure S8 Gene ontology analysis of differentially expressed genes in iMSCs compared to MSCs. Figure S9 Expression of cell aging genes. Figure S10 Senescence-associated β-galactosidase (β-Gal) staining and bromodeoxyuridine (BrdU) incorporation. Figure S11 Calcium and phosphate deposition by iMSCe2-FV clones. Figure S12 Gene-targeted iPSCs form bone in teratomas. Figure S13 Teratoma formation by differentiated iPSCs. Table S1 Reprogramming frequencies. Table S2 Gene-targeted iPSC lines. Table S3 Surface marker expression on iMSCs. Table S4 Genes with more than fourfold difference in expression levels in iMSCs vs MSCs. Table S5 Genes more than fourfold upregulated in senescent MSCs. Table S6 PCR primers.
Acknowledgments
This research was funded by NIH grants AR48328, AR48328-09S1, DK55759, GM86497, HL53750, and AR53917. We thank Erik Olson, Roli K. Hirata, and Yi Li at the University of Washington for technical assistance. D.W.R. is on the scientific advisory board of Horizon Discovery. The authors declared no conflict of interest.
Supplementary Material
Reprogramming vectors.
iPSCs derived from OI MSCs targeted at COL1A1.
Global and pluripotency gene expression analysis of ESCs, iPSCs, MSCs, and iMSCs.
Trilineage teratoma assay of iPSCe2-LVc4.
Normal karyotypes of iPSCe2-FV cell lines.
Expression of reprogramming transgenes in iPSCs.
Collagen thermostability.
Gene ontology analysis of differentially expressed genes in iMSCs compared to MSCs.
Expression of cell aging genes.
Senescence-associated β-galactosidase (β-Gal) staining and bromodeoxyuridine (BrdU) incorporation.
Calcium and phosphate deposition by iMSCe2-FV clones.
Gene-targeted iPSCs form bone in teratomas.
Teratoma formation by differentiated iPSCs.
Reprogramming frequencies.
Gene-targeted iPSC lines.
Surface marker expression on iMSCs.
Genes with more than fourfold difference in expression levels in iMSCs vs MSCs.
Genes more than fourfold upregulated in senescent MSCs.
PCR primers.
REFERENCES
- Byers PH.2000Disorders of collagen biosynthesis and structure Scriver CR, Beaudet AL, Sly WS., and, Valle, D.eds.). The Metabolic and Molecular Basis of Inherited Disease McGraw-Hill: New York; 5241–5286. [Google Scholar]
- Schwarze U, Hata R, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE.et al. (2004Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway Am J Hum Genet 74917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing MC, Deschenes SP, Scott DA, Byers PH, Slayton RL, Pitts SH.et al. (1994Osteogenesis imperfecta type I: molecular heterogeneity for COL1A1 null alleles of type I collagen Am J Hum Genet 55638–647. [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM.et al. (2004Gene targeting in stem cells from individuals with osteogenesis imperfecta Science 3031198–1201. [DOI] [PubMed] [Google Scholar]
- Chamberlain JR, Deyle DR, Schwarze U, Wang P, Hirata RK, Li Y.et al. (2008Gene targeting of mutant COL1A2 alleles in mesenchymal stem cells from individuals with osteogenesis imperfecta Mol Ther 16187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY.et al. (2002Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone Proc Natl Acad Sci USA 998932–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K.et al. (2007Induction of pluripotent stem cells from adult human fibroblasts by defined factors Cell 131861–872. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S.et al. (2007Induced pluripotent stem cell lines derived from human somatic cells Science 3181917–1920. [DOI] [PubMed] [Google Scholar]
- Hwang NS, Varghese S, Lee HJ, Zhang Z, Ye Z, Bae J.et al. (2008In vivo commitment and functional tissue regeneration using human embryonic stem cell-derived mesenchymal cells Proc Natl Acad Sci USA 10520641–20646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberi T, Willis LM, Socci ND., and, Studer L. Derivation of multipotent mesenchymal precursors from human embryonic stem cells. PLoS Med. 2005;2:e161. doi: 10.1371/journal.pmed.