

Abstract
The completely N1-selective Pd-catalyzed arylation of unsymmetric imidazoles with aryl halides and triflates is described. This study showed that imidazoles have a strong inhibitory effect on the in situ formation of catalytically-active Pd(0)-ligand complex. The efficacy of the N-arylation reaction was improved drastically by the use of pre-activated solution of Pd2(dba)3 and L1. From these findings it is clear that while imidazoles can prevent binding of L1 to the Pd, once the ligand is bound to the metal, these heterocycles do not displace it. The utility of the present catalytic system was demonstrated by the regioselective synthesis of clinically important tyrosine kinase inhibitor nilotinib.
Keywords: Palladium, Arylation, Imidazole, Benzimidazole, Aryl Halide, Nilotinib
Introduction
4-Substituted N1-arylimidazoles represent key structural motifs in medicinally important compounds including tyrosine kinase inhibitors,1 γ-secretase modulators,2 serotonin receptor antagonists3 and glycine transporter type-1 inhibitors (Figure 1).4
Figure 1.

Biologically active compounds containing N-arylimidazole motif
Traditionally, N-arylimidazoles have been prepared via an SNAr process or classical Ullmann-type coupling with stoichiometric copper. In the former case, aryl donors are limited to aryl halides with strong electron-withdrawing groups or activated heteroaryl halides.5 The classical Ullmann coupling has a broader substrate scope; however, the reaction requires long reaction time under high temperature (150–200 °C), which limits its functional group tolerance. Over the past decade, there has been remarkable progress in the development of Cu-catalyzed N-arylation methods of imidazoles with aryl halides6 or aryl boronic acids.7 Due to its broad scope under mild conditions, along with low-cost and low-toxicity of copper salts, the Cu-catalyzed N-arylation method quickly found many applications in medicinal chemistry and material science fields.8 However, due to the tautomeric nature of unsymmetric 1H-imidazoles, the Cu-catalyzed arylation and SNAr reactions of the 4-substituted imidazoles oftentimes give poor to moderate regioselectivities.9 In addition, the similar physical properties of the N1-aryl and N3-aryl regioisomers oftentimes make separation of products difficult. Therefore, development of general catalytic methods that selectively produce the N1-arylated product from tautomeric 4-substituted imidazoles, while challenging, would be particularly valuable.10
During the past few decades, extensive effort from different groups has led to the discovery of a wide variety of Pd-catalyzed methods for the formation of C-N bonds at aromatic systems.11 Although the Pd-catalyzed N-arylation of a variety of nitrogen nucleophiles including amines/anilines,12 imines,13 amides,14 indoles,15 hydrazine,16 hydrazones,17 ammonia,18 and nitrite19 has been well documented, the corresponding N-arylation of imidazoles is still not satisfactory; only a few reports of the Pd-catalyzed N-arylation of imidazoles have appeared, and these have mostly been applied to a small number of activated aryl bromides and chlorides.20 In 2006, our group reported the Pd-catalyzed N-arylation of imidazole with unactivated aryl bromide (4-bromotoluene) using biaryl phosphine L1.21 However, the reaction required relatively high catalyst loadings (5 mol % Pd and 10 mol % L1) and the reaction gave N-arylated product in only a moderate yield even after 24 h. In addition, reactions of 4-substituted imidazoles, functionalized aryl halides and heteroaryl halides were not disclosed. Herein, we report a full account of our investigations of completely N1-selective arylation of imidazoles and disclose a significantly improved Pd-catalyzed protocol. Usefulness of the completely N1-selective arylation of unsymmetric 4-substituted imidazoles was demonstrated by the regioselective syntheses of medicinally important compounds GSK2137305 and nilotinib.
Results and Discussion
Design and development of the completely N1-selective arylation of 4-methylimidazole
Scheme 1 shows some of the presumed intermediates for the Pd-catalyzed N-arylation of 4-substituted imidazoles.
Scheme 1.

