

Graphical abstract
Highlights
► Phylogenetics has revolutionized the study of disparate fields from genomics to ecology. ► Phylogenetics has revealed the origin of both HIV-1 and HIV-2. ► HIV it is a model to the study of infectious diseases. ► Phylogenetics has been applied to problems such as resistance and reservoirs. ► Phylogenetic methods have been also exploited to follow ongoing epidemics.
Keywords: Phylogenetics, Virus evolution, Phylodynamics, HIV, Molecular epidemiology, HIV origins, Drug resistance, Vaccine resistance, Reservoir, Viral transmission
Abstract
Molecular phylogenetics has revolutionized the study of not only evolution but also disparate fields such as genomics, bioinformatics, epidemiology, ecology, microbiology, molecular biology and biochemistry. Particularly significant are its achievements in population genetics as a result of the development of coalescent theory, which have contributed to more accurate model-based parameter estimation and explicit hypothesis testing. The study of the evolution of many microorganisms, and HIV in particular, have benefited from these new methodologies. HIV is well suited for such sophisticated population analyses because of its large population sizes, short generation times, high substitution rates and relatively small genomes. All these factors make HIV an ideal and fascinating model to study molecular evolution in real time. Here we review the significant advances made in HIV evolution through the application of phylogenetic approaches. We first examine the relative roles of mutation and recombination on the molecular evolution of HIV and its adaptive response to drug therapy and tissue allocation. We then review some of the fundamental questions in HIV evolution in relation to its origin and diversification and describe some of the insights gained using phylogenies. Finally, we show how phylogenetic analysis has advanced our knowledge of HIV dynamics (i.e., phylodynamics).
1. Introduction
AIDS is one of the most serious modern diseases (Leeper and Reddi, 2010, UNAIDS, 2010) and the Human Immunodeficiency Virus (HIV) is the causative agent (Barre-Sinoussi et al., 1983, Gallo et al., 1983, Popovic et al., 1984, Sarngadharan et al., 1984). According to World Health Organization (WHO), 33.4 million people (31.1–35.8 million) were living with HIV worldwide as of 2009. In the same year, 2 million infected people died and the disease grew at a rate of 7400 new infections per day, more than 97% of which occurred in low- and middle-income countries. To date, sub-Saharan Africa is the most affected region on earth, with 67% of the world’s HIV infections (UNAIDS, 2010). Host restriction factors in the form of proteins such as TRIM5/22, APOBEC, and Tetherin have been to some extent ineffective in blocking early HIV-1 infection (Neil et al., 2008, Sakuma et al., 2007, Stopak et al., 2003, Tissot and Mechti, 1995). In addition, Highly Active Antiretroviral Therapy (HAART) has been very effective at reducing viral loads within patients and thereby significantly prolonging life expectancy for HIV infected individuals, particularly in those countries where HAART is accessible. However, even when HAART is available, effective control remains elusive due to the number of evolved mechanisms that HIV uses to evade the host immune system (Fischer et al., 2010, Wei et al., 2003), the evolution of drug resistance (Price et al., 2011, Shi et al., 2010), and the isolation of viral reservoirs from drug treatments (Chomont et al., 2009, Finzi et al., 1997). The extraordinary genetic diversity observed among circulating populations of HIV has hampered the development of a vaccine to provide immunity or control of AIDS. Despite much research, phase III trials of HIV vaccines have been few and have failed to provide full protection, which is probably due to the extensive variation of HIV isolates (McBurney and Ross, 2008). Although some partial protection was observed in the Thai RV144 Phase III HIV vaccine trial (Rerks-Ngarm et al., 2009), an effective vaccine against HIV remains elusive.
While our knowledge of HIV biology is still limited, we have gained significant insights through the application of phylogenetics to HIV diversity. For example, phylogenetic analyses have elucidated the origins of HIV-1 and -2 epidemic (Gao et al., 1994, Gao et al., 1999, Korber et al., 2000, Lemey et al., 2003, Salemi et al., 2001, Sharp et al., 2000), the relationships of HIV to other simian lentiviruses (Bailes et al., 2003, Essex, 1994, Gao et al., 1992, Wertheim and Worobey, 2007), and the classification of HIV diversity within HIV-1 (Kosakovsky Pond et al., 2009). Cross-species transmissions (Bailes et al., 2003, Beer et al., 1999, Gao et al., 1999, Hahn et al., 2000, Plantier et al., 2009, Takehisa et al., 2009, Wertheim and Worobey, 2007, Worobey et al., 2004) have been identified and characterized through the use of phylogenetic approaches. Such methods have been used to test hypotheses of transmission events of HIV between individuals (Hillis and Huelsenbeck, 1994, Leitner et al., 1996, Xin et al., 1995), including their use as evidence of transmission in legal settings (Bernard et al., 2007, Crandall, 1995, Metzker et al., 2002, Ou et al., 1992).
Phylogenetics has been key to the identification of drug-resistance mutational pathways (Buendia et al., 2009, Crandall et al., 1999) and the mechanisms of drug resistance (Carvajal-Rodríguez et al., 2008, Lemey et al., 2005a, Machado et al., 2009). Moreover, phylogenetic approaches have been used to assess within- and among-host HIV diversity and population dynamics (i.e., phylodyanamics) (Grenfell et al., 2004), co-divergence (Beer et al., 1999, Bibollet-Ruche et al., 2004, Chen et al., 1996, Wertheim and Worobey, 2007), and the role of recombination in the diversification process (Carvajal-Rodríguez et al., 2007, Jobes et al., 2006, Schlub et al., 2010). This exceptional diversity has been examined to infer geographical distribution and dispersion patterns (Lemey et al., 2003, Robbins et al., 2003) and thereby test hypotheses associated with molecular epidemiology (Holmes et al., 1995, Salemi et al., 2008). Phylogenetics has been key to identifying patterns and mechanisms of natural selection (Lemey et al., 2007, Pond et al., 2008, Poon et al., 2010, Templeton et al., 2004), including the intra- and inter-host adaptive forces that shape the evolution of the virus (Carvajal-Rodríguez et al., 2008, Keele et al., 2008a, Salazar-González et al., 2008, Shankarappa et al., 1999), which is essential for effective vaccine development (Frahm et al., 2008).
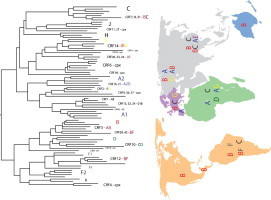
The first applications of phylogenetics to the study of HIV date from the early 1990s and were aimed at inferring the origins of HIV-1 and the classification of HIV into different types (1 and 2), groups (M, N, O within HIV-1), and subtypes (A–D, F–H, and J and K within Group M of HIV-1) (Huet et al., 1990; Fig. 1 ). Today, phylogenetic analysis has become a common practice of many HIV/AIDS research programs, due mainly to the many insights these analyses can provide and the novel questions they can address over a variety of topics related to HIV biology. Over the past two decades, HIV data have accumulated rapidly in public and specialized databases thereby creating one of the richest datasets we have for a single entity in terms of sequence tallies and epidemiological information (e.g., sampling locality, drug resistance mutations, tissue allocation, etc.). For instance, the number of available sequences in the Los Alamos database (http://www.hiv.lanl.gov) has exploded to 339,306 sequences, a 45% increase over the preceding year, with 2576 complete genomes (Kuiken et al., 2010).
Fig. 1.
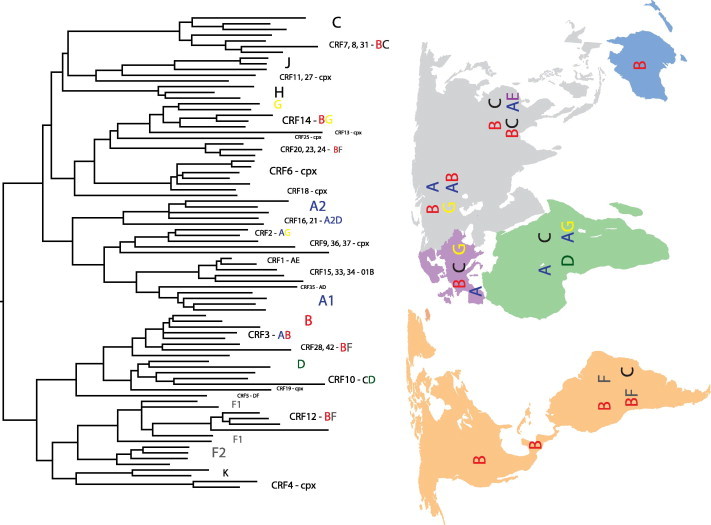
Phylogenetic tree representation of HIV-1 recombinants and discrete subtypes. Note the lack of genetic structure due to the presence of recombinant sequences. Also note that the existence of discrete subtypes is questionable due to the high evolutionary rates the virus exhibits. This tree should be regarded as a snapshot of part of the observable diversity. CRF = circulating recombinant form; cpx = complex recombinant pattern. A–D, F–H, J and K denote HIV-1 subtypes.
Here, we review the application of phylogenies to the study of HIV. Although because of the nature of the subject, no article-size review can be comprehensive, we hope to instructively show how phylogenetic approaches have influenced our current understanding of the emergence, evolution of drug resistance, epidemiology and dynamics of HIV and assist with the problem of its eradication (Grenfell et al., 2004, Holmes and Grenfell, 2009, Stack et al., 2010).
2. Molecular evolution of HIV
The defining feature of HIV is its exceptional genetic diversity. This high diversity stems from at least four sources; namely, high substitution rates, a rather small genome, short generation times, and high recombination frequency. HIV-1 substitution rates [∼0.002 substitutions/site/year (Korber et al., 2000)] are related, among others, to high mutation rates, which in HIV-1 has been estimated to be 0.1–0.2 mutations/genome/generation (Mansky and Temin, 1995), that is ∼33 times more than Neurospora crassa, but around 10-fold lower than that of Influenza A virus (Drake et al., 1998). The HIV genome, as well as of other RNA viruses, is small (exceptions are coronaviruses and roniviruses with >25 kb) with ∼9.8 kb in length (see Fig. 2 for genome structure). Genome size in HIV, as in other RNA viruses, is apparently limited by the error-prone nature of its replication machinery: the longer the genome, the more mutations produced, most of them being deleterious (Holmes, 2009). In terms of HIV diversity, such a small genome size impacts on generation time (1.2 days for HIV-1) with ∼1010 virions produced daily in an infected individual (Rodrigo et al., 1999). In addition to the staggering numbers above, HIV-1 recombines at a frequency of 1–3 recombination events/genome/generation (Jetzt et al., 2000, Shriner et al., 2004). Altogether, this represents a tremendous amount of raw material for evolution. However, variation is distributed unevenly across the genome. The HIV-1 genome structure is composed of three main genes, gag, pol and env, plus accessory genes, tat, rev, vif, vpr, vpu and nef, flanked by Long-Terminal Repeats (LTRs). All genes are coded over the three forward reading frames, including frame shifts in the case of tat and rev (Fig. 2). Variation in substitution rates is known to happen genome-wide and within specific genes, which has been interpreted as evidence of functional and structural constrains acting upon nucleotide sequences (Ngandu et al., 2008, Pond and Muse, 2005). As described below, this variation leads to divergent patterns in HIV evolution, whether we look at population or within-host data, with consequences to disease progression, natural selection, and drug resistance.
Fig. 2.
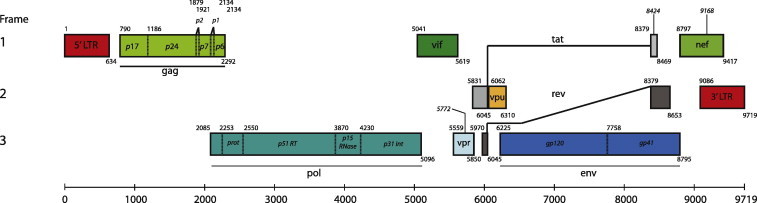
Schematic representation of HIV-1 genome organization. The three coding reading frames are depicted along with their open reading frames (rectangles). Genome position numbering is based on theHXB2 reference strain. The small number in the upper left corner of each rectangle indicates the gene start, while the number in the lower right indicates the last position of the stop codon. Trans-spliced rev and tat forms are represented by black connecting lines between the third and second, and second and first open reading frames, respectively.
2.1. Substitution rates
Phylogenies allow researchers to determine patterns of the extensive genetic diversity of HIV to examine human-scale ecological and epidemiological processes. Divergent patterns of HIV evolution are observed when comparing intra- and inter-host phylogenies. Ladder-like intra-host phylogenies are evidence of a continuous immune-driven selection [similar to inter-host influenza phylogenies (Buonagurio et al., 1986, Bush et al., 1999], in which there is no high genetic diversity at any given time point; rather, there are few lineages with sequential replacement of strains over time. In contrast, inter-host phylogenies do not exhibit this pattern; instead multiple lineages coexist at any given time. This is probably the product of major bottlenecks at transmission (Brown, 1997). Whether drift, selection or both govern these bottlenecks is not clear (Edwards et al., 2006, Keele et al., 2008a, Salazar-González et al., 2008), but the impact of random genetic drift on the population dynamics, genetic diversity, and clinical outcome is well studied (Tazi et al., 2011). Thus, HIV-1 possesses intrinsic mutational properties that prompt it to exhibit different patterns of substitution depending upon whether we look at within-host or inter-host genetic data.
2.1.1. Disease progression
The relationship between disease progression and substitution rates (or genetic diversity) was recognized early in HIV studies. The hypothesis is that host processes that determine HIV pathogenesis, also determine viral replication rates. Thus, by looking at substitution rates we can infer what is happening with HIV within patients. In a thorough study, Shankarappa et al. (1999) monitored nine patients over a 6–12 years period. They distinguished three phases of disease progression associated with diversity in ∼600 bp of env. In the first phase, a linear increase of diversity was associated with initial features of infection and X5 HIV populations. During the second phase, diversity leveled off or even decreased, which was correlated with the appearance of X4 HIV populations. Finally, the third phase was related to a decline in CD4 + T cells and with the failure of T cell homeostasis and a reduction in diversity. Although, these results might not be general for other patients, as these may have been infected with phenotypically different strains (different rates), and different genes may generate different patterns, other studies have somewhat supported this hypothesis. Using the previous dataset along with others, Lemey et al. (2007) found a positive association between disease progression and HIV-1 synonymous substitution rates. When analyzing HIV-2 sequences, they observed an overall low substitution rate that might reflect the reduced virulence that this viral type exhibits. On the other hand, non-synonymous rate changes have been associated with immune pressure. Consequently, a decrease in selective pressure would correlate with the breakdown of the immune system. However, with a different approach and on a different dataset, Carvajal-Rodríguez et al. (2008) reported no relationship between disease progression and substitution rates when analyzed separately into adaptive and neutral categories of variation. An opposite pattern of synonymous and non-synonymous substitutions were observed through time when dividing the dataset in rapid (RP) and non-rapid progressors (NRP), with RP showing a slow increase in non-synonymous substitutions and NRP showing a fast increase. These results could be a result of the different methodologies used; Lemey et al. were comparing absolute rates of substitution while Carvajal-Rodríguez et al. were comparing relative rates. Moreover, the former study accounted for deleterious mutations, but not for recombination and the latter did the opposite. It remains unknown if both datasets would converge on similar conclusions if the same methodologies were used. It is worth mentioning that animal models can provide an optimal environment to account for subject variability, sample size, and HIV variation (Berges et al., 2010), thus, providing a well-defined and structured opportunity to test the hypothesis above.
