

Abstract
We describe an effective approach for the covalent immobilization of antimicrobial peptides (AMPs) to bioinert substrates via CuI-catalyzed azide–alkyne cycloaddition (CuAAC). The bioinert substrates were prepared by surface hydrosilylation of oligo-(ethylene glycol) (OEG) terminated alkenes on hydrogen-terminated silicon surfaces. To render the OEG monolayers “clickable”, mixed monolayers were prepared using OEG-alkenes with and without a terminal alkyne protected by a trimethylgermanyl (TMG) group. The mixed monolayers were characterized by X-ray photoelectron spectroscopy (XPS), elliposometry and contact angle measurement. The TMG protecting group can be readily removed to yield a free terminal alkyne by catalytic amounts of CuI in an aqueous media. This step can then be combined with the subsequent CuAAC reaction. Thus, the immobilization of an azide modified AMP (N3-IG-25) was achieved in a one-pot de-protection/coupling reaction. Varying the ratio of the two alkenes in the deposition mixture allowed for control over the density of the alkynyl groups in the mixed monolayer, and subsequently the coverage of the AMPs on the monolayer. These samples allowed for study of the dependence of antimicrobial activities on the AMP density. The results show that a relative low coverage of AMPs (~1.6×1013 molecule per cm2) is sufficient to significantly suppress the viability of Pseudomonas aeruginosa, while the surface presenting the highest density of AMPs (~2.8×1013 molecule per cm2) is still cyto-compatible. The remarkable antibacterial activity is attributed to the long and flexible linker and the site-specific “click” immobilization, which may facilitate the covalently attached peptides to interact with and disrupt the bacterial membranes.
Keywords: antibacterial activity, antimicrobial peptides, click chemistry, hydrosilylation, organic monolayer
Introduction
Despite the wide use of antibiotics and sterile procedures, biomaterials associated infection remains a major barrier to the long-term use of medical devices, especially for implants.[1,2] Infection starts with bacterial adhesion, followed by bacterial colonization and formation of biofilms that can serve as reservoirs to protect the bacteria from antibiotic remediation and other adverse environmental factors.[3,4] Consequently, there is a clear need to modify substrates with antimicrobial agents that prevent the colonization of bacteria while not being harmful to the host cells. In this article, we present a novel method for covalent attachment of antimicrobial peptides (AMPs) to model surfaces based on “click” reaction, and the study of their antibacterial activity and cytotoxicity.
AMPs are found in most organisms as a first line of defense against invading pathogens. These cationic peptides target bacteria with a negatively charged surface, and kill them by disruption of the cell membrane.[5–12] Compared to conventional antimicrobial agents, AMPs have several unique advantages, including a broad spectrum of antimicrobial activity, a low susceptibility of developing bacterial resistance and a short contact time to induce killing.[13–15] These unique properties have recently attracted research activities on covalent attachment of AMPs and their synthetic mimics onto various substrates.[16–32] The proposed mechanistic models suggest that the orientation and flexibility of the immobilized AMPs may affect their antimicrobial activities. Indeed, Gabriel et al. reported that the attachment of an AMP to titanium surfaces selectively at the N-terminal of the peptide via a flexible poly(ethylene glycol) spacer exhibited the highest bactericidal activity as compared to random attachment or without the use of flexible linkers.[27] However, most of the reported immobilization methods for AMPs containing multiple amino and carboxylic acid groups were based on amidation reaction which does not allow for control over the location and number of molecules binding to the substrate.[29–32] Moreover, the involvement of amino groups in the covalent modification reduces the net positive charge of the peptide, which may compromise the activity of the surface bound cationic AMP.[32] To enable the selective bonding, Glinel et al. have attached an AMP to maleimide-containing polymer brushes via site-specific thiol-maleimide immobilization.[28] This approach has the limitation that maleimide groups may not be compatible or difficult to introduce to some substrates, and the regioselectivity may not be realized for a large family of AMPs containing (multiple) internal cysteine residues.[33] Thus, there is a strong need to develop new methods for the facile covalent immobilization of AMPs in a chemo- and regioselective fashion. In this paper, we report a novel method to attach AMPs to a bioinert platform via site-specific “click” reaction.
The bioinert platform was prepared by surface hydrosilylation of oligo(ethylene glycol) (OEG) terminated alkenes on the hydrogen-terminated silicon substrate. We chose this platform because of its remarkable long-term stability under physiological conditions and the high resistance to non-specific adsorption of proteins.[34,35] To render the platform “clickable” for attaching azido-labeled biomolecules, we recently developed a method for selective hydrosilylation of α,ω-alkenynes on hydrogen-terminated silicon surfaces.[36] The selective attachment of the vinyl end to the surface was achieved by masking the alkyne with a trimethylgermanyl (TMG) protecting group as in compound TMG-EG10 (Scheme 1). The TMG protecting group can be rapidly removed in aqueous media with catalytic amounts of CuI.[37,38] Remarkably, this condition coincides with the subsequent CuI-catalyzed azide–alkyne cycloaddition (CuAAC, a “click” reaction).[39–43] Therefore, the deprotection and coupling steps can be combined in a one-pot reaction. We now use this strategy to attach large biomolecules (an AMP in the present study) in a site-specific manner to silicon substrates. Thus, the aim of the present work is twofold: first, to generate a mixed monolayer on silicon substrates with bioinert background presenting various density of alkynyl groups for “click” immobilization, and second, to use the immobilized AMP with controlled densities as a model to study the bioactivity of the surface bound AMPs.
Scheme 1.

Surface hydrosilylation using a mixture of the TMG-protected alkenyne TMG-EG10 and the alkene EG7 on H-terminated Si(111) surfaces, followed by attachment of IG-25 with an azido tag (N3-IG-25). The mole fraction of TMG-EG10 was 1.0, 0.4, 0.2, 0.1, 0.05 or 0, to afford the corresponding surface 1a–f, respectively. The corresponding peptide-presenting surfaces 2a–e are formed upon CuAAC reaction of N3-IG-25 with the TMG-alkyne surfaces 1a–e.
In our system, the AMP (N3-IG-25) to be immobilized consists of two parts: the peptide with antibacterial activity and a flexible inert OEG linker terminated with an azido group. To control the density of the AMP on surface, we prepared a series of mixed monolayers (1a–f) using the mixture of OEG-alkene (EG7) and OEG-alkenyne (TMG-EG10) with varied ratios. As illustrated in Scheme 1, AMP is then attached to the mixed monolayers via CuAAC reaction, leading to the surfaces (2a–e) presenting various density of IG-25. The mixed monolayers prepared from a given ratio of TMG-EG10/EG7 and the resulting surfaces presenting IG-25 upon CuAAC reaction are designated in Table 1.
Table 1.
Designaiton of the mixed monolayers prepared from the mixtures of TMG-EG10 and EG7 at various ratios, and the corresponding IG-25-presenting surface upon the CuAAC reaction with N3-IG-25.
| Code for the mixed monolayers | TMG-EG10/EG7 | Code for the IG-25 presenting surfaces |
|---|---|---|
| 1a | 100:0 | 2a |
| 1b | 40:60 | 2b |
| 1c | 20:80 | 2c |
| 1d | 10:90 | 2d |
| 1e | 5:95 | 2e |
| 1f | 0:100 | – |
The films were characterized by X-ray photoelectron spectroscopy (XPS), ellipsometry and contact angle measurement. The subsequent bioactivity tests demonstrated that the immobilized IG-25 exhibited a remarkable antibacterial activity (>80%) when the peptide density was above ~1.6×1013 cm2. Significantly, they were also cyto-compatible to mammalian cells.
