

Abstract
Nicotinamidases catalyze the hydrolysis of nicotinamide to nicotinic acid and ammonia. Nicotinamidases are absent in mammals but function in NAD+ salvage in many bacteria, yeast, plants, protozoa, and metazoans. We have performed structural and kinetic investigations of the nicotinamidase from S. cerevisiae (Pnc1). Steady-state product inhibitor analysis revealed an irreversible reaction where ammonia is the first product released, followed by nicotinic acid. A series of nicotinamide analogs acting as inhibitors or substrates were examined revealing that the nicotinamide carbonyl oxygen and ring nitrogen are critical for binding and reactivity. X-ray structural analysis revealed a covalent adduct between nicotinaldehyde and Cys167 of Pnc1 and coordination of the nicotinamide ring nitrogen to the active-site zinc ion. Using this structure as a guide, the function of several residues was probed via mutagenesis and primary 15N and 13C kinetic isotope effects (KIE) on V/K for amide bond hydrolysis. The KIE values of almost all variants were increased indicating that C-N bond cleavage is at least partially rate limiting; however, a decreased KIE for D51N was observed indicative of a higher commitment to catalysis. In addition, KIE values using slower alternate substrates indicated that C-N bond cleavage is at least partially rate limiting with nicotinamide to highly rate limiting with thionicotinamide. A detailed mechanism is discussed involving nucleophilic attack of Cys167, followed by elimination of ammonia and then hydrolysis to liberate nicotinic acid. These results will aid design of mechanism-based inhibitors to target pathogens that rely on nicotinamidase activity.
Nicotinamidases (EC 3.5.1.19) are amidohydrolases that catalyze the hydrolysis of nicotinamide to nicotinic acid and ammonia (Scheme 1). Nicotinamidases play a central role in the NAD+ salvage pathway (1) of multiple species of bacteria, yeast (2), protozoa (3), and plants (4) and are present in many metazoans such as Drosophila melanogaster (5) and Caenorhabditis elegans (6, 7). However, mammals do not encode a nicotinamidase but instead use nicotinamide phosphoribosyltransferase to convert nicotinamide directly to nicotinamide mononucleotide, which is then recycled to NAD+ (8).
Scheme 1.

General reaction catalyzed by nicotinamidases.
The importance of nicotinamidase activity in the NAD+ salvage pathways of human pathogens, combined with the absence of a nicotinamidase in human NAD+ salvage pathways, suggests that nicotinamidases are potential drug targets. Indeed, nicotinamidase was shown to be an essential enzyme for the infectious phenotype of Borrelia burgdorferi, the bacteria that cause Lyme disease (9). In Brucella abortus, which causes abortion in domestic animals and undulant fever in humans, the B. abortus nicotinamidase is essential for replication (10). Furthermore, erythrocytes infected with Plasmodium falciparum, a parasite that causes malaria in humans, displayed increased nicotinamidase activity and NAD+ synthesis (3). It is likely that other human pathogens require nicotinamidase activity for viability, because many, including P. falciparum, do not possess the genes necessary for de novo NAD+ synthesis.
Current tuberculosis treatments target the nicotinamidase (PncA) of Mycobacterium tuberculosis, which hydrolyzes the prodrug pyrazinamide to the active form, pyrazinoic acid. Pyrazinamide, when administered in combination with isoniazid and rifampin, forms the current short-course treatment recommended by the World Health Organization (11) and shortens tuberculosis treatment from 9- to 6-months. Pyrazinoic acid displays its toxicity by inhibiting M. tuberculosis trans-translation through binding the ribosomal protein S1 (12). Mutations in the M. tuberculosis PNCA gene are associated with clinical resistance to pyrazinamide (13–16).
Nicotinamidases have also been implicated in increasing the lifespan of Saccharomyces cerevisiae (17–20), C. elegans (7), and D. melanogaster (5). The observed lifespan extension in these organisms is potentially mediated through increasing the activity of sirtuin NAD+-dependent deacetylases by decreasing cellular nicotinamide levels and increasing NAD+ levels. In support of this hypothesis, sirtuin overexpression was reported to increase lifespan in S. cerevisiae (21, 22), C. elegans (23), and D. melanogaster (24, 25) and nicotinamide is a potent sirtuin product inhibitor (19, 26–28). However, a recent report revealed that the apparent lifespan extension through sirtuin overexpression in C. elegans and D. melanogaster was abolished when a more appropriate genetic background was used (29).
Despite the importance of nicotinamidases in diverse biological processes, their precise mechanism of catalysis has yet to be fully elucidated (30–33). Understanding the chemical mechanism and nature of the transition state in the reaction catalyzed by nicotinamidases would aid the design of inhibitors or prodrugs (such as pyrazinamide) that target nicotinamidases for anti-microbial applications. Here, we establish the overall kinetic mechanism of the eukaryotic nicotinamidase from S. cerevisiae (Pnc1) through product inhibition analysis. We then show that ketone and aldehyde containing nicotinamide analogs are Pnc1 inhibitors. Using this knowledge we obtained the first crystal structure of a eukaryotic nicotinamidase with a nicotinamide analog bound in the active site. This structure suggested several residues potentially involved in catalysis, and the steady-state kinetic parameters of several Pnc1 mutants were determined. Steady-state kinetic parameters with alternate nicotinamide analog substrates were also measured. We then further delineated the mechanism by determining the primary 15N and 13C kinetic isotope effects (KIE) of the C-N bond breaking and the pH dependence of the reaction. To determine if C-N bond cleavage is partially or fully rate limiting, we compared the 15N and 13C KIE for nicotinamide hydrolysis by Pnc1 with those determined using slow substrate analogs. In addition, the KIE values for nicotinamide hydrolysis by several Pnc1 mutants were compared, allowing for the validation of the proposed catalytic function of active-site residues. These results suggest specific roles for several Pnc1 residues during catalysis as well as structure activity relationships for Pnc1 catalyzed hydrolysis of nicotinamide analogs.
Experimental Procedures
Expression and purification of Pnc1
Yeast Pnc1 cloned into pET-16b or pET-17b (20, 32) was transformed into E. coli strain BL21(DE3). A single transformant colony was grown in 1.0 L 2XYT medium at 37°C to OD600nm ~0.6–0.8. IPTG was added to 0.5 mM, and expression was continued for 6–8 hr at 25°C. Cells were harvested by centrifugation and stored at −20 °C. Cellular pellets were resuspended in buffer A (50 mM NaH2PO4, pH 7.5, 300 mM NaCl, 1 mM β-mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride) containing 30 mM imidazole and lysed by sonication. Recombinant Pnc1 was purified using immobilized metal affinity chromatography with a Ni2+-nitrilotriacetic column. The column was washed with buffer A containing 30 mM imidazole and bound proteins were eluted with a gradient of 30 to 500 mM imidazole in buffer A at pH 7.5. Pooled fractions containing Pnc1 were concentrated and protein concentrations were determined using the Bradford method (34) using bovine serum albumin as a standard.
Mutagenesis of Pnc1
Pnc1 mutagenesis was performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene) according to the manufacturers protocol. The primers used for mutagenesis are included in supplemental Table S1.
Determination of kinetic parameters
Nicotinamidase activity was measured continuously using an enzyme-coupled assay with glutamate dehydrogenase using a Multiskan Ascent microplate reader (LabSystems; Franklin, MA, USA). This assay is slightly modified from that described by Su et al. (35). Typical assay mixtures contained 1.25 µM to 3.2 mM nicotinamide or analog, 0.2 mM NADPH, 3.3 mM α-ketoglutarate, 50 nM to 10 µM Pnc1 WT or mutant, and 3 units of glutamate dehydrogenase from bovine liver in 50 mM sodium phosphate at pH 7.5. Assays were carried out in a final volume of 300 µL per well in a clear, flat-bottomed, 96-well plate. All assay components except Pnc1 were preincubated at 25 °C for 5 min or until absorbance at 340 nm stabilized, and the reaction was initiated by the addition of Pnc1. The rates were monitored continuously for NADPH consumption at 340 nm. Rates were determined from the slopes of the initial linear portion of each curve using an extinction coefficient for NADPH of 6.22 mM−1cm−1 and a pathlength of 0.9 cm for 300 µL reactions. The background rates of reactions lacking Pnc1 resulting from the spontaneous formation of ammonia were subtracted from the initial velocities of the Pnc1-catalyzed reactions.