2.1.2. Natural selection
Substitution rates are logically tied to the identification of natural selection and site-based molecular adaptation. Development and refinement of methods have opened a plethora of questions that biologists can address with these sophisticated methods (Mens et al., 2007, Pond and Muse, 2005). Several studies have addressed questions regarding natural selection in HIV at the molecular level. It is generally accepted that nucleotide changes that do not change the amino acid composition (synonymous; dS) are more likely to be neutral than changes that affect amino acid composition (non-synonymous; dN) (Sharp, 1997). Therefore, rate ratio changes between both types of substitutions (dN/dS) could predict whether purifying (dN/dS < 1), positive (dN/dS > 1) or neutral (dN/dS = 1) selection is present at the gene and/or codon level. Under this paradigm and using phylogenies to make sense of nucleotide changes, several studies have tried to identify molecular determinants of selection in HIV. For instance, these studies have been instrumental in demonstrating that the switch from X5 to X4 tropic populations is highly positively selective, and so is the switch from non-syncytium forming HIV strains to the ones able to form syncytia (Templeton et al., 2004). Using these approaches, others have attempted to study selection dynamics within- and among- hosts (even inter-populations) to ascertain the extent to which HIV variation is maintained and passed between individuals (Pond et al., 2006, Poon et al., 2007). At the inter-population level, several within-host adaptations seem to be transient while persistent substitutions are subject to stronger selective pressures, which ultimately fix these variants in different populations (Pond et al., 2006). More ambitious studies have analyzed large quantities of data in order to come up with novel drug-resistant and high-fitness mutations that exhibit signatures of positive selection (Chen et al., 2004). Recently, a method well suited for analyzing adaptive rates (adaptations/codon/year) in large datasets has been published (Bhatt et al., 2011); some of its virtues are computation-tractability and robustness to biases introduced by synonymous mutations and RNA secondary structures. When using serially sampled data, a previous time point can be used as a homologous outgroup to the more contemporaneous large ingroup dataset. In doing so, the algorithm determines which sites are ancestral or derived. Sites are classified as silent or replacement and/or high-, medium- and low- frequency polymorphisms. These values are then combined so that the output reflects the proportion of fixed sites that have undergone adaptive change. For additional details on detecting selection, see the following (McDonald and Kreitman, 1991, Pérez-Losada et al., 2007c, Smith and Eyre-Walker, 2002).
2.2. Drug resistance
Drug therapy was once envisioned as a potential cure for HIV infected patients (Perelson et al., 1997, Wain-Hobson, 1997). With the emergence of the first drugs during the mid of the 1980s, antivirals seemed to control viral infection readily (Mitsuya et al., 1985). However, it took just 4 years following the introduction of Zidovudine (AZT) before the first mono-resistant HIV-1 strains were found and officially reported (Larder and Kemp, 1989). HIV drug resistance has risen considerably in resource-rich countries, perhaps due to widespread treatment access, although resistance is also present in low- and middle-income countries. Mathematical predictions failed to account for certain biological characteristics that impact the population dynamics of HIV, including, (i) extremely fast evolutionary rates and within host population structure by specific cell types (Perelson et al., 1997); (ii) high within-host population sizes, reaching 107–108 productively infected cells in lymphoid tissue; (iii) high substitution rates due to an error-prone reverse transcriptase (RT) (see Mansky and Temin, 1995); and (iv) the once-neglected recombination process that seems to play a major role in HIV evolution and, consequently, in drug resistance (Carvajal-Rodríguez et al., 2006). In fact, recombination is of prime importance in HIV, accounting for much of the observed diversity, and at times exceeding the mutation rate by 5.5 times (Shriner et al., 2004). Furthermore, cells can harbor different proviruses, (Keele et al., 2008a) and multiple infections can occur simultaneously, (Jobes et al., 2006, Xin et al., 1995).
Several gene products have been targeted for drug treatment as these are involved in key stages of the viral replication cycle. Those include RT, protease, the envelope glycoprotein complex (gp120–gp41) and lately the virion infectivity factor (vif) (see Coffin, 1999) (Fig. 2). Inhibitors include nucleoside and nucleotide analogs as well as non-nucleoside analogs that are able to impair some parts of reverse transcription (analog incorporation, analog removal), inhibitors of protease activity, and inhibitors of fusion to plasma membrane (Clavel and Hance, 2004). There is an extensive database (http://hivdb.stanford.edu/) of mutations conferring drug resistance spread throughout the HIV genome, some of them conferring cross-resistance, i.e., resistance to drugs that the patient has never been exposed to and mutations that have compensatory effects on fitness lost by the primary mutations (Rhee et al., 2003). Without doubt, antiviral treatments such as HAART have greatly improved the quality of life and life expectancy of those infected. However, it is far from being a cure as, in part, evidenced by the appearance of drug resistance mutations in treated and untreated patients (Lataillade et al., 2010).
2.3. Recombination
Recombination in retroviruses was described a few decades ago with mechanistic details (Coffin, 1979, Goodrich and Duesberg, 1990, Temin, 1991). However, through the mid 1990s, recombination in HIV-1 was regarded as almost non-existent mainly because it was thought that multiple infections within the same individual were rather unlikely. This led to the general thought that recombination could not contribute to HIV-1 evolution. Retroviral recombination was demonstrated experimentally in feline and murine species, for which recombinant viruses possessed altered tropism, host ranges or virulence (Golovkina et al., 1994, Tumas et al., 1993); but again evidence for recombination in HIV was rare. However, in 1995 and using a phylogenetic incongruence method, recombination was detected in HIV and it was suggested to be underestimated (Robertson et al., 1995a). Initially, recombination was detected readily in Africa, probably due to the high genetic divergence of HIV-1 strains co-circulating in that country. This allowed for the opportunity for co-infection of different subtypes, which made recombination detection easier. Soon more evidence of recombination in HIV-1 (Diaz et al., 1995, Robertson et al., 1995b, Zhu et al., 1995) and for HIV-2 was detected using phylogenies (Gao et al., 1994). Evidence of intra-subtype recombination within-host was also detected through phylogenetic and substitution rate analyses (Jobes et al., 2006). There are now a variety of methods available for detecting recombination in HIV sequences and estimating recombination rates (Posada et al., 2002) and it is apparent that recombination plays a significant role in HIV evolution (Rambaut et al., 2004).
HIV recombination has some unique characteristics that resembles sexual reproduction in multicellular organisms (Temin, 1991). HIV is essentially diploid in that it possesses two full-length replication-capable genome copies within the protein capsid that can have different evolutionary histories and thus be viewed as a heterozygous virion. It is not diploid though in the sense that just one genome copy gets replicated and finally segregated when the virus infects another cell; instead just one allele is passed onto the progeny (Onafuwa-Nuga and Telesnitsky, 2009). However, different HIV genomes within the same cell can recombine and yield offspring carrying genetic material from both “parents.” The costs of sexual reproduction have been reviewed extensively (Fox et al., 2001), though it is widely accepted that sex can put together “good mutations” and it can purge bad mutations out of the gene pool. Moreover, when a super-infection occurs, i.e., an infection by a second strain in a patient already infected, the likelihood of recombination or recombination detection should increase. Thus, this process could accelerate the emergence of multi-drug resistance recombinant forms. However, this view has been challenged by research groups that have found negative correlations between the appearance of drug resistance mutations and super-infection under computational genetic models (Bretscher et al., 2004).
Regardless of these models, inter-subtype recombinants, i.e., circulating recombinant forms (CRFs), have been described since 1996 and now occur worldwide encompassing at least 49 variants (Fig. 1 and http://www.hiv.lanl.gov/). These recombinant forms reflect successful recombination events that have been fixed in populations and may represent higher fitness forms; nevertheless this is debatable because of a lack of fitness measurements in vivo (Holmes, 2009). Therefore, evidence of recombination has been now found at every virus level, i.e., inter- and intra-subtypes, among HIV groups and among primate lentiviruses. In fact, recombination is so pervasive and has such an impact on genetic structure, that it is questionable whether HIV occurs as discrete subtypes (Holmes, 2009).
The effect of recombination on drug resistance evolution seems to be dependent on the intensity of selection pressure. By using simulated data, Carvajal-Rodríguez et al. (2007)painted a more detailed picture of this process. They found that, under high selection pressures, recombination would favor the appearance of drug resistant HIV variants. This would be dependent on population size; the larger the population size, the more likely drug resistance recombinants will appear and become fixed in the population. We expect this prediction to be met in, for instance, patients under drug treatment and/or experiencing a strong adaptive immune response.
Given that recombination is such an important factor in the evolution of HIV, it is important to test for recombination in DNA sequence data prior to phylogenetic analyses (Posada and Crandall, 2002). Phylogenetic recombination detection methods are the most popular if the goal is to analyze considerable amounts of data, e.g., bootscanning algorithm (Lole et al., 1999), although it has been shown that they do not perform as well as others, e.g., Runs test (Posada and Crandall, 2001). On the other hand, experimental detection of recombination relies on laborious and time-consuming assays based on single-round replication cycles. Typically, these use pairs of vectors which reconstitute a selectable marker when recombination occurs, or they use single vectors when the goal is to assess intra-strand recombination (Onafuwa-Nuga and Telesnitsky, 2009). The information you can draw from experimental studies allows for detecting average recombination frequencies or hotspots. However, the estimation of recombination rates, breakpoints, and the identification of parental sequences at the population level is not likely achievable in this framework.
On the other hand, statistical methods are well suited for HIV inter-, intra-, and host population studies. The literature contains numerous methodologies for detecting recombination breakpoints (reviewed in Posada et al. (2002). Based on relevant evidence for recombination, they have been tentatively classified as distance methods, phylogenetic methods, compatibility methods and substitution distribution methods (Posada and Crandall, 2001). By far, phylogenetic methods are the most commonly used (again, not always the best choice). Despite the plethora of methodological alternatives at hand, recombination detection is not an easy task. One reason for this is that it depends on several factors, including the amount of divergence among sequences, and where and how frequently the event is occurring (Lewis-Rogers et al., 2004). In addition, modern recombination rate estimation involves the use of coalescent-based methods that account for evolutionary history and uncertainty in the estimates (Kuhner, 2006). Although efforts have been made to implement more complex and realistic models, caution should be exercised when using them because of their sensitivity to assumption violations (e.g., deviations from neutrality and population stability, which are likely to occur in natural populations, thus frequently violated (Carvajal-Rodríguez et al., 2006, Kuhner, 2009).
In HIV research, recombination methods have been used to explore many different aspects of HIV biology. For instance, intra-host recombination has been studied in post mortem tissues exhibiting normal and abnormal histopathology in patients who received HAART. Tissues with abnormal histopathology show higher numbers of recombinant sequences. Likewise, these tissues display increased macrophage proliferation and it is well known that this cell type is involved in hiding HIV from HAART. Thus, macrophages may contribute to elusive recombinant forms evidenced by extensive recombination in non-lymphoid populations (Lamers et al., 2009). Nora et al. (2007) provided additional evidence supporting the role of recombination in drug resistance evolution. Based on phylogenetic incongruence of samples taken before and after patient treatment change, they showed that resistant HIV strains, after the treatment change, likely originated through recombination of strains carrying previously existing resistance mutations in a novel combination. It is worth noting that the observed patterns were apparently not consistent with convergent evolution because potential donors could be identified due to the extensive sampling performed. Moreover, the pattern of substitutions observed in the multidrug resistant variants present after treatment change suggests that this variation arises by recombination and not likely by the accumulation of mutations. However, it would be interesting to see whether observed recombination patterns are congruent when applying more stringent statistical and phylogenetic methods of detection (Martin et al., 2005).
2.4. The viral reservoir
A viral reservoir refers to a specific cell type or anatomical compartment where (i) HIV is protected from antiviral drugs and the immune system, (ii) shows greater stability than virus in the active replicating pool, (iii) possesses greater genetic diversity than non-reservoir virus due to the presence of archival strains, and (iv) remains replication-competent. HAART effectively reduces viral loads to <50 genome-copies/mL, the detection limit of most approved assays. The importance of virus in reservoirs is underscored by the observation that once therapy is withdrawn, viral loads increase within a few weeks to the levels of drug-naïve individuals (e.g., Imamichi et al., 2001). Taking into account the average half-life of long-term, latently infected cells, Siliciano et al. (2003) estimated that it would take around 60 years to deplete the main viral reservoir. This estimate is based on current antiretroviral therapy and does not consider drug resistance, drug toxicity and tolerance, or treatment costs. Thus, although novel methods for inducing proliferation of latently infected cells and subsequent elimination have been proposed (Marsden and Zack, 2009), the problem of eradication still persists and is not likely to be eliminated under existing therapies.
Phylogenetic methods are especially helpful in characterizing HIV reservoirs. By sampling a suspected reservoir over time and inferring evolutionary relationships, answers to questions such as whether HIV replicates in a particular compartment or not can be answered by looking at the branch lengths (i.e., genetic changes over time) of specific phylogenies. Depending on the amount of change and data richness present in the collected dataset, more specific inferences can be drawn such as divergence times and changes in population size over the time. A reservoir is then expected to have greater genetic diversity than other compartments (e.g., blood). Genotype networks are particularly suited to this aim since ancestral genotypes are located “center-wise” in the network from which “founder viruses” branch off (Crandall and Templeton, 1993), migrating into other compartments (Fig. 3 ). This is also true if the aim is to test whether archival strains are present in the bloodstream. To a great extent, phylogenies are also instrumental to estimate parameters that can describe diversity in within-host HIV populations. Modern estimates of genetic diversity, expressed as substitution rate-scaled effective population size (θ = 4Neμ), rely on coalescent simulations for which genealogical reconstructions and phylogenetic models are essential.
Fig. 3.
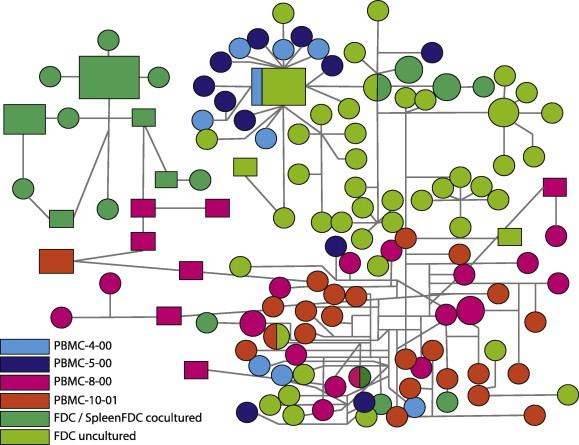
HIV-1 intra-host Statistical Parsimony network. Strains were isolated from different tissue types. Note how Follicular Dendritic Cell (FDC) derived sequences cluster in the center (ancestral) portion of the network; whereas, Peripheral Blood Mononuclear Cell (PBMC) derived sequences surround FDC-derived HIV sequences. This suggests an active role for the FDCs as an HIV reservoir.
Several cell types have been identified that play a role in hosting HIV at different time points during the course of drug therapy. The main cell type that plays host to HIV after initiation of HAART is activated CD4+ T cells (Chun et al., 1997, Delobel et al., 2005), which rapidly die within 2–3 days. Then, dendritic cells (DC), partially activated CD4+ T cells, and macrophages are thought to contribute to persistence due to their susceptibility to HIV infection, less vulnerability to cytopathic effects, and half-life up to several weeks (Dahl et al., 2010). However, long-lived, memory CD4+ T cells bearing latent integrated provirus contribute the most to the problem of persistence. Populations of this cell type are maintained by the intrinsic long-term survival and homeostatic proliferation of infected cells (Chomont et al., 2009).