Presumed intermediates for the Pd-catalyzed N-arylation of 4-substituted imidazoles
We reasoned that selective formation of the N1-arylated product might be achievable due to unfavorable steric interactions in B′ relative to B. The idea was supported by density functional theory (DFT) calculations with L1; complex B was favored over complex B′ and the transition states for the reductive elimination (B-TS and B′-TS) were also significantly different (ΔΔG‡= 5.1 kcal/mol) in favor of the transition state to the N1-arylated product (Figure 2).
Figure 2.

Energy diagram for reductive elimination with L1
To test the above hypothesis, we investigated the N-arylation of 4-methylimidazole with bromobenzene with a variety of phosphine ligands (Table 1). An examination of reaction conditions revealed that the use of Pd2(dba)3 (0.75 mol %), L1 (1.8 mol %) and K3PO4 selectively produced N1-arylated product, albeit only in 6% yield (entry 1). The examination of other structurally related biaryl phosphine ligands (L2–L5) and bases (Cs2CO3, K2CO3, NaOtBu) provided no improvement in reaction efficacy (entries 2–8). Similarly, we obtained no N-arylation product with L6, L7 or L8 (entries 9–11), which were previously employed for the N-arylation of unsubstituted imidazole with activated aryl bromides20a and chlorides20b in the presence of Pd(OAc)2 using the reported protocol published for each of these ligands. Further experimentations using L1 revealed that an improved yield (66%) could be obtained by employing higher catalyst loading (5 mol % Pd and 10 mol % L1), however, the reaction was sluggish and full conversion of bromobenzene was not achieved even after 20 h (entry 12). Since imidazoles are known to be ligands in Pd(0)-catalyzed reactions,22 we suspected that the ineffectiveness of the reaction might be due to prevention of the in situ formation of the catalytically-active phosphine-ligated Pd(0) complex by excess 4-methylimidazole. Thus, we heated a premixed solution of Pd2(dba)3 and L1 in the solvent for 3 min at 120 °C prior to the injection to other reagents. With this protocol, the preactivated catalyst solution (Pd2(dba)3/L1) showed high activity and the N1-arylated product was obtained in 97% GC yield (95% isolated yield) in 5 h with 1.5 mol % Pd and 1.8 mol % L1 (entry 13).23,24 To further understand the source of inhibition of the reaction (entry 1 vs 13), imidazoles and each of the reaction components (K3PO4 and PhBr) were, in separate experiments, added to the premixed solution of Pd2(dba)3 and L1 (Scheme 2). When the catalyst premixing was peformed in the presence of 4-methylimidazole, the reaction was significantly less efficient and the product was obtained only in 4% yield (Scheme 2, entry 1). Similarly, 1-methylimidazole showed inhibitory effect on the reaction (entry 2). On the other hand, addition of 1-methylpyrrole, bromobenzene or K3PO4 to the premixing solution did not significantly affect the reaction outcome and the product was obtained in high yields (entries 3–5). To observe the effect of 4-methylimidazole on the in situ formation of active catalyst, 31P NMR signal of L1 was monitored at the catalyst premixing step (Figure 3). New phosphorus signals at 88 ppm and 91 ppm appeared after heating L1 and Pd2(dba)3 in toluene at 120 °C, for 3 min, 25 while only free L1 was observed when L1 and Pd2(dba)3 were heated in the presence of excess 4-methylimidazole, suggesting that 4-methylimidazole prevents in situ ligand binding to Pd(0). Overall, our investigation led to the conclusions that (1) L1 is the highly effective ligand for the Pd-catalyzed N-arylation of 4-methylimidazole and (2) imidazoles have an inhibitory effect on the in situ formation of catalytically-active Pd(0)-ligand complex, and therefore, L1 and Pd2(dba)3 should be pre-heated together in the solvent before they were exposed to the imidazoles. Of great significance is that while 4-methylimidazole prevents the binding of L1 to the Pd center, once the ligand is bound it is not displaced by the imidazole.
Table 1.
