

Rap1 acts together with the subtelomere-binding protein Tbf1 and inhibits localization of Mre11 complex to DNA ends. Depletion of Tbf1 protein stimulates checkpoint activation in cells containing short telomeres. The results suggest that Tbf1 and Rap1 collaborate to maintain genomic stability of short telomeres.
Abstract
Chromosome ends, known as telomeres, have to be distinguished from DNA double-strand breaks that activate DNA damage checkpoints. In budding yeast, the Mre11-Rad50-Xrs2 (MRX) complex associates with DNA ends and promotes checkpoint activation. Rap1 binds to double-stranded telomeric regions and recruits Rif1 and Rif2 to telomeres. Rap1 collaborates with Rif1 and Rif2 and inhibits MRX localization to DNA ends. This Rap1-Rif1-Rif2 function becomes attenuated at shortened telomeres. Here we show that Rap1 acts together with the subtelomere-binding protein Tbf1 and inhibits MRX localization to DNA ends. The placement of a subtelomeric sequence or TTAGGG repeats together with a short telomeric TG repeat sequence inhibits MRX accumulation at nearby DNA ends in a Tbf1-dependent manner. Moreover, tethering of both Tbf1 and Rap1 proteins decreases MRX and Tel1 accumulation at nearby DNA ends. This Tbf1- and Rap1-dependent pathway operates independently of Rif1 or Rif2 function. Depletion of Tbf1 protein stimulates checkpoint activation in cells containing short telomeres but not in cells containing normal-length telomeres. These data support a model in which Tbf1 and Rap1 collaborate to maintain genomic stability of short telomeres.
INTRODUCTION
Double-strand breaks (DSBs) are induced by exogenous DNA-damaging agents and carcinogens or by endogenous byproducts including reactive oxygen species. The repair of DSBs is crucial for maintaining genome stability (Pierce et al., 2001; O'Driscoll and Jeggo, 2006). Linear eukaryotic chromosomes have specialized structures at their ends called telomeres (Vega et al., 2003; Smogorzewska and de Lange, 2004; Louis and Vershinin, 2005). Telomeres prevent the recognition of chromosome ends as DSBs, but dysfunctional or short telomeres behave like DSB lesions and initiate the DNA damage response (Longhese, 2008; Palm and de Lange, 2008). The cellular responses to DSBs consist of activation of checkpoint signaling and DNA repair processes (Zhou and Elledge, 2000; Harrison and Haber, 2006).
Checkpoint signals are initiated through two large protein kinases—ataxia-telangiectasia mutated (ATM) and ATM-Rad3-related (ATR) (Zhou and Elledge, 2000; Abraham, 2001). ATM and ATR are highly conserved among eukaryotes. In budding yeast, ATM and ATR correspond to Tel1 and Mec1, respectively (Harrison and Haber, 2006). Several lines of evidence have established that the Mre11-Rad50-Nbs1 (Xrs2 in budding yeast) complex is the primary sensor that recruits ATR/Mec1 and ATM/Tel1 to DSBs (Nakada et al., 2003a, 2004; Falck et al., 2005; You et al., 2005). In budding yeast, the Mre11-Rad50-Xrs2 (MRX) complex recruits Tel1 to DNA ends through its interaction with Xrs2 (Nakada et al., 2003a) and modulates Tel1 catalytic activity at DNA ends (Fukunaga et al., 2011). MRX collaborates with Sae2 and initiates the generation of single-stranded DNA (ssDNA) at DSB ends (Krogh and Symington, 2004). The heterotrimeric replication protein A (RPA) binds ssDNA with high affinity and plays a pivotal role in homologous recombination repair of DSBs (Wold, 1997; Krogh and Symington, 2004). Mec1 accumulates at DNA ends through its interaction with the RPA–ssDNA complex (Zou and Elledge, 2003; Nakada et al., 2005). Mec1 and Tel1 subsequently phosphorylate the downstream kinase, Rad53, and contribute to full activation of the kinase (Schwartz et al., 2002; Sweeney et al., 2005). Activation of the Mec1/Tel1-Rad53 pathway leads to transient cell-cycle arrest and transcriptional activation of genes involved in DNA repair (Harrison and Haber, 2006).