Determination of Ki values
Assay mixtures contained 6.7 µM to 200 µM nicotinamide, 0.2 mM NADPH, 1 or 3.3 mM α-ketoglutarate, 100 nM to 0.5 µM Pnc1 WT, and 3 units of glutamate dehydrogenase from bovine liver in 50 mM sodium phosphate at pH 7.5. For nicotinic acid inhibition 200 µM to 1.2 mM nicotinic acid was used. For nicotinaldehyde inhibition 1 µM to 6 µM was used. For 3-acetylpyridine inhibition 300 µM to 1.8 mM was used. Benzaldehyde and pyrazinoic acid were initially dissolved in DMSO and 5 to 10 mM was used (10% final v/v DMSO). Assays were run and analyzed as detailed under determination of kinetic parameters. Initial velocity data were fitted in Kinetasyst (Intellikinetics, State College, PA) to competitive inhibition patterns (eq 1 or 2) based on the algorithms defined by Cleland (36). All data were displayed using Kaleidagraph (Synergy Software, Reading, PA).
| (1) |
| (2) |
Protein crystallization and structure determination
For crystallization trials, the protein was purified as described above, with the addition of a Sephacryl S-300 size-exclusion step. Protein was dialyzed against buffer containing 15 mM Tris pH 7.5, 50 mM NaCl, 4 mM MgCl2, 10 mM Na citrate and 5% glycerol and the inhibitor nicotinaldehyde was added at a 4:1 molar ratio. The protein (5 mg/ml) was mixed with mother liquor (1.6 M NaOAc, 10% ethylene glycol, 0.1 M HEPES pH 7.4) at a 1:1 (vol) ratio. Crystals were formed by hanging drop vapor diffusion. Crystals were transferred to a cryoprotectant solution (1.5 M NaOAc, 20% ethylene glycol, 0.1 M HEPES pH 7.4) and flash-frozen in liquid nitrogen.
Diffraction data were indexed and scaled using HKL2000 (37). The structure of Pnc1p with the inhibitor nicotinaldehyde was solved by molecular replacement (Phaser) (38) using the apo Pnc1p structure (32) as a search model. The structure was improved by rounds of manual fitting using Coot (39) and refinement using REFMAC5 (40). Coordinate and structure factor files have been deposited in the Protein Data Bank (PDB ID XXXX).
15N kinetic isotope effects
Reactions for isotope effect analysis were carried out in 7.5–10 mL of 20 mM potassium phosphate (pH 7.5) containing 10 mM nicotinamide or analog and 0.75–36 µM Pnc1 WT or mutant. Reactions were quenched by addition of 8 M HCl to a final concentration of 40 mM. The enzyme was removed by Amicon filtration (10,000 MWCO) and the solution was diluted to 100 mL with distilled water. The pH was adjusted to 6 and the solution was loaded onto an AG1X8 resin column (Cl− form, 2.5 cm × 30 cm) at 1 mL/min. Fraction collection (8.5 mL/fraction) was started at the time of loading. When the loading was complete, the products were eluted with water at 1 mL/min. NH4Cl eluted between fractions 3–18 and was detected using 50 µL samples of each fraction added to 50 µL of Nessler’s reagent in 96-well plates. A yellow color indicated the presence of NH4Cl. The residual nicotinamide or analog generally eluted between fractions 25–50 and was detected by spotting on UV active TLC plates. The product, nicotinic acid, was eluted with 1 M HCl from the column and found by UV at 262 nm. Product NH4Cl was reduced to 50 mL by rotary evaporation and purified by steam distillation with 12 mL of 13 M KOH. The NH3 was trapped in 10 mL of 100 mN H2SO4 and collected until the total volume reached 50 mL.
The residual substrate was pooled and concentrated to 50 mL. It was then steam distilled at high pH, hydrolyzing the amide, and the resultant NH3 was trapped in acid. The hydrolysis of nicotinamide (40–50 µmoles) required the addition of 12 mL of 13N NaOH to a 50 mL solution. The other analogs required 13N NaOH in the following amounts: 10 mL for thionicotinamide, 8 mL for 5-methylnicotinamide, and 3 mL for pyrazinamide. Considerable time was spent determining the correct amount of base to be used since too much caused the decomposition of the pyridine ring and production of non-amide NH3. This was especially true for pyrazinamide where the pyrazine is not as stable as pyridine.
The ammonia concentration for both the product and residual substrate samples was determined by UV-Vis spectrometry using Nessler’s reagent to establish that no product NH4Cl had been lost. The (NH4)2SO4 was then reduced to ~1 mL by rotary evaporation and transferred to a flask with a side arm. The side arm was filled with ~4 mL of NaOBr. The system was sealed with a stopcock and the solution was frozen at −78 °C and taken through three freeze-pump-thaw cycles to remove all gases. After the final thaw, the two solutions were mixed slowly and the liquid was again frozen at −78 °C. The freshly produced N2 was distilled through two −78 °C traps, one −196 °C trap, and then collected on molecular sieves at −196 °C. The N2 was analyzed by IRMS to give the 15N/14N ratio.
13C kinetic isotope effects
This methodology is a modification of previous work by Scott and others (41–43) (Scheme 2). The solution of nicotinic acid product was reduced to dryness and dissolved in 2 mL of HPLC grade methanol. The methanol/nicotinic acid solution was added to a quartz glass tube (25cm, 9mm o.d., 7mm i.d.) and the methanol was removed under vacuum at slightly elevated temperature (40 °C) for several hours. To this, 0.5 g of dry copper chromite was added and the tube was evacuated and flame sealed. The sample was placed in a furnace and heated at 250 °C for nicotinic acid (232 °C for pyrazinoic acid, 235 °C for thionicotinic acid, 240 °C for 5-methylnicotinic acid), for 3 hours and then cooled in a room temperature water bath. The tube was cracked and the CO2 was distilled through two −130 °C liquid N2/pentane traps and finally collected in a third trap at −196 °C. Liquid N2/pentane traps were necessary because −78 °C traps were inefficient and allowed pyridine, a product of the decarboxylation of nicotinic acid, to contaminate the CO2 sample. The collected CO2 (~80% of the total sample) was isolated in the trap and was not removed from the line at this time.
Scheme 2.

The decarboxylation of nicotinic acid by copper chromite.
The final 20% of the CO2 was collected by acidification of the copper chromite powder. The copper chromite and a small stir bar were placed in a 30 mL flask equipped with a glass joint, side arm with a stopcock, and septum. The flask was fitted to a glass stopcock and placed on the high vacuum line and evacuated for at least 15 min. After closing the top stopcock, 2.5 mL of 1M H2SO4 was added via syringe through the septum. CO2 was evolved immediately and the solution was stirred for 5 min. A −78 °C bath was used to freeze the sample and the CO2 was collected in the same manner as above and trapped with the first portion of gas. The entire gas sample was isolated and the −196 °C trap was removed and replaced with a −78 °C trap. The entire CO2 sample was collected in a gas sample tube at −196 °C and was then analyzed by IRMS. IRMS analysis of the two CO2 portions individually showed a difference of 1–1.5 δ so collection of both was deemed necessary for the most accurate measurement.
When this technique was used to decarboxylate thionicotinic acid the delta values were not reproducible. It was assumed that the sulfur had not been completely removed from the compound, even though it was heavily acidified. Also these samples were not easily evacuated from the IRMS and a small residual impurity had to be frozen out of the system at −196 °C onto molecular sieves. Thus we were unable to reliably determine the 13C KIE for thionicotinamide.
Fraction of Reaction
The fraction of reaction (f value) was determined by using a known amount of substrate for the enzymatic reaction (50 µmoles) and then comparing that to the amount of product NH3 recovered after the reaction. To double check the f value the amount of product NH3 was also compared to the amount of NH3 recovered in 0.1 N H2SO4 after steam distillation under basic conditions of the residual substrate. In all cases the amount of NH3 was determined by comparison of the sample to a standardized curve of NH4Cl by UV at 425 nm. The fraction of reaction determined for the NH3 prod vs the initial substrate concentration and the fraction of reaction for NH3 prod vs NH3 res sub were generally within 1–3% of one another.
Kinetic isotope effects
Isotope effects were determined from changes in the natural abundance ratio of 15N/14N (or 13C/12C) in the compound during the reaction. The product and residual substrate are separated, purified, and converted to N2 (or CO2) gas. Each gas sample is analyzed individually by IRMS to determine its isotopic ratio compared to a known standard to give δ, defined as:
| (3) |
To determine the isotope effect the samples are converted to an R value defined as:
| (4) |
R-values for the reaction product, Rp, and residual substrate, Rs, along with the isotopic ratio of the starting material, Ro, and f, the fraction of reaction, are used in the following equations to arrive at the
| (5) |
| (6) |
| (7) |
Dependence of activity on pH
Reactions contained 0.1–3 µM Pnc1 and varying concentrations of nicotinamide (2.5 µM to 1 mM) or pyrazinamide (20 µM to 2 mM) in 100 µL at 25 °C. TBA buffer (50 mM Tris, 50 mM BisTris, 100 mM sodium acetate) was used for the pH range 4.0–8.5. ATE buffer (100 mM ACES, 52 mM Tris, 52 mM ethanolamine) was used for the pH range 8.0–10.5. These buffer mixtures are designed to give a constant ionic strength over a wide pH range (44). Reactions were quenched with 20 µL 6% v/v TFA before 10% of the substrate was converted to products at intervals of 60–120 seconds. Percent product conversion was determined spectrophotometrically by coupling to glutamate dehydrogenase (see previous paper) or using a previously published HPLC assay (45). Plots of kcat versus pH were fitted to equation 8:
| (8) |
using KinetAsyst (IntelliKinetics, State College, PA), where C is the pH-independent value, H is the proton concentration, and Ka and Kb are the ionization constants of the groups involved in the reaction.