Whether or not HAART impairs viral replication and then HIV evolution within CD4+ cell reservoirs is not entirely clear, yet replication in reservoirs is thought to be low (Hermankova et al., 2001, Kieffer et al., 2004, Parera et al., 2004, Persaud et al., 2007, Tobin et al., 2005). For instance, some studies have looked at different CD4+ memory cells and found almost no drug resistance mutations in this reservoir and short genetic distances when inferring phylogenetic trees after 8.3 years of uninterrupted HAART treatment (Nottet et al., 2009). Although the authors chose a less-reliable method of phylogenetic analysis and substitution model (Sullivan and Joyce, 2005, Susko et al., 2004), their inferences seem robust since other studies have reached similar conclusions (Mens et al., 2007). Nottet et al. (2009) hypothesized that viral replication in reservoirs would be indicated the appearance of drug resistance mutations, indicative of evolution within the reservoir. They concluded that due to the lack of drug resistance mutations and short tree branch lengths observed in the HIV viruses stored in reservoirs, replication and evolution have been halted in the blood compartment. Bailey et al. (2006) also inspected CD4+ T cells with a thorough sampling strategy and found limited evolution in the CD4+ reservoir. This conclusion was reached even when a greater diversity of pol genes was found in the reservoir than in plasma samples and that some sequences isolated from plasma were also found in the reservoir. Of course these results do not preclude the possibility that other reservoirs are the source of residual viremia. Although lack of evolution within an organism or entity is conceptually difficult to imagine, particularly since evolution can be defined in its simplest form as genetic change over time, irrespective of the observed magnitude, it seems like HIV hiding in reservoirs evolves at a slow rate and/or that the viruses released from the reservoir are subjected to strong purifying selection in the plasma.
Diversity between reservoirs and peripheral blood cells or within specific tissues has been studied in other reservoirs as well. For example, Follicular Dendritic Cells (FDCs) also act as a viral reservoir; although less is known about this reservoir compared to latent CD4+ cells and macrophages. In the mouse, FDC-trapped HIV has a half-life of about 2 months and it remains replication-competent for at least 9 months (Burton et al., 2002). In contrast to the other common reservoirs of HIV, the FDC is not infected, but contains only trapped extracellular HIV. Because the studies cited above were performed in mice, it was unclear how FDC-virus could contribute to HIV persistence (Smith et al., 2001). However, using experimental and phylogenetic approaches with human tissues and cells, Keele et al. (2008b) confirmed that HIV trapped on FDCs was replication-competent. More importantly, HIV on FDCs had greater genetic diversity than viruses in other tissues and cells examined, including CD4+ T cells. Importantly, within the FDC trapped viruses, drug resistance variants were found and these were not identified in other sites (Fig. 3). Moreover, with an elegant network approach, they showed the existence of archival viral variants from various time points of infection that were trapped on FDCs. Altogether these findings indicate that FDCs can act as reservoir and hold viruses for years (Fig. 3).
Phylogenetic analysis has been used to explore the contribution of HIV to tumorigenesis in reservoir cell types. Recently, Salemi et al. (2009) explored the dynamics of HIV-infected macrophages using p24 staining and prediction of co-receptor usage in tumor and non-tumor postmortem tissues from patients that died of AIDS-related lymphoma (ARL). They observed a high degree of compartmentalization between HIV from macrophages found in tumor and non-tumor tissues and an intermixing of HIV strains obtained from auxiliary lymph nodes. Viral effective population size was 100-fold greater in tumor tissues than in non-tumor tissues and, strikingly, the onset of lymphoma correlate with viral expansion. Moreover, evidence of gene flow to/from lymph nodes and tumor tissues indicates that lymph nodes might facilitate the movement of metastatic cells to different parts of the body.
Poor penetration of HAART or properties such as immune privileged tissues can drive anatomical compartments to act as “sanctuary sites”, places where HIV keeps replicating. Some suggested sanctuaries include the central nervous system (CNS), gut-associated lymphoid tissue (GALT) and the genitourinary tract (Dahl et al., 2010). Evidence of compartmentalization of HIV sequences from different tissue types has been typically inferred from monophyletic assemblages of those sequences in phylogenetic trees (Wang et al., 2001, Wong et al., 1997). Even further sub-compartmentalization has been found in GALT tissue throughout the gastrointestinal tract (van Marle et al., 2007). When coupled with differential HIV gene expression, this can indicate that GALT has the capacity to host different HIV replicating strains. Hence, GALT tissue should also be considered when screening for drug resistant variants. While these conclusions are insightful for HIV genetic structure in GALT, overlooking nucleotide substitution model of selection, not using an optimality criterion for tree inference that accounts for phylogenetic uncertainty, and lacking powerful statistical methods such as the coalescent that account for historical patterns of divergence (Wakeley, 2004) (all issues with theses studies), can bias conclusions.
3. Origin and timing of HIV
Phylogenetic analyses can bring to light dimensions of HIV evolution such as “where and when” and even “how” infections are spreading across the globe that are impossible to assess with other approaches (Grenfell et al., 2004, Holmes, 2004, Holmes, 2007, Holmes, 2009, Holmes and Drummond, 2007, Moya et al., 2004, Welch et al., 2005). In the following sections, we discuss these dimensions and how phylogenetics has led to our current understanding of HIV origin and geographic spread.
3.1. Geographic and host origins, where and how?
The origins of HIV have been controversial since the beginning of the epidemic. HIV-1 and HIV-2, both species belonging to the genus Lentivirus (Retroviridae), are distinguished on the basis of their genome organization and phylogenetic relationships, clinical characteristics, virulence, infectivity and geographic distribution. During the beginning of the HIV-1 epidemic, serological evidence pointed to African green monkeys (Chlorocebus spp.; agm) as carriers of an HIV-1-like virus, simian T-cell lymphotrophic virus 3 [STLV-3, now Simian Immunodeficiency Virus (SIV) agm; Kanki et al. (1987)]. Serum from HIV-1 infected patients cross-reacted with STLV-3 proteins (Hirsch et al., 1986, Kanki et al., 1985a, Kanki et al., 1985b) and STLV-3-infected African green monkeys had an overlapping geographic distribution with the HIV-1 epidemic, which led researchers to believe that HIV-1 jumped to humans from African green monkeys (Kanki et al., 1987). In contrast to the serological evidence, phylogenetics argued for a chimpanzee origin; however, scientists distrusted the results, taking the analyses with caution and concluding that their evidence was not enough to prove a chimpanzee cross-species infection (Huet et al., 1990). The justifications stated for ignoring the phylogenetic evidence included the observations that high vpu gene divergence occurred between HIV-1 and SIVcpz, few lentiviruses had been isolated from simian hosts, and SIV had low prevalence in chimpanzees (Huet et al., 1990).
Evidence of a simian origin is now clear, as similar lentiviruses have been found in more than 40 species of African primates (Bibollet-Ruche et al., 2004, Hahn et al., 2000, Van Heuverswyn and Peeters, 2007) and geographical correlation exists between SIVs hosted in different primate species and HIV (Peeters et al., 2008). Phylogenetic evidence has shed light on this subject, showing that HIV-1 and HIV-2 are the product of several cross-species transmission events between chimpanzee (Pan troglodytes troglodytes) SIV (SIVcpz) and sooty mangabey SIV (Cercocebus atys; SIVsm) with humans (Gao et al., 1992, Gao et al., 1999, Hahn et al., 2000, Huet et al., 1990, Plantier et al., 2009, Van Heuverswyn and Peeters, 2007) (Fig. 4 ). Moreover, SIVcpz and SIVsm geographic range distributions correlate well with African regions where HIV-1 and HIV-2 show great endemicity, e.g., sooty mangabeys are most abundant in the regions of West Africa where HIV-2 is highly prevalent and diverse; thus, HIV-2 likely emerged there (Chen et al., 1996, Chen et al., 1997, Santiago et al., 2005). The HIV-2 simian origin was rapidly established, since the only species naturally infected with a closely related virus is C. atys (Chen et al., 1996). Furthermore, SIVsm cross-species transmission has occurred in multiple occasions demonstrated by phylogenetic analyses (Chen et al., 1997, Gao et al., 1992, Gao et al., 1994, Lemey et al., 2003).
Fig. 4.
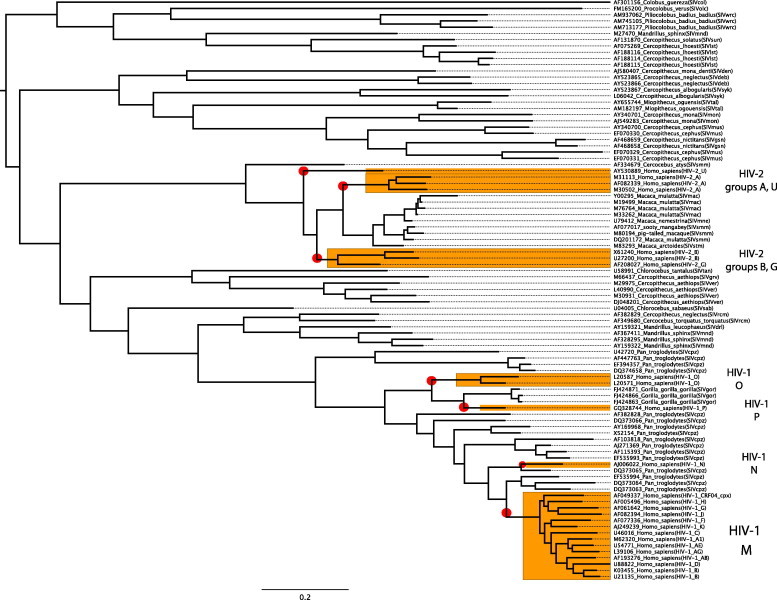
Phylogenetic tree showing HIV cross-species transmission. The tree was built using 93 pol gene amino acid sequences from Los Alamos database and the Bayesian approach implemented in MrBayes 3.1.2. Taxon names represent accession number, host species name and isolate designation. A red circle highlights potential cross-species transmissions. Representative sequences from HIV-1 and 2 types are also shown. The scale bar denotes amino acid changes as substitutions per site. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Indications of natural chimpanzee infections have been increasingly corroborated by phylogenetic analyses. Support for “the chimpanzee hypothesis” began to accumulate when SIV was found in natural populations in West Africa, initially in extremely low prevalence (Santiago et al., 2002). Subsequently, scientists sequenced the entire genome of SIVcpz from a fecal sample of a wild chimpanzee in Tanzania (Santiago et al., 2003) and confirmed previous phylogenetic inferences based on the gag, pol and env genes. Moreover, epidemiological evidence has shown that SIVcpz can reach prevalence rates of up to 29–35% in some African communities (Keele et al., 2006). Recently, also in Tanzania, Keele et al. (2009) followed populations of chimpanzees over 9 years and found typical AIDS-like features, e.g., increased death hazard for animals having SIVcpz, CD4+ T-cell depletion with high viral replication and histopathological findings consistent with end-stage AIDS. Altogether, epidemiological, physiological and clinical evidence support early phylogenetic predictions establishing chimpanzee cross-species transmission to humans, HIV-1 origin and chimpanzees as natural reservoir for the virus.
3.2. Timing, when?
The timescale of the evolution of HIV-1 and -2 has been estimated and it has allowed us to understand the circumstances surrounding the emergence of HIV and to test the hypothesis regarding natural or artificial means of cross-species transmission. Initially, several hypothesis of “when” HIV came into human populations could not be tested in a reductionist framework. Among these, the so-called Oral Polio Vaccine (OPV) hypothesis (Hooper, 2003) stated that HIV was introduced into human populations by the use of inadvertently infected monkeys (advocates claimed chimpanzees) as means for polio vaccine production. In fact, around 1960 African green monkeys were used to produce an attenuated polio vaccine (Plotkin, 2001). Thus, if the molecular timing of HIV “jump” in humans matched this date, the result would be consistent with this hypothesis. On the other hand, if the timing of the HIV “jump” significantly predated 1960, then OPV hypothesis would seem less likely.
As increasing amounts of data and more powerful computational/statistical approaches became available, the time to the most recent common ancestor (TMRCA) of HIV-1, whether it was in a human or a chimpanzee host, was estimated with increasing confidence (Korber et al., 2000, Salemi et al., 2001, Sharp et al., 2000). These studies have used different tactics, yet obtained similar estimates for the HIV-1M group radiation (strict and relaxed molecular clock analyses). Applications of these methods have led scientists to estimate that the M group originated near 1930 with a range, depending on the study, of 10–20 years on either side. It is worth noting that, although most estimates are consistent, they can be biased partly by recombination and few historical samples. Recombination, apart from violating the single ancestry assumption, may increase apparent variation in rates among nucleotide sites and also has a decreasing effect upon genetic distances between sequences (Posada, 2001). On the other hand, the partial lack of historical samples makes the calibration of such methods a hard task resulting in estimates with wide confidence intervals.
The only archival samples available, DRC60 and ZR59, suggest extensive genetic diversity of HIV-1 in West Africa by 1960. In turn, divergence time estimates date to the 1920s, depending upon the coalescent tree model chosen (Worobey et al., 2008). Since divergence time estimates represent the TMRCA of just the isolates included in the analysis, it is likely that new diverse archival sequences will yield even older divergence time estimates for the HIV-1M group radiation. Hence, the OPV hypothesis can be ruled out because the molecular data suggest that HIV-1 group M isolates originated 30 years prior to the use of primates in OPV preparation (Korber et al., 2000, Worobey et al., 2008). In addition, it has not been possible to detect chimpanzee DNA in archival stocks of OPV (Berry et al., 2001, Blancou et al., 2001). Most likely, cross-species transmission can be explained by invoking socio-cultural factors during the postcolonial period in Africa (Chitnis et al., 2000).
By similar means, the origin of HIV-2 has been dated to 1940 ± 16 for subtype A and to 1945 ± 14 for subtype B (Lemey et al., 2003). Moreover, phylogenetic inference has dated the introduction of HIV-1 clade B in North America to 1968 (1966–1970) (Gilbert et al., 2007, Pérez-Losada et al., 2010), which is consistent with the earliest known retrospective studies (Robbins et al., 2003).
It is worth mentioning that time estimation of deep (old) viral divergent nodes based on molecular clock analyses can be biased towards the present if no external calibrations are used. This could arise from extremely high saturation problems and constraints that would be hard to account for with substitution models, as it has been shown using an island biogeography approach to SIV dating (Worobey et al., 2010).
The study of the origin of HIV viruses is far from being a resolved issue; rather, phylogenetics is currently providing more insights as new isolates are being analyzed, especially isolates from other non-human primates. The study of HIV relatives shows that similar processes shape their natural history. For instance, SIVcpz itself is the product of recombination between SIVrcm (red-capped mangabeys, Cercocebus torquatus) and SIVgsn (greater spot-nosed monkeys, Cercopithecu snictitans). This has been revealed by strong discordance between topologies that suggested a hybrid origin for SIVcpz (Bailes et al., 2003). Likewise, western gorilla SIV (SIVgor, from Gorilla gorilla gorilla) also seems to be the product of cross-species transmission (Fig. 4). Despite the small number of samples available, phylogenetic analyses suggest that SIVgor is closely related to SIVcpz from Pan troglodytes troglodytes being also sister taxa to HIV-1 group O (Takehisa et al., 2009). However, due to the yet poor sampling of SIVgor, whether chimpanzees infected gorillas and humans, or humans were infected first and then humans infected gorillas or even gorillas to humans, is yet to be determined (Fig. 4). Interestingly, a new HIV-1 group P, has been proposed based on one isolate found in a Cameroonian woman. The phylogenetic placement of group P as the sister taxon to all SIVgor but distinct from HIV-1 group O, could play a key role in testing hypotheses of human–gorilla transmissions (Fig. 4) (Plantier et al., 2009).