Ligand effects on Pd-catalyzed N-arylation of 4-methylimidazole.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | Pd source (mol %) | ligand (mol %) | base | time (h) | GC conv. (%) | GC yield (%) |
| 1 | Pd2(dba)3 (0.75) | L1 (1.8) | K3PO4 | 20 | 8 | 6 |
| 2 | Pd2(dba)3 (0.75) | L2 (1.8) | K3PO4 | 20 | 6 | 3 |
| 3 | Pd2(dba)3 (0.75) | L3 (1.8) | K3PO4 | 20 | 5 | 0 |
| 4 | Pd2(dba)3 (0.75) | L4 (1.8) | K3PO4 | 20 | 5 | 0 |
| 5 | Pd2(dba)3 (0.75) | L5 (1.8) | K3PO4 | 20 | 3 | 0 |
| 6 | Pd2(dba)3 (0.75) | L1 (1.8) | Cs2CO3 | 20 | 3 | 0 |
| 7 | Pd2(dba)3 (0.75) | L1 (1.8) | K2CO3 | 20 | 3 | 0 |
| 8 | Pd2(dba)3 (0.75) | L1 (1.8) | NaOtBu | 20 | 9 | 0 |
| 9b | Pd(OAc)2 (5) | L6 (10) | KOtBu | 0.25 | 8 | 0 |
| 10c | Pd(OAc)2 (2) | L7(2) | NaOtBu | 24 | 6 | 0 |
| 11d | Pd(OAc)2 (2) | L8 (2) | NaOtBu | 12 | 9 | 0 |
| 12 | Pd2(dba)3 (2.5) | L1 (10) | K3PO4 | 20 | 69 | 66 |
| 13e | Pd2(dba)3 (0.75) | L1 (1.8) | K3PO4 | 5 | 100 | 97 (95)f |
Conditions for entries 1–8 and 12–13: bromobenzene (1 mmol), 4-methylimidazole (1.2 mmol), base (2 mmol), Pd2(dba)3 (0.75 or 2.5 mol%), ligand (1.8 or 10 mol%), toluene-dioxane (5:1, 1.0 mL), 120 °C, 5 or 20 h.
Reaction was performed in DMF (2 mL) under microwave heating (180 °C for 15 min).
Reaction was performed in o-xylene (20 mL) at 120 °C for 24 h.
Reaction was performed in THF (20 mL) at room temperature for 12 h.
Pd2(dba)3 and L1 were premixed in the solvent at 120 °C for 3 min.
Isolated yield.
Scheme 2.

Inhibitory effect of imidazoles on the Pd-catalyzed N-arylation of 4-methylimidazole
Figure 3.
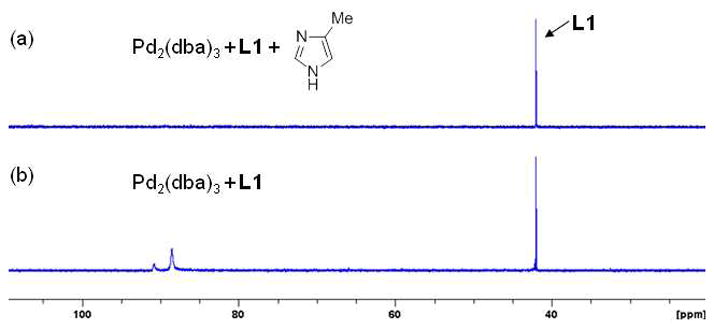
31P NMR spectra of the solution of L1 and Pd2(dba)3 in the presence (a) or absence (b) of 4-methylimidazole (16 equiv to Pd)
Substrate scope of N-arylation of 4-substituted imidazole
With an optimized protocol in hand, we next investigated the scope of the N-arylation of unsymmetric imidazoles and benzimidazoles (Table 2). The Pd-catalyzed N1-selective arylation was highly general and a variety of functionalized aryl bromides and chlorides could be employed for the arylation of 4-methylimidazole (entries 1–9), 4-phenylimidazole (entries 12–13), N-acetylhistamine (entry 14) and 4-cyanomethylimidazole (entries 15–16) using 0.5–2.5 mol % Pd. In addition, aryl triflates were suitable substrates (entries 10–11). To the best of our knowledge, this is the first example of N-arylation of imidazole derivatives with aryl triflates. No N3-arylated regioisomers were detected in all cases examined. The N-arylation of 4-substituted imidazoles with 3-halopyridines (entries 4 and 12), 2-bromopyridines (entries 8 and 15) and chloropyrazine (entry 16) demonstrates the utility of the method with heteroaryl substrates. Lastly, the N-arylation of 4-substituted benzimidazoles exclusively occurred at less sterically hindered N1 position to give N1-arylated benzimidazoles in good yields (entries 17–18).