Results and Discussion
Grafting of organic monolayer on the Si(111) surface
As depicted in Scheme 1, in order to prepare organic monolayers on Si(111) via Si–C bonds, the native SiO2 layer on the substrate is removed by etching with NH4F to form a hydrogen-terminated silicon. This surface then reacts with the alkenes by UV induced hydrosilylation. The apparatus for surface hydrosilylation were previously reported.[34] The main advantages of this setup are: 1) only very small amount of the reactants (~1 mg cm−2) are required; 2) the possibility of oxidation and contamination is greatly reduced by the high vacuum (<1×10−4 mbar) during the monolayer formation.
XPS spectrum of the single-component monolayer from neat TMG-EG10 was recorded. As expected, the survey spectra indicate the presence of Si, C, O and Ge elements (Figure S1, see Supporting Information).
In order to vary the density of the TMG-protected alkynyl groups on the monolayer platform, TMG-EG10 was pre-mixed with the inert EG7 in different molar ratios (Table 1) and allowed to react with the hydrogen-terminated silicon surfaces. All of the surfaces shown in Table 1 were prepared in the same apparatus under identical reaction conditions. Figure 1 demonstrates the non-linear relationship between the actual mole fractions of the two components in the monolayer (Xsurf) and the mole fraction in the deposition solution (Xsoln). The result was obtained according to the ratio of the integrated areas of deconvoluted C1s signals for etheric and alkyl/alkynyl carbon atoms. The equations used to calculate the mole fractions of TMG-EG10 in the mixed monolayers and the corresponding XPS spectra are given in the Supporting Information (Figure S2 and Table S1).
Figure 1.
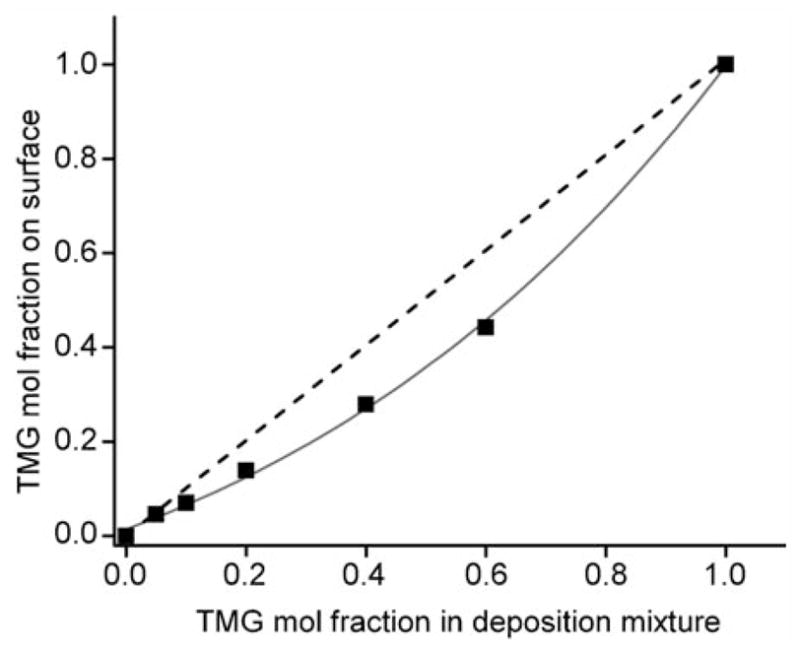
Correlation between TMG-EG10 mole fraction in the deposition mixture and the fraction in the resulting organic monolayers as derived from the ratio of integrated areas of deconvoluted C1s signals for etheric and alkyl/alkynyl carbon atoms. Dashed line represents equal molar fractions in the deposition mixture. Solid line connects the data points measured on the resulting films, showing the preferential binding of the smaller alkene EG7 over the larger alkene TMG-EG10 on the silicon surfaces.
Static water contact angles provide an alternative estimate of the composition of the mixed monolayers. The single component films prepared from TMG-EG10 and EG7 exhibited water contact angles of approximately 62 and 49°, respectively. The higher contact angle of TMG-EG10 as compared to EG7 can be rationalized by the presence of hydrophobic TMG groups on the surface. The contact angles of the mixed monolayers generally fall between that of surface 1a and 1f. As for surface 1b, a water contact angle of 53° was obtained. Since the surface contact angle was contributed by these two alkenes with different polarity, a surface composition of roughly 31% TMG-EG10 was estimated, assuming that the mixed organic film was homogenous.[44] Within experimental uncertainty, this value is in satisfactory agreement with the percentage calculated from the XPS spectra (28%).
Taken together, our result shows that the mol fraction of active component TMG-EG10 in the mixed monolayer is lower than that of the mol fraction in solution during the monolayer formation, which can be attributed to the higher rate of surface hydrosilylation for the smaller alkene (EG7) than the larger one (TMG-EG10). This observation was different from the one reported by Wayner and co-workers.[45] In their system, the ratios of the two components on the films were approximately equal to the ratio of the two alkenes in the deposition solution. It may suggest that the extent of preferential alkene binding on silicon substrate during binary monolayer formation is sensitive to the chain length and/or terminal groups, and thus cannot be generalized. Regardless, we were able to reliably identify and control the surface density of TMG-alkynyl groups on the OEG monolayers, which is critical for the following attachment of AMPs via CuAAC reaction.
“Click” immobilization of AMP to the mixed OEG monolayers presenting alkynyl groups
The AMP we used (N3-IG-25) is a truncated version of LL-37, which is an extensively studied human AMP in general adapting a helical conformation in physiological conditions.[46,47] The sequences of the peptides are shown in Table 2. The choice of IG-25 was based on a previous study showing similar antimicrobial activity of this shorter peptide as its parent LL-37.[46] In addition, it was also reported that the removal of N-terminal hydrophobic amino acid residues from LL-37 decreases its cytotoxicity to host cells as well as its inhibition by serum.[48] An azido tag is attached to the N-terminal of the peptide via a long OEG linker to provide flexibility to peptide upon immobilization.
Table 2.
Peptide sequence.
| Peptide | Sequence |
|---|---|
| IG-25 | I-G-K-E-F-K-R-I-V-Q-R-I-K-D-F-L-R-N-L-V-P-R-T-E-S |
| LL-37 | L-L-G-D-F-F-R-K-S-K-E-K-I-G-K-E-F-K-R-I-V-Q-R-I-K-D-F-L-R-N-L-V-P-R-T-E-S |
The immobilization of N3-IG-25 onto the functionalized silicon substrates was achieved by a one-pot deprotection/coupling reaction. The degermanylation of the films was the prerequisite for the subsequent click coupling, and can be easily quantified by the distinct Ge 3d signal. As for the de-protection process, the wafer was incubated in an aqueous solution containing CuSO4 (2.5 mM) and ascorbic acid (10 mM). As demonstrated in Figure 2a, the intensity of the Ge 3d signal for the 100% TMG-alkynyl terminated film 1a was decreased by over 95% after incubation in the CuI solution for 6 h. It is conceivable that the degermanylation on films 1b–e should be faster than on the film 1a due to the lower density of the TMG groups and thus lower steric hindrance for the deprotection reaction. For the subsequent CuAAC reaction with the peptide N3-IG-25, the appearance of the N 1s signal, which was absent in the precursor monolayer, was used to characterize the formation of triazole and the efficiency of the surface modification. Figure 2b shows the narrow scan of N 1s for surface 1a before and after CuAAC reaction in the presence of CuSO4 (2.5 mM) and ascorbic acid (10 mM) for 6 h. The broad signal of N 1s around 400.5 eV was contributed by the peptide and triazole linkages. Physisorbed peptides were largely removed from the surface by thoroughly washing with 0.01 M phosphate buffered saline (PBS) buffer after the reaction, as indicated by the absence of an azido signal near 403 eV.[49,50] However, this surface reaction was slow and the yield was low. According to the estimation by N/C ratio,[51] only about 5% alkynyl groups on surface 1a were coupled with N3-IG-25 in 10 h under the above conventional CuAAC reaction condition.