Results
Product inhibition and irreversibility of reaction
To establish the kinetic mechanism for S. cerevisiae nicotinamidase (Pnc1), steady-state product inhibition analysis was performed. Double-reciprocal analysis of nicotinamidase inhibition by nicotinic acid and pyrazinoic acid displayed competitive inhibition patterns with Ki values of 120 µM and 6.7 mM, respectively (Table 1; Figure S1). The competitive inhibition displayed by nicotinic acid is consistent with that shown by Gadd et al. for a nicotinamidase from Micrococcus lysodeikticus (46) but in contrast to French et al. who observed no inhibition with nicotinic acid for S. cerevisiae Pnc1 (30). Double-reciprocal analysis of nicotinamidase inhibition by ammonium ion using a previously published HPLC assay (45) exhibited non-competitive inhibition patterns (data not shown). However, greater than 10 mM ammonium chloride was required for inhibition, consistent with several previous reports on nicotinamidases from M. lysodeikticus and yeast (46, 47). Therefore, it was unclear if the ammonium ion added was acting as a product inhibitor or as a denaturant as non-competitive inhibition patterns would be predicted in either case. To determine if the weak inhibition by ammonium ion could be explained by low reversibility of the reaction, 100 mM ammonium chloride and 5 mM nicotinic acid were incubated with 5 µM Pnc1 for 150 minutes at pH 7.5 and assayed for nicotinamide formation using an HPLC assay (45). No nicotinamide was detected under these conditions leading to an upper estimate of the reverse reaction rate at < 10−5 s−1, at least four orders of magnitude slower than the forward reaction rate with nicotinamide (kcat = 0.69 s−1). Therefore, the Pnc1 catalyzed reaction is essentially irreversible.
Table 1.
Ki values of Pnc1 competitive inhibition by nicotinamide analogs. Ki values were determined as described under Experimental Procedures. All reactions were performed at pH 7.5.
| Inhibitor | Structure | Ki value (µM) |
|---|---|---|
| nicotinic acid | 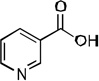 |
120 ± 19 |
| pyrazinoic acid | 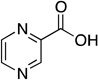 |
6700 ± 1700 |
| nicotinaldehyde | 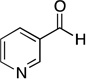 |
0.94 ± 0.35 |
| benzaldehyde | 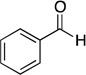 |
20600 ± 5700 |
| 3-acetylpyridine | 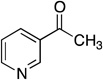 |
316 ± 101 |
Inhibition by nicotinamide analogs
The product inhibition analysis above suggested that nicotinic acid was the second product released and therefore that an enzyme intermediate might exist between Pnc1 and nicotinic acid. We hypothesized that non-hydrolyzable nicotinamide analogs might trap this intermediate and display potent nicotinamidase inhibition. All analogs tested displayed competitive inhibition with Ki values ranging from high nM to low mM (Table 1). The competitive inhibition observed by 3-acetylpyridine (Ki = 316 µM; Table 1) was consistent with several previous reports on nicotinamidases from Mycobacterium phlei (48), M. lysodeikticus (Ki = 160 µM) (49), Tortula cremoris (Ki = 305 µM) (50), Fleischmann’s yeast (Ki = 65 µM) (47), Flavobacterium peregrinum (51), and S. cerevisiae (Ki = 46 µM) (30). Nicotinaldehyde displayed by far the most potent inhibition among the analogs tested with a Ki value of 940 nM (Table 1; Figure S1), 10-fold below the Km value of 9.6 µM for nicotinamide. Potent inhibition by nicotinaldehyde was previously observed for nicotinamidases from M. lysodeikticus (Ki = 18 nM) (49), S. cerevisiae (Ki = 1.4 µM) (30), and Mycobacterium tuberculosis (Ki = 290 nM) (33). Benzaldehyde was the weakest inhibitor tested with a Ki value of 20.6 mM, similar to the previously observed benzaldehyde inhibition of M. lysodeikticus nicotinamidase (Ki = 1.8 mM) (49).
Structure of nicotinaldehyde inhibited Pnc1
The potent inhibition displayed by nicotinaldehyde inspired us to co-crystallize Pnc1 with nicotinaldehyde. Pnc1 crystallized with the nicotinaldehyde inhibitor at a 4:1 (nicotinaldehyde:Pnc1) molar ratio in the same space group and unit cell parameters as the apo structure (Table 2) (32). The crystals diffracted to 2.7-Å resolution and the structure was determined by molecular replacement using the apo Pnc1 structure (32) as a search model (38). Fo−Fc electron density maps revealed the location of the inhibitor covalently bound to C167, which was modeled into the electron density as a modified amino acid (Figure 1A). Overall, the structure is similar to the apo structure with a root mean square deviation (rmsd) of 0.3 Å2, and no major structural changes were induced by complex formation with the inhibitor.
Table 2.
X-ray data collection and structure determination statistics
| Data Collection | |
|---|---|
| Wavelength, Å | 0.97872 |
| Resolution Range (high resolution bin), Å | 30-2.70 (2.75-2.70) |
| Space Group | R3 |
| Unit Cell (a, b, c (Å)) | 298.72, 298.72, 112.65 |
| (α, β, γ (°)) | 90, 90, 120 |
| Completeness, % | 100.0 (100.0) |
| Total/Unique Reflections | 578,608/101,147 |
| Redundancy | 5.7 (5.5) |
| <I/σI> | 19.8 (5.3) |
| Rsyma, % | 11.3 (34.8) |
| Refinement | |
| Resolution, Å | 30-2.70 |
| Rwork/Rfreeb, % | 18.9/21.4 |
| Rms deviations | |
| Bonds, Å | 0.009 |
| Angles, ° | 1.23 |
| Ramachandran statistics, % | |
| Most favored | 89.8 |
| Allowed | 9.9 |
| Generously allowed | 0.3 |
| Disallowed | 0.0 |
| # atoms | |
| Protein | 12,439 |
| Water | 296 |
| Ligandc | 14 |
| <B factor>, Å2 | |
| Protein | 27.5 |
| Water | 23.9 |
| Ligandc | 82.7 |
Rsym = ΣΣj|Ij − 〈I〉|ΣIj, where Ij is the intensity measurement for reflection j and 〈I〉 is the mean intensity for multiply recorded reflections.
Rwork/Rfree = Σ‖Fobs| − |Fcalc‖/|Fobs|, where the working and free R factors are calculated by using the working and free reflection sets, respectively. The free R reflections (5% of the total) were held aside throughout refinement.
Ligand refers to 7 zinc and 7 magnesium ions (one zinc and one magnesium for each of 7 monomers in the asymmetric unit).
Figure 1.

Structure of nicotinaldehyde covalently bound in the active site of yeast Pnc1. A. Fo−Fc electron density map (3.5 σ) showing the inhibitor and Zn2+ in cross-eyed stereo view. The inhibitor (nicotinaldehyde) covalently attached to Cys167 was modeled into the density. Additional residues that coordinate the Zn2+ (Asp51, His53, and His94) are also shown. B. A cross-eyed stereo view of the active site of Pnc1 with nicotinaldehyde covalently bound. C. A cross-eyed stereo view of ordered water within the nicotinaldehyde-bound Pnc1 active site.
Within the crystal structure, nicotinaldehyde is covalently bound within the Pnc1 active site through a thiohemiacetal linkage to C167. The residues D8, K122, and C167 are all within hydrogen bond distance from one another (Figure 1B) and form a putative catalytic triad that is conserved throughout all known nicotinamidases (Figure S2). In addition to the covalent linkage to C167, nicotinaldehyde is ligated to the active-site zinc through the pyridine ring nitrogen (Figure 1). This zinc ligation is consistent with recent structures of nicotinamidases from Acinetobacter baumanii and Streptococcus pneumoniae (31, 52). The active-site zinc is also ligated by D51, H53, and H94, consistent with the previous S. cerevisiae Pnc1 apo-structure (32). Furthermore, we observed evidence for two water molecules that also coordinate the zinc; one of these is within hydrogen bonding distance of one conformation of E129 (E129 is present in two conformations in the structure) (Figure 1C). The carbonyl oxygen of nicotinaldehyde forms hydrogen bonds to the backbone amide nitrogens of A163 and C167 consistent with recent structures of S. pneumoniae PncA (52). The pyridine ring of nicotinaldehyde is also bound within an aromatic cage consisting of F13, W91, Y131, and Y166 (Figure 1B).