Table 2.
Substrate scope of N1-selective arylation of unsymmetric imidazolesa
 | ||||
|---|---|---|---|---|
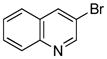
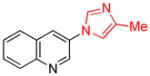
| entry | Ar-X | product | Pd (mol%) | yield (%) |
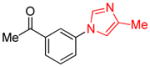
| 1 |
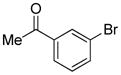
|
|
1.5 | 84 |
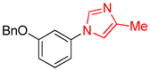
| 2 |
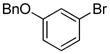
|
|
0.5 | 93 |
| 3 |
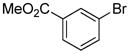
|
|
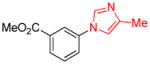
0.5 | 91 |
| 4 |
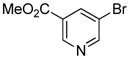
|
|
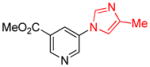
1.5 | 85 |
| 5 |
|
|
0.5 | 94 |
| 6 |
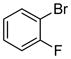
|
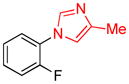
|
1.5 | 94 |
| 7 |
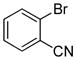
|
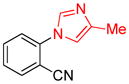
|
2 | 88 |
| 8 |
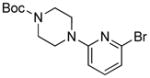
|
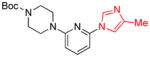
|
2 | 80 |
| 9 |
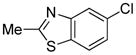
|
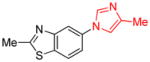
|
1.5 | 79 |
| 10 |
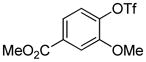
|
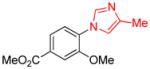
|
1.5 | 77 |
| 11 |
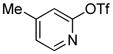
|
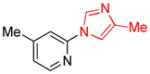
|
1.5 | 80 |
| 12 |
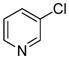
|
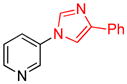
|
1.5 | 89 |
| 13 |
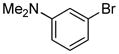
|
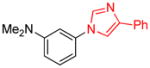
|
1.5 | 96 |
| 14 |
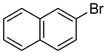
|
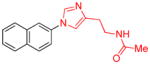
|
1.5 | 95 |
| 15 |
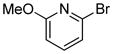
|
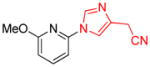
|
1.5 | 90 |
| 16 |
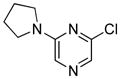
|
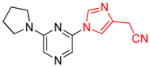
|
2.5 | 92 |
| 17 |
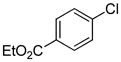
|
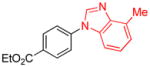
|
1 | 90 |
| 18 |
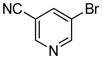
|
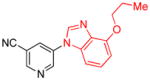
|
1.5 | 86 |
Ar-X (1 mmol), 4-substituted imidazole (1.1–1.2 mmol), K3PO4 (2 mmol), Pd2(dba)3 (0.25–1.25 mol %), L1 (0.5–2.5 mol %), toluene/dioxane (5:1, 1.0 mL), 120 °C, 5 h. Pd2(dba)3 and L1 were premixed in the solvent at 120 °C for 3 min. Isolated yield, average of two runs.
Synthesis of GSK2137305
GSK2137305 is a potent glycine transporter inhibitor developed by GlaxoSmithKline for the treatment of neurological disorders. Previously, the N1-aryl-4-methylimidazole 2 was synthesized by the Cu-catalyzed N-arylation of 4-methylimidazole with aryl bromide 1a. Although the Cu-catalyzed N-arylation proceeded under ligand-free condition, approximately 4:1 mixture of N1- and N3-arylated product was formed and yield of only 54% was obtained.4 Using 0.1 mol % Pd2(dba)3 and 0.2 mol % L1, N-arylation of 4-methylimidazole with aryl bromide 1a gave 2 in 93% yield as a single regioisomer (Scheme 3). Alternatively, aryl chloride substrate 1b, which was prepared from less expensive 4-chlorobenzaldehyde,26 could be employed for the N-arylation reaction with 0.15 mol % Pd2(dba)3 and 0.3 mol % L1. Subsequent N-alkylation of 2 afforded GSK2137305 (3) in 82% yield.