Figure 2.

Narrow scan of a) Ge 3d and b) N 1s regions for substrate 1a before (bottom spectra) and after (top spectra) click reaction in solution containing CuSO4 (2.5 mM) and ascorbic acid (10 mM) for 6 h.
Recent studies reported that CuAAC reaction could be greatly accelerated by appropriate CuI ligand, especially tris-triazole ligands.[41–43] However, we observed that for some surface CuAAC reactions, the mono-triazole ligands, such as L1 shown in Figure 3, performed better than the tris-triazole ligands, probably due to reduced steric hindrance.[52] Therefore, we employed mono-triazole ligand in this study and monitored the surface CuAAC reaction on the TMG-alkynyl terminated surface 1b with N3-IG-25 by the N/C ratio. Figure 3 shows the reaction profiles for the immobilization of N3-IG-25 (30 μM) onto surface 1b using two different protocols, including CuSO4 + ascorbic acid and Cu(CH3CN)4PF6 + ligand L1 + ascorbic acid in an aqueous solution. The use of [Cu(CH3CN)4]PF6 plus ligand L1 clearly gave superior results as compared to the conventional catalytic system for the immobilization of N3-IG-25, completing the reaction in 5 h. The experiment was carried out in N2 atmosphere in the presence of excess ligands, which is critical to reduce the formation of oxy radicals and electrophiles that are detrimental to peptides.[42] In addition, a relatively small amount of CuI salt (1.25 mM) was required to catalyze the reaction in the presence of a ligand, thereby facilitating the removal of the harmful copper residue by EDTA solution (Figure S3 in Supporting Information).
Figure 3.
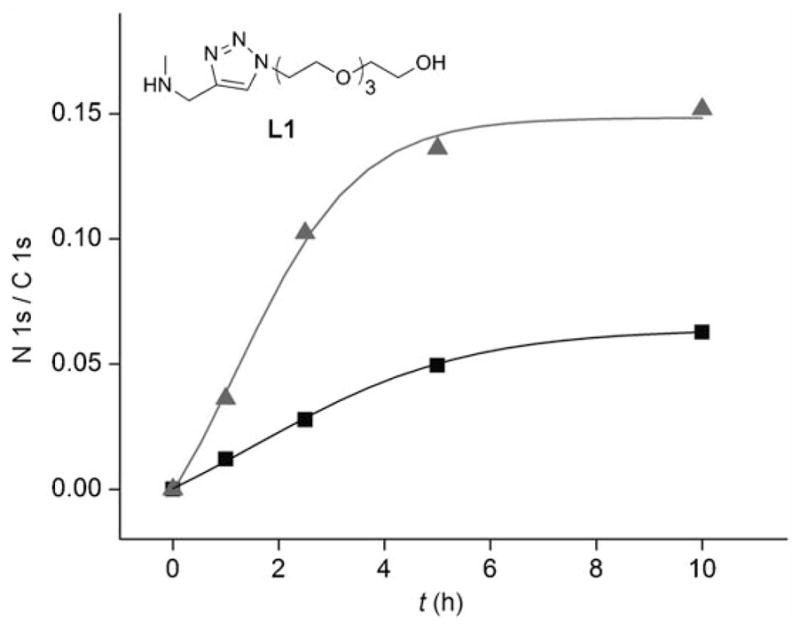
N/C ratio vs the duration of the CuAAC reactions of films 1b with N3 -IG-25 (30 μM) under different reaction conditions: Cu-(MeCN)4PF6 (1.25 mM), ascorbic acid (10 mM) plus ligand L1 (10 mM) (triangle) or CuSO4 (2.5 mM) and ascorbic acid (10 mM) (square).
Previous studies suggest that the density of the reactive sites on surfaces significantly influenced the efficiency of the CuAAC immobilization.[44] In our case, the correlation between the alkynyl composition on the surface and the integration of the N 1s signal (proportional to the peptide density) is presented in Figure 4. It shows a near linear increase of the peptide attached to the films with low TMG-alkyne coverage, but further increase of the density of alkynyl groups led to an attenuation of the peptide attachment. This result may be attributed to the existence of a steric limit to the immobilization of peptides. For the surface 1a with densely packed TMG-alkynyl groups, after a threshold density of the peptide was reached, accessing the underlying alkynyl groups by additional azido tagged peptide was blocked. A similar phenomenon has also been observed for the immobilization of ferrocene on Au (111) surface,[44] which shows that the steric limit of ferrocene attached on Au (111) is about 55% coverage of the thio-azide functional groups. Since the size of IG-25 is much larger than ferrocene, it is reasonable that the steric limit appeared at a lower coverage.
Figure 4.
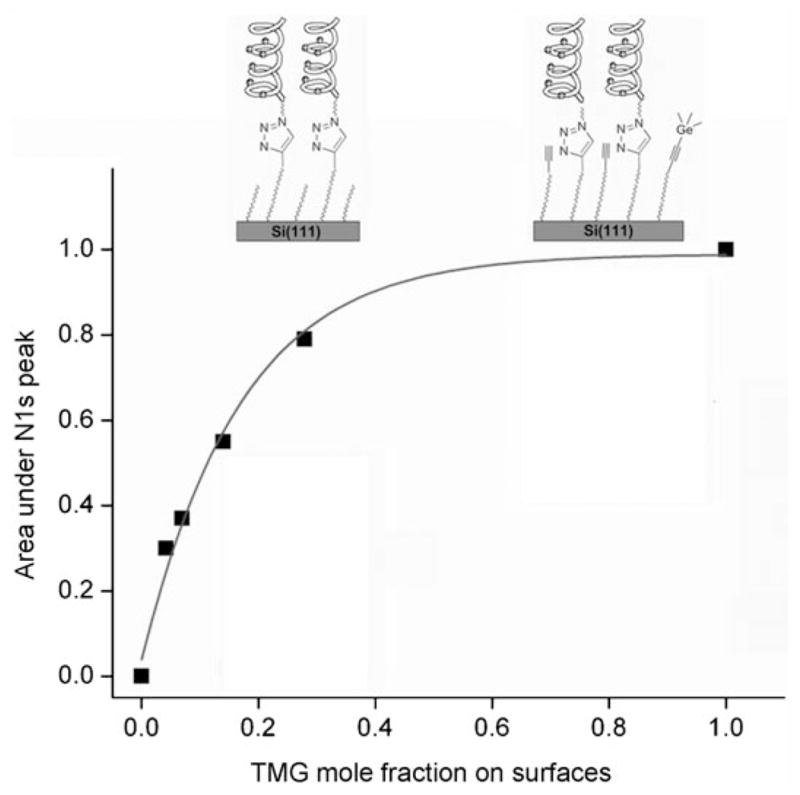
Correlation between the TMG-EG10 mole fraction on the surface and the area under N 1s peak of XPS spectra after CuAAC reaction for 5 h. The intensity was normalized to the integrated area of N 1s on surface 2a and the inset pictures illustrate the steric limit of the immobilization due to the large size of IG-25.