Kinetic parameters of Pnc1 active-site mutants
The structure with nicotinaldehyde bound within the Pnc1 active site and a sequence alignment of prokaryotic and eukaryotic nicotinamidases (Figure S2) suggested several conserved residues that may be involved in catalysis. Therefore, we determined the steady-state kinetic parameters for Pnc1 mutants of D8, D51, H53, H94, K122, and C167 (Table 3). The putative base D8 was substituted with Ala, Asn, or Glu, yielding mutants that displayed 103–104 fold lower kcat values compared to Pnc1 WT (Table 3). The D8E mutant exhibited slightly faster rates compared to either D8A or D8N. The equivalent D8E mutant in M. tuberculosis nicotinamidase was previously shown to harbor a 100-fold lower specific activity compared to wild type (53). When individually substituted with Ala, the zinc-binding residues D51, H53, and H94 yielded enzymes with 10–50 fold lower kcat values. Similar losses of specific activity (10–3000 fold) were observed when the corresponding zinc-binding residues were mutated to Ala in the M. tuberculosis nicotinamidase (53, 54). The D51N mutant was slightly more active (3-fold) than the D51A mutant. The zinc-binding mutants displayed Km values that were similar to or lower than those for Pnc1 WT (Table 3). The nicotinamidase active-site Zn2+ is tightly bound, as addition of 10 mM EDTA did not inhibit Pnc1 (data not shown) consistent with a previous report on nicotinamidases from T. cremoris (50), M. lysodeikticus (46), and M. tuberculosis (33). Addition of 2 mM ZnCl2 failed to increase activity of wild type (consistent with a previous report (30)) or rescue activity of D51A, D51N, H53A, or H94A mutants (data not shown) further indicating that Zn2+ is tightly ligated by D51, H53, and H94 and that these residues are critical for catalysis. When K122 was replaced with Ala, the kcat was decreased by 16-fold, whereas the K122R mutant displayed a dramatic 770-fold drop in kcat compared to Pnc1 WT. When the proposed nucleophile C167 was replaced with Ala, the observed rate was below the detection limit of the coupled assay (0.0005 s−1), the lowest of any mutant. A complete loss of activity was also observed when the active-site Cys was mutated to Ala in the nicotinamidase from M. tuberculosis (53, 54).
Table 3.
Steady-state kinetic parameters of Pnc1 mutants. Assays were performed as described under Experimental Procedures. All reactions were performed at pH 7.5.
| Pnc1 mutant | kcat (s−1)a | Km (µM)a | kcat/Km (M−1s−1) | 15N KIE | 13C KIE |
|---|---|---|---|---|---|
| wild type | 0.69 ± 0.04 | 9.6 ± 2.1 | (7.2 ± 1.2) × 104 | 1.0122 ± 0.0002 | 1.0125 ± 0.0004 |
| D8A | 0.0009 ± 0.0001 | NAb | NAb | NDc | NDc |
| D8N | 0.0006 ± 0.0001 | NAb | NAb | NDc | NDc |
| D8E | 0.0070 ± 0.0001 | NAb | NAb | 1.0218 ± 0.0015 | 1.0258 ± 0.0012 |
| D51A | 0.020 ± 0.005 | < 2.4 | > 8.3 × 103 | 1.0166 ± 0.0018 | 1.0075 ± 0.0015 |
| D51N | 0.064 ± 0.012 | < 1.8 | > 3.5 × 104 | 1.0047 ± 0.0006 | 1.0058 ± 0.0006 |
| H53A | 0.014 ± 0.002 | 6.5 ± 3.6 | (2.2 ± 0.9) × 103 | NDc | NDc |
| H94A | 0.020 ± 0.002 | < 2.7 | > 7.4 × 103 | NDc | NDc |
| K122A | 0.044 ± 0.023 | < 6.5 | > 6.7 × 103 | NDc | NDc |
| K122R | 0.0009 ± 0.0002 | NAb | NAb | 1.0127 ± 0.0003 | 1.0172 ± 0.0001 |
| C167A | < 0.0005 | NAb | NAb | NDc | NDc |
kcat and Km values were determined from fitting the data to the Michaelis-Menten equation using Kaleidagraph (Synergy Software, Reading, PA). Errors represent the error of the fit to the data.
Not available. Measured rates were too slow for accurate Km values to be determined.
Not determined.
Kinetic parameters of nicotinamide analogs as alternate substrates
Kinetic values for the reaction using substrate analogs produced a wide variation in kcat/Km values (Table 4). The Km value of 9.6 µM observed for nicotinamide was within the range of 6–10 µM obtained from previous reports on yeast nicotinamidases (30, 47). Substitution of the nicotinamide pyridine ring at the 4-position with nitrogen (pyrazinamide) or addition of a methyl group to the 5-position resulted in 3.7- and 2.5-fold faster kcat values but lower kcat/Km values due to 16- and 6-fold higher Km values, respectively. The Km value of 157 µM determined for pyrazinamide was similar to previously determined values for nicotinamidases from S. cerevisiae (Km = 200 µM) (2) and M. tuberculosis (Km = 300 µM) (33). The Km value of 61 µM determined for 5-methylnicotinamide was 5-fold lower than a previously determined value of 360 µM for a nicotinamidase from M. lysodeikticus (46). Perturbation of the pyridine nitrogen of nicotinamide by methylation (1-methylnicotinamide) or substitution with carbon (benzamide) resulted in 8400- and 200-fold decreased kcat/Km values compared to nicotinamide. The activity observed for 1-methylnicotinamide was in contrast to French et al. who observed no activity with S. cerevisiae Pnc1 (30). We were unable to accurately determine the kcat value for 1-methylnicotinamide as saturation could not be obtained, but benzamide resulted in a 78-fold lower kcat value compared to nicotinamide. Substitution of the amide oxygen of nicotinamide with a sulfur (thionicotinamide) resulted in a 1400-fold lower kcat/Km value. Finally, no activity could be detected above the detection limit of the HPLC assay (10−5 s−1) for nicotinamide mononucleotide (NMN+) or NAD+.
Table 4.
Steady-state kinetic parameters and 15N and 13C KIE for the reaction catalyzed by Pnc1 with alternate substrates. Assays were performed as described under Experimental Procedures at pH 7.5.
| Substrate | Structure | kcat (s−1)a | Km (µM)a | kcat/Km (M−1s−1) | 15N KIE | 13C KIE |
|---|---|---|---|---|---|---|
| nicotinamide | 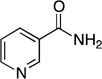 |
0.69 ± 0.04 | 9.6 ± 2.1 | (7.2 ± 1.2) × 104 | 1.0122 ± 0.0002 | 1.0125 ± 0.0004 |
| pyrazinamide | 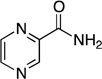 |
2.56 ± 0.10 | 157 ± 16 | (1.6 ± 0.1) × 104 | 1.0231 ± 0.0007 | 1.0196 ± 0.0023 |
| 5-methylnicotinamide | 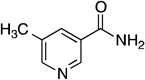 |
1.75 ± 0.05 | 61 ± 6 | (2.9 ± 0.2) × 104 | 1.0151 ± 0.0009 | 1.0269 ± 0.0037 |
| 1-methylnicotinamide | 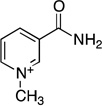 |
NAb | NAb | (8.6 ± 1.7) × 100 | NDd | NDd |
| benzamide | 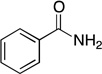 |
0.0088 ± 0.0004 | 25 ± 7 | (3.5 ± 0.9) × 102 | NDd | NDd |
| thionicotinamide | 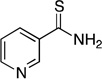 |
NAb | NAb | (5.0 ± 1.7) × 101 | 1.0262 ± 0.0019 | NDd |
| nicotinamide mononucleotide (NMN+) | < 10−5 c | |||||
| nicotinamide adenine dinucleotide (NAD+) | < 10−5 c |
kcat and Km values were determined from fitting the data to the Michaelis-Menten equation using Kaleidagraph (Synergy Software, Reading, PA). Errors represent the error of the fit to the data.
Saturation was not obtained, therefore only kcat/Km values are reported.
No activity was observed above the detection limit of the assay of 10−5 s−1.
Not determined.