Scheme 3.

Synthesis of GSK2137305
Reagents and conditions; a) NaCN (1.1 equiv), NH4OAc (3.0 equiv), aq. NH4OH, EtOH, rt, 4 h then, cyclopentanone (2.5 equiv), NaOEt (0.1 equiv), n-BuOH, 80 °C, 12 h, 38% (X = Br), 32% (X = Cl). b) DDQ (1.1 equiv), EtOAc, 60 °C, 1 h, 89% (X = Br), 90% (X = Cl). c) Chloroacetyl chloride (1.1 equiv), 3-aminobenzotrifluoride (1.15 equiv), KOH (2.5 equiv), NMP, 82%.
Synthesis of nilotinib (Tasigna®)
The usefulness of the Pd-catalyzed N1-selective arylation of 4-substituted imidazoles was further demonstrated by the synthesis of the clinically important anti-cancer drug nilotinib (Scheme 4). Nilotinib (Tasigna®) is a second-generation BCR-ABL tyrosine kinase inhibitor that shows greater efficacy compared to imatinib (Gleevec®) in the treatment of chronic myelogenous leukemia (CML), including Gleevec-resistant patients.1 In the previous syntheses of nilotinib, 4-methylimidazole was introduced by moderately regioselective reactions such as the SNAr reaction with an aryl fluoride27 or Cu-catalyzed N-arylation with an aryl bromide.28,29 Using 0.25 mol % Pd2(dba)3 and 0.5 mol % L1, key intermediate 5 was prepared from aryl bromide 4 in 90% yield as a single regioisomer. Use of excess (2.4 eq.) 4-methylimidazole, toluene-tBuOH mixed solvent and lower concentration (0.5 M ArBr) were found to be important to supress undesirable N-arylation of aniline nitrogen of 4 and 5. Ester 8 could be prepared from aminopyrimidine 6 and aryl bromide 7 using 0.3 mol% BrettPhos (L3) precatalyst.12b Combining 5 and 8 gave nilotinib base 9 in 90% yield. Alternatively, 4-methylimidazole could be introduced at the last step of the synthesis using aryl bromide 10, 1 mol% Pd2(dba)3 and 2.2 mol% L1. Again, the N-arylated product 9 was obtained as single regioisomer. This late-stage N-arylation route could potentially provide facile access to a large variety of nilotinib analogues.
Scheme 4.

Synthesis of nilotinib base
Reagents and conditions; a) KOtBu (5.5 equiv), THF, rt, 12 h, 90%, b) 4 (1.05 equiv), KOtBu (5.5 equiv), THF, rt, 12 h, 85%.
Conclusion
We have established a catalytic method, based on mechanistic considerations, for the completely N1-selective arylation of unsymmetric imidazoles with aryl bromides, chlorides and triflates. This study showed that imidazoles have a strong inhibitory effect on the in situ formation of catalytically-active Pd(0)-ligand complex. By heating Pd2(dba)3 and L1 in the absence of imidazoles before the reaction, no inhibitory effect was observed and the efficacy of the N-arylation of imidazoles was improved drastically. From these findings it is clear that while imidazoles can prevent binding of L1 to the Pd, once the ligand is bound to the metal, these heterocycles do not displace it. They also point to the importance, in general, of allowing the preparation of a catalyst (or more accurately the ligand-metal complex) to take place prior to exposure of it to substrates that otherwise would tightly binding the metal in question. The utility of this method was demonstrated by the regiocontrolled syntheses of GSK2137305 and nilotinib. Highly N1-selective couplings of 4-substituted imidazoles with aryl bromides, chlorides and triflates with SNAr or Cu-based systems have not yet been reported. Thus, the present Pd-catalyst system complements existing SNAr-based and Cu-catalyzed N-arylation methods. Studies on the Pd-catalyzed arylation of other 5-membered nitrogen heterocycles are currently underway in our laboratory.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health (GM58160). S.U. thanks the Japan Society for the Promotion of Sciences (JSPS) for a Postdoctral Fellowship for Research Abroad. We thank Dr. Andrew T. Parsons for helpful discussion and help with preparation of this manuscript.