The grafting density of the precursor TMG-alkynyl monolayer can be estimated by the ellipsometeric thickness according to the published literatures,[53,54] detailed in the Supporting Information. Subsequently, the surface density of AMPs presenting on the substrate was obtained by multiplying the yield of CuAAC coupling determined by N/C ratio. Table 3 shows the correlation between the density of the TMG-alkynyl groups and the density of IG-25 attached to the surfaces. It was found that the density of the peptide was significantly lower than that of the alkynyl groups on the precursor surface. On the surface 2a with entire coverage of TMG-EG10, the yield of the click immobilization is less than 10%. The low yield was likely due to the steric hindrance induced by the large AMP moieties and the densely-packed TMG-alkynyl functionalities. Indeed, the dilution of TMG functional groups by EG7 considerably increased the efficiency for the surface immobilization. For instance, surface 1e with about 5% coverage of TMG-EG10 achieved more than 50% conversion to triazole. The higher yield of immobilization on mixed monolayers was attributable to the increased accessibility of the reactants in solution to the alkynyl groups on the substrate.
Table 3.
Correlation of the TMG-EG10 density on the mixed monolayer and the corresponding peptide density upon CuAAC reaction.
| Surface | Density of TMG-EG10 before CuAAC reaction (molecule per cm2)[a] | Density of IG-25 (molecule per cm2)[b] | Yield of immobilization [%] |
|---|---|---|---|
| 2a | 3.3×1014 | 2.8×1013 | 9 |
| 2b | 9.9×1013 | 2.2×1013 | 22 |
| 2c | 5.1×1013 | 1.6×1013 | 31 |
| 2d | 2.6×1013 | 1.0×1013 | 44 |
| 2e | 1.5×1013 | 8.6×1012 | 51 |
The density of TMG-EG10 on surface 1a was calculated from the ellipsometric thickness of the monolayer according the previous literature,[53,54] and the density of alkynyl groups on the mixed monolayers was derived from the area of corresponding Ge 3d signal compared to that of the surface 1a in XPS spectra.
The density of the IG-25 on surface 2a was obtained by the yield of the reaction (according to N/C ratio) times the density of alkynyl groups, and the density of the following mixed surface was derived from the ratio of the corresponding N 1s area compared to that of the surface 2a.
Antibacterial properties of “click” immobilized IG-25
The bactericidal properties of the IG-25 were evaluated using a strain of Pseudomonas aeruginosa (PA, PA01). PA is a common Gram-negative bacterium, and a major pathogen causing infections on many biomedical implants, including contact lenses and urinary catheter.[55,56] Our previous study indicates that both LL-37 and the modified peptide N3-IG-25 was active against PA01 in solution, showing EC50 equal to 19.5±1.2 and 35.5±2.8 μg mL−1, respectively. While these values are useful predictors for antibacterial coatings, the measurements do not directly correlate to the amount required for surface applications because interactions between the bacteria and the immobilized AMPs are expected to differ from the interactions when peptides are free in solution.
To facilitate the antimicrobial testing on surfaces, the bacteria used were transformed to express green fluorescent protein (GFP-PA). The adhesion of PA onto the IG-25 modified surfaces was first investigated. Substrates presenting 100% EG7 (1f) and the unmodified silicon wafer were used as controls. In a typical experiment, after 2 h of incubation with the bacteria at 37°C, the sample was taken out and briefly rinsed with PBS buffer before characterization by fluorescence microscopy. As shown in Figure 5, PA adhered more on the IG-25 presenting surfaces with increasing peptide density. The bacteria also adhered strongly on the cationic surfaces and could not be removed by simple agitation. On the contrary, fewer bacteria were observed on the OEG-presenting surface 1f than on the unmodified silicon wafer and other AMP-presenting surfaces, indicating the substrate derived from neat antifouling OEG moieties efficiently resisted the bacterial attachment. Interestingly, the bacteria on the surfaces with a low density of AMPs (surface 2b–e) tended to colonize on certain restricted areas rather than being evenly distributed as in the image of unmodified silicon wafer, likely suggesting that AMPs may aggregate and were unevenly presented on the monolayers with a bioinert background.
Figure 5.
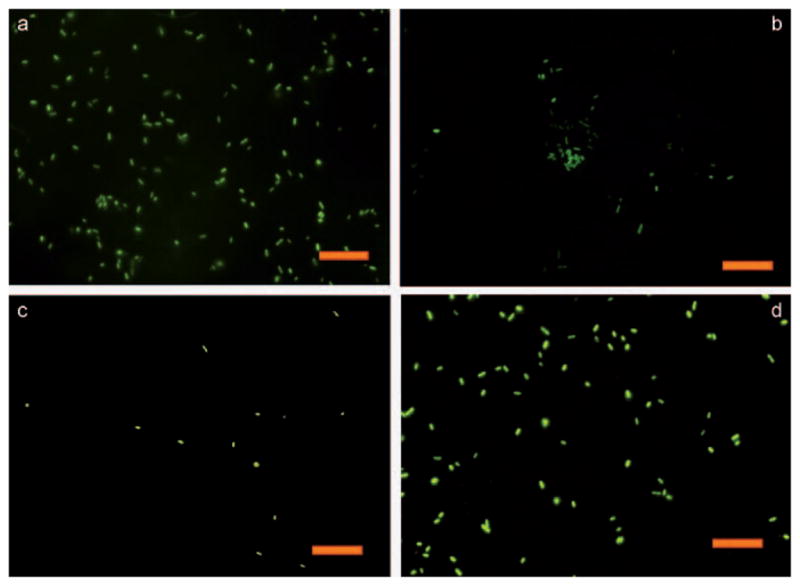
Fluorescence images of GFP-PA adhered on different substrates: a) Surface 2a, b) surface 2e, (c) surface 1f and (d) the unmodified silicon wafer. The scale bar is 20 μm.
The viability of the bacteria adhered on the surfaces was quantified using propidium iodide (PI) as an indicator.[57] Bacteria with a compromised membrane fluoresce red (PI-positive) due to the uptake of PI. As an example, Figure 6 shows that among all of the green fluorescent bacteria absorbed on the IG-25 presenting surface 2b (Figure 6a), most of them fluoresced red under the TRITC filter (Figure 6b). This can be better viewed by the overlay of the two images, clearly showing the membrane-compromised (orange) and the live (green) bacteria (Figure 6c and Figure S4 in Supporting Information).
Figure 6.
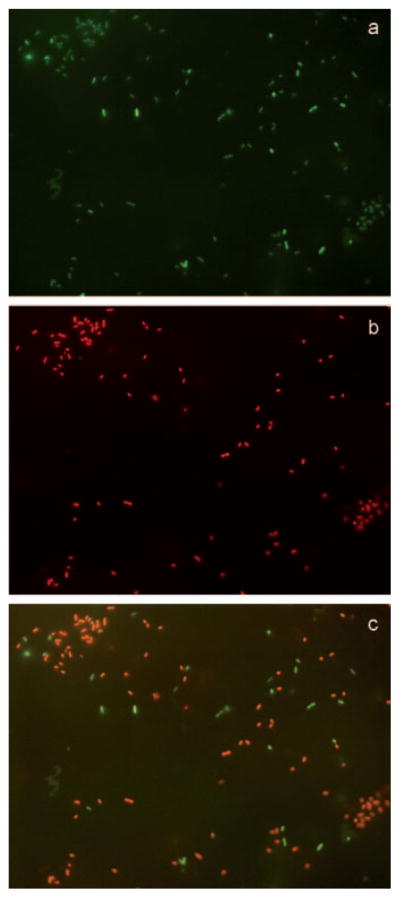
Fluorescence images (600X magnification) showing a) all adsorbed GFP-PA on surface 2b, b) with a TRITC filter showing the PI-positive PA on the same area, and c) overlay of images a) and b), revealing the PI-negative PA (green) and PI-positive PA (orange).