Telomeres contain a double-stranded DNA region of tandem repeats (e.g., vertebrates and most higher eukaryotes, T2AG3; budding yeast, TG1-3) and a 3′ protruding ssDNA region of the G-rich strand (Louis and Vershinin, 2005; Palm and de Lange, 2008). Single-stranded tails on telomeres are bound by sequence-specific ssDNA binding proteins, such as Cdc13 in budding yeast (Lin and Zakian, 1996; Nugent et al., 1996). Cdc13 forms a complex with Stn1 and Ten1 and acts as a telomere cap to protect telomeres from degradation (Garvik et al., 1995; Nugent et al., 1996; Grandin et al., 2001; Pennock et al., 2001; Petreaca et al., 2007; Xu et al., 2009). Cdc13-mediated telomere capping inhibits RPA recruitment and subsequent Mec1 accumulation (Rouse and Jackson, 2002; Hirano and Sugimoto, 2007) but does not affect accumulation of Tel1 or the MRX complex at DNA ends (Hirano and Sugimoto, 2007). Several lines of evidence have established that Tel1 associates preferentially with short telomeres and promotes telomere addition (Bianchi and Shore, 2007; Chang et al., 2007; Hector et al., 2007; Sabourin et al., 2007; Viscardi et al., 2007). Short telomeric TG repeat acts as a seed for the addition of telomere sequence at DNA ends (Diede and Gottschling, 1999). MRX or Tel1 associates efficiently with DNA breaks adjacent to a short (81 base pair) telomeric TG repeat, whereas little or no Mre11 or Tel1 binding is detected at DNA ends next to long telomere tracts (Negrini et al., 2007; Hirano et al., 2009). Double-stranded telomeric DNA repeats are bound by the sequence-specific binding protein Rap1, which recruits Rif1 and Rif2 proteins via its C-terminal domain (Conrad et al., 1990; Lustig et al., 1990; Hardy et al., 1992; Wotton and Shore, 1997). Rif1 and Rif2 inhibit Tel1 association, but not MRX association, with DNA ends (Hirano et al., 2009). Rap1 inhibits MRX localization to DNA ends, but inhibition is not effective in the absence of Rif1 and Rif2 (Negrini et al., 2007; Hirano et al., 2009). If Tel1 is absent, however, Rap1 does not require Rif1 or Rif2 function to represses MRX localization (Hirano et al., 2009). Thus Rap1 collaborates with Rif1 and Rif2 and inhibits MRX accumulation at telomeres. Rif2 competes with Tel1 for binding to the C-terminus of Xrs2 (Hirano et al., 2009), but the functions of Rap1 or Rif1 remain to be determined.
Telomeres are connected to blocks of homologous sequences termed subtelomeres. Subtelomeric elements include the X core, the subtelomeric repeat element (STR), and the Y′ repeat (Louis and Vershinin, 2005). All telomeres contain the X core element, most contain STR, and two-thirds of chromosome ends contain the Y′ repeat (Louis, 1995). The STR sequence consists of multiple copies of the vertebrate telomeric motif, TTAGGG (Louis, 1995). In budding yeast, TTAGGG is recognized by Tbf1 protein, which contains a Myb domain at its C-terminus (Brigati et al., 1993; Koering et al., 2000; Lavoie et al., 2010). Y′ elements also contain the Tbf1-binding TTAGGG sequence (Louis, 1995). Overall, most telomere–subtelomere junctions contain multiple Tbf1-binding sites. Previous studies showed that targeting of Tbf1 to telomeres causes reduction in telomere length (Brevet et al., 2003; Berthiau et al., 2006). It is not precisely understood, however, how Tbf1 controls telomere length. Because Tbf1 binds to subtelomeric regions immediately adjacent to telomeres, it is possible that Tbf1 works together with Rap1 or its partners Rif1 and Rif2.