Phylogenetic analysis can be very informative, but the accuracy of phylogenetic conclusions is highly dependent on the method chosen and sampling strategy. As more lentivirus sequences from different locations and archival sequences become available, the issue of the origin of HIV should converge to more reliable conclusions. Before we explore different aspects of HIV dynamics and its applications, we should add a cautionary note on genetic marker choice to capture transmission and other desired signals. Extensive debate exists concerning the gene(s) choice in HIV phylogenetics (Hué et al., 2005a). The env gene is sometimes preferable on the basis of high genetic variability; however, indications of convergent evolution on this region would preclude its use since it violates the unique evolutionary history assumption made by phylogenetic methods. On the other hand, the pol gene has been suggested as a candidate as well, however some researchers have been reluctant to use it given the number of drug resistance mutations associated with this region. Lemey et al. (2005b) showed that phylogenetic trees based on pol sequences, after excluding codons associated with resistance, were congruent with independent data on epidemiology and with trees based on env sequences. Similarly, criminal cases of HIV transmission that rely solely on phylogenetic evidence are precarious. Besides the inherent issues about model selection and phylogenetic inference, data availability also plays a major role. Some of the concerns are related to the direction of transmission or who infected whom, availability of all involved sexual contacts, and interpretation of the phylogeny given that certain individuals could be infected with more than one strain. Finally, issues of convergent evolution can erroneously link individuals in the absence of any other independent source of evidence (Pillay et al., 2007).
4. Phylodynamics and HIV
The term phylodynamics was coined in reference to “the melding of immunodynamics, epidemiology, and evolutionary biology […]” in particular to pathogens such as viruses and bacteria in the whole breadth from within-host variation and immunity through transmission events, bottlenecks and global epidemiological dynamics (Grenfell et al., 2004). This is one of the most exciting and insightful ongoing fields in which phylogenetics is contributing to our understanding of virus evolution and in particular to HIV. Viral populations dynamics can be explored using phylogenetic, coalescent and other statistical methods to make historical inferences about their temporal and spatial distributions. This is possible, basically, by taking advantage of certain attributes, such as high mutation rates, large population sizes, short generation times and the realization that genetic changes occur so fast that ecological and epidemiological processes leave marks on their genomes. The basic idea behind this new approach is that phylogenies are modulated by immune selection, viral population sizes and spatial dynamics and thus, together with experimental data, it would be possible to tear apart individual contributions and identify forces dominating pathogen evolution and behavior. Although mainly focused on RNA viruses (Amore et al., 2010, Bennett et al., 2010, Holmes and Grenfell, 2009, Kerr et al., 2009, Mondini et al., 2010, Pérez-Losada et al., 2010, Pérez-Losada et al., 2011, Rambaut et al., 2008, Siebenga et al., 2010), phylodynamic approaches have been also applied to DNA viruses (Zehender et al., 2010a) and bacteria (Conlan et al., 2007, Pérez-Losada et al., 2007a, Pérez-Losada et al., 2007b, Pérez-Losada et al., 2005, Tazi et al., 2010).
Most of the methods available (see: Kuhner, 2009; Pérez-Losada et al., 2007c) take advantage of the coalescent theory developed by Kingman based on previous work made by Wright and others (Kingman, 2000). Although the idea of a coalescent theory was used several times in population genetics [reviewed in Tavaré (1984)], the development is credited to Kingman (1982) and independently to Hudson, 1983, Tajima, 1983 [see Wakeley (2008) for a thorough discussion of coalescent theory]. The basic idea behind coalescent theory, as opposed to summary statistics or classic population genetics, is that coalescence tries to explain the present of a population by taking a look into its past. It is a realization of the Wright–Fisher neutral model of evolution, recording the genealogical relationships among a random sample of population genetic data. The model has been further generalized to account for varying population size, different time scales, structure, recombination and selection (Nordborg, 2004).
Within a coalescent framework, statistical advances in Bayesian inference regarding the use of time-stamped data (Drummond et al., 2002), models of population dynamics and relaxation of molecular clock assumptions (Drummond et al., 2005, Drummond et al., 2006, Drummond and Rambaut, 2007) have greatly helped to understand better HIV patterns and processes on a temporal scale. Recent advances in sequence dating provide tools to estimate unknown sequence ages as these can be jointly or individually estimated under a full probabilistic framework (Shapiro et al., 2011). In addition, this temporal framework can be enhanced by explicitly modeling spatial dispersion rates in a phylogeographic context (Lemey et al., 2009, Lemey et al., 2010). Using phylogenetic diffusion models, one can infer ancestral state locations for the sequences sampled under a discrete or continuous context. Moreover, the most parsimonious explanation for the diffusion process is obtained under this Bayesian framework by the implementation of Bayesian Stochastic Search Variable Selection (BSSVS). This new methodology has several advantages over previous maximum likelihood and maximum parsimony methods such as fitting a diffusion model simultaneously with a substitution model, incorporation of branch lengths in ancestral state reconstruction, and accommodation of uncertainty in both the phylogeny and the diffusion process. Applications of this method are rapidly increasing as recent studies in dengue (Raghwani et al., 2011), influenza (Nelson et al., 2011), Staphylococcus aureus (Gray et al., 2011), and, of course, HIV-1 (Esbjörnsson et al., 2011, Skar et al., 2011) indicate. More recently, new methods have been adapted from systematic studies to estimate the basic reproductive number (R 0) (Stadler, 2010, Stadler et al., 2011). The R 0 parameter has been traditionally used in epidemics to determine whether or not an infectious agent can spread in a population, i.e., if R 0 > 1 the infectious agent will spread in the population and if R 0 < 1 the infection will die out. Given the amount of viral sequence data and the ease of data acquisition, estimating R 0 from genetic data can become a novel tool for molecular epidemiologists. The model uses a Birth–Death process (as in species phylogenetics) instead of a coalescent model, in which a birth event is equivalent to a new infection and a death event to various phenomena such as death, treatment, eradication, etc.
In order to exploit most of these methods, sampling strategy is paramount and arbitrary sequence collection from GenBank or the Los Alamos database is probably not adequate (Stack et al., 2010). Next-gen sequencing approaches, e.g. (Bybee et al., 2011), allow for more comprehensive sampling at efficient costs for future studies of HIV diversity rather than half-hazard sampling of sequences available from other studies in public databases. Clearly, for greatest utility in studying HIV, sequence data submitted to public databases should include geographic, clinical, and, especially, temporal information (see http://datadryad.org for storage options for such data).
4.1. Transmission dynamics
Transmission dynamics have been studied thoroughly, in particular regarding transmission network reconstructions and inspecting the loss of diversity at the transmission event. Popular examples of HIV transmission are cases involving legal issues such as the Florida dentist case (Ou et al., 1992) and the Louisiana attempted murder trial (Metzker et al., 2002). In both of these cases, phylogenetic evidence was concordant with the transmission hypothesis from the defendant to the victims. In fact, the Louisiana case constituted the first case in the US in which phylogenetic analyses were used in a criminal court case. In this case, different substitution models, genes (env and pol), and optimality criteria were used in linking the defendant with the victim’s HIV-1 variants. Additionally, drug resistance genetic signatures were also used as indications of transmission events. Some other examples of transmission reconstruction include a Swedish rape case (Albert et al., 1994), and a healthcare related case in Baltimore (Holmes et al., 1993). It is also interesting to highlight a recent report of highly divergent HIV variants transmitted by a donor to another two individuals on the same evening (English et al., 2011).
Transmission dynamics have also been studied across transmissions because of the opportunity for treatment due to a reduction in genetic diversity (Edwards et al., 2006, Fischer et al., 2010). Most of the work has focused on monitoring discordant couples (i.e., couples in which one partner is HIV positive and the other is not) and to test whether there is a reduction in genetic diversity down to one virus at the transmission event. Edwards et al. (2006) showed using phylodynamic methods that the reduction in genetic diversity (<1%) is no different between horizontal (homo- or heterosexual) and vertical transmission (mother-to-child). Understanding genetic diversity at transmission events has therapeutic implications as less diverse populations of small size are strongly influenced by genetic drift, decreasing the chance of transmission of high fitness variants. Indications that single virus variants were transmitted horizontally came first from studies using Sanger sequencing and single-genome amplification coupled with phylogenetic and mathematical modeling (Keele et al., 2008a, Salazar-González et al., 2008), and were further supported by the enhanced capabilities of ultra-deep-sequencing (UDS). UDS revealed that early HIV variants explored extensive sequence space within epitope regions. Interestingly, as the infection proceeds, reversion to the canonical subtype sequence occurred in positions under immune pressure, but not in positions that were not under pressure even in the earliest samples, suggesting that immune pressure is present earlier than previously known (Fischer et al., 2010). Thus, phylogenetic analyses can be very insightful regarding practical situations such as court trials, and also in situations of medical importance such as characterizing genetic diversity for potential therapy development.
4.2. Population dynamics
While the population dynamics of HIV has been well characterized (Coffin, 1995), phylogenetic studies have added greatly to our understanding, especially of the dynamic nature of genetic diversity over the course of infection within a host individual and across transmission events. Much of the phylodynamic research has focused on associations between clinical/epidemiological aspects and genetic diversity such as transmission/spread dynamics between men having sex with men [MSM; Lewis et al. (2008)] revealing episodic clusters of transmission, and between heterosexual patients in which transmission dynamics appeared to be slower compared to MSM (Hughes et al., 2009). Note that these studies used large sample sizes to draw their conclusions, a desirable feature to capture the phylodynamic signature from the data.
Phylodynamic studies have focused on temporal dynamics of transmission and its frequency. In Italy, a recent study showed similar conclusions to that of Hué et al. (2005b) in the UK in that currently circulating subtype B HIV was introduced multiple times into MSM populations. Similarly, when comparing inter-node intervals between transmission clades in Lewis et al., 2008, Zehender et al., 2010b the time between transmission events differed with medians of 14 months and 30 months for UK and Italian study, respectively. These results could reflect actual differences in transmission dynamics or could be due to an artifact because of the small sample size and the restricted area sampled.
Other studies have explored dissemination patterns and possible transmission of particular HIV strains between risk groups. Recently, Liao et al. (2009) looked at spatial and temporal patterns to explain the distribution of the CRF01_AE variant in Vietnam. Their results suggested that CRF01_AE came from Thailand and that, within Vietnamese population, it has been transmitted from heterosexual patients to Intravenous Drug Users (IDUs). Similar work was done with other subtypes and recombinant forms in Asia (Tee et al., 2008), South America (Bello et al., 2010), Africa (Gray et al., 2009, Lemey et al., 2003) and North America (Gilbert et al., 2007), among others.
Patterns of diversity among populations and how they compare to epidemiological data have been studied. Pérez-Losada et al. (2010) studied HIV-1 envelope gene sequence variation in cohorts of vaccinated and placebo-treated patients in North America. Their phylodynamic analysis showed that genetic diversity remained nearly constant from approximately the 1970s to date, suggesting that viral populations had already expanded around ten years before HIV was detected in the US (Fig. 5 ). In addition, despite a drop in the number of cases since the 90’s, genetic diversity has remained high across time. Previously, Robbins et al. (2003) showed similar results in a different cohort of HIV-1 positive US subjects. Although they used parametric and non-parametric methods that did not account for phylogenetic uncertainty and a smaller dataset, they reached similar demographic conclusions. In a similar study, phylodynamic analyses revealed that CRF01_AE was cryptically circulating in the Thai HIV-1 virus population for 3–10 years before it was detected in 1989 [Fig. 5; (Pérez-Losada et al., 2011)]. In both the North American and Thai studies, historical estimates of genetic diversity correlate well with known epidemiological data.
Fig. 5.
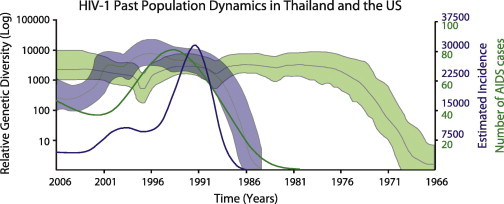
HIV-1 past population dynamics in North America (green) and Thailand (gray). Plots were built using the env gene and the Bayesian Skyline Plot model. The analyses primarily revealed that genetic diversity (thick lines) has remained high through time despite the number of AIDS cases (thin lines) dropping considerably and thatHIV was circulating years before the first AIDS cases were detected. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4.3. Within-host dynamics
Within-host evolution in HIV has proven to be important in understanding clinical processes associated with disease progression. Within-host, HIV genetic diversity of plasma isolates is reduced at any time point, but increasing over the course of infection, similar to that observed in the population phylogenies of influenza virus (Grenfell et al., 2004, Rambaut et al., 2008, Shankarappa et al., 1999). HIV evolution could be also different in specific tissues. For example, phylodynamic analyses of post mortem brain tissues have revealed that HIV is evolving at different rates at different brain compartments. However, this is apparently not related to selective pressure, but rather to inherent drift associated with macrophage-tropic viral expansion after immune failure (Salemi et al., 2005).
Within-host variation, commonly misnamed as quasispecies (Holmes, 2009), has been addressed under a phylodynamic framework for co-receptor usage. For example, Salemi et al. (2007) explored co-receptor usage dynamics in tissue and peripheral blood mononuclear cells (PBMC) and found a temporal structure between CCR5-tropic (R5) virus and the appearance of CXCR4 (X4) variants, i.e., majority of X4 virus found in thymus tissue seemed to come from PBMC viruses. Conversely, substitution rates between R5 and X4 sequences were not significantly different; supporting that X4 amplification could be due to the availability of target cells. As recognized by the authors, phylodynamic studies of HIV subpopulations can be greatly enlightening, but they have some limitations as human tissue samples are not easy to obtain and could involve ethical issues. As new animal models (e.g., new humanized mice) for HIV infections become available (Denton and Garcia, 2009), viral subpopulations can be studied over the course of 1–2 years infection (Berges et al., 2010). Nonetheless, despite the potential of these models, little has been done in experimental HIV evolution due to little interaction between evolutionary and molecular biologists.
Although the field of phylodynamics is young, its statistical tools are key in linking epidemiological and evolutionary information (Tibayrenc, 2005). Surveillance programs would greatly benefit from the implementation of these approaches, as it would be possible to study the impact of vaccines/chemotherapeutic treatments in population genetic diversity. Likewise, such studies would allow for the identification of novel risk groups and indicate changes (or not) in population dynamics as a result of intervention strategies. At the same time, the creation of specialized databases to collect phylodynamic informative data (randomly sampled HIV sequences across a broad target area of HIV infection to monitor HIV diversity and associated changes) would greatly aid the implementation of these approaches.