Figure 7 summarizes the number of live/dead bacteria attached on the surfaces during 2 h incubation in PA01 solution. It shows that the number of absorbed bacteria as well as the percentage of killing increases with higher density of IG-25 presenting on the surface. It is well recognized that the bacteria with negative charges on the membrane surface are attracted to the cationic peptides, thus are more likely to accumulate on the surface with higher density of AMPs through ionic interactions.[58,59] However, as demonstrated in Figure 7, the number of bacteria adhered to surface 2d presenting cationic peptides is only slightly less than the hydrophilic unmodified silicon wafer, which suggests that the anti-fouling background formed by EG7 may still be effective even after click immobilization, at least at the low concentration of AMPs, to reduce the initial bacterial adhesion. In other words, the substrate fouling is suppressed by the presence of OEG monolayer as the base.
Figure 7.
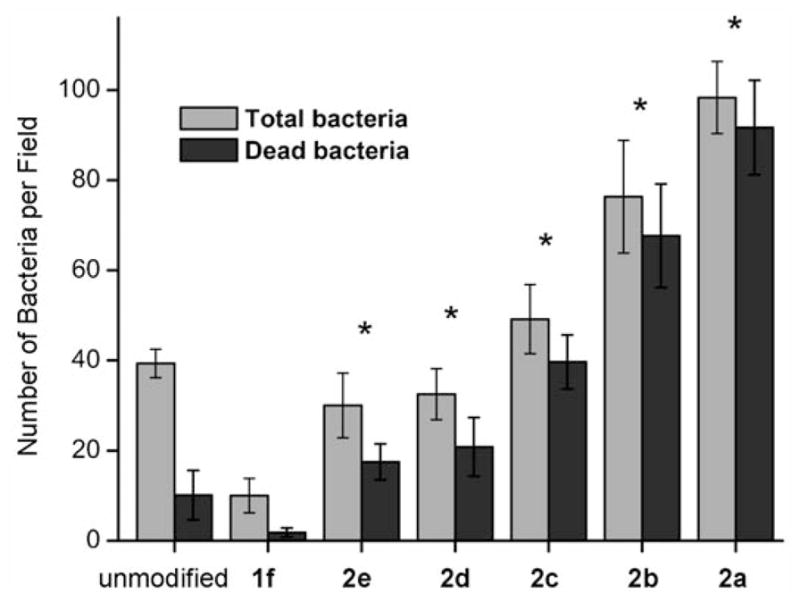
Comparison of the number of bacteria adhered on the unit area of surfaces (1 mm2×1 mm2) during the 2 h incubation in PA solution and briefly washed by PBS buffer. Values are averages with standard deviations of 10 pictures conducted on a minimum of three separate experiments. * denotes significant difference in terms of the antibacterial rate compared to the unmodified control (p<0.01, T test).
Minimization of the non-specific adsorption of bacteria to silicon wafer was not the primary aim of this work; nevertheless, this is a key consideration for the practical application of biomaterials, since the adhesion and accumulation of dead cells on the biomaterial via hydrophobic interaction could contaminate the antibacterial coatings, thereby making the coatings ineffective at preventing the growth of pathogens and subsequent bacterial infection after the long term use of the biomaterial.[60] It should be noted that Geith et al. recently have developed antifouling copolymer brushes that were subsequently used as platforms to covalently immobilize natural antibacterial peptide. Low coverage of bacteria (less than 1% of the surface) was observed for the modified substrates.[28] In our experiment, the versatility arising from a combination of protein resistant OEG background and the “clickable” reactive site using hydrosilylation chemistry also allows us to control the adsorption of bacteria, which suggested that the combination of biocidal substances on an antifouling coating could be a desirable approach to prevent initial bacterial adhesion and the subsequent formation of bacterial colonies.
The ability of the differently modified surfaces to kill the bacteria was quantified by counting the total number of bacteria and the PI-positive bacteria binding per unit area in the fluorescence images. Figure 8 plots the percentage of the PI-positive bacteria (red) as a function of surface density of IG-25 determined by the XPS, which indicates that the bacterial killing rate was strongly related to AMP density but is not strictly linear. Substrate 2a with the highest AMP density of ~2.81×1013 molecule per cm2 can suppress the viability of most bacteria attached. Within a certain range of AMP surface density (substrate 2a–c), the killing efficiencies of these substrates were similar (within the statistical range). Further decreasing the density of AMP to ~1.0×1013 cm2 (surface 2d), the antibacterial efficiency drops from ~80 to ~60%, but still much higher than neat OEG surface and the unmodified silicon substrate which can be perceived as a negative control. The observation that the antibacterial activity of immobilized AMP was particularly enhanced when the coverage was beyond ~1.6×1013 cm2, probably suggests that an “effective local concentration” may exist for the surface bound AMPs to act cooperatively and dramatically increase the bactericidal efficiency.
Figure 8.

Correlation between the density of IG-25 presenting on the substrate and the percentage of non-viable PA (PI-positive) attached on the modified silicon surface.
A parallel approach to evaluate antimicrobial activity of the functionalized silicon surfaces relies on spreading the bacteria adhered on the wafers on an agar plate and counting the bacterial colonies. Bacterial growth inhibition was determined as a percentage of the colonies on the control sample (unmodified silicon substrate), as shown in Figures S5 and S6 in the Supporting Information. The results are in good agreement with the above analysis of counting the live/dead bacteria in fluorescence images.
Several models, such as the “carpet model” and “barrel-stave model”, have been put forth to explain how AMPs ultimately lead to cell death in solution.[61] However, the exact mechanistic action of the immobilized AMPs (IG-25) for killing the adsorbed bacteria is not fully understood. Based on previous studies,[26–31] it seems that the maintenance of peptide amphipathicity and mobility are important to retain the antibacterial activity of the tethered AMPs. Both properties were achieved in our strategy by tethering the peptides selectively at the N-terminus via a long flexible OEG linker. Thus, our model platform facilitates the multiple charges on the peptide to interact simultaneously with the bacterial membrane, which probably renders the behavior of immobilized AMPs compatible with current models of transmembrane actions in solution.
Cytotoxicity of the IG-25 immobilized substrates
A basic requirement for useful antimicrobial coating on biomaterials is low cytotoxicity to mammalian cells. AMPs become cytotoxic in solution when the concentration is slightly higher than the effective concentration needed to rapidly kill the bacteria. This is especially the case for some synthetic peptides.[62,63] Therefore, for the immobilized AMPs with a very high local concentration, the cytotoxicity to host cells is a major concern, which has rarely been addressed by previous studies.
To address this concern, we measured the cytotoxicity of the surface 2a presenting the highest density of IG-25, the precursor film 1a and the degermanylated alkynyl surface, using MTS cell proliferation assay with NIH3T3 fibroblast cells. An unmodified silicon substrate was used for negative control, and for positive control the hydrogen peroxide was used. In the MTS assay, MTS is chemically reduced by viable cells into soluble formazan that can be quantified by measurement of its absorbance at 495 nm.[64] Since the production of formazan is proportional to the number of living cells, the absorbance provides an estimate of the viability of the cells. Figure 9 presents the viability of the cells after incubation with the samples for 24 h. The data are normalized to the absorbance obtained on the unmodified silicon surface that is nontoxic to the cells.[65] Significantly, the result shows about 80% survival of the cells adhered on the IG-25 surface 2a with a high density of the peptide after 24 h incubation. Thus, the attachment of the AMP on the alknyl surface did not noticeably increase the toxicity of the surface to the NIH3T3 cells.
Figure 9.