In this study, we investigated how the subtelomere-binding protein Tbf1 contributes to telomere homeostasis. We show that Tbf1 and Rap1 act together and inhibit MRX localization to DNA ends independently of Rif1 or Rif2. The presence of TTAGGG repeats reduces MRX and Tel1 accumulation at nearby DNA ends mimicking short telomeres. Moreover, tethering of both Tbf1 and Rap1 proteins decreases MRX and Tel1 accumulation at adjacent DNA ends. In both cases, inhibition of MRX accumulation occurs independently of Rif1 and Rif2. We also show that Tbf1 and Rap1 suppress checkpoint activation at short telomeres. Depletion of Tbf1 protein increases checkpoint activation in cells containing short telomeres but not in those containing normal-length telomeres. Moreover, Tbf1 is required to repress checkpoint activation at DNA ends where a subtelomeric sequence is placed together with a short TG repeat. These data support a model in which Tbf1 and Rap1 collaborate to protect DNA ends at short telomeres.
RESULTS
Effect of a subtelomeric sequence on localization of MRX and Tel1 to DNA ends
Rap1 collaborates with Rif1 and Rif2 to inhibit MRX localization to DNA ends (Hirano et al., 2009). Subtelomere-binding proteins Reb1 and Tbf1 have been shown to negatively control telomere elongation (Berthiau et al., 2006). Tbf1 binding, but not Reb1 binding, shortens telomere length in the presence of Tel1 (Berthiau et al., 2006). We thus tested a hypothesis that Tbf1 stimulates the Rap1 function that inhibits MRX localization to DNA ends.
Tbf1 has been identified as a protein that binds to the vertebrate telomeric TTAGGG repeat sequence (Brigati et al., 1993; Koering et al., 2000), and recent chip-on-chip studies showed that Tbf1 has a well-defined specificity for the TAGGG(C/T) motif (Lavoie et al., 2010). The sequence information for telomere–subtelomere junctions is available for 22 chromosome ends in the Saccharomyces Genomic Database. Of these, 21 sequences contain one to eight copies of the TAGGG(C/T) sequence with random spacing. The XIII-R subtelomere region contains six TAGGG(C/T) motifs within 40 base pairs of the telomeric TG sequence. We placed the 40–base pair XIII-R subtelomeric sequence (XIIIR) near the TG81-HO sequence or just the HO cleavage site generating the XIIIR-TG81-HO or XIIIR-HO cassette, respectively (Figure 1A). We then examined whether the subtelomere sequence XIIIR decreases MRX accumulation near TG81 or nontelomeric HO ends (Figure 1, B and C). TG81 ends have been shown to behave similarly to a short telomere; the 81–base pair TG81 sequence acts as a seed for the addition of telomere sequence (Diede and Gottschling, 1999) and allows MRX or Tel1 to accumulate at nearby DNA ends (Hirano et al., 2009). Cells transformed with the GAL-HO plasmid were grown in sucrose to prevent activation of the GAL1 promoter and arrested with nocodazole at G2/M. After arrest, cells were incubated with galactose to induce HO expression. Aliquots of cells were collected at the indicated times after HO expression and subjected to chromatin immunoprecipitation (ChIP) assay to monitor Mre11 accumulation at HO-induced DNA ends. The introduction of XIIIR decreased Mre11 binding at adjacent TG81 ends (XIIIR-TG81 ends; Figure 1B). Tel1 localizes to telomeric or nontelomeric DNA ends in an Xrs2-dependent manner (Nakada et al., 2003a; Sabourin et al., 2007). Similar inhibition was observed for Tel1 binding (Figure 1B). The introduction of XIIIR, however, did not affect Mre11 or Tel1 binding at nontelomeric DNA ends (XIIIR-HO ends; Figure 1C). These results raised the possibility that subtelomere-binding Tbf1 protein stimulates Rap1-mediated inhibition of MRX binding at DNA ends.