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya A, Rodríguez-Pizà I, Guenechea G, Vassena R, Navarro S, Barrero MJ.et al. (2009Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells Nature 46053–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM.et al. (2011Genomic safe harbors permit high ß-globin transgene expression in thalassemia induced pluripotent stem cells Nat Biotechnol 2973–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden SE, Gore A, Li Z, Fung HL, Nisler BS, Nie J.et al. (2011Genetic correction and analysis of induced pluripotent stem cells from a patient with gyrate atrophy Proc Natl Acad Sci USA 1086537–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Suzuki K, Qu J, Sancho-Martinez I, Yi F, Li M.et al. (2011Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs Cell Stem Cell 8688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Hanna J, Saha K, Gao Q, Mitalipova M.et al. (2009Reprogramming of murine and human somatic cells using a single polycistronic vector Proc Natl Acad Sci USA 106157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge G, Josephson N, Vassilopoulos G, Mac J., and, Russell DW. Improved foamy virus vectors with minimal viral sequences. Mol Ther. 2002;6:321–328. doi: 10.1006/mthe.2002.0672. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yin X, Qin H, Zhu F, Liu H, Yang W.et al. (2008Two supporting factors greatly improve the efficiency of human iPSC generation Cell Stem Cell 3475–479. [DOI] [PubMed] [Google Scholar]
- Bonadio J, Holbrook KA, Gelinas RE, Jacob J., and, Byers PH. Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J Biol Chem. 1985;260:1734–1742. [PubMed] [Google Scholar]
- Baxter MA, Wynn RF, Jowitt SN, Wraith JE, Fairbairn LJ., and, Bellantuono I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells. 2004;22:675–682. doi: 10.1634/stemcells.22-5-675. [DOI] [PubMed] [Google Scholar]
- Wagner W, Horn P, Castoldi M, Diehlmann A, Bork S, Saffrich R.et al. (2008Replicative senescence of mesenchymal stem cells: a continuous and organized process PLoS ONE 3e2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss PG, Closs EI, Schmidt J., and, Erfle V. Gene expression during osteogenic differentiation in mandibular condyles in vitro. J Cell Biol. 1990;110:1369–1378. doi: 10.1083/jcb.110.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyle DR, Li Y, Olson EM., and, Russell DW. Nonintegrating foamy virus vectors. J Virol. 2010;84:9341–9349. doi: 10.1128/JVI.00394-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Schuldiner M., and, Benvenisty N. Modeling for Lesch-Nyhan disease by gene targeting in human embryonic stem cells. Stem Cells. 2004;22:635–641. doi: 10.1634/stemcells.22-4-635. [DOI] [PubMed] [Google Scholar]
- Zwaka TP., and, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Wang PR, Li Y, Kho J, Nelson A.et al. (2010Engineering of human pluripotent stem cells by AAV-mediated gene targeting Mol Ther 181192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, Suzuki K, Aizawa E, Kawase E, Suemori H, Nakatsuji N.et al. (2009Gene targeting in human pluripotent stem cells with adeno-associated virus vectors Biochem Biophys Res Commun 388711–717. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Mitsui K, Aizawa E, Hasegawa K, Kawase E, Yamagishi T.et al. (2008Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors Proc Natl Acad Sci USA 10513781–13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA.et al. (2007Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery Nat Biotechnol 251298–1306. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC.et al. (2009Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases Nat Biotechnol 27851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petek LM, Fleckman P., and, Miller DG. Efficient KRT14 targeting and functional characterization of transplanted human keratinocytes for the treatment of epidermolysis bullosa simplex. Mol Ther. 2010;18:1624–1632. doi: 10.1038/mt.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG.et al. (2009Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors Cell 136964–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem HP, Kaul R.et al. (2006Foamy virus vector integration sites in normal human cells Proc Natl Acad Sci USA 1031498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrie PC, Huo Y, Stolitenko RB., and, Russell DW. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol Ther. 2008;16:534–540. doi: 10.1038/sj.mt.6300398. [DOI] [PubMed] [Google Scholar]
- Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II.et al. (2009Human induced pluripotent stem cells free of vector and transgene sequences Science 324797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaki N, Ban H, Nishiyama A, Saeki K., and, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad, Ser B, Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubov E, Rechavi G, Rozenblatt S., and, Givol D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem Biophys Res Commun. 2010;394:189–193. doi: 10.1016/j.bbrc.2010.02.150. [DOI] [PubMed] [Google Scholar]
- Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F.et al. (2010Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA Cell Stem Cell 7618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS.et al. (2009Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins Cell Stem Cell 4472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R.et al. (2011Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations Cell 146318–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digirolamo CM, Stokes D, Colter D, Phinney DG, Class R., and, Prockop DJ. Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br J Haematol. 1999;107:275–281. doi: 10.1046/j.1365-2141.1999.01715.x. [DOI] [PubMed] [Google Scholar]
- De Ugarte DA, Alfonso Z, Zuk PA, Elbarbary A, Zhu M, Ashjian P.et al. (2003Differential expression of stem cell mobilization-associated molecules on multi-lineage cells from adipose tissue and bone marrow Immunol Lett 89267–270. [DOI] [PubMed] [Google Scholar]
- Hua J, Yu H, Dong W, Yang C, Gao Z, Lei A.et al. (2009Characterization of mesenchymal stem cells (MSCs) from human fetal lung: potential differentiation of germ cells Tissue Cell 41448–455. [DOI] [PubMed] [Google Scholar]
- Sessarego N, Parodi A, Podestà M, Benvenuto F, Mogni M, Raviolo V.et al. (2008Multipotent mesenchymal stromal cells from amniotic fluid: solid perspectives for clinical application Haematologica 93339–346. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P.et al. (2010Epigenetic memory in induced pluripotent stem cells Nature 467285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Wang Y, Dausman J., and, Jaenisch R. A transgenic mouse strain expressing four drug-selectable marker genes. Nucleic Acids Res. 1997;25:3745–3746. doi: 10.1093/nar/25.18.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramezani A, Hawley TS., and, Hawley RG. Lentiviral vectors for enhanced gene expression in human hematopoietic cells. Mol Ther. 2000;2:458–469. doi: 10.1006/mthe.2000.0190. [DOI] [PubMed] [Google Scholar]
- Pace JM, Kuslich CD, Willing MC., and, Byers PH. Disruption of one intra-chain disulphide bond in the carboxyl-terminal propeptide of the proalpha1(I) chain of type I procollagen permits slow assembly and secretion of overmodified, but stable procollagen trimers and results in mild osteogenesis imperfecta. J Med Genet. 2001;38:443–449. doi: 10.1136/jmg.38.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger RL, Tucker AH., and, Wolfe MR. Differentiation and characterization of human MSCs. Methods Mol Biol. 2008;449:93–107. doi: 10.1007/978-1-60327-169-1_7. [DOI] [PubMed] [Google Scholar]
- Koch TG, Heerkens T, Thomsen PD., and, Betts DH. Isolation of mesenchymal stem cells from equine umbilical cord blood. BMC Biotechnol. 2007;7:26. doi: 10.1186/1472-6750-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharwan H, Hirata RK, Wang P, Richard RE, Wang L, Olson E.et al. (2007Transduction of human embryonic stem cells by foamy virus vectors Mol Ther 151827–1833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reprogramming vectors.
iPSCs derived from OI MSCs targeted at COL1A1.
Global and pluripotency gene expression analysis of ESCs, iPSCs, MSCs, and iMSCs.
Trilineage teratoma assay of iPSCe2-LVc4.
Normal karyotypes of iPSCe2-FV cell lines.
Expression of reprogramming transgenes in iPSCs.
Collagen thermostability.
Gene ontology analysis of differentially expressed genes in iMSCs compared to MSCs.
Expression of cell aging genes.
Senescence-associated β-galactosidase (β-Gal) staining and bromodeoxyuridine (BrdU) incorporation.
Calcium and phosphate deposition by iMSCe2-FV clones.
Gene-targeted iPSCs form bone in teratomas.
Teratoma formation by differentiated iPSCs.
Reprogramming frequencies.
Gene-targeted iPSC lines.
Surface marker expression on iMSCs.
Genes with more than fourfold difference in expression levels in iMSCs vs MSCs.
Genes more than fourfold upregulated in senescent MSCs.
PCR primers.