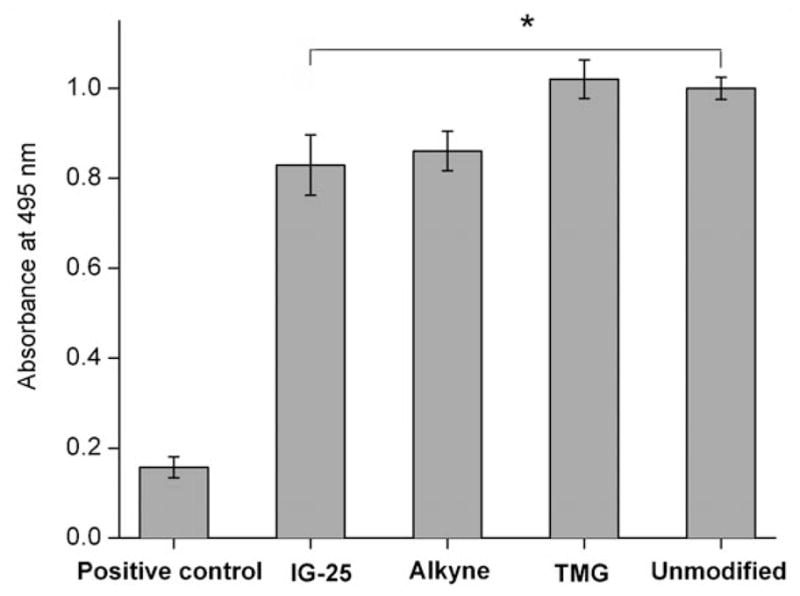
MTS assay of the cytotoxicity of the substrates presenting IG-25 (surface 2a), TMG protected alkyne (surface 1a), deprotected alkyne and unmodified silicon wafer on NIH3T3 cells. The intensity of 495 nm absorbance was normalized to the unmodified silicon wafer. Hydrogen peroxide-treated NIH3T3 was used as positive control. Results depicted are the medians of three experiments with three parallel readings per substrate per experiment. Error bars denote standard deviation. * Denotes significant difference compared to positive control (p<0.001, T test).
Conclusion
We have presented an efficient, site-specific attachment of azido-labeled antimicrobial peptides onto a “clickable”, bio-inert monolayer platform via CuAAC reaction. The platform was generated by selective hydrosilylation of TMG-protected α,ω-alkenynes on H-terminated silicon substrates. The surface density of alkynyl groups can be controlled by the co-deposition of an inert OEG-alkene. Azido-labeled AMPs were directly tethered to these platforms with varied densities by a one-pot deprotection/CuAAC reaction. The immobilized peptides exhibited a remarkable bactericidal activity when their density was above ~1.6×1013 cm2, while remaining cyto-compatible to mammalian cells.
The results support the potential use of AMPs for antimicrobial coating of biomaterials and demonstrate an alternative method for site-specific tethering of AMPs on surfaces. Further investigation on the long-term stability of immobilized peptides is necessary for their prolonged use on biomedical implants. The “clickable”, bioinert platform may also be used for tethering a wide range of azido-labeled biomolecules to study many other biological events, such as sensing and indentifying cells.
Experimental Section
General
The silicon wafer was purchased from North East silicon technologies Inc. The antimicrobial peptide, IG-25 tethering an EG12-N3 group at the N-terminus was purchased from Alpha Diagnostic International (San Antonio, TX). The Pseudomonas aeruginosa (PA) strain expressing green-fluorescent protein and a carbenicillin resistance gene (GFP-PA01) was a generous gift from Dr. Alice Prince (Columbia University, NY). NIH3T3 cells were a generous gift from Dr. Albee Messing (University of Wisconsin, Madison). Other reagents and solvents were purchased from commercial suppliers (Adrich, Fluka, or Alfa Aesar) and were used as supplied without any further purification. The synthesis of EG7[34] and L1[52] was described previously. The synthesis of TMG-EG10 will be reported elsewhere.[36]
Photo-induced surface hydrosilylation
H-terminated silicon(111) surfaces were prepared similar to previous described procedures.[35] Briefly, single-sided polished silicon (111) wafers were cut into pieces of ca. 2×2 cm2, cleaned with piranha solution H2SO4/H2O2 (v/v 3.5:1) at 80 °C for 20 min. Then the wafer was thoroughly washed with Millipore water, etched in 40% NH4F solution for 10 min under N2 purge, and dried with a flow of nitrogen. The setup and procedures for photo-induced surface hydrosilylation of H-terminated silicon substrate with alkenes were described elsewhere.[34,35] Briefly, a freshly prepared H-Si(111) substrate was placed inside a vacuum chamber, facing a droplet (~0.2 mg) of the alkene (TMG-EG10 or EG7) that was placed on fresh cleaned and dried quartz. After the system was degassed at vacuum for 10 min, the wafer was allowed to contact with the droplet, sandwiching a thin and homogeneous layer of the alkene between the substrate and the quartz wall. The substrate was then illuminated with a hand-held 254 nm UV lamp (Model UVLS-28, UVP, 9 W) for 2 h, followed by washing sequentially with CH2Cl2 and ethanol, and finally dried with a stream of N2.
WARNING
Piranha solution reacts violently, even explosively, with organic materials. It should be used with extremely care and should not be stored.
X-ray photoelectron spectroscopy (XPS)
A PHI 5700 X-ray photoelectron spectrometer was equipped with a monochromatic AlKα X-ray source (hν =1486.7 eV) incident at 90° relative to the axis of a hemispherical energy analyzer. The spectrometer was operated at a photoelectron take off angle of 45° from the surface, and an analyzer spot diameter of 1.1 mm. The survey spectra were collected from 0 to 1400 eV, and the high resolution spectrum were obtained for photoelectrons emitted from C 1s, O 1s, Si 2p, N 1s and Ge 3d. All spectra were collected at room temperature with a base pressure of less than 1×10−8 Torr. Electron binding energies were calibrated with respect to the C 1s line at 284.8 eV for C–C bond. A free software XPSpeak41, developed by Dr. Raymond Kwok (The Chinese University of Hong Kong), was used for all data processing. The high resolution data were analyzed first by background subtraction using the Shirley routine and a subsequent non-linear fitting by mixed Gaussian-Lorentzian functions. Atomic compositions of the prepared surface samples were corrected for the number of scans accumulated and for the atomic sensitivity of the element.
Contact angle measurements
Contact angles of water were measured at room temperature and ambient relative humidity using a Ramé-Hart model 100 contact angle goniometer. The data were collected and averaged over at least three separate slides using three drops per slide for each substrate.
Ellipsometry measurement
The ellipsometer (Rudolph Research, Auto EL III) was employed for thickness measurement. It was operated with a 632.8 nm He/Ne laser at an incident angle of 70°. At least three measurements were taken for each sample, and the mean values were reproducible within ± 1 Å.
Immobilization of IG-25 on the TMG-terminated Si(111) surface via click chemistry
The two-step/one-pot CuAAC reaction was performed in two procedures. In the first procedure (which generated all samples for antibacterial and cytotoxicity assay), ligand L1 (3.02 mg, 10.0 μmol), ascorbic acid (1.76 mg, 10.0 μmol), and N3-IG-25 (1.1 mg, 0.03 μmol) were combined in a reaction vial containing 0.9 mL PBS buffer (pH 7.4) and the TMG-terminated silicon surface (1×1 cm2). Then 0.1 mL DMSO solution containing Cu(MeCN)4PF6 (0.46 mg, 0.13 mmol) was added to the mixture to a final concentration of 1.25 mM CuI. In the second procedure, CuSO4 (2.5 mM) and ascorbic acid (10 mM) was used to generate a catalytically active CuI species in aqueous solution. For monitoring progress of the reaction, the reaction was terminated at the specified time points. Otherwise, the reaction lasted for 5 h under N2 atmosphere. Then the substrate was taken out and immersed in EDTA solution (5 mM) with shaking for 20 min to remove copper residue, followed by sonication cleaning for 2 min in PBS buffer and a thorough wash with Milli Q water. The substrate was dried under a gentle stream of argon.