15N and 13C kinetic isotope effects with substrate analogs
We next investigated the first portion of the nicotinamidase mechanism up to and including the first irreversible step (C-N bond cleavage) using kinetic isotope effects (KIEs). Using slow substrates with Pnc1 allowed us to determine if and when C-N bond cleavage is rate limiting for catalysis. The 15N and 13C KIE values were determined for Pnc1 with nicotinamide and three substrate analogs: 5-methylnicotinamide, pyrazinamide, and thionicotinamide (Table 4). With nicotinamide as the substrate the 15N and 13C isotope effects are ~1.2%. Since the upper theoretical limit for 15N and 13C effects is 3–4%, these primary effects indicate that C-N bond cleavage is at least partially rate-limiting. When 5-methylnicotinamide, pyrazinamide, or thionicotinamide were utilized as substrates, all of the KIE are more fully expressed than those with nicotinamide (Table 4). Also, in comparing the 15N effects versus the kcat/Km data, there is good correlation between the kinetic data and the KIE showing that the slower substrates exhibit higher primary KIEs. The trend is not as well defined for the 13C data, but the KIE for 5-methylnicotinamide is roughly the same as pyrazinamide when the standard error is taken into consideration.
15N and 13C kinetic isotope effects of Pnc1 mutants
The same KIEs determined with the substrate analogs were also determined for four Pnc1 mutants: D8E, D51N, D51A, and K122R, with nicotinamide as the substrate (Table 3). These Pnc1 mutants were chosen because of the differences observed in the determined kinetic parameters and their ability to achieve adequate product conversion to determine KIEs. These KIEs permitted us to investigate whether the mutated residues are responsible for the actions discussed here and by others (30, 31, 33, 52, 55). For the Zn2+ ligation mutant, D51N, the KIE values were both lower than for Pnc1 WT, whereas a higher 15N KIE and a lower 13C KIE was observed for D51A compared to Pnc1 WT. For the K122R mutant, the 15N KIE is exactly the same as that determined for the wild-type enzyme while the 13C effect is only ~0.4% higher. Finally, the D8E mutant displayed increased KIEs that were very close to the maximum observed with the substrate analogs.
pH dependence of kinetic parameters
Several residues within the Pnc1 active site may require a particular ionization state for catalysis. Within Pnc1, these include D8, K122, and C167. To provide evidence for the involvement of these residues during catalysis, the effect of pH on the kcat values of Pnc1 was determined. First, the stability of Pnc1 at pH extremes was determined to ensure any changes in rate was due to the protonation states of active-site residues and not global protein unfolding. Determination of Pnc1 kcat values with saturating nicotinamide over a pH range from 4.5 to 10.5 showed no critical ionizations but did show a slight pH-dependent rate change that varied only 4-fold between the pH extremes (0.45 s−1 at low pH and 1.87 s−1 at high pH) (Figure 2A). Similarly, the kcat/Km profile did not reveal any critical ionizations (data not shown). The lack of a critical ionization using nicotinamide as the substrate was consistent with previous reports on nicotinamidases from Torula cremoris and M. tuberculosis (33, 50).
Figure 2.

pH rate profiles. Reaction rates were determined as described under Experimental Procedures. (A) Effect of pH on kcat of Pnc1 catalyzed hydrolysis of nicotinamide. (B) Effect of pH on kcat of Pnc1 catalyzed hydrolysis of pyrazinamide.
The higher KIE values and slower kcat/Km values for pyrazinamide compared to nicotinamide (Table 4) indicated that C–N bond cleavage was only partially rate limiting for nicotinamide but nearly fully rate limiting for pyrazinamide. Therefore, we hypothesized that the pH profile for pyrazinamide would display critical ionizations involved in C–N bond cleavage. Indeed, Pnc1 kcat values for pyrazinamide revealed two critical ionizations over a pH range from 4 to 10, one with an apparent pKa value of 5.1 ± 0.2 that must be unprotonated for activity and another with an apparent pKb value of 9.0 ± 0.2 that must be protonated for activity (Figure 2B). The residues responsible for these ionizations are discussed below.
Discussion
Nicotinamidase kinetic mechanism
Although several nicotinamidase chemical mechanisms have been proposed (30–33), the kinetic mechanism had not been investigated in detail. Here, we determined that Pnc1 follows an ordered Uni Bi kinetic mechanism (Figure 3) in which nicotinamide binding is followed by formation of a thioester intermediate between C167 and nicotinic acid. Evidence supporting this thioester intermediate is discussed below. Ammonia is released and then the thioester intermediate is hydrolyzed to form nicotinic acid, which is then released. The product inhibition pattern expected for an ordered Uni Bi mechanism is that the second product released acts as a competitive inhibitor versus nicotinamide and the first product released acts as a non-competitive inhibitor versus nicotinamide (56). Therefore, nicotinic acid is the second product released based on the competitive inhibition patterns displayed versus nicotinamide (Figure S1). The non-competitive inhibition displayed by ammonium chloride is also consistent with ammonia as the first product released, but we were unable to confirm that the inhibition was specific to the Pnc1 active site or that the inhibition was caused by NH3 rather than NH4+.
Figure 3.

Proposed kinetic mechanism of the nicotinamidase reaction. E represents the unmodified Pnc1 enzyme, F represents the covalent thioester intermediate between nicotinic acid and C167, NAM represents nicotinamide, and NA represents nicotinic acid.
Nicotinamide binding may proceed through a hydrophobic tunnel
The Pnc1 active site is largely sequestered from solvent. However, a small tunnel exists from the active site to the protein surface that is constricted by L20, Y70, and I192 (Figure 4A). I192 appears to be especially important in the opening and closing of this tunnel as I192 resides on a dynamic C-terminal loop in Pnc1. The dynamic nature of the C-terminal loop and helix is exemplified by the high B factors of this region in the crystal structure (Figure 4B). Furthermore, the C-terminal helix displayed greater variance than any other portion of the structure when aligned with previous nicotinamidase structures (31, 52, 53, 55) (Figure 4C). However, Pnc1 does not undergo significant conformational changes outside of this C-terminal loop and helix upon nicotinamide binding as shown by the low rmsd values (rmsd 0.23–0.38 Å) between the apo- (32) and nicotinaldehyde-bound Pnc1 structures. Once nicotinamide proceeds through this tunnel, binding is stabilized within the active site through edge-face aromatic interactions between the aromatic cage residues F13, W91, Y131, and Y166 and the pyridine ring of nicotinamide (Figure 1B).
Figure 4.

Putative route of nicotinamide access to the Pnc1 active site. (A) A narrow tunnel exists in the Pnc1 structure from the active site to the protein surface. This tunnel was discovered using Caver (66, 67). (B) The C-terminal loop and helix of the nicotinaldehyde-Pnc1 structure is the most dynamic portion of the protein. All seven Pnc1 monomers in the asymmetric unit were aligned and represented using the b factor putty preset of Pymol (68). The range of b factors in the structure are represented as thin (low b factor) to thick (high b factor) main chains and a color gradient from blue (low b factor) to red (high b factor). (C) Alignment of all published nicotinamidase structures reveals that the C-terminal loop and helix containing I192 is the most dynamic portion of the structures. The nicotinamidases shown are from Pyrococcus horikoshii (PDB 1IM5; yellow), Mycobacterium tuberculosis (PDB 3PL1; orange), Streptococcus pneumoniae (PDB 3O91; blue), Acinetobacter baumannii (PDB 2WT9; pink), and Saccharomyces cerevisiae (this work; green).
Zinc is involved in both binding and activation of substrate
The Zn2+ ligated by D51, H53, and H94 also stabilizes nicotinamide binding within the active site. Our structure of nicotinaldehyde bound to Pnc1 revealed that the pyridyl nitrogen is ligated to the active-site Zn2+ at a distance of 2.6 Å. This Zn2+ ligation is important for binding and/or activating nicotinamide for catalysis as mutation of D51, H53, or H94 decreased the kcat value 11- to 49-fold. The importance of the Zn2+ ligation is further shown by the alternate substrate benzamide where the pyridyl nitrogen is replaced with a carbon. Using benzamide as an alternate substrate reduced the kcat/Km value 200-fold compared to nicotinamide (Table 4). This rate difference can partially be explained by the observed 7-fold slower pseudo-first-order rate of benzamide hydrolysis in basic solutions compared to nicotinamide (0.00408 versus 0.0301 hr−1 at 25 °C in 0.1 N NaOH) (57). In addition, benzaldehyde was a 22,000-fold less potent inhibitor compared to nicotinaldehyde (Table 1) indicating that the pyridyl nitrogen plays a significant role in substrate affinity through Zn2+ ligation. Besides nicotinamide binding, the Zn2+ also acts as a Lewis acid to activate nicotinamide towards hydrolysis. Interestingly, this method of Lewis acid catalysis appears unique among zinc hydrolases, which typically facilitate hydrolysis through binding to the carbonyl oxygen of the substrate (58, 59). In nicotinamidases, the Zn2+ instead acts as a ‘vinylogous Lewis acid’ activating the amide carbon for nucleophilic attack by C167 through the electron-withdrawing character of Zn2+, which acts through the conjugation of the pyridine ring instead of directly at the amide oxygen.