Footnotes
Supporting Information Available: Complete ref 1a and 20d, experimental procedures, product characterization and copies of 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kantarjian H, et al. N Eng J Med. 2006;354:2542. [Google Scholar]; (b) Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. Br J Cancer. 2006;94:1765. doi: 10.1038/sj.bjc.6603170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Kimura T, Kawano K, Doi E, Kitazawa N, Takaishi M, Ito K, Kaneko T, Sasaki T, Miyagawa T, Hagiwara H, Yoshida Y. 20070117839. US. 2006; (b) Huang X, Aslanian R, Zhou W, Zhu X, Qin J, Greenlee W, Zhu Z, Zhang L, Hyde L, Chu I, Cohen-Williams M, Palani A. ACS Med Chem Lett. 2010;1:184. doi: 10.1021/ml1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harada K, Aota M, Inoue T, Matsuda R, Mihara T, Yamaji T, Ishibashi K, Matsuoka N. Eur J Pharmacol. 2006;553:171. doi: 10.1016/j.ejphar.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 4.Graham JP, Langlade N, Northall JM, Roberts AJ, Whitehead AJ. Org Process Res Dev. 2011;15:44. [Google Scholar]
- 5.See, footnote 1 in ref 6a.
- 6.Kiyomori A, Marcoux JF, Buchwald SL. Tetrahedron Lett. 1999;40:2657.Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727. doi: 10.1021/ja016226z.Antilla JC, Baskin JM, Barder TE, Buchwald SL. J Org Chem. 2004;69:5578. doi: 10.1021/jo049658b.Cristau HJ, Cellier PP, Spindler JF, Taillefer M. Chem Eur J. 2004;10:5607. doi: 10.1002/chem.200400582.Ma D, Cai Q. Synlett. 2004:128.Zhang H, Cai Q, Ma D. J Org Chem. 2005;70:5164. doi: 10.1021/jo0504464.Liu L, Frohn M, Xi N, Dominguez C, Hungate R, Reider PJ. J Org Chem. 2005;70:10135. doi: 10.1021/jo051640t.Jerphagnon T, van Link GPM, de Vries JG, van Koten G. Org Lett. 2005;7:5241. doi: 10.1021/ol052113z.Altman RA, Buchwald SL. Org Lett. 2006;8:2779. doi: 10.1021/ol0608505.Xie YX, Pi SF, Wang J, Yin DL, Li JH. J Org Chem. 2006;71:8324. doi: 10.1021/jo061572q.Altman RA, Koval ED, Buchwald SL. J Org Chem. 2007;72:6190. doi: 10.1021/jo070807a.Zhu L, Cheng L, Zhang Y, Xie R, You J. J Org Chem. 2007;72:2737. doi: 10.1021/jo062059f.Zhu L, Guo P, Li G, Lan J, Xie R, You J. J Org Chem. 2007;72:8535. doi: 10.1021/jo0712289.Lv X, Bao W. J Org Chem. 2007;72:3863. doi: 10.1021/jo070443m.Taillefer M, Xia N, Ouali A. Angew Chem Int Ed. 2007;46:934. doi: 10.1002/anie.200603173.Zhu L, Li G, Luo L, Guo P, Lan J, You J. J Org Chem. 2009;74:2200. doi: 10.1021/jo802669b.Liang L, Li Z, Zhou X. Org Lett. 2009;11:3294. doi: 10.1021/ol9010773.Chen H, Wang D, Wang X, Huang W, Cai Q, Ding K. Synthesis. 2010:1505.Correction: Chen H, Wang D, Wang X, Huang W, Cai Q, Ding K. Synthesis. 2011:2684.