Antimicrobial activity of IG-25 immobilized Si(111) surfaces
One single isolated GFP-PA01 colony was used to inoculate 5 mL of LB with 300 μg mL−1 carbenicillin (for positive selection) overnight at 37°C. A bacterial suspension (1 mL) was inoculated to fresh LB (50 mL) with 300 μg mL−1 carbenicillin, which was then incubated for 2 h with shaking at 37°C to achieve mid-log phase growth. Twenty-five milliliters of the warm PA culture were centrifuged at 3000 rpm for 10 min, and the bacterial cell pellet was resuspended in cold phosphate buffer (8.2 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4). Optical density of the suspension was adjusted to 0.26 at 620 nm by adding an appropriate volume of PB. Bacterial suspension (100 μL, approximately 107 CFU mL−1) was added to each well with modified silicon substrates. The incubation lasted for about 2 h at 37°C. Then the substrates were taken out and were quickly transferred to a freshly prepared PBS buffer under gentle shaking for another 2 min to remove the non-adsorbed bacteria to facilitate imaging of the strongly adhered bacteria. A 1–2 μL aliquot of propidium iodide (PI) was then placed on the silicon wafers as the viability indicator before imaging with a florescence microscope.
Fluorescence images were obtained by acquiring 10 images from random location on each substrate using a Nikon 80i fluorescence microscope with a 60× water-immersion objective. Filter FITC was used for exciting GFP, and TRITC filter was used to capture the PI-positive PA, and the overlay of the FITC image and TRITC image reveals the PI-negative PA. The number of bacteria adhered per unit area was analyzed with a computer imaging system (NIS-Elements AR 3.00).
The antibacterial property of the functionalized silicon substrates was also checked by plating out decimal dilutions of the bacterial suspension (after 2 h incubation with the samples in each well) on an agar plate and counting the number of bacterial colonies. Bacterial growth inhibition was determined as a percentage of the colonies on the control sample (unmodified silicon substrate).
MTS assay
The cytotoxicity of the modified silicon substrates was performed using CellTiter 96 AQueous (Promega). Assay kits containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) and an electron coupling reagent, phenazine methosulfate (PMS). The cells were maintained in Dulbecco’s modified Eagle medium (DMEM) media with 10% fetal bovine serum (FBS). Culture medium was removed from the flask and approximately 5 mL of trypsin EDTA solution were then added to the flask and incubated at 37°C for a few minutes. Modified silicon substrates were placed on the bottom of a 24-well culture plate and then cells were seeded onto the samples with a seeding density of 2.5×104 cells cm−2. The plate was incubated at 37°C and 5% CO2 in humidified air for 24 h. The cell culture medium was then aspirated from the wells and the substrates were gently rinsed with DMEM to remove any non-adherent cells. The adherent cells were evaluated for their viability using MTS assay as described in the Promega (Promega, Madison, WI, USA) technical bulletin. Briefly, MTS and PMS detection reagents were mixed, using a ratio of MTS/PMS 20:1, immediately prior to addition to the cell culture media DMEM at a ratio of detection reagents/cell culture medium 1:5. Then the aspirated wells containing the samples were incubated for 2 h at 37°C in a 5% CO2 atmosphere. A sample of culture medium was used as a “medium-only” control. The same cell suspension with unmodified silicon wafer was also used as a negative control. For the positive control, hydrogen peroxide (0.1 mM) was added to the cell grown on the plate with unmodified silicon wafer. The absorbance of the formazan was read by a HTS 7000 Bio Assay microplate reader (PERKIN ELMER) at wavelength of 495 nm.
Supplementary Material
Acknowledgments
This work was supported by the NSF grant DMR-0706627, NIH R21 HD058985, The Welch Foundation (E-1498), and the Texas Center for Superconductivity at the University of Houston.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201001533.
References
- 1.Gristina AG. Science. 1987;237:1588–1595. doi: 10.1126/science.3629258. [DOI] [PubMed] [Google Scholar]
- 2.Hall-Stoodley L, Costerton JW, Stoodley P. Nat Rev Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 4.Costerton JW. Int J Antimicrob Agents. 1999;11:217–221. doi: 10.1016/s0924-8579(99)00018-7. [DOI] [PubMed] [Google Scholar]
- 5.Zasloff M. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 6.Bagheri M, Beyermann M, Dathe M. Antimicrob Agents Chemother. 2009;53:1132. doi: 10.1128/AAC.01254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haynie SL, Crum GA, Doele BA. Antimicrob Agents Chemother. 1995;39:301–307. doi: 10.1128/aac.39.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuzaki K, Sugishita K, Fujii N, Miyajima K. Biochemistry. 1995;34:3423–3429. doi: 10.1021/bi00010a034. [DOI] [PubMed] [Google Scholar]
- 9.Oren Z, Shai Y. Biopolymers. 1998;47:451–463. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 10.Wang G. J Biol Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- 11.Chromek M, Brauner A. J Mol Med. 2008;86:37–47. doi: 10.1007/s00109-007-0256-4. [DOI] [PubMed] [Google Scholar]
- 12.Ding JL, Ho B. Drug Dev Res. 2004;62:317–335. [Google Scholar]
- 13.Chromek M, SlamovQ Z, Bergman P, KovQcs L, PodrackQ L, Ehrén I, Hökfelt T, Gudmundsson GH, Gallo RL, Agerberth B, Brauner A. Nat Med. 2006;12:636–641. doi: 10.1038/nm1407. [DOI] [PubMed] [Google Scholar]
- 14.Jenssen H, Hamill P, Hancock REW. Clin Microbiol Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon YJ, Romanowski EG, McDermott AM. Curr Eye Res. 2005;30:505–515. doi: 10.1080/02713680590968637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madkour AE, Dabkowski JM, Nusslein K, Tew GN. Langmuir. 2009;25:1060–1067. doi: 10.1021/la802953v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tew GN, Scott RW, Klein ML, DeGrado WF. Acc Chem Res. 2010;43:30–39. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Statz AR, Park JP, Chongsiriwatana NP, Barron AE, Messersmith PB. Biofouling. 2008;24:439–448. doi: 10.1080/08927010802331829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilpert K, Elliott M, Jenssen H, Kindrachuk J, Fjell CD, Körner J. Chem Biol. 2009;16:58–69. doi: 10.1016/j.chembiol.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Humblot V, Yala J, Thebault P, Boukerma K, Hequet A, Berjeaud J, Pradier C. Biomaterials. 2009;30:3503–3512. doi: 10.1016/j.biomaterials.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Matsuzaki K, Sugishita K, Nobutaka F, Koichiro M. Biochemistry. 2002;41:3423–3429. [Google Scholar]