FIGURE 1:

Effect of a subtelomeric sequence on localization of MRX and Tel1 to DNA ends with telomeric sequences. (A) Schematic of the HO cleavage site, the 81–base pair telomeric TG repeat, the XIII-R subtelomeric region, the 22–base pair telomeric TG repeat, and the TTAGGG repeats at the ADH4 locus on chromosome VII-L. In HO cells, the ADH4 locus was replaced with a DNA fragment containing the KanMX gene and an HO cleavage site (inverted triangle). The 81–base pair TG81 sequence (three repetitive arrowheads) or the XIII-R subtelomeric (XIIIR) sequence (trapezoid) was placed centromere-proximal to the HO cleavage site, generating the TG81-HO or XIIIR-HO strain, respectively. The XIIIR or TTAGGG repeat (wedge) sequence was integrated centromere-proximal to the TG repeat in TG81-HO cells, creating the XIIIR-TG81-HO or TAGn-TG81-HO strain, respectively. The 22–base pair TG22 (one arrowhead) sequence was inserted centromere-proximal to the HO cleavage site to generate the XIIIR-TG22-HO-CA or TG22-HO-CA strain. The inverted 81–base pair TG81 (CA) sequence was placed centromere-distal to the HO cleavage site for the XIIIR-TG22-HO-CA, TG22-HO-CA, or XIIIR-HO-CA strain. Centromere is shown as a circle on the left (CEN). The HO primer pair was designed to amplify a region 1 kbp away from the HO site, and the cutting-site primer pair was used to determine the HO cutting efficiency. (B) Effect of the chromosome XIIIR subtelomeric sequence on the association of Mre11 and Tel1 with DNA ends. TG81-HO (KSC2849) or XIIIR-TG81-HO (KSC2850) cells expressing myc-tagged Mre11 (Mre11-myc) and HA-tagged Tel1 (Tel1-HA) were transformed with the GAL-HO plasmid. Transformed cells were grown in sucrose and synchronized at G2/M with nocodazole. After arrest, galactose was added to the culture to induce HO expression. Aliquots of cells were collected at the indicated times after HO expression and subjected to ChIP assay to monitor the association of Mre11 or Tel1 with DNA ends. Coprecipitated DNA was analyzed by real-time PCR using the HO primer pair shown in A and the SMC2 primer pair for the control SMC2 locus on chromosome VI. Relative enrichment was determined from three independent experiments, and bars represent averages with standard deviations indicated by lines above. HO cutting efficiency for each construct is shown in square brackets. (C) Effect of the chromosome XIII-R subtelomeric sequence on the association of Mre11 and Tel1 with nontelomeric DNA ends. HO (KSC2851) or XIIIR-HO (KSC2852) cells expressing Mre11-myc and Tel1-HA were transformed with the GAL-HO plasmid, and association of Mre11 or Tel1 with DNA ends was analyzed by ChIP assay as in B.
To address whether Tbf1 controls MRX association with DNA ends, we used a heat-inducible tbf1 degron (tbf1-d) construct to examine the effect of Tbf1 depletion. The TBF1 gene is essential for cell proliferation (Brigati et al., 1993). Correspondingly, tbf1-d mutants did not proliferate efficiently at 37°C on galactose medium (Figure 2A). The degron-fused Tbf1 protein was rapidly degraded at high temperatures when the Ubr1 protein was overexpressed from the GAL1 promoter; Tbf1-d protein was undetectable 3 h after incubation with galactose at 37°C (Figure 2B). Consistent with the Tbf1 degradation patterns, tbf1-d mutants essentially stopped proliferation 6 h after incubation with galactose at 37°C (Figure 2C). DNA flow cytometry studies showed that Tbf1 depletion did not result in cell cycle–specific arrest (Figure 2D). We then tested the effect of Tbf1 depletion on MRX or Tel1 accumulation at XIIIR-TG81 ends. Tbf1 depletion was found to restore both Mre11 and Tel1 binding at XIIIR-TG81 ends (Figure 2, E and F). Thus Tbf1 deficiency reversed the effect of the subtelomeric sequence fusion, indicating that subtelomeric Tbf1 binding decreases MRX localization to TG81 ends.