- 7.(a) Collman JP, Zhong M. Org Lett. 2000;2:1233. doi: 10.1021/ol000033j. [DOI] [PubMed] [Google Scholar]; (b) Collman JP, Zhong M, Zeng Li, Costanzo S. J Org Chem. 2001;66:1528. doi: 10.1021/jo0016780. [DOI] [PubMed] [Google Scholar]; (c) Collman JP, Zhong M, Zhang C, Costanzo S. J Org Chem. 2001;66:7892. doi: 10.1021/jo010615u. [DOI] [PubMed] [Google Scholar]; (d) Yu XQ, Yamamoto Y, Miyaura N. Chem Asian J. 2008;3:1517. doi: 10.1002/asia.200800135. [DOI] [PubMed] [Google Scholar]
- 8.Reviews, see; Bellina F, Rossi R. Adv Synth Catal. 2010;352:1223.Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400. doi: 10.1002/anie.200300594.Monnier F, Taillefer M. Angew Chem Int Ed. 2009;48:6954. doi: 10.1002/anie.200804497.
- 9.Excellent N1-selectivities (N1/N3 = 17/1 and higher) were reported only for a few aryl iodide substrates, aryl bromides with bulky ortho-substituents and 4-arylimidazoles, see ref. 6b, 6k and 6p. N-Arylation of 4-methylimidazoles with ortho-unsubstituted aryl bromides showed moderate N1-selectivity (N1/N3 = 4.1–5.7/1), see, ref 4, 6k, 6m, 6r and 28.
- 10.A rare example of Cu-catalyzed N1-selective arylation of 4-substituted imidazoles was reported using aryllead (IV) reagents as aryl donor; Elliott GI, Konopelski JP. Org Lett. 2000;2:3055. doi: 10.1021/ol006271w.
- 11.(a) Surry DS, Buchwald SL. Chem Sci. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Torborg C, Beller M. Adv Synth Catal. 2009;351:3027. [Google Scholar]; (c) Hartwig JF. Nature. 2008;455:314. doi: 10.1038/nature07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selected examples; Fors BP, Buchwald SL. J Am Chem Soc. 2010;132:15914. doi: 10.1021/ja108074t.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552. doi: 10.1021/ja8055358.Shen Q, Ogata T, Hartwig JF. J Am Chem Soc. 2008;130:6586. doi: 10.1021/ja077074w.Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J Am Chem Soc. 2006;128:4101. doi: 10.1021/ja057704z.Shen Q, Shekhar S, Stambuli JP, Hartwig JF. Angew Chem Int Ed. 2005;44:1371. doi: 10.1002/anie.200462629.
- 13.Wolfe JP, Åhman J, Sadighi JP, Singer RA, Buchwald SL. Tetrahedron Lett. 1997;38:6367. [Google Scholar]
- 14.(a) Hicks JD, Hyde AM, Martinez Cuezva A, Buchwald SL. J Am Chem Soc. 2009;131:16720. doi: 10.1021/ja9044357. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ikawa T, Barder TE, Biscoe MR, Buchwald SL. J Am Chem Soc. 2007;129:13001. doi: 10.1021/ja0717414. [DOI] [PubMed] [Google Scholar]; (c) Ghosh A, Sieser JE, Riou M, Cai W, Rivera-Ruiz L. Org Lett. 2003;5:2207. doi: 10.1021/ol034428p. [DOI] [PubMed] [Google Scholar]; (d) Yin J, Buchwald SL. J Am Chem Soc. 2002;124:6043. doi: 10.1021/ja012610k. [DOI] [PubMed] [Google Scholar]
- 15.(a) Mann G, Hartwig JF, Driver MS, Fernández-Rivas C. J Am Chem Soc. 1998;120:827. [Google Scholar]; (b) Old DW, Harris MC, Buchwald SL. Org Lett. 2000;2:1403. doi: 10.1021/ol005728z. [DOI] [PubMed] [Google Scholar]
- 16.Lundgren RJ, Stradiotto M. Angew Chem Int Ed. 2010;49:8686. doi: 10.1002/anie.201003764. [DOI] [PubMed] [Google Scholar]
- 17.(a) Wagaw S, Yang BH, Buchwald SL. J Am Chem Soc. 1998;120:6621. [Google Scholar]; (b) Wagaw S, Yang BH, Buchwald SL. J Am Chem Soc. 1999;121:10251. [Google Scholar]; (c) Arterburn JB, Rao KV, Ramdas R, Dible BR. Org Lett. 2001;3:1351. doi: 10.1021/ol015731y. [DOI] [PubMed] [Google Scholar]; (d) Mauger C, Mignani G. Adv Synth Catal. 2005;347:773. [Google Scholar]; (e) Thiel OR, Achmatowicz MM, Reichelt A, Larsen RD. Angew Chem Int Ed. 2010;49:8395. doi: 10.1002/anie.201001999. [DOI] [PubMed] [Google Scholar]