5. Future prospects
Phylogenetics has a new vigor. The development of new robust statistical frameworks such as Bayesian inference (Huelsenbeck et al., 2001) has allowed the testing of complex hypothesis accounting for the inherent uncertainties of historical and unrepeatable processes. Currently, implementation of these methods under biologist-accessible software has empowered scientists to test biologically meaningful scenarios. To a certain extent, the great phylogenetic questions in HIV evolution have been undertaken, e.g., HIV origin, evolutionary driving forces, within and among host variation. However, phylogenetics opens the door to new questions and new insights in HIV. For example, are recombination/substitution rates impacted by antiviral treatments? Do transmission routes influence the outcome of infection? How does compartmentalization of HIV strains evolve within an individual? These are all questions motivated by phylogenetic approaches. Similarly, large collaborative efforts such as the UK HIV Drug Resistance Collaboration provide opportunities for addressing key questions in phylodynamics and drug resistance through the study of longitudinal and/or retrospective data from large cohorts. In summary, phylogenetics is an ever-evolving field that promises to give more insights into pathogen evolution, mainly in pathogens such as HIV that form measurably evolving populations (Drummond et al., 2003).
Acknowledgments
E.C.N. would like to thank Justin Bagley for revising an early version of this manuscript. We thank anonymous reviewers for their helpful comments in greatly improving our manuscript. E.C.N. was funded by Comisión Nacional de Investigación Científica y Tecnológica (CONICYT), Gobierno de Chile – Becas Chile and by the BYU Graduate Mentoring Award 2011–12. K.A.C. would like to thank Brigham Young University for continuous support and NIH Grant R01GM66276. G.F.B. was funded by NIH grant R56AI39963 and by College of Physical and Mathematical Sciences CHIRP grant.
References
- Albert J., Wahlberg J., Leitner T., Escanilla D., Uhlen M. Analysis of a rape case by direct sequencing of the human immunodeficiency virus type 1 pol and gag genes. J. Virol. 1994;68:5918–5924. doi: 10.1128/jvi.68.9.5918-5924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amore G., Bertolotti L., Hamer G.L., Kitron U.D., Walker E.D., Ruiz M.O., Brawn J.D., Goldberg T.L. Multi-year evolutionary dynamics of West Nile virus in suburban Chicago, USA, 2005–2007. Philos. Trans. Roy. Soc. B. 2010;365:1871–1878. doi: 10.1098/rstb.2010.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailes E., Gao F., Bibollet-Ruche F., Courgnaud V., Peeters M., Marx P.A., Hahn B.H., Sharp P.M. Hybrid origin of SIV in chimpanzees. Science. 2003;300:1713. doi: 10.1126/science.1080657. [DOI] [PubMed] [Google Scholar]
- Bailey J.R., Sedaghat A.R., Kieffer T., Brennan T., Lee P.K., Wind-Rotolo M., Haggerty C.M., Kamireddi A.R., Liu Y., Lee J., Persaud D., Gallant J.E., Cofrancesco J., Quinn T.C., Wilke C.O., Ray S.C., Siliciano J.D., Nettles R.E., Siliciano R.F. Residual human immunodeficiency virus type 1 viremia in some patients on Antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4(+) T cells. J. Virol. 2006;80:6441–6457. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barre-Sinoussi F., Chermann J., Rey F., Nugeyre M., Chamaret S., Gruest J., Dauguet C., Axler-Blin C., Vezinet-Brun F., Rouzioux C., Rozenbaum W., Montagnier L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- Beer B., Bailes E., Goeken R., Dapolito G., Coulibaly C. Simian immunodeficiency virus (SIV) from sun-tailed monkeys (Cercopithecus solatus): evidence for host-dependent evolution of SIV within the C. Lhoesti superspecies. J. Virol. 1999;73:7734. doi: 10.1128/jvi.73.9.7734-7744.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello G., Aulicino P.C., Ruchansky D., Guimaraes M.L., Lopez-Galindez C., Casado C., Chiparelli H., Rocco C., Mangano A., Sen L., Morgado M.G. Phylodynamics of HIV-1 circulating recombinant forms 12_BF and 38_BF in Argentina and Uruguay. Retrovirology. 2010;7 doi: 10.1186/1742-4690-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S.N., Drummond A.J., Kapan D.D., Suchard M.A., Munoz-Jordan J.L., Pybus O.G., Holmes E.C., Gubler D.J. Epidemic dynamics revealed in dengue evolution. Mol. Biol. Evol. 2010;27:811–818. doi: 10.1093/molbev/msp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berges B.K., Akkina S.R., Remling L., Akkina R. Humanized Rag2–/–[gamma]c–/–(RAG-hu) mice can sustain long-term chronic HIV-1 infection lasting more than a year. Virology. 2010;397:100–103. doi: 10.1016/j.virol.2009.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard E., Azad Y., Vandamme A., Weait M., Geretti A. HIV forensics: pitfalls and acceptable standards in the use of phylogenetic analysis as evidence in criminal investigations of HIV transmission. HIV Med. 2007;8:382–387. doi: 10.1111/j.1468-1293.2007.00486.x. [DOI] [PubMed] [Google Scholar]
- Berry N., Davis C., Jenkins A., Wood D., Minor P., Schild G., Bottiger M., Holmes H., Almond N. Vaccine safety: analysis of oral polio vaccine CHAT stocks. Nature. 2001;410:1046–1047. doi: 10.1038/35074176. [DOI] [PubMed] [Google Scholar]
- Bhatt S., Holmes E.C., Pybus O.G. The genomic rate of molecular adaptation of the human influenza A virus. Mol. Biol. Evol. 2011;28:2443–2451. doi: 10.1093/molbev/msr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibollet-Ruche F., Bailes E., Gao F., Pourrut X., Barlow K. New simian immunodeficiency virus infecting De Brazza’s monkeys (Cercopithecus neglectus): evidence for a Cercopithecus monkey virus clade. J. Virol. 2004;78:7748. doi: 10.1128/JVI.78.14.7748-7762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancou P., Vartanian J.-P., Christopherson C., Chenciner N., Basilico C., Kwok S., Wain-Hobson S. Polio vaccine samples not linked to AIDS. Nature. 2001;410:1045–1046. doi: 10.1038/35074171. [DOI] [PubMed] [Google Scholar]
- Bretscher M.T., Althaus C.L., Muller V., Bonhoeffer S. Recombination in HIV and the evolution of drug resistance. for better or for worse? Bioessays. 2004;26:180–188. doi: 10.1002/bies.10386. [DOI] [PubMed] [Google Scholar]
- Brown A.J.L. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad. Sci. USA. 1997;94:1862–1865. doi: 10.1073/pnas.94.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buendia P., Cadwallader B., DeGruttola V. A phylogenetic and Markov model approach for the reconstruction of mutational pathways of drug resistance. Bioinformatics. 2009;25:2522–2529. doi: 10.1093/bioinformatics/btp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonagurio D., Nakada S., Parvin J., Krystal M., Palese P., Fitch W. Evolution of human influenza A viruses over 50 years: rapid, uniform rate of change in NS gene. Science. 1986;232:980–982. doi: 10.1126/science.2939560. [DOI] [PubMed] [Google Scholar]
- Burton G.F., Keele B.F., Estes J.D., Thacker T.C., Gartner S. Follicular dendritic cell contributions to HIV pathogenesis. Semin. Immunol. 2002;14:275–284. doi: 10.1016/s1044-5323(02)00060-x. [DOI] [PubMed] [Google Scholar]
- Bush R.M., Bender C.A., Subbarao K., Cox N.J., Fitch W.M. Predicting the evolution of human influenza A. Science. 1999;286:1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Bybee S.M., Bracken-Grissom H., Haynes B.D., Hermansen R.A., Byers R.L., Clement M.J., Udall J.A., Wilcox E.R., Crandall K.A. Targeted amplicon sequencing (TAS): a scalable next-gen approach to multi-locus, multi-taxa phylogenetics. Gen. Biol. Evol. 2011 doi: 10.1093/gbe/evr106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Rodríguez A., Crandall K.A., Posada D. Recombination estimation under complex evolutionary models with the coalescent composite-likelihood method. Mol. Biol. Evol. 2006;23:817–827. doi: 10.1093/molbev/msj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Rodríguez A., Crandall K.A., Posada D. Recombination favors the evolution of drug resistance in HIV-1 during antiretroviral therapy. Infect. Genet. Evol. 2007;7:476–483. doi: 10.1016/j.meegid.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Rodríguez A., Posada D., Pérez-Losada M., Keller E., Abrams E.J., Viscidi R.P., Crandall K.A. Disease progression and evolution of the HIV-1 env gene in 24 infected infants. Infect. Genet. Evol. 2008;8:110–120. doi: 10.1016/j.meegid.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Chen Z., Telfier P., Gettie A., Reed P., Zhang L., Ho D., Marx P. Genetic characterization of new West African simian immunodeficiency virus SIVsm: geographic clustering of household-derived SIV strains with human immunodeficiency virus type 2 subtypes and genetically diverse viruses from a single feral sooty mangabey troop. J. Virol. 1996;70:3617–3627. doi: 10.1128/jvi.70.6.3617-3627.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Luckay A., Sodora D., Telfer P., Reed P., Gettie A., Kanu J., Sadek R., Yee J., Ho D., Zhang L., Marx P. Human immunodeficiency virus type 2 (HIV-2) seroprevalence and characterization of a distinct HIV-2 genetic subtype from the natural range of simian immunodeficiency virus-infected sooty mangabeys. J. Virol. 1997;71:3953–3960. doi: 10.1128/jvi.71.5.3953-3960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.M., Perlina A., Lee C.J. Positive selection detection in 40,000 human immunodeficiency virus (HIV) type 1 sequences automatically identifies drug resistance and positive fitness mutations in HIV protease and reverse transcriptase. J. Virol. 2004;78:3722–3732. doi: 10.1128/JVI.78.7.3722-3732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis A., Rawls D., Moore J. Origin of HIV Type 1 in Colonial French Equatorial Africa? AIDS Res. Hum. Retrov. 2000;16:5–8. doi: 10.1089/088922200309548. [DOI] [PubMed] [Google Scholar]
- Chomont N., El-Far M., Ancuta P., Trautmann L., Procopio F.A., Yassine-Diab B., Boucher G., Boulassel M.-R., Ghattas G., Brenchley J.M., Schacker T.W., Hill B.J., Douek D.C., Routy J.-P., Haddad E.K., Sekaly R.-P. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun T.-W., Carruth L., Finzi D., Shen X., DiGiuseppe J.A., Taylor H., Hermankova M., Chadwick K., Margolick J., Quinn T.C., Kuo Y.-H., Brookmeyer R., Zeiger M.A., Barditch-Crovo P., Siliciano R.F. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- Clavel F., Hance A.J. HIV drug resistance. New Engl. J. Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Coffin J.M. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 1979;42:1–26. doi: 10.1099/0022-1317-42-1-1. [DOI] [PubMed] [Google Scholar]
- Coffin J.M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- Coffin J.M. Molecular biology of HIV. In: Crandall K.A., editor. The Evolution of HIV. Johns Hopkins University Press; Baltimore, MD: 1999. pp. 3–40. [Google Scholar]
- Conlan A.J.K., Coward C., Grant A.J., Maskell D.J., Gog J.R. Campylobacter jejuni colonization and transmission in broiler chickens: a modelling perspective. J. Roy. Soc. Interface. 2007;4:819–829. doi: 10.1098/rsif.2007.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall K.A. Intraspecific phylogenetics – support for dental transmission of human-immunodeficiency-virus. J. Virol. 1995;69:2351–2356. doi: 10.1128/jvi.69.4.2351-2356.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall, K.A., Templeton, A.R., 1993. Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. pp. 959–969. [DOI] [PMC free article] [PubMed]
- Crandall K.A., Kelsey C.R., Imamichi H., Lane H.C., Salzman N.P. Parallel evolution of drug resistance in HIV: failure of nonsynonymous/synonymous substitution rate ratio to detect selection. Mol. Biol. Evol. 1999;16:372–382. doi: 10.1093/oxfordjournals.molbev.a026118. [DOI] [PubMed] [Google Scholar]
- Dahl V., Josefsson L., Palmer S. HIV reservoirs, latency, and reactivation: prospects for eradication. Antivir. Res. 2010;85:286–294. doi: 10.1016/j.antiviral.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Delobel P., Sandres-Saune K., Cazabat M., L’Faqihi F.E., Aquilina C., Obadia M., Pasquier C., Marchou B., Massip P., Izopet J. Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. AIDS. 2005;19:1739–1750. doi: 10.1097/01.aids.0000183125.93958.26. [DOI] [PubMed] [Google Scholar]
- Denton P., Garcia J. Novel humanized murine models for HIV research. Current HIV/AIDS Rep. 2009;6:13–19. doi: 10.1007/s11904-009-0003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz R., Sabino E., Mayer A., Mosley J., Busch M. Dual human immunodeficiency virus type 1 infection and recombination in a dually exposed transfusion recipient. The Transfusion Safety Study Group. J. Virol. 1995;69:3273–3281. doi: 10.1128/jvi.69.6.3273-3281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J.W., Charlesworth B., Charlesworth D., Crow J.F. Rates of spontaneous mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Nicholls G.K., Rodrigo A.G., Solomon W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics. 2002;161:1307–1320. doi: 10.1093/genetics/161.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Pybus O.G., Rambaut A., Forsberg R., Rodrigo A.G. Measurably evolving populations. Trends Ecol Evol. 2003;18(personal edition):481–488. [Google Scholar]
- Drummond A.J., Rambaut A., Shapiro B., Pybus O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- Drummond, A.J., Ho, S.Y.W., Phillips, M.J., Rambaut, A., 2006. Relaxed phylogenetics and dating with confidence. PloS Biol 4, 699–710. [DOI] [PMC free article] [PubMed]
- Edwards C.T.T., Holmes E.C., Wilson D.J., Viscidi R.P., Abrams E.J., Phillips R.E., Drummond A.J. Population genetic estimation of the loss of genetic diversity during horizontal transmission of HIV-1. BMC Evol. Biol. 2006;6:28. doi: 10.1186/1471-2148-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English S., Katzourakis A., Bonsall D., Flanagan P., Duda A., Fidler S., Weber J., McClure M., Investigators S.T., Phillips R., Frater J. Phylogenetic analysis consistent with a clinical history of sexual transmission of HIV-1 from a single donor reveals transmission of highly distinct variants. Retrovirology. 2011;8:54. doi: 10.1186/1742-4690-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esbjörnsson J., Mild M., Månsson F., Norrgren H., Medstrand P. HIV-1 molecular epidemiology in guinea-bissau, West Africa: origin, demography and migrations. PLoS ONE. 2011;6:e17025. doi: 10.1371/journal.pone.0017025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essex M. Simian immunodeficiency virus in people. New Engl. J. Med. 1994;330:209–210. doi: 10.1056/NEJM199401203300313. [DOI] [PubMed] [Google Scholar]