FIGURE 2:

Effect of Tbf1 depletion on localization of MRX and Tel1 to DNA ends with telomeric or subtelomeric sequences. (A) Effect of tbf1-degron mutation on colony formation on galactose medium at 37°C. Wild-type or tbf1-degron mutant (tbf1-d; KSC2853) cells were transformed with YCpU-TBF1 or the control vector. Cells were serially diluted and spotted on sucrose or galactose plates selectable for the plasmids. Plates were incubated at the indicated temperatures. (B) UBR1-induced Tbf1 protein degradation at 37°C. tbf1-degron mutant (tbf1-d) (KSC2853) cells were initially grown in sucrose at 30°C and were then incubated with galactose and concomitantly shifted to 37°C. Aliquots of cells were collected at the indicated times after incubation with galactose at 37°C and subjected to immunoblotting analysis with anti-myc antibodies. The degron cassette is fused to a myc epitope tag. (C, D) Proliferation of tbf1-degron mutants after incubation with galactose at 37°C. tbf1-degron mutant (tbf1-d) (KSC2853) cells were initially grown in sucrose at 30°C and were then incubated with galactose and concomitantly shifted to 37°C. Aliquots of cells were collected at the indicated times after incubation with galactose at 37°C to determine the cell division rate (C) and DNA content of cells (D). DNA content was determined by flow cytometric analysis. (E, F) Effect of Tbf1 depletion on Mre11 or Tel1 localization to XIIIR-TG81 ends. XIIIR-TG81-HO tbf1-d cells expressing Mre11-HA (KSC2854) or Tel1-HA (KSC2855) were transformed with the YCpU-TBF1 plasmid or the control vector, together with the GAL-HO plasmid. Transformed cells were treated as in A and analyzed by ChIP assay as in Figure 1B to monitor the accumulation of Mre11 (E) or Tel1 (F) at DNA ends. HO cutting efficiency of cells carrying each plasmid is shown in square brackets.
Effect of copy number of TTAGGG on MRX binding at TG81 ends
Subtelomeres contain varying numbers of Tbf1 binding motifs, as well as sequences targeted by other proteins. To further evaluate Tbf1 function, we placed various numbers of TTAGGG repeats adjacent to the TG81 sequence (TAGn-TG81-HO; Figure 1A). The introduction of the 2x, 4x, or 10x TTAGGG sequences decreased Mre11 or Tel1 localization to nearby TG81 ends in a copy number–dependent manner (Figure 3). Similar to the XIIIR sequence, those repeat sequences did not affect Mre11 or Tel1 binding at HO ends (data not shown). These results strongly suggest that subtelomere-binding protein Tbf1 controls the localization of MRX complex to telomeric DNA ends in a dosage-dependent manner.
FIGURE 3:

Effect of TTAGGG repeats on association of MRX and Tel1 with TG81 ends. TG81-HO (KSC2849), TAG2-TG81-HO (KSC2856), TAG4-TG81-HO (KSC2857), TAG10-TG81-HO (KSC2858), TAG10-TG81-HO rif1Δ rif2Δ (KSC2859), or CTA10-TG81-HO (KSC2860) cells expressing Mre11-myc and Tel1-HA were transformed with the GAL-HO plasmid, and accumulation of Mre11 or Tel1 was analyzed by ChIP assay as Figure 1B.