- 18.(a) Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:10028. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]; (b) Surry DS, Buchwald SL. J Am Chem Soc. 2007;129:10354. doi: 10.1021/ja074681a. [DOI] [PubMed] [Google Scholar]; (c) Vo GD, Hartwig JF. J Am Chem Soc. 2009;131:11049. doi: 10.1021/ja903049z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schulz T, Torborg C, Enthaler S, Schäffner B, Dumrath A, Spannenberg A, Neumann H, Börner A, Beller M. Chem Eur J. 2009;15:4528. doi: 10.1002/chem.200802678. [DOI] [PubMed] [Google Scholar]
- 19.Fors BP, Buchwald SL. J Am Chem Soc. 2009;131:12898. doi: 10.1021/ja905768k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Wan Y, Alterman M, Hallberg A. Synthesis. 2002:1597. [Google Scholar]; (b) Andrus MB, Mettath SN, Song C. J Org Chem. 2002;67:8284. doi: 10.1021/jo026217o. [DOI] [PubMed] [Google Scholar]; (c) Burns CJ, Harte MF, Palmer JT. 2008058341. WO. 2008; (d) Lee C, et al. 2010027236. WO. 2010
- 21.Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Angew Chem Int Ed. 2006;45:6523. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 22.(a) Mathews CJ, Smith PJ, Welton T. J Mol Catal A. 2003;206:77. [Google Scholar]; (b) Haneda S, Ueba C, Eda K, Hayashi M. Adv Synth Catal. 2007;349:833. [Google Scholar]
- 23.Benefical effects of the catalyst premixing were previously reported. Wolfe JP, Buchwald SL. J Org Chem. 2000;65:1144. doi: 10.1021/jo9916986.Ueda S, Su M, Buchwald SL. Angew Chem Int Ed. 2011;50:8944. doi: 10.1002/anie.201103882.See also ref 12c. Toluene-dioxane mixed solvent gave better result than toluene probably due to higher solubility of imidazole derivative in the mixed solvent.
- 24.Single-component Pd-ligand precatalysts have proven to be an ideal source for the in situ production of catalytically active Pd(0)-ligand complex. However, preparation of Pd-L1 precatalyst was not successful. For single-component palladium precatalysts; Biscoe MR, Fors BP, Buchwald SL. J Am Chem Soc. 2008;130:6686. doi: 10.1021/ja801137k.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799.
- 25.The two peaks might correspond to the comformers that arise from rotation around aromatic C-P bond. The C-P bond rotamers were previously observed in the Pd-biarylphosphine complex, see; ref 12b. Although isolation of the Pd(0)-L1 complex was not successful, formation of Pd(0)-L1 complex was confirmed by ESI-MS analysis of the crude mixture. See the supporting information.
- 26.4-Bromobenzaldehyde: 154.5 USD/100 g; 4-chlorobenzaldehyde: 38.7 USD/250 g. (Sigma-Aldrich, 2011)
- 27.Breitenstein W, Furet P, Jacob S, Manley PW. 2004005281. WO. 2004
- 28.These reactions gave 5.5:1 or 5:1 mixture of N1- and N3-arylated products; Huang WS, Shakespeare WC. Synthesis. 2007:2121.See also ref. 6r.
- 29.Although regioisomeric ratio was not disclosed, Cu-catalyzed N-arylation of 4-methylimidazole with 4 or 10 was reported. Yeori A, Wang Y, Li J, Zhu J, Lifshitz-Liron R, He X. 2010060074. WO. 2010Wang Y, Li K, Vinod K, Zhu J, Lifshitz-Liron R, Mistry DN, Vasoya SL, Ariyamuthu S, Pilarski G, He X. 2010009402. WO. 2010
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.