- Finzi D., Hermankova M., Pierson T., Carruth L.M., Buck C., Chaisson R.E., Quinn T.C., Chadwick K., Margolick J., Brookmeyer R., Gallant J., Markowitz M., Ho D.D., Richman D.D., Siliciano R.F. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- Fischer W., Ganusov V.V., Giorgi E.E., Hraber P.T., Keele B.F., Leitner T., Han C.S., Gleasner C.D., Green L., Lo C.-C., Nag A., Wallstrom T.C., Wang S., McMichael A.J., Haynes B.F., Hahn B.H., Perelson A.S., Borrow P., Shaw G.M., Bhattacharya T., Korber B.T. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS ONE. 2010;5:e12303. doi: 10.1371/journal.pone.0012303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox C.W., Roff D.A., Fairbairn D.J. Oxford University Press; USA: 2001. Evolutionary Ecology: Concepts and Case Studies. [Google Scholar]
- Frahm N., Nickle D.C., Linde C.H., Cohen D.E., Zuñiga R., Lucchetti A., Roach T., Walker B.D., Allen T.M., Korber B.T., Mullins J.I., Brander C. Increased detection of HIV-specific T cell responses by combination of central sequences with comparable immunogenicity. AIDS. 2008;22:447–456. doi: 10.1097/QAD.0b013e3282f42412. [DOI] [PubMed] [Google Scholar]
- Gallo R., Sarin P., Gelmann E., Robert-Guroff M., Richardson E., Kalyanaraman V., Mann D., Sidhu G., Stahl R., Zolla-Pazner S., Leibowitch J., Popovic M. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS) Science. 1983;220:865–867. doi: 10.1126/science.6601823. [DOI] [PubMed] [Google Scholar]
- Gao F., Yue L., White A.T., Pappas P.G., Barchue J., Hanson A.P., Greene B.M., Sharp P.M., Shaw G.M., Hahn B.H. Human infection by genetically diverse SIVSM-related HIV-2 in West Africa. Nature. 1992;358:495–499. doi: 10.1038/358495a0. [DOI] [PubMed] [Google Scholar]
- Gao F., Yue L., Robertson D.L., Hill S.C., Hui H., Biggar R.J., Neequaye A.E., Whelan T.M., Ho D.D., Shaw G.M. Genetic diversity of human immunodeficiency virus type 2: evidence for distinct sequence subtypes with differences in virus biology. J. Virol. 1994;68:7433–7447. doi: 10.1128/jvi.68.11.7433-7447.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Bailes E., Robertson D.L., Chen Y., Rodenburg C.M., Michael S.F., Cummins L.B., Arthur L.O., Peeters M., Shaw G.M., Sharp P.M., Hahn B.H. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397:436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- Gilbert M.T.P., Rambaut A., Wlasiuk G., Spira T.J., Pitchenik A.E., Worobey M. The emergence of HIV/AIDS in the Americas and beyond. Proc. Natl. Acad. Sci. USA. 2007;104:18566–18570. doi: 10.1073/pnas.0705329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovkina T.V., Jaffe A.B., Ross S.R. Coexpression of exogenous and endogenous mouse mammary tumor virus RNA in vivo results in viral recombination and broadens the virus host range. J. Virol. 1994;68:5019–5026. doi: 10.1128/jvi.68.8.5019-5026.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich D.W., Duesberg P.H. Retroviral recombination during reverse transcription. Proc. Natl. Acad. Sci. 1990;87:2052–2056. doi: 10.1073/pnas.87.6.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R.R., Tatem A.J., Lamers S., Hou W., Laeyendecker O., Serwadda D., Sewankambo N., Gray R.H., Wawer M., Quinn T.C., Goodenow M.M., Salemi M. Spatial phylodynamics of HIV-1 epidemic emergence in east Africa. AIDS. 2009;23:F9–F17. doi: 10.1097/QAD.0b013e32832faf61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R.R., Tatem A.J., Johnson J.A., Alekseyenko A.V., Pybus O.G., Suchard M.A., Salemi M. Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a Bayesian framework. Mol. Biol. Evol. 2011;28:1593–1603. doi: 10.1093/molbev/msq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell B., Pybus O., Gog J., Wood J., Daly J. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- Hahn, B.H., Shaw, G.M., De, K.M., Cock, Sharp, P.M., 2000. AIDS as a zoonosis: scientific and public health implications. Science 287, 607–614. [DOI] [PubMed]
- Hermankova M., Ray S.C., Ruff C., Powell-Davis M., Ingersoll R., D’Aquila R.T., Quinn T.C., Siliciano R.F., Persaud D. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/mL receiving combination therapy. JAMA. 2001;286:196–207. doi: 10.1001/jama.286.2.196. [DOI] [PubMed] [Google Scholar]
- Hillis D.M., Huelsenbeck J.P. Support for dental HIV transmission. Nature. 1994;369:24–25. doi: 10.1038/369024a0. [DOI] [PubMed] [Google Scholar]
- Hirsch V., Riedel N., Kornfeld H., Kanki P.J., Essex M., Mullins J.I. Cross-reactivity to human T-lymphotropic virus type III/lymphadenopathy-associated virus and molecular cloning of simian T-cell lymphotropic virus type III from African green monkeys. Proc. Natl. Acad. Sci. USA. 1986;83:9754–9758. doi: 10.1073/pnas.83.24.9754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.C. The phylogeography of human viruses. Mol. Ecol. 2004;13:745. doi: 10.1046/j.1365-294x.2003.02051.x. [DOI] [PubMed] [Google Scholar]
- Holmes E.C. Viral evolution in the genomic age. PLoS Biol. 2007;5:e278. doi: 10.1371/journal.pbio.0050278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.C. Oxford University Press; New York, NY, USA: 2009. The Evolution and Emergence of RNA Viruses. [Google Scholar]
- Holmes E.C., Drummond A.J. The evolutionary genetics of viral emergence. Curr. Top. Microbiol. 2007;315:51–66. doi: 10.1007/978-3-540-70962-6_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.C., Grenfell B.T. Discovering the phylodynamics of RNA viruses. PloS Comput. Biol. 2009;5 doi: 10.1371/journal.pcbi.1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.C., Zhang L.Q., Simmonds P., Rogers A.S., Brown A.J.L. Molecular investigation of human-immunodeficiency-virus (HIV) infection in a patient of an HIV-infected surgeon. J. Infect. Dis. 1993;167:1411–1414. doi: 10.1093/infdis/167.6.1411. [DOI] [PubMed] [Google Scholar]
- Holmes E.C., Zhang L.Q., Robertson P., Cleland A., Harvey E., Simmonds P., Brown A.J.L. The molecular epidemiology of human immunodeficiency virus type 1 in Edinburgh. J. Infect. Dis. 1995;171:45–53. doi: 10.1093/infdis/171.1.45. [DOI] [PubMed] [Google Scholar]
- Hooper E. Penguin; London: 2003. The River: a Journey Back to the Source of HIV and AIDS. [Google Scholar]
- Hudson R.R. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 1983;23:183–201. doi: 10.1016/0040-5809(83)90013-8. [DOI] [PubMed] [Google Scholar]
- Hué S., Clewley J.P., Cane P.A., Pillay D. Investigation of HIV-1 transmission events by phylogenetic methods: requirement for scientific rigour. AIDS. 2005;19:449–450. doi: 10.1097/01.aids.0000161778.15568.a1. [DOI] [PubMed] [Google Scholar]
- Hué S., Pillay D., Clewley J.P., Pybus O.G. Genetic analysis reveals the complex structure of HIV-1 transmission within defined risk groups. Proc. Natl. Acad. Sci. USA. 2005;102:4425–4429. doi: 10.1073/pnas.0407534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J.P., Ronquist F., Nielsen R., Bollback J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Huet T., Cheynier R., Meyerhans A., Roelants G., Wain-Hobson S. Genetic organization of a chimpanzee lentivirus related to HIV-1. Nature. 1990;345:356–359. doi: 10.1038/345356a0. [DOI] [PubMed] [Google Scholar]
- Hughes, G.J., Fearnhill, E., Dunn, D., Lycett, S.J., Rambaut, A., Brown, A.J.L., Collaboration, U.H.D.R., 2009. Molecular phylodynamics of the heterosexual HIV epidemic in the United Kingdom. PloS Pathog. 5. [DOI] [PMC free article] [PubMed]
- Imamichi H., Crandall K.A., Natarajan V., Jiang M.K., Dewar R.L., Berg S., Gaddam A., Bosche M., Metcalf J.A., Davey R.T. Human immunodeficiency virus type 1 quasi species that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasi species present before initiation of therapy. J. Infect. Dis. 2001;183:36–50. doi: 10.1086/317641. [DOI] [PubMed] [Google Scholar]
- Jetzt A.E., Yu H., Klarmann G.J., Ron Y., Preston B.D., Dougherty J.P. High rate of recombination throughout the human immunodeficiency virus Type 1 genome. J. Virol. 2000;74:1234–1240. doi: 10.1128/jvi.74.3.1234-1240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobes D.V., Daoust M., Nguyen V.T., Padua A., Sinangil F., Pérez-Losada M., Crandall K.A., Oliphant T., Posada D., Rambaut A., Fuchs J., Berman P.W. Longitudinal population analysis of dual infection with recombination in two strains of HIV type 1 subtype B in an individual from a phase 3 HIV vaccine efficacy trial. AIDS Res. Hum. Retrov. 2006;22:968–978. doi: 10.1089/aid.2006.22.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki P.J., Kurth R., Beckerg W., Dreesman G., McLane F., Essex M. Antibodies to simian T-lymphotropic retrovirus type Ill in African green monkeys and recognition of STLV-Ill viral proteins by AIDS and related sera. Lancet. 1985;1:1330–1332. doi: 10.1016/s0140-6736(85)92818-1. [DOI] [PubMed] [Google Scholar]
- Kanki P.J., McLane M.F., King N.W., Letvin N.L., Hunt R.D., Sehgal P., Daniel M.D., Desrosiers R.C., Essex M. Serologic identification and characterization of a macaque T-lymphotropic retrovirus closely related to HTLV-III. Science. 1985;228:1199–1201. doi: 10.1126/science.3873705. [DOI] [PubMed] [Google Scholar]
- Kanki P.J., Hopper J.R., Essex M. The Origins of HIV-1 and HTLV-4/HIV-2. Ann. NY Acad. Sci. 1987;511:370–375. doi: 10.1111/j.1749-6632.1987.tb36265.x. [DOI] [PubMed] [Google Scholar]
- Keele B.F., Van Heuverswyn F., Li Y., Bailes E., Takehisa J., Santiago M.L., Bibollet-Ruche F., Chen Y., Wain L.V., Liegeois F., Loul S., Ngole E.M., Bienvenue Y., Delaporte E., Brookfield J.F.Y., Sharp P.M., Shaw G.M., Peeters M., Hahn B.H. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele B.F., Giorgi E.E., Salazar-González J.F., Decker J.M., Pham K.T., Salazar M.G., Sun C., Grayson T., Wang S., Li H., Wei X., Jiang C., Kirchherr J.L., Gao F., Anderson J.A., Ping L.-H., Swanstrom R., Tomaras G.D., Blattner W.A., Goepfert P.A., Kilby J.M., Saag M.S., Delwart E.L., Busch M.P., Cohen M.S., Montefiori D.C., Haynes B.F., Gaschen B., Athreya G.S., Lee H.Y., Wood N., Seoighe C., Perelson A.S., Bhattacharya T., Korber B.T., Hahn B.H., Shaw G.M. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele B.F., Tazi L., Gartner S., Liu Y.L., Burgon T.B., Estes J.D., Thacker T.C., Crandall K.A., McArthur J.C., Burton G.F. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J. Virol. 2008;82:5548–5561. doi: 10.1128/JVI.00124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keele B.F., Jones J.H., Terio K.A., Estes J.D., Rudicell R.S., Wilson M.L., Li Y., Learn G.H., Beasley T.M., Schumacher-Stankey J., Wroblewski E., Mosser A., Raphael J., Kamenya S., Lonsdorf E.V., Travis D.A., Mlengeya T., Kinsel M.J., Else J.G., Silvestri G., Goodall J., Sharp P.M., Shaw G.M., Pusey A.E., Hahn B.H. Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature. 2009;460:515–519. doi: 10.1038/nature08200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr P.J., Kitchen A., Holmes E.C. Origin and phylodynamics of rabbit hemorrhagic disease virus. J. Virol. 2009;83:12129–12138. doi: 10.1128/JVI.01523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer T.L., Finucane M.M., Nettles R.E., Quinn T.C., Broman K.W., Ray S.C., Persaud D., Siliciano R.F. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 2004;189:1452–1465. doi: 10.1086/382488. [DOI] [PubMed] [Google Scholar]
- Kingman J.F.C. On the genealogy of large populations. J. Appl. Prob. 1982;19:27–43. [Google Scholar]
- Kingman J.F.C. Origins of the coalescent: 1974–1982. Genetics. 2000;156:1461–1463. doi: 10.1093/genetics/156.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Muldoon M., Theiler J., Gao F., Gupta R., Lapedes A., Hahn B.H., Wolinsky S., Bhattacharya T. Timing the ancestor of the HIV-1 pandemic strains. Science. 2000;288:1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond S.L., Posada D., Stawiski E., Chappey C., Poon A.F.Y., Hughes G., Fearnhill E., Gravenor M.B., Leigh Brown A.J., Frost S.D.W. An evolutionary model-based algorithm for accurate phylogenetic breakpoint mapping and subtype prediction in HIV-1. PloS Comput. Biol. 2009;5:e1000581. doi: 10.1371/journal.pcbi.1000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner M.K. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006;22:768–770. doi: 10.1093/bioinformatics/btk051. [DOI] [PubMed] [Google Scholar]
- Kuhner M.K. Coalescent genealogy samplers: windows into population history. Trends Ecol. Evol. 2009;24:86–93. doi: 10.1016/j.tree.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken C., Foley B., Leitner T., Apetrei T., Hahn B., Mizrachi I., Mullins J., Rambaut A., Wolinsky S., Korber B.E. Los Alamos National Laboratory, Theoretical Biology and Biophysics; Los Alamos, New Mexico: 2010. HIV Sequence Compendium. 2010. [Google Scholar]
- Lamers S.L., Salemi M., Galligan D.C., de Oliveira T., Fogel G.B., Granier S.C., Zhao L., Brown J.N., Morris A., Masliah E., McGrath M.S. Extensive HIV-1 intra-host recombination is common in tissues with abnormal histopathology. PLoS ONE. 2009;4:e5065. doi: 10.1371/journal.pone.0005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larder B.A., Kemp S.D. Multiple mutations in Hiv-1 reverse-transcriptase confer high-level resistance to zidovudine (Azt) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- Lataillade M., Chiarella J., Yang R., Schnittman S., Wirtz V., Uy J., Seekins D., Krystal M., Mancini M., McGrath D., Simen B., Egholm M., Kozal M. Prevalence and clinical significance of HIV drug resistance mutations by ultra-deep sequencing in antiretroviral-naïve subjects in the CASTLE study. PLoS ONE. 2010;5:e10952. doi: 10.1371/journal.pone.0010952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeper S.C., Reddi A. United States global health policy: HIV/AIDS, maternal and child health, and The President’s Emergency Plan for AIDS Relief (PEPFAR) AIDS. 2010;24:2145–2149. doi: 10.1097/QAD.0b013e32833cbb41. [DOI] [PubMed] [Google Scholar]
- Leitner T., Escanilla D., Franzén C., Uhlén M., Albert J. Accurate reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proc. Natl. Acad. Sci. USA. 1996;93:10864–10869. doi: 10.1073/pnas.93.20.10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Pybus O.G., Wang B., Saksena N.K., Salemi M., Vandamme A.-M. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl. Acad. Sci. USA. 2003;100:6588–6592. doi: 10.1073/pnas.0936469100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Derdelinckx I., Rambaut A., Van Laethem K., Dumont S., Vermeulen S., Van Wijngaerden E., Vandamme A.-M. Molecular footprint of drug-selective pressure in a human immunodeficiency virus transmission chain. J. Virol. 2005;79:11981–11989. doi: 10.1128/JVI.79.18.11981-11989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Van Dooren S., Van Laethem K., Schrooten Y., Derdelinckx I., Goubau P., Brun-Vézinet F., Vaira D., Vandamme A.-M. Molecular testing of multiple HIV-1 transmissions in a criminal case. AIDS. 2005;19:1649–1658. doi: 10.1097/01.aids.0000187904.02261.1a. [DOI] [PubMed] [Google Scholar]