Effect of Tbf1 tethering on MRX localization to nearby TG81 ends
Because Tbf1 protein binds to the TTAGGG repeat sequence, Tbf1 function may depend on sequence-specific DNA binding. To address this possibility, we set up a system to tether Tbf1 protein to non-TTAGGG sequences adjacent to the TG81 repeat (Figure 4A). We constructed a LacI-Tbf1 fusion gene that fully rescues the proliferation defect of tbf1Δ mutants (data not shown). To deploy LacI-Tbf1 protein to non-TTAGGG sequence, we placed eight copies of the lacI-binding sequence (lacO) adjacent to the TG81 sequence. We examined the effect of LacI-Tbf1 tethering on Mre11 or Tel1 binding to nearby TG81 or HO ends (Figure 4, B and C). LacI-Tbf1 expression inhibited Mre11 or Tel1 binding to TG81 ends but not HO ends. Expression of LacI by itself did not affect Mre11 or Tel1 binding to TG81 ends (Figure 4B). Thus sequence-specific DNA binding is dispensable for the Tbf1 function. The foregoing findings also support the idea that the Tbf1-mediated inhibition of MRX localization does not depend on direction-specific Tbf1 binding. Consistent with this idea, the 10xCCCTAA repeat inhibited Mre11 or Tel1 localization to TG81 ends similarly to the 10xTTAGGG repeat (Figure 3).
FIGURE 4:

Effect of Tbf1 or Rap1 tethering on MRX association with nearby DNA ends. (A) Schematic of the HO cleavage site, the LacO8 array, the TetO4 array, or the TG81 repeat at the ADH4 locus on chromosome VII-L. In LacO8-TG81-HO or LacO8-HO cells, eight copies of the lacO sequence (gray rectangle) were placed centromere-proximal to the TG81-HO cassette or the HO cleavage site, respectively. In LacO8-TetO4-HO cells, eight copies of the lacO sequence and four copies of the tetO sequence (barrel) were placed centromere-proximal to the HO cleavage site. Centromere is shown as a circle on the left (CEN). The HO primer pair was designed to amplify a region 1 kbp away from the HO site, and the cutting-site primer pair was used to determine the HO cutting efficiency. (B) Effect of Tbf1 tethering on association of Mre11 or Tel1 with nearby TG81 ends. LacO8-TG81-HO (KSC2861) cells expressing Mre11-myc and Tel1-HA were transformed with placI-TBF1 or the control vector, together with the GAL-HO plasmid. Cells were analyzed by ChIP assay as in Figure 1B to monitor accumulation of Mre11 or Tel1 at DNA ends. (C) Effect of Tbf1 tethering on association of Mre11 or Tel1 with nearby HO ends. LacO8-HO (KSC2862) cells expressing Mre11-myc and Tel1-HA were transformed with placI-TBF1 or the control vector, together with the GAL-HO plasmid. Cells were analyzed by ChIP assay as in B to monitor accumulation of Mre11 or Tel1 at DNA ends.
Rap1-dependent, but Rif1- and Rif2-independent, inhibition of MRX localization to DNA ends
Because telomeric TG repeats are covered with Rap1, it seems likely that Tbf1 function is mediated through Rap1. However, it remains possible that Tbf1 acts on telomeric TG repeat-binding proteins other than Rap1. To address whether Tbf1 collaborates with Rap1, we inserted four copies of the TetR-binding sequence (tetO4) between the lacO8 sequence and the HO cleavage, site generating the lacO8-tetO4-HO cassette (Figure 4A). We then examined the synergistic effect of LacI-Tbf1 and TetR-Rap1 expression on MRX and Tel1 localization to a DNA end near the lacO8-tetO4 sequence (LacO8-TetO4) ends (Figure 5). LacI-Tbf1 expression alone did not inhibit Mre11 or Tel1 localization to LacO8-TetO4 ends, and TetR-Rap1 expression moderately decreased Mre11 or Tel1 localization. In contrast, coexpression of LacI-Tbf1 and TetR-Rap1 nearly shut out Mre11 or Tel1 localization. Rap1 interacts with Rif1 and Rif2 through the C-terminus (Conrad et al., 1990; Lustig et al., 1990; Hardy et al., 1992; Wotton and Shore, 1997). Rap1 collaborates with Rif1 and Rif2 to decreases MRX and Tel1 localization to DNA ends (Hirano et al., 2009). Consistent with this view, TetR-Rap1 expression failed to decrease Mre11 or Tel1 localization to LacO8-TetO4 ends in rif1Δ rif2Δ double mutants. However, coexpression of LacI-Tbf1 and TetR-Rap1 inhibited Mre11 or Tel1 localization in rif1Δ rif2Δ double mutants as effectively as in RIF1 RIF2 wild-type cells. No effect of rif1Δ rif2Δ double mutation was observed for LacI-Tbf1 expression alone. We also investigated the effect of rif1Δ rif2Δ double mutation on Mre11 or Tel1 accumulation at TAG10-TG81 ends (Figure 3). The rif1Δ rif2Δ double mutation did not affect Mre11 or Tel1 accumulation. Thus the Tbf1-Rap1 pathway inhibits MRX localization to DNA ends through a Rif1- and Rif2-independent mechanism.