- 22.Boulmedais F, Frisch B, Etienne O, Lavalle P, Picart C, Ogier JA. Biomaterials. 2004;25:2003–2011. doi: 10.1016/j.biomaterials.2003.08.039. [DOI] [PubMed] [Google Scholar]
- 23.Etienne O, Gasnier C, Taddei C, Voegel JC, Aunis D, Schaaf P. Biomaterials. 2005;26:6704–6712. doi: 10.1016/j.biomaterials.2005.04.068. [DOI] [PubMed] [Google Scholar]
- 24.Etienne O, Picart C, Taddei C, Haikel Y, Dimarcq J, Schaaf P. Antimicrob Agents Chemother. 2004;48:3662–3669. doi: 10.1128/AAC.48.10.3662-3669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guyomard AD, Jouenne T, Malandain J, Muller G, Glinel K. Adv Funct Mater. 2008;18:758–765. [Google Scholar]
- 26.Sridhar S, Smitha B, Mayor S, Prathab B, Aminabhavi TM. Adv Exp Med Biol. 2009;611:241–242. [Google Scholar]
- 27.Gabriel M, Nazmi K, Veerman EC, Amerongen AVN, Zentner A. Bioconjugate Chem. 2006;17:548–550. doi: 10.1021/bc050091v. [DOI] [PubMed] [Google Scholar]
- 28.Glinel K, Jonas AM, Jouenne T, Leprince J, Galas L, Huck WTS. Bioconjugate Chem. 2009;20:71–77. doi: 10.1021/bc800280u. [DOI] [PubMed] [Google Scholar]
- 29.Willcox MDP, Hume EBH, Aliwarga Y, Kumar N, Cole N. J Appl Microbiol. 2008;105:1817–1825. doi: 10.1111/j.1365-2672.2008.03942.x. [DOI] [PubMed] [Google Scholar]
- 30.Appendini P, Hotchkiss JH. J Appl Polym Sci. 2001;81:609–616. [Google Scholar]
- 31.Steven MD, Hotchkiss JH. J Appl Polym Sci. 2008;110:2665–2670. [Google Scholar]
- 32.Mannoor MS, Zhang S, Link AJ, McAlpine MC. Proc Natl Acad Sci USA. 2010;107:19207–19212. doi: 10.1073/pnas.1008768107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato Y, Komatsu S. J Biol Chem. 1996;271:30493–30498. doi: 10.1074/jbc.271.48.30493. [DOI] [PubMed] [Google Scholar]
- 34.Gu J, Yam CM, Li S, Cai C. J Am Chem Soc. 2004;126:8098–8099. doi: 10.1021/ja048405x. [DOI] [PubMed] [Google Scholar]
- 35.Yam CM, Lopez-Romero JM, Gu J, Cai C. Chem Commun. 2004:2510–2511. doi: 10.1039/b401499e. [DOI] [PubMed] [Google Scholar]
- 36.Qin G, Santos C, Zhang W, Li Y, Kumar A, Erasquin UJ, Liu K, Muradov P, Trautner BW, Cai C. J Am Chem Soc. 2010;132:16432–16441. doi: 10.1021/ja1025497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ernst A, Gobbi L, Vasella A. Tetrahedron Lett. 1996;37:7959–7962. [Google Scholar]
- 38.Cai C, Vasella A. Helv Chim Acta. 1995;78:732–757. [Google Scholar]
- 39.Kolb HC, Finn MG, Sharpless KB. Angew Chem. 2001;113:2056–2075. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 41.Lewis WG, Magallon FG, Fokin VV, Finn MG. J Am Chem Soc. 2004;126:9152–9153. doi: 10.1021/ja048425z. [DOI] [PubMed] [Google Scholar]
- 42.Hong V, Presolski SI, Ma C, Finn MG. Angew Chem. 2009;121:10063–10067. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 44.Collman JP, Devaraj NK, Eberspacher TPA, Chidsey CED. Langmuir. 2006;22:2457–2464. doi: 10.1021/la052947q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boukherroub R, Wayner DDM. J Am Chem Soc. 1999;121:11513–11515. [Google Scholar]
- 46.Li X, Li Y, Han H, Miller DW, Wang G. J Am Chem Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]
- 47.Burton MF, Steel PG. Nat Prod Rep. 2009;26:1572–1584. doi: 10.1039/b912533g. [DOI] [PubMed] [Google Scholar]
- 48.Ciornei CD, Sigurdardottir T, Schmidtchen A, Bodelsson M. Antimicrob Agents Chemother. 2005;49:2845–2850. doi: 10.1128/AAC.49.7.2845-2850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ciampi S, Bocking T, Kilian KA, James M, Harper JB, Gooding JJ. Langmuir. 2007;23:9320–9329. doi: 10.1021/la701035g. [DOI] [PubMed] [Google Scholar]
- 50.Devadoss A, Chidsey CED. J Am Chem Soc. 2007;129:5370–5371. doi: 10.1021/ja071291f. [DOI] [PubMed] [Google Scholar]
- 51.We assume that the yield of CuAAC reaction on the surface is X. Then, there is a mixture of IG-25 and bare alkyne on the substrates where the mole fraction of IG-25 is X and the mole fraction of the alkyne is (1–X), assuming that all TMG groups have been removed. Since there are 164 carbon atoms and 45 nitrogen atoms in N3-IG-25, the yield X can be calculated from the equation 45X/[199X+35-(1–X)]=N/C, where N/C is measured by XPS. Since most of the nitrogen atoms were present in the AMP molecules that were on the top of the monolayer, the reported yields represent an overestimation because the attenuation of C 1s signal is higher than N 1s signal. However, this systematic error does not affect the trend for the antibacterial efficiency vs surface density.
- 52.Santos CM, Kumar A, Zhang W, Cai C. Chem Commun. 2009:2854–2856. doi: 10.1039/b821148e. [DOI] [PubMed] [Google Scholar]
- 53.Sofia ESJ, Premnath V, Merrill EW. Macromolecules. 1998;31:5059–5070. doi: 10.1021/ma971016l. [DOI] [PubMed] [Google Scholar]
- 54.Zhu XY, Jun Y, Staarup DR, Major RC, Danielson S, Boiadjiev V, Gladfelter WL, Bunker BC, Guo A. Langmuir. 2001;17:7798–7803. [Google Scholar]
- 55.Donlan RM. Emerging Infect Dis. 2002;8:881–890. doi: 10.3201/eid0809.020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Delden C, Iglewski BH. Emerging Infect Dis. 1998;4:551–560. doi: 10.3201/eid0404.980405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sauch JF, Flanigan D, Galvin ML, Berman D, Jakubowski W. Appl Environ Microbiol. 1991;57:3243–3247. doi: 10.1128/aem.57.11.3243-3247.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kulagina NV, Lassman ME, Ligler FS, Taitt CR. Anal Chem. 2005;77:6504–6508. doi: 10.1021/ac050639r. [DOI] [PubMed] [Google Scholar]
- 59.Gregory K, Mello CM. Appl Environ Microbiol. 2005;71:1130–1134. doi: 10.1128/AEM.71.3.1130-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng G, Xue H, Zhang Z, Chen S, Jiang S. Angew Chem. 2008;120:8963–8966. doi: 10.1002/anie.200803570. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:8831–8834. doi: 10.1002/anie.200803570. [DOI] [PubMed] [Google Scholar]
- 61.Alcantar NA, Aydil ES, Israelachvili JN. J Biomed Mater Res. 2000;51:343–351. doi: 10.1002/1097-4636(20000905)51:3<343::aid-jbm7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 62.Mader JS, Hoskin DW. Expert Opin Invest Drugs. 2006;15:933–946. doi: 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- 63.Pacorl S, Giangaspero A, Bacac M, Sava G, Tossi A. J Antimicrob Chemother. 2002;50:339–348. doi: 10.1093/jac/dkf141. [DOI] [PubMed] [Google Scholar]
- 64.Malich G, Markovic B, Winder C. Toxicology. 1997;124:179–192. doi: 10.1016/s0300-483x(97)00151-0. [DOI] [PubMed] [Google Scholar]
- 65.Voskerician G, Shive MS, Shawgo RS, Recum HV, Anderson JM, Cimac MJ, Langer R. Biomaterials. 2003;24:1959–1967. doi: 10.1016/s0142-9612(02)00565-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.