The Zn2+ ligand mutants also displayed the most interesting KIE results. For Pnc1 D51N, both the 15N and 13C KIE are ~0.5% compared to ~1.25% for those of Pnc1 WT (Table 3). We believe this is due to an increase in one of the back reaction off rates. A truncated derivation demonstrating how this is possible is included in the supporting information. Another intriguing KIE result is that for the Zn2+ ligand mutant, D51A, which has a 15N KIE that is larger than the wild type, while the 13C KIE is smaller. One feasible explanation is that the carbon is bonded more stiffly than the nitrogen at the transition state in the D51A mutant. This may be due to the inability of the alanine residue in the mutant enzyme to be liganded to the Zn2+, which affects the position of the Zn2+ and nicotinamide substrate in the active site compared to the wild type. If true, the orientation of the substrate has changed before the nucleophilic attack by C167, possibly affecting the bond stiffness of carbon to nitrogen in the transition state (Scheme 3). One final possibility for the difference in KIE for the D51N and D51A mutants compared to wild type is the change of charge at the active site upon mutation of a negatively charged aspartate residue to the neutral asparagine or alanine. There is precedence for the requirement of certain charges in and around the active site for proper substrate binding and catalysis (60). With Pnc1 it is unclear how loss of a negative charge may cause changes in the KIE and kinetic data but it may be due to the same factors discussed with the pH profiles.
Scheme 3.

Possible transition states depicting a stiffer bonded carbon (lower KIE) and a less stiffly bond nitrogen (higher KIE) in the D51A mutant compared to WT.
C167 is activated by Pnc1 active-site residues
Our Pnc1 structure suggested D8, K122, and C167 form a catalytic triad, which resembles the catalytic triad of the nitrilase superfamily (61). These three residues are universally conserved among all nicotinamidases (Figure S2) and an active-site Cys residue (C167 in Pnc1) was previously proposed to act as a nucleophile during catalysis (30, 31, 46, 47, 50–52, 55) attacking the carbonyl carbon of nicotinamide to form a thioester intermediate. Here, we present several lines of evidence that are highly consistent with this nucleophilic role of C167. First, nicotinamide analog aldehydes and ketones are competitive inhibitors versus nicotinamide. Although these aldehydes and ketones are predicted to form covalent adducts with C167, classical competitive inhibition patterns and no time dependent loss of activity are expected as reaction with C167 is completely reversible (49, 50, 62) and infinite concentrations of nicotinamide would preclude inhibitor binding to Pnc1. Nicotinaldehyde is a particularly potent inhibitor (Ki value of 940 nM), and the weaker inhibition of 3-acetylpyridine (Ki = 316 µM) is consistent with the greater reactivity of aldehydes compared to ketones (63). We co-crystallized nicotinaldehyde with Pnc1 and continuous electron density was observed between C167 and nicotinaldehyde (Figure 1A) indicating a covalent thiohemiacetal adduct was present in the crystals. The importance of C167 was confirmed through mutagenesis as the Pnc1 C167A mutant lost all detectable nicotinamidase activity (Table 3).
The Pnc1 pH profile using pyrazinamide as a substrate revealed a critical ionization with an apparent pKa value of 5.1 that must be unprotonated and a critical ionization with an apparent pKa value of 9.0 that must be protonated for activity (Figure 3B). The group that must be unprotonated for activity might reflect C167, consistent with a previously determined pKa of 6.6 for cysteine alkylation by iodoacetamide with the nicotinamidase from M. tuberculosis (33). The lowered pKa of C167 is likely due to the positively charged K122 ε-amino group within 3.1 Å of the C167 sulfur. The group that must be protonated (pKa ~ 9) may reflect the requirement for protonated K122. The precise positioning of the K122 ε-amine within the Pnc1 active site is critical as both the Pnc1 K122A and K122R mutants displayed significantly diminished kcat values 16- and 767-fold below those of Pnc1 WT, respectively. For the K122R mutant, the 15N KIE is exactly the same as that determined for Pnc1 WT while the 13C effect is only ~0.4% higher. This may reflect a classic case of non-productive substrate binding, which affects the kcat, but not the kcat/Km value. Arginine likely sterically interferes with nicotinamide binding causing a greater degree of non-productive binding. However, once the substrate achieves the proper near attack conformation the reaction proceeds normally resulting in a minimal increase in the KIE. Another possibility is that the positively charged K122 might lower the pKa values of D8 and C167 (31, 33). If so, the KIEs of K122R suggest that the positive charge of arginine is sufficient to maintain the lowered pKa values of D8 and C167 in the transition state. If not, the KIE would be expected to increase since an increased pKa value of either residue would slow proton abstraction and thus the C-N bond cleavage.
Within the Pnc1 active site, D8 is likely the base that initiates catalysis by abstracting the proton from C167 leading to the attack at the nicotinamide amide carbon (30, 31, 33, 52). The Pnc1 D8A, D8N, and D8E mutants revealed the importance of both the charge and positioning of D8 within the Pnc1 active site. Removal of the charge (D8A and D8N) resulted in 767- and 1150-fold reductions in kcat value compared to Pnc1 WT. Repositioning the negative charge (D8E) increased both the 15N and the 13C KIE to very close to the maximum (~2.7% for 15N; Table 3) observed with the substrate analogs and resulted in a 100-fold reduction in kcat value. Therefore, the extra methylene group of glutamate likely pushes the carboxyl group out of position to substantially slow proton abstraction, resulting in slower C-N bond cleavage and the fullest expression of the KIEs. Finally, nucleophilic attack by C167 is further favored by the presence of a putative oxyanion hole (Figure 1B) originally proposed by Fyfe et al. (31) consisting of the amide backbone nitrogens of the conserved A163 and invariant C167 (Figure S2). These nitrogens form hydrogen bonds to the oxygen of the trapped thiohemiacetal adduct in the structure of nicotinaldehyde inhibited Pnc1.
Structure-activity relationships and rate-limiting steps of nicotinamide analogs
The pH dependence of nicotinamide hydrolysis displayed no critical ionizations over the range 4.5–10.5 (Figure 2A), indicating a step other than C167 nucleophilic attack and C–N bond cleavage is rate-limiting for nicotinamide, such as tetrahedral intermediate breakdown or product release. In addition, the KIEs determined for nicotinamide were significantly less than the maximum observed for the substrate analogs indicating that C–N bond cleavage is only partially rate-limiting with respect to the kcat/Km value. The two critical features of proper nicotinamide binding and orientation are the ring nitrogen, which is liganded to the zinc atom and the nicotinamide carbonyl oxygen, which is held in place by backbone amide interactions. Replacing the oxygen with sulfur (i.e. thionicotinamide) leads to a >103 drop in kcat/Km value and a large 1.4% increase in the 15N KIE value compared to nicotinamide. Therefore, the overall rate limiting step likely shifts from product release for nicotinamide, to C–N bond cleavage for thionicotinamide. Oxygen is more electronegative than sulfur (3.44 vs 2.58, Pauling Scale) (64), which can affect the reaction in two possible ways. First, the hydrogen bonding strength is less for a thionyl group than a carbonyl leading to a more loosely bound substrate (i.e. the interaction with the backbone amides of A163 and C167). Secondly, the drop in electronegativity directly impacts the electrophilicity of the thionyl amide carbon thus diminishing its reactivity when attacked by C167. In addition, the larger size of sulfur and the longer bond length of C=S versus C=O bonds may also sterically clash with the enzyme and result in non-optimal substrate positioning. Any of these reasons would lead to a decreased forward commitment factor of the reaction and thus an increase in the observed 15N and 13C KIE.