FIGURE 5:
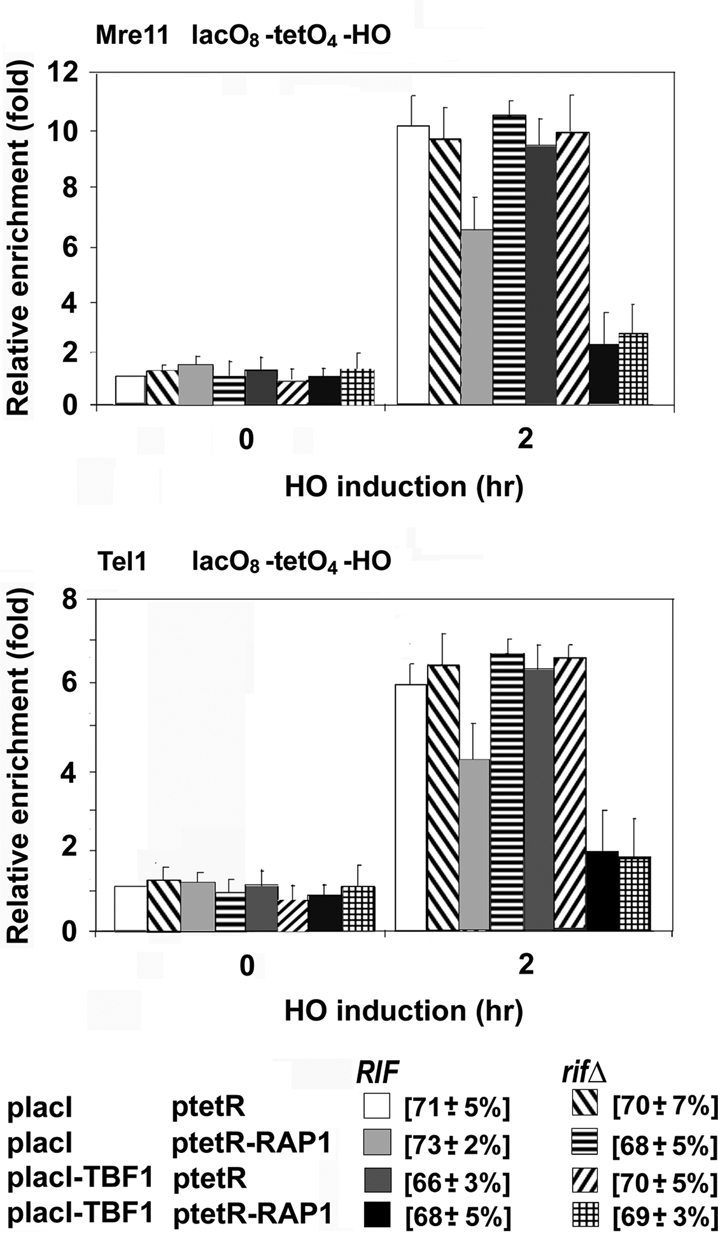
Effect of Tbf1 and Rap1 cotethering on MRX association with nearby DNA ends. LacO8-TetO4-HO (KSC2863) and LacO8-TetO4-HO rif1Δ rif2Δ (KSC2864) cells expressing Mre11-myc and Tel1-HA were transformed with placI-TBF1, ptetR-RAP1, or the control vector, together with the GAL-HO plasmid. Cells were analyzed for Mre11 binding or Tel1 binding by ChIP assay as in Figure 1B. The LacO8-TetO4-HO construct is shown in Figure 4A. The strains used here contained a sir2Δ mutation to suppress inefficient HO cleavage resulting from Rap1 tethering nearby (Hirano et al., 2009).