- Lemey P., Pond S.L.K., Drummond A.J., Pybus O.G., Shapiro B., Barroso H., Taveira N., Rambaut A. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PloS Comput. Biol. 2007;3:282–292. doi: 10.1371/journal.pcbi.0030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Drummond A.J., Suchard M.A. Bayesian phylogeography finds its roots. PloS Comput. Biol. 2009;5 doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Welch J.J., Suchard M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010;27:1877–1885. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis F., Hughes G.J., Rambaut A., Pozniak A., Brown A.J.L. Episodic sexual transmission of HIV revealed by molecular phylodynamics. PloS Med. 2008;5:392–402. doi: 10.1371/journal.pmed.0050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis-Rogers N., Crandall K.A., Posada D. Evolutionary analyses of genetic recombination. Dynam. Genet. 2004:408. [Google Scholar]
- Liao H.A., Tee K.K., Hase S., Uenishi R., Li X.J., Kusagawa S., Pham H.T., Nguyen T.H., Pybus O.G., Takebe Y. Phylodynamic analysis of the dissemination of HIV-1 CRF01_AE in Vietnam. Virology. 2009;391:51–56. doi: 10.1016/j.virol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Lole K.S., Bollinger R.C., Paranjape R.S., Gadkari D., Kulkarni S.S., Novak N.G., Ingersoll R., Sheppard H.W., Ray S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado E.S., Afonso A.O., Nissley D.V., Lemey P., Cunha S.M., Oliveira R.H., Soares M.A. Emergency of primary NNRTI resistance mutations without antiretroviral selective pressure in a HAART-treated child. PLoS ONE. 2009;4:e4806. doi: 10.1371/journal.pone.0004806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansky L.M., Temin H.M. Lower in-vivo mutation-rate of human-immunodeficiency-virus type-1 than that predicted from the fidelity of purified reverse-transcriptase. J. Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden M.D., Zack J.A. Eradication of HIV: current challenges and new directions. J. Antimicrob. Chemother. 2009;63:7–10. doi: 10.1093/jac/dkn455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.P., Williamson C., Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–262. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- McBurney S.P., Ross T.M. Viral sequence diversity: challenges for AIDS vaccine designs. Exp. Rev. Vacc. 2008;7:1405–1417. doi: 10.1586/14760584.7.9.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald J.H., Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mens H., Pedersen A.G., Jorgensen L.B., Hue S., Yang Y.Z., Gerstoft J., Katzenstein T.L. Investigating signs of recent evolution in the pool of proviral HIV type 1 DNA during years of successful HAART. AIDS Res. Hum. Retrov. 2007;23:107–115. doi: 10.1089/aid.2006.0089. [DOI] [PubMed] [Google Scholar]
- Metzker M.L., Mindell D.P., Liu X.M., Ptak R.G., Gibbs R.A., Hillis D.M. Molecular evidence of HIV-1 transmission in a criminal case. Proc. Natl. Acad. Sci. USA. 2002;99:14292–14297. doi: 10.1073/pnas.222522599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya H., Weinhold K.J., Furman P.A., St Clair M.H., Lehrman S.N., Gallo R.C., Bolognesi D., Barry D.W., Broder S. 3′-Azido-3′-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA. 1985;82:7096–7100. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondini A., Bronzoni R.V.D., Nunes S.H.P., Chiaravalloti-Neto F., Massad E., Alonso W.J., Zanotto P.M.D., Nogueira M.L. Spatio-temporal tracking and phylodynamics of a DENV-3 outbreak in a city from Brazil. Cladistics. 2010;26:218. [Google Scholar]
- Moya A., Holmes E.C., González-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2004;2:279. doi: 10.1038/nrmicro863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil S.J.D., Zang T., Bieniasz P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- Nelson M.I., Lemey P., Tan Y., Vincent A., Lam T.T.-Y., Detmer S., Viboud C.C., Suchard M.A., Rambaut A., Holmes E.C., Gramer M. Spatial dynamics of human-origin H1 influenza A virus in North American swine. PloS Pathog. 2011;7:e1002077. doi: 10.1371/journal.ppat.1002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngandu N., Scheffler K., Moore P., Woodman Z., Martin D., Seoighe C. Extensive purifying selection acting on synonymous sites in HIV-1 Group M sequences. Virol. J. 2008;5:160. doi: 10.1186/1743-422X-5-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora T., Charpentier C., Tenaillon O., Hoede C., Clavel F., Hance A.J. Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 2007;81:7620–7628. doi: 10.1128/JVI.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M. John Wiley & Sons, Ltd; 2004. Coalescent Theory. Handbook of Statistical Genetics. [Google Scholar]
- Nottet H.S.L.M., van Dijk S.J., Fanoy E.B., Goedegebuure I.W., de Jong D., Vrisekoop N., van Baarle D., Boltz V., Palmer S., Borleffs J.C.C., Boucher C.A.B. HIV-1 can persist in aged memory CD4(+) T lymphocytes with minimal signs of evolution after 8.3 years of effective highly active antiretroviral therapy. JAIDS. 2009;50:345–353. doi: 10.1097/QAI.0b013e318197eb04. [DOI] [PubMed] [Google Scholar]
- Onafuwa-Nuga A., Telesnitsky A. The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol. Mol. Biol. Rev. 2009;73:451–480. doi: 10.1128/MMBR.00012-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, C.-Y., Ciesielski, C.A., Myers, G., Bandea, C.I., Luo, C.-C., Korber, B.T.M., Mullins, J.I., Schochetman, G., Berkelman, R.L., Economou, A.N., Witte, J.J., Furman, L.J., Satten, G.A., Maclnnes, K.A., Curran, J.W., Jaffe, H.W., Laboratory Investigation Group, Epidemiologic Investigation Group, 1992. Molecular epidemiology of HIV transmission in a dental practice. Science 256, 1165–1171. [DOI] [PubMed]
- Parera M., Ibanez A., Clotet B., Martinez M.A. Lack of evidence for protease evolution in HIV-1-infected patients after 2 years of successful highly active antiretroviral therapy. J. Infect. Dis. 2004;189:1444–1451. doi: 10.1086/382485. [DOI] [PubMed] [Google Scholar]
- Peeters M., Chaix M.L., Delaporte E. Genetic diversity and phylogeographic distribution of SIV: how to understand the origin of HIV. M S-Med. Sci. 2008;24:621–628. doi: 10.1051/medsci/20082467621. [DOI] [PubMed] [Google Scholar]
- Perelson A.S., Essunger P., Cao Y., Vesanen M., Hurley A., Saksela K., Markowitz M., Ho D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- Pérez-Losada M., Viscidi R.P., Demma J.C., Zenilman J., Crandall K.A. Population genetics of Neisseria gonorrhoeae in a high-prevalence community using a hypervariable outer membrane porB and 13 slowly evolving housekeeping genes. Mol. Biol. Evol. 2005;22:1887–1902. doi: 10.1093/molbev/msi184. [DOI] [PubMed] [Google Scholar]
- Pérez-Losada M., Crandall K.A., Bash M.C., Dan M., Zenilman J., Viscidi R.P. Distinguishing importation from diversification of quinolone-resistant Neisseria gonorrhoeae by molecular evolutionary analysis. BMC Evol. Biol. 2007;7:84. doi: 10.1186/1471-2148-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M., Crandall K.A., Zenilman J., Viscidi R.P. Temporal trends in gonococcal population genetics in a high prevalence urban community. Infect. Genet. Evol. 2007;7:271–278. doi: 10.1016/j.meegid.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M., Porter M.L., Tazi L., Crandall K.A. New methods for inferring population dynamics from microbial sequences. Infect. Genet. Evol. 2007;7:24–43. doi: 10.1016/j.meegid.2006.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M., Jobes D.V., Sinangil F., Crandall K.A., Posada D., Berman P.W. Phylodynamics of HIV-1 from a phase-III AIDS vaccine trial in North America. Mol. Biol. Evol. 2010;27:417–425. doi: 10.1093/molbev/msp254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M., Jobes D.V., Sinangil F., Crandall K.A., Arenas M., Posada D., Berman P.W. Phylodynamics of HIV-1 from a phase III AIDS vaccine trial in Bangkok, Thailand. PLoS ONE. 2011;6:e16902. doi: 10.1371/journal.pone.0016902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud D., Ray S.C., Kajdas J., Ahonkhai A., Siberry G.K., Ferguson K., Ziemniak C., Quinn T.C., Casazza J.P., Zeichner S., Gange S.J., Watson D.C. Slow human immunodeficiency virus type 1 evolution in viral reservoirs in infants treated with effective antiretroviral therapy. AIDS Res. Hum. Retrov. 2007;23:381–390. doi: 10.1089/aid.2006.0175. [DOI] [PubMed] [Google Scholar]
- Pillay D., Rambaut A., Geretti A.M., Brown A.J.L. HIV phylogenetics. BMJ. 2007;335:460–461. doi: 10.1136/bmj.39315.398843.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantier J.-C., Leoz M., Dickerson J.E., De Oliveira F., Cordonnier F., Lemee V., Damond F., Robertson D.L., Simon F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009;15:871–872. doi: 10.1038/nm.2016. [DOI] [PubMed] [Google Scholar]
- Plotkin S.A. Untruths and consequences: the false hypothesis linking CHAT type 1 polio vaccination to the origin of human immunodeficiency virus. Philos. Trans. Roy. Soc. Lond. B: Biol. Sci. 2001;356:815–823. doi: 10.1098/rstb.2001.0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond S.K., Muse S.V. Site-to-site variation of synonymous substitution rates. Mol. Biol. Evol. 2005;22:2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Pond S.L.K., Frost S.D.W., Grossman Z., Gravenor M.B., Richman D.D., Brown A.J.L. Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PloS Comput. Biol. 2006;2:530–538. doi: 10.1371/journal.pcbi.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond S.L.K., Poon A.F.Y., Zarate S., Smith D.M., Little S.J., Pillai S.K., Ellis R.J., Wong J.K., Brown A.J.L., Richman D.D., Frost S.D.W. Estimating selection pressures on HIV-1 using phylogenetic likelihood models. Stat. Med. 2008;27:4779–4789. doi: 10.1002/sim.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon A.F.Y., Pond S.L.K., Bennett P., Richman D.D., Brown A.J.L., Frost S.D.W. Adaptation to human populations is revealed by within-host polymorphisms in HIV-1 and hepatitis C virus. PloS Pathog. 2007;3 doi: 10.1371/journal.ppat.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon A.F.Y., Swenson L.C., Dong W.W.Y., Deng W.J., Pond S.L.K., Brumme Z.L., Mullins J.I., Richman D.D., Harrigan P.R., Frost S.D.W. Phylogenetic analysis of population-based and deep sequencing data to identify coevolving sites in the nef gene of HIV-1. Mol. Biol. Evol. 2010;27:819–832. doi: 10.1093/molbev/msp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic M., Sarngadharan M., Read E., Gallo R. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science. 1984;224:497–500. doi: 10.1126/science.6200935. [DOI] [PubMed] [Google Scholar]
- Posada D. Unveiling the molecular clock in the presence of recombination. Mol. Biol. Evol. 2001;18:1976–1978. doi: 10.1093/oxfordjournals.molbev.a003738. [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K.A. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. USA. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D., Crandall K.A. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 2002;54:396–402. doi: 10.1007/s00239-001-0034-9. [DOI] [PubMed] [Google Scholar]
- Posada D., Crandall K.A., Holmes E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2002;36:75–97. doi: 10.1146/annurev.genet.36.040202.111115. [DOI] [PubMed] [Google Scholar]
- Price M.A., Wallis C.L., Lakhi S., Karita E., Kamali A., Anzala O., Sanders E.J., Bekker L.G., Twesigye R., Hunter E., Kaleebu P., Kayitenkore K., Allen S., Ruzagira E., Mwangome M., Mutua G., Amornkul P.N., Stevens G., Pond S.L.K., Schaefer M., Papathanasopoulos M.A., Stevens W., Gilmour J., Study I.E.I.C. Transmitted HIV Type 1 drug resistance among individuals with recent HIV infection in east and Southern Africa. AIDS Res. Hum. Retrov. 2011;27:5–12. doi: 10.1089/aid.2010.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghwani J., Rambaut A., Holmes E.C., Hang V.T., Hien T.T., Farrar J., Wills B., Lennon N.J., Birren B.W., Henn M.R., Simmons C.P. Endemic dengue associated with the co-circulation of multiple viral lineages and localized density-dependent transmission. PloS Pathog. 2011;7:e1002064. doi: 10.1371/journal.ppat.1002064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Posada D., Crandall K.A., Holmes E.C. The causes and consequences of HIV evolution. Nat. Rev. Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- Rambaut A., Pybus O.G., Nelson M.I., Viboud C., Taubenberger J.K., Holmes E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008;453:U612–U615. doi: 10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rerks-Ngarm S., Pitisuttithum P., Nitayaphan S., Kaewkungwal J., Chiu J., Paris R., Premsri N., Namwat C., de Souza M., Adams E., Benenson M., Gurunathan S., Tartaglia J., McNeil J.G., Francis D.P., Stablein D., Birx D.L., Chunsuttiwat S., Khamboonruang C., Thongcharoen P., Robb M.L., Michael N.L., Kunasol P., Kim J.H. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. New Engl. J. Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- Rhee S.-Y., Gonzales M.J., Kantor R., Betts B.J., Ravela J., Shafer R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucl. Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins K.E., Lemey P., Pybus O.G., Jaffe H.W., Youngpairoj A.S., Brown T.M., Salemi M., Vandamme A.-M., Kalish M.L. US human immunodeficiency virus type 1 epidemic: date of origin, population history, and characterization of early strains. J. Virol. 2003;77:6359–6366. doi: 10.1128/JVI.77.11.6359-6366.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D.L., Hahn B.H., Sharp P.M. Recombination in AIDS viruses. J. Mol. Evol. 1995;40:249–259. doi: 10.1007/BF00163230. [DOI] [PubMed] [Google Scholar]
- Robertson D.L., Sharp P.M., McCutchan F.E., Hahn B.H. Recombination in HIV-1. Nature. 1995;374:124–126. doi: 10.1038/374124b0. [DOI] [PubMed] [Google Scholar]
- Rodrigo A.G., Shpaer E.G., Delwart E.L., Iversen A.K.N., Gallo M.V., Brojatsch J.r., Hirsch M.S., Walker B.D., Mullins J.I. Coalescent estimates of HIV-1 generation time in vivo. Proc. Natl. Acad. Sci. 1999;96:2187–2191. doi: 10.1073/pnas.96.5.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma R., Noser J.A., Ohmine S., Ikeda Y. Rhesus monkey TRIM5[alpha] restricts HIV-1 production through rapid degradation of viral Gag polyproteins. Nat. Med. 2007;13:631–635. doi: 10.1038/nm1562. [DOI] [PubMed] [Google Scholar]