Addition of a methyl group on the ring nitrogen (i.e. 1-methylnicotinamide) and replacement of N1 with carbon (i.e. benzamide) both block the interaction with the bound zinc atom resulting in a dramatic drop in kcat and kcat/Km values (see above). Methylation at the C5 position (i.e. 5-methylnicotinamide) is tolerated, yielding a substrate that displays modest effects on kcat/Km values but not of kcat values compared to nicotinamide. The higher KIE values for 5-methylnicotinamide (and all the analogs tested) compared to nicotinamide indicated that 5-methylnicotinamide is not as “sticky” as nicotinamide (i.e. the ratio of substrate dissociation to forward reaction is greater for the analogs). Thus, the C-N bond cleavage is almost entirely rate-limiting in regard to the kcat/Km value. Therefore, the extra methyl group of 5-methylnicotinamide is most likely sterically interacting with a nearby residue (possibly Y166) hindering achievement of the proper orientation for optimal catalysis. However, the steric hindrance of the methyl group of 5-methylnicotinamide also results in a more loosely bound substrate (higher Km) and a higher kcat value likely due to faster product release. Therefore, 5-methylnicotinic acid release, not C-N bond breaking, is the overall rate-limiting step reflected in the kcat value. Consistent with the high specificity of nicotinamidases toward nicotinamide, NAD+ salvage pathway metabolites NMN+ and NAD+ are not substrates.
Pyrazinamide displayed a 4.5-fold lower kcat/Km value compared to nicotinamide, but a 3.7-fold higher kcat value (Table 4). The size and geometry of the pyrazine ring are nearly identical to the pyridine ring of nicotinamide, so the additional nitrogen in the pyrazine ring slows the binding and chemical steps reflected in the kcat/Km value (substrate binding, nucleophilic attack of C167, and release of ammonia) through inductive effects or unfavorable interaction of the unshared pair of electrons with nearby aromatic residues involved in substrate binding and positioning leading to a more loosely bound substrate. However, the higher kcat value indicates that pyrazinamide reacts faster in the chemical steps after release of ammonia such as thioester hydrolysis and pyrazinoic acid release. Indeed, the 56-fold higher Ki value of pyrazinoic acid compared to nicotinic acid (Table 1) is consistent with a higher off rate constant for pyrazinoic acid and an overall rate enhancement due to an increased rate of rate-limiting product release. The 4.5-fold lower kcat/Km value for pyrazinamide is due to slower and nearly fully rate-limiting amide C–N bond as revealed by the higher 15N and 13C KIE values of 2.3% and 2.0% determined for pyrazinamide, compared to 1.2% and 1.3% determined for nicotinamide.
Nicotinamidase chemical mechanism
Previous nicotinamidase studies indicate that there are three residues that directly chelate the Zn2+ at the active site (D51, H53, and H94 in Pnc1) and three that are involved as a catalytic triad (C167, D8, and K122 in Pnc1) (31, 32, 55). Here, our data suggests specific roles for each of these residues and other active-site residues during catalysis (Scheme 4). In the first step of the mechanism, the substrate binds at the active site replacing one of two equatorial water molecules ligated to the Zn2+ and is stabilized by edge face interactions with the aromatic residues F13, W91, Y131 and Y166. The nitrogen of the pyridine ring is liganded to the Zn2+ at the active site and the proton is removed from C167 by D8, forming a thiolate that then attacks the amide carbonyl carbon creating a tetrahedral intermediate. The recently abstracted proton is transferred to the amino portion of the amide, which is then ejected as NH3 as the intermediate collapses and the C–N bond is broken. Although this final step before C-N scission is shown as concerted in Scheme 4, proton transfer and C-N cleavage may be in fact stepwise.
Scheme 4.

Proposed Pnc1 chemical mechanism.
At the next stage of the mechanism there is some disagreement on how the reaction moves forward. It has been hypothesized that the Zn2+ ligated water is in proper position to attack the thiol ester (52), but our crystal structure indicates the water is too far away (~5–7 Å) from the carbonyl carbon to be effective without a significant conformational rearrangement or dissociation from the Zn2+. Another option is that the Zn2+ ligated water is deprotonated and can then deprotonate a bulk solvent water molecule that attacks the thiol ester (30). The final possibility is that a bulk solvent water molecule is deprotonated by D8 and then acts as the nucleophile reforming a tetrahedral intermediate (31, 33). We favor the third option as D8 is in proper position to accomplish this deprotonation. Finally, the intermediate collapses to form nicotinic acid and the cysteine thiolate abstracts the proton from D8 and the catalytic cycle is complete.
It is possible that if the thiol pKa in Pnc1 is lowered to the same level as in PncA (pKa 6.6) (33), presumably by the interaction with the positive charge of K122, then under the conditions of our experiments (pH 7.5) ~90% of the cysteine would exist in its thiolate form and thus would not require deprotonation by D8. Accordingly, D8 would only be involved in the hydrolytic portion of the reaction, removing a proton from a water molecule and reforming the tetrahedral intermediate. The glutamate in the D8E mutant is out of position relative to the substrate for forming the tetrahedral intermediate, thus retarding the reaction so that the KIE is fully expressed in the D8E mutant because the forward commitment has become very small.
Conclusion
This detailed characterization of a eukaryotic nicotinamidase provides critical information on the structure and mechanism of nicotinamidases. In particular, this work provides fundamental understanding for the development of mechanism-based inhibitors and prodrugs that target nicotinamidases to treat fungal, bacterial, or parasite infections. In addition, results like those reported here, along with theoretical calculations of transition-state bond lengths, have been used by others to design excellent transition-state inhibitors for other enzymes (65). Similarly, we anticipate that these findings can lead to the development of nicotinamidase inhibitors to treat infections in which nicotinamidase activity is critical for viability or the infectious phenotype.
Supplementary Material
Acknowledgement
We thank Advanced Photon Source staff (LS-CAT beamline) and Katrina Forest for assistance with data collection, and Ken Satyshur and Nick George for help with structure determination. We also thank Dr. Tonya Zeczycki for her helpful comments and critique of this manuscript.
Abbreviations
- EDTA
ethylenediaminetetraacetic acid
- KIE
kinetic isotope effect
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high performance liquid chromatography
- IRMS
isotope ratio mass spectrometry
- NAD+
β-nicotinamide adenine dinucleotide
- NADPH
β-nicotinamide adenine dinucleotide 2′-phosphate reduced form
- NMN+
nicotinamide mononucleotide
- Pnc1
nicotinamidase from S. cerevisiae
- WT
wild type
Footnotes
This work was supported by National Institutes of Health grants GM065386 (to J.M.D.) and GM18938 (to W.W.C.), by National Institutes of Health Biotechnology Training Grant NIH 5 T32 GM08349 (to B.C.S.), and by an NIH training grant in Molecular Biosciences GM07215 (to K.A.H.).
All amino acid numbering used is based on the Pnc1 S. cerevisiae nicotinamidase sequence.