- Salazar-González J.F., Bailes E., Pham K.T., Salazar M.G., Guffey M.B., Keele B.F., Derdeyn C.A., Farmer P., Hunter E., Allen S., Manigart O., Mulenga J., Anderson J.A., Swanstrom R., Haynes B.F., Athreya G.S., Korber B.T.M., Sharp P.M., Shaw G.M., Hahn B.H. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 2008;82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M., Strimmer K., Hall W.W., Duffy M., Delaporte E., Mboup S., Peeters M., Vandamme A.-M. Dating the common ancestor of SIVcpz and HIV-1 group M and the origin of HIV-1 subtypes using a new method to uncover clock-like molecular evolution. FASEB J. 2001;15:276–278. doi: 10.1096/fj.00-0449fje. [DOI] [PubMed] [Google Scholar]
- Salemi M., Lamers S.L., Yu S., de Oliveira T., Fitch W.M., McGrath M.S. Phylodynamic analysis of human immunodeficiency virus type 1 in distinct brain compartments provides a model for the neuropathogenesis of AIDS. J. Virol. 2005;79:11343–11352. doi: 10.1128/JVI.79.17.11343-11352.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M., Burkhardt B.R., Gray R.R., Ghaffari G., Sleasman J.W., Goodenow M.M. Phylodynamics of HIV-1 in lymphoid and non-lymphoid tissues reveals a central role for the thymus in emergence of CXCR4-using quasispecies. PLoS ONE. 2007;2 doi: 10.1371/journal.pone.0000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M., de Oliveira T., Ciccozzi M., Rezza G., Goodenow M.M. High-resolution molecular epidemiology and evolutionary history of HIV-1 subtypes in Albania. PLoS ONE. 2008;3:e1390. doi: 10.1371/journal.pone.0001390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi M., Lamers S.L., Huysentruyt L.C., Galligan D., Gray R.R., Morris A., McGrath M.S. Distinct patterns of HIV-1 evolution within metastatic tissues in patients with non-Hodgkins lymphoma. PLoS ONE. 2009;4 doi: 10.1371/journal.pone.0008153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago M.L., Rodenburg C.M., Kamenya S., Bibollet-Ruche F., Gao F., Bailes E., Meleth S., Soong S.-J., Kilby J.M., Moldoveanu Z., Fahey B., Muller M.N., Ayouba A., Nerrienet E., McClure H.M., Heeney J.L., Pusey A.E., Collins D.A., Boesch C., Wrangham R.W., Goodall J., Sharp P.M., Shaw G.M., Hahn B.H. SIVcpz in wild chimpanzees. Science. 2002;295:465. doi: 10.1126/science.295.5554.465. [DOI] [PubMed] [Google Scholar]
- Santiago M.L., Bibollet-Ruche F., Bailes E., Kamenya S., Muller M.N., Lukasik M., Pusey A.E., Collins D.A., Wrangham R.W., Goodall J., Shaw G.M., Sharp P.M., Hahn B.H. Amplification of a complete simian immunodeficiency virus genome from fecal RNA of a wild chimpanzee. J. Virol. 2003;77:2233–2242. doi: 10.1128/JVI.77.3.2233-2242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago M.L., Range F., Keele B.F., Li Y., Bailes E., Bibollet-Ruche F., Fruteau C., Noe R., Peeters M., Brookfield J.F.Y., Shaw G.M., Sharp P.M., Hahn B.H. Simian immunodeficiency virus infection in free-ranging sooty mangabeys (Cercocebus atys atys) from the Tai Forest, Cote d’Ivoire: implications for the origin of epidemic human immunodeficiency virus type 2. J. Virol. 2005;79:12515–12527. doi: 10.1128/JVI.79.19.12515-12527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarngadharan M., Popovic M., Bruch L., Schupbach J., Gallo R. Antibodies reactive with human T-lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science. 1984;224:506–508. doi: 10.1126/science.6324345. [DOI] [PubMed] [Google Scholar]
- Schlub T.E., Smyth R.P., Grimm A.J., Mak J., Davenport M.P. Accurately measuring recombination between closely related HIV-1 genomes. PloS Comput. Biol. 2010;6 doi: 10.1371/journal.pcbi.1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R., Margolick J.B., Gange S.J., Rodrigo A.G., Upchurch D., Farzadegan H., Gupta P., Rinaldo C.R., Learn G.H., He X., Huang X.L., Mullins J.I. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 1999;73:10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B., Ho S.Y.W., Drummond A.J., Suchard M.A., Pybus O.G., Rambaut A. A Bayesian phylogenetic method to estimate unknown sequence ages. Mol. Biol. Evol. 2011;28:879–887. doi: 10.1093/molbev/msq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P.M. In search of molecular darwinism. Nature. 1997;385:111–112. doi: 10.1038/385111a0. [DOI] [PubMed] [Google Scholar]
- Sharp P.M., Bailes E., Gao F., Beer B.E., Hirsch V.M., Hahn B.H. Origins and evolution of AIDS viruses: estimating the time-scale. Biochem. Soc. Trans. 2000;28:275–282. doi: 10.1042/bst0280275. [DOI] [PubMed] [Google Scholar]
- Shi B., Kitchen C., Weiser B., Mayers D., Foley B., Kemal K., Anastos K., Suchard M., Parker M., Brunner C., Burger H. Evolution and recombination of genes encoding HIV-1 drug resistance and tropism during antiretroviral therapy. Virology. 2010;404:5–20. doi: 10.1016/j.virol.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner D., Rodrigo A.G., Nickle D.C., Mullins J.I. Pervasive genomic recombination of HIV-1 in vivo. Genetics. 2004;167:1573–1583. doi: 10.1534/genetics.103.023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebenga J.J., Lemey P., Pond S.L.K., Rambaut A., Vennema H., Koopmans M. Phylodynamic reconstruction reveals norovirus GII. 4 epidemic expansions and their molecular determinants. PloS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano J.D., Kajdas J., Finzi D., Quinn T.C., Chadwick K., Margolick J.B., Kovacs C., Gange S.J., Siliciano R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4(+) T cells. Nature Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Skar H., Axelsson M., Berggren I., Thalme A., Gyllensten K., Liitsola K., Brummer-Korvenkontio H., Kivela P., Spangberg E., Leitner T., Albert J. Dynamics of two separate but linked HIV-1 CRF01_AE outbreaks among injection drug users in Stockholm, Sweden, and Helsinki. Finland J. Virol. 2011;85:510–518. doi: 10.1128/JVI.01413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N.G.C., Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- Smith B.A., Gartner S., Liu Y., Perelson A.S., Stilianakis N.I., Keele B.F., Kerkering T.M., Ferreira-Gonzalez A., Szakal A.K., Tew J.G., Burton G.F. Persistence of infectious HIV on follicular dendritic cells. J. Immunol. 2001;166:690–696. doi: 10.4049/jimmunol.166.1.690. [DOI] [PubMed] [Google Scholar]
- Stack J.C., Welch J.D., Ferrari M.J., Shapiro B.U., Grenfell B.T. Protocols for sampling viral sequences to study epidemic dynamics. J. Roy. Soc. Interface. 2010;7:1119–1127. doi: 10.1098/rsif.2009.0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T. Sampling-through-time in birth–death trees. J. Theor. Biol. 2010;267:396–404. doi: 10.1016/j.jtbi.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Stadler T., Kouyos R., von Wyl V., Yerly S., Böoni J., Bürgisser P., Klimkait T., Joos B., Rieder P., Xie D., Günthard H.F., Drummond A.J., Bonhoeffer S., Study T.S.H.C. Estimating the basic reproductive number from viral sequence data. Mol. Biol. Evol. 2011:11–12. doi: 10.1093/molbev/msr217. [DOI] [PubMed] [Google Scholar]
- Stopak K., de Noronha C., Yonemoto W., Greene W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Sullivan J., Joyce P. Model selection in phylogenetics. Ann. Rev. Ecol. Evol. Syst. 2005;36:445–466. [Google Scholar]
- Susko E., Inagaki Y., Roger A.J. On inconsistency of the neighbor-joining, least squares, and minimum evolution estimation when substitution processes are incorrectly modeled. Mol. Biol. Evol. 2004;21:1629–1642. doi: 10.1093/molbev/msh159. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehisa J., Kraus M.H., Ayouba A., Bailes E., Van Heuverswyn F., Decker J.M., Li Y., Rudicell R.S., Learn G.H., Neel C., Ngole E.M., Shaw G.M., Peeters M., Sharp P.M., Hahn B.H. Origin and biology of simian immunodeficiency virus in wild-living western gorillas. J. Virol. 2009;83:1635–1648. doi: 10.1128/JVI.02311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. Line-of-descent and genealogical processes, and their applications in population genetics models. Theor. Popul. Biol. 1984;26:119–164. doi: 10.1016/0040-5809(84)90027-3. [DOI] [PubMed] [Google Scholar]
- Tazi L., Pérez-Losada M., Gu W.M., Yang Y., Xue L., Crandall K.A., Viscidi R.P. Population dynamics of Neisseria gonorrhoeae in Shanghai, China: a comparative study. BMC Infect. Dis. 2010;10 doi: 10.1186/1471-2334-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazi L., Imamichi H., Hirschfeld S., Metcalf J., Orsega S., Pérez-Losada M., Posada D., Lane H.C., Crandall K. HIV-1 infected monozygotic twins: a tale of two outcomes. BMC Evol. Biol. 2011;11:62. doi: 10.1186/1471-2148-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee K.K., Pybus O.G., Li X.J., Han X.X., Shang H., Kamarulzaman A., Takebe Y. Temporal and spatial dynamics of human immunodeficiency virus type 1 circulating recombinant forms 08_BC and 07_BC in Asia. J. Virol. 2008;82:9206–9215. doi: 10.1128/JVI.00399-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temin H.M. Sex and recombination in retroviruses. Trends Genet. 1991;7:71–74. doi: 10.1016/0168-9525(91)90272-R. [DOI] [PubMed] [Google Scholar]
- Templeton A.R., Reichert R.A., Weisstein A.E., Yu X.F., Markham R.B. Selection in context: patterns of natural selection in the glycoprotein 120 region of human immunodeficiency virus 1 within infected individuals. Genetics. 2004;167:1547–1561. doi: 10.1534/genetics.103.023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M. Bridging the gap between molecular epidemiologists and evolutionists. Trends Microbiol. 2005;13:575–580. doi: 10.1016/j.tim.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Tissot C., Mechti N. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J. Biol. Chem. 1995;270:14891–14898. doi: 10.1074/jbc.270.25.14891. [DOI] [PubMed] [Google Scholar]
- Tobin N.H., Learn G.H., Holte S.E., Wang Y., Melvin A.J., McKernan J.L., Pawluk D.M., Mohan K.M., Lewis P.F., Mullins J.I., Frenkel L.M. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J. Virol. 2005;79:9625–9634. doi: 10.1128/JVI.79.15.9625-9634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumas K.M., Poszgay J.M., Avidan N., Ksiazek S.J., Overmoyer B., Blank K.J., Prystowsky M.B. Loss of antigenic epitopes as the result of Env gene recombination in retrovirus-induced leukemia in immunocompetent mice. Virology. 1993;192:587–595. doi: 10.1006/viro.1993.1075. [DOI] [PubMed] [Google Scholar]
- UNAIDS, 2010. Global Report: UNAIDS Report on the Global AIDS Epidemic 2010. Joint United Nations Programme on HIV/AIDS (UNAIDS).
- Van Heuverswyn F., Peeters M. The origins of HIV and implications for the global epidemic. Current Infectious Disease Reports. 2007;9:338–346. doi: 10.1007/s11908-007-0052-x. [DOI] [PubMed] [Google Scholar]
- van Marle G., Gill M.J., Kolodka D., McManus L., Grant T., Church D.L. Compartmentalization of the gut viral reservoir in HIV-1 infected patients. Retrovirology. 2007;4 doi: 10.1186/1742-4690-4-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain-Hobson S. Down or out in blood and lymph? Nature. 1997;387:123–124. doi: 10.1038/387123a0. [DOI] [PubMed] [Google Scholar]
- Wakeley J. Recent trends in population genetics: more data! More math! Simple models? J. Hered. 2004;95:397–405. doi: 10.1093/jhered/esh062. [DOI] [PubMed] [Google Scholar]
- Wakeley J. Roberts & Company Publishers; Greenwood Village, CO: 2008. Coalescent Theory: An Introduction. [Google Scholar]
- Wang T.H., Donaldson Y.K., Brettle R.P., Bell J.E., Simmonds P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 2001;75:11686–11699. doi: 10.1128/JVI.75.23.11686-11699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X., Decker J.M., Wang S., Hui H., Kappes J.C., Wu X., Salazar-Gonzalez J.F., Salazar M.G., Kilby J.M., Saag M.S., Komarova N.L., Nowak M.A., Hahn B.H., Kwong P.D., Shaw G.M. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- Welch D., Nicholls G.K., Rodrigo A., Solomon W. Integrating genealogy and epidemiology: the ancestral infection and selection graph as a model for reconstructing host virus histories. Theor. Popul. Biol. 2005;68:65. doi: 10.1016/j.tpb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Wertheim J., Worobey M. A challenge to the ancient origin of SIVagm based on African green monkey mitochondrial genomes. PLoS Pathog. 2007;3:e95. doi: 10.1371/journal.ppat.0030095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J., Ignacio C., Torriani F., Havlir D., Fitch N., Richman D. In vivo compartmentalization of human immunodeficiency virus: evidence from the examination of pol sequences from autopsy tissues. J. Virol. 1997;71:2059–2071. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey, M., Santiago, M.L., Keele, B.F., Ndjango, J.-B.N., Joy, J.B., Labama, B.L., Dhed’a, B.D., Rambaut, A., Sharp, P.M., Shaw, G.M., Hahn, B., H., 2004. Origin of AIDS: contaminated polio vaccine theory refuted. Nature 428, 820–820. [DOI] [PubMed]
- Worobey M., Gemmel M., Teuwen D.E., Haselkorn T., Kunstman K., Bunce M., Muyembe J.-J., Kabongo J.-M.M., Kalengayi R.M., Van Marck E., Gilbert M.T.P., Wolinsky S.M. Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature. 2008;455:661–664. doi: 10.1038/nature07390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey M., Telfer P., Souquiere S., Hunter M., Coleman C.A., Metzger M.J., Reed P., Makuwa M., Hearn G., Honarvar S., Roques P., Apetrei C., Kazanji M., Marx P.A. Island biogeography reveals the deep history of SIV. Science. 2010;329:1487. doi: 10.1126/science.1193550. [DOI] [PubMed] [Google Scholar]
- Xin K.Q., Ma X.H., Crandall K.A., Bukawa H., Ishigatsubo Y., Kawamoto S., Okuda K. Dual infection with Hiv-1 Thai subtype-B and subtype-E. Lancet. 1995;346:1372–1373. doi: 10.1016/s0140-6736(95)92391-8. [DOI] [PubMed] [Google Scholar]
- Zehender G., De Maddalena C., Canuti M., Zappa A., Amendola A., Lai A., Galli M., Tanzi E. Rapid molecular evolution of human bocavirus revealed by Bayesian coalescent inference. Infect. Genet. Evol. 2010;10:215–220. doi: 10.1016/j.meegid.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Zehender G., Ebranati E., Lai A., Santoro M.M., Alteri C., Giuliani M., Palamara G., Perno C.F., Galli M., Lo Presti A., Ciccozzi M. Population dynamics of HIV-1 subtype B in a cohort of men-having-sex-with-men in Rome, Italy. JAIDS. 2010;55:156–160. doi: 10.1097/QAI.0b013e3181eb3002. [DOI] [PubMed] [Google Scholar]
- Zhu T., Wang N., Carr A., Wolinsky S., Ho D. Evidence for coinfection by multiple strains of human immunodeficiency virus type 1 subtype B in an acute seroconvertor. J. Virol. 1995;69:1324–1327. doi: 10.1128/jvi.69.2.1324-1327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]