Supporting Information Available
Derivation to explain KIE observed for Pnc1 D51N. Table of primers used for Pnc1 mutagenesis. Double-reciprocal inhibition plots. Sequence alignment of nicotinamidases. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Denu JM. Vitamins and aging: pathways to NAD+ synthesis. Cell. 2007;129:453–454. doi: 10.1016/j.cell.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 2.Ghislain M, Talla E, Francois JM. Identification and functional analysis of the Saccharomyces cerevisiae nicotinamidase gene, PNC1. Yeast. 2002;19:215–224. doi: 10.1002/yea.810. [DOI] [PubMed] [Google Scholar]
- 3.Zerez CR, Roth EF, Jr, Schulman S, Tanaka KR. Increased nicotinamide adenine dinucleotide content and synthesis in Plasmodium falciparum-infected human erythrocytes. Blood. 1990;75:1705–1710. [PubMed] [Google Scholar]
- 4.Wang G, Pichersky E. Nicotinamidase participates in the salvage pathway of NAD biosynthesis in Arabidopsis. Plant J. 2007;49:1020–1029. doi: 10.1111/j.1365-313X.2006.03013.x. [DOI] [PubMed] [Google Scholar]
- 5.Balan V, Miller GS, Kaplun L, Balan K, Chong ZZ, Li F, Kaplun A, VanBerkum MF, Arking R, Freeman DC, Maiese K, Tzivion G. Life span extension and neuronal cell protection by Drosophila nicotinamidase. J Biol Chem. 2008;283:27810–27819. doi: 10.1074/jbc.M804681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vrablik TL, Huang L, Lange SE, Hanna-Rose W. Nicotinamidase modulation of NAD+ biosynthesis and nicotinamide levels separately affect reproductive development and cell survival in C. elegans. Development. 2009;136:3637–3646. doi: 10.1242/dev.028431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Horst A, Schavemaker JM, Pellis-van Berkel W, Burgering BM. The Caenorhabditis elegans nicotinamidase PNC-1 enhances survival. Mech Ageing Dev. 2007;128:346–349. doi: 10.1016/j.mad.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Kurokawa D, Watanabe K, Makino S, Shirahata T, Watarai M. Brucella abortus nicotinamidase (PncA) contributes to its intracellular replication and infectivity in mice. FEMS Microbiol Lett. 2004;234:289–295. doi: 10.1016/j.femsle.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Mitchison D. The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis. 2003;7:6–21. [PubMed] [Google Scholar]
- 12.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, 3rd, Wang H, Zhang W, Zhang Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science. 2011;333:1630–1632. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konno K, Feldmann FM, McDermott W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis. 1967;95:461–469. doi: 10.1164/arrd.1967.95.3.461. [DOI] [PubMed] [Google Scholar]
- 14.Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med. 1996;2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 15.Hirano K, Takahashi M, Kazumi Y, Fukasawa Y, Abe C. Mutation in pncA is a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:117–122. doi: 10.1016/s0962-8479(98)80004-x. [DOI] [PubMed] [Google Scholar]
- 16.Sreevatsan S, Pan X, Zhang Y, Kreiswirth BN, Musser JM. Mutations associated with pyrazinamide resistance in pncA of Mycobacterium tuberculosis complex organisms. Antimicrob Agents Chemother. 1997;41:636–640. doi: 10.1128/aac.41.3.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandmeier JJ, Celic I, Boeke JD, Smith JS. Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD(+) salvage pathway. Genetics. 2002;160:877–889. doi: 10.1093/genetics/160.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 20.Gallo CM, Smith DL, Jr, Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol Cell Biol. 2004;24:1301–1312. doi: 10.1128/MCB.24.3.1301-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 23.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 24.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 26.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J Biol Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 27.Landry J, Slama JT, Sternglanz R. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem Biophys Res Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- 28.Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, Vazquez-Manrique RP, Orfila AM, Ackerman D, Au C, Vinti G, Riesen M, Howard K, Neri C, Bedalov A, Kaeberlein M, Soti C, Partridge L, Gems D. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.French JB, Cen Y, Vrablik TL, Xu P, Allen E, Hanna-Rose W, Sauve AA. Characterization of nicotinamidases: steady state kinetic parameters, classwide inhibition by nicotinaldehydes, and catalytic mechanism. Biochemistry. 2010;49:10421–10439. doi: 10.1021/bi1012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fyfe PK, Rao VA, Zemla A, Cameron S, Hunter WN. Specificity and mechanism of Acinetobacter baumanii nicotinamidase: implications for activation of the frontline tuberculosis drug pyrazinamide. Angew Chem Int Ed Engl. 2009;48:9176–9179. doi: 10.1002/anie.200903407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu G, Taylor AB, McAlister-Henn L, Hart PJ. Crystal structure of the yeast nicotinamidase Pnc1p. Arch Biochem Biophys. 2007;461:66–75. doi: 10.1016/j.abb.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seiner DR, Hegde SS, Blanchard JS. Kinetics and inhibition of nicotinamidase from Mycobacterium tuberculosis. Biochemistry. 2010;49:9613–9619. doi: 10.1021/bi1011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 35.Su S, Chaykin S. Nicotinamide deamidase. Methods Enzymol. 1971;18:185–192. [Google Scholar]
- 36.Cleland WW. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Vol 276. New York: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 41.Hankes LV, Henderson LM. The metabolism of carboxyl-labeled 3-hydroxyanthranilic acid in the rat. J Biol Chem. 1957;225:349–354. [PubMed] [Google Scholar]
- 42.Scott TA. A method for the degradation of radioactive nicotinic acid. Biochem J. 1967;102:87–93. doi: 10.1042/bj1020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watson GK, Cain RB. Microbial metabolism of the pyridine ring. Metabolic pathways of pyridine biodegradation by soil bacteria. Biochem J. 1975;146:157–172. doi: 10.1042/bj1460157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellis KJ, Morrison JF. Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 1982;87:405–426. doi: 10.1016/s0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
- 45.Oishi M, Ogasawara Y, Ishii K, Tanabe S. Assay of nicotinamide deamidase activity using high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1998;720:59–64. doi: 10.1016/s0378-4347(98)00266-7. [DOI] [PubMed] [Google Scholar]
- 46.Gadd REA, Johnson WJ. Kinetic studies of nicotinamide deamidase from Micrococcus lysodeikticus. Int. J. Biochem. 1974;5:397–407. [Google Scholar]
- 47.Calbreath DF, Joshi JG. Inhibition of nicotinamidase by nicotinamide adenine dinucleotide. J Biol Chem. 1971;246:4334–4339. [PubMed] [Google Scholar]
- 48.Grossowicz N, Halpern YS. Inhibition of nicotinamidase activity in cell-free extracts of Mycobacterium phlei by 3-acetylpyridine. Biochim Biophys Acta. 1956;20:576–577. doi: 10.1016/0006-3002(56)90365-1. [DOI] [PubMed] [Google Scholar]
- 49.Johnson WJ, Gadd REA. Inhibition of nicotinamide deamidase from Micrococcus lysodeikticus by analogs of nicotinamide. Int. J. Biochem. 1974;5:633–641. [Google Scholar]
- 50.Joshi JG, Handler P. Purification and properties of nicotinamidase from Torula cremoris. J Biol Chem. 1962;237:929–935. [PubMed] [Google Scholar]
- 51.Tanigawa Y, Shimoyama M, Dohi K, Ueda I. Purification and properties of nicotinamide deamidase from Flavobacterium peregrinum. J Biol Chem. 1972;247:8036–8042. [PubMed] [Google Scholar]
- 52.French JB, Cen Y, Sauve AA, Ealick SE. High-resolution crystal structures of Streptococcus pneumoniae nicotinamidase with trapped intermediates provide insights into the catalytic mechanism and inhibition by aldehydes. Biochemistry. 2010;49:8803–8812. doi: 10.1021/bi1012436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrella S, Gelus-Ziental N, Maudry A, Laurans C, Boudjelloul R, Sougakoff W. Crystal structure of the pyrazinamidase of Mycobacterium tuberculosis: insights into natural and acquired resistance to pyrazinamide. PLoS One. 2011;6:e15785. doi: 10.1371/journal.pone.0015785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Deng JY, Bi LJ, Zhou YF, Zhang ZP, Zhang CG, Zhang Y, Zhang XE. Characterization of Mycobacterium tuberculosis nicotinamidase/pyrazinamidase. FEBS J. 2008;275:753–762. doi: 10.1111/j.1742-4658.2007.06241.x. [DOI] [PubMed] [Google Scholar]
- 55.Du X, Wang W, Kim R, Yakota H, Nguyen H, Kim SH. Crystal structure and mechanism of catalysis of a pyrazinamidase from Pyrococcus horikoshii. Biochemistry. 2001;40:14166–14172. doi: 10.1021/bi0115479. [DOI] [PubMed] [Google Scholar]
- 56.Cleland WW. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. II. Inhibition: nomenclature and theory. Biochim Biophys Acta. 1963;67:173–187. doi: 10.1016/0006-3002(63)91815-8. [DOI] [PubMed] [Google Scholar]
- 57.Kakemi K, Sezaki H, Nakano M, Ohsuga K, Mitsunaga T. Hydrolytic and associative behavior of aromatic amides in aqueous solution. Chem. Pharm. Bull. (Tokyo) 1969;17:901–905. [Google Scholar]
- 58.Hernick M, Fierke CA. Zinc hydrolases: the mechanisms of zinc-dependent deacetylases. Arch Biochem Biophys. 2005;433:71–84. doi: 10.1016/j.abb.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 59.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011 doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lodi PJ, Chang LC, Knowles JR, Komives EA. Triosephosphate isomerase requires a positively charged active site: the role of lysine-12. Biochemistry. 1994;33:2809–2814. doi: 10.1021/bi00176a009. [DOI] [PubMed] [Google Scholar]
- 61.Thuku RN, Brady D, Benedik MJ, Sewell BT. Microbial nitrilases: versatile, spiral forming, industrial enzymes. J Appl Microbiol. 2009;106:703–727. doi: 10.1111/j.1365-2672.2008.03941.x. [DOI] [PubMed] [Google Scholar]
- 62.Yan C, Sloan DL. Purification and characterization of nicotinamide deamidase from yeast. J Biol Chem. 1987;262:9082–9087. [PubMed] [Google Scholar]
- 63.Wolfenden R. Transition state analog inhibitors and enzyme catalysis. Annu Rev Biophys Bioeng. 1976;5:271–306. doi: 10.1146/annurev.bb.05.060176.001415. [DOI] [PubMed] [Google Scholar]
- 64.Huheey JE. Inorganic Chemistry: Principles of Structure and Reactivity. 3rd ed. New York: Harper & Row; 1983. [Google Scholar]
- 65.Schramm VL. Enzymatic transition state theory and transition state analogue design. J Biol Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 66.Damborsky J, Petrek M, Banas P, Otyepka M. Identification of tunnels in proteins, nucleic acids, inorganic materials and molecular ensembles. Biotechnol J. 2007;2:62–67. doi: 10.1002/biot.200600208. [DOI] [PubMed] [Google Scholar]
- 67.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.