Requirement of the N-terminus of Tbf1 protein for complex formation and regulation of MRX localization to DNA ends
The family of telomere-repeat binding factors, including TRF1, TRF2, and Taz1, shares a common architecture with two separate structural domains (Palm and de Lange, 2008; Moser and Nakamura, 2009). An N-terminal domain is required for the formation of homodimers, whereas a C-terminal Myb domain recognizes double-stranded TTAGGG repeats (Figure 6A). Tbf1 contains a C-terminal Myb-type DNA-binding domain but does not share similarities with TRF1, TRF2, and Taz1 at the N-terminus (Cockell et al., 2009). It remains possible, however, that Tbf1 functions as a homodimer or homooligomer similar to TRF1, TRF2, and Taz1. We therefore examined protein–protein interaction of Tbf1 protein by coimmunoprecipitation experiments (Figure 6B). Extracts were prepared from cells expressing FLAG-tagged Tbf1 (Tbf1-FLAG) or hemagglutinin (HA)-tagged Tbf1 protein (Tbf1-HA) and subjected to immunoprecipitation with anti-HA antibodies. Immunoprecipitates were then analyzed by immunoblotting with anti-FLAG or anti-HA antibodies. Coprecipitation of Tbf1-HA and Tbf1-FLAG was detected only in cells expressing both tagged proteins (Figure 6B). Thus Tbf1 proteins interact with each other.
FIGURE 6:

Role of the N-terminus of Tbf1 protein in inhibiting MRX localization to DNA ends. (A) Deletion or substitution mutations of TBF1. The budding yeast Saccharomyces cerevisiae Tbf1 protein, consisting of 562 amino acid residues, contains a Myb-related DNA-binding domain at the C-terminus. The N-terminal conserved domain was shaded. Amino acid alignment shows the N-terminal conserved domains of S. cerevisiae, Kluyveromyces lactis, Candida albicans, and Schizosaccharomyces pombe Tbf1 proteins. Identical or related amino acid residues are boxed in black or gray, respectively. The tbf1-ΔN mutation lacks the N-terminal 111–210 amino acid residues. The tbf1-25 or tbf1-26 mutation replaces the conserved amino acid residues at 177 and 180 or 187 and 190 (indicated by asterisks) with alanine, respectively. (B) Protein–protein interaction of Tbf1. tbf1Δ mutants carrying YCp33-TBF1-HA, YCp22-TBF1-FLAG, or the respective control vector were grown in medium selectable for the plasmids and subjected to immunoprecipitation with anti-HA antibodies. Immunoprecipitates (IP) and whole-cell extracts (WCE) were subjected to immunoblotting analysis with anti-HA or anti-FLAG antibodies. All the cells used here are haploid, which are derived from a TBF1/tbf1Δ diploid strain (KSC2505). (C) Requirement of the N-terminus for protein–protein interaction. tbf1-d (KSC2756) cells were transformed with pRS316-TBF1-HA and pRS314-TBF1-FLAG, or pRS316-tbf1ΔN-HA and pRS314-tbf1ΔN-FLAG. Cells were grown in sucrose at 30°C and synchronized at G2/M with nocodazole. After synchronization, the culture was incubated with galactose and concomitantly shifted to 37°C. Aliquots of cells were collected after 3 h of incubation with galactose at 37°C and analyzed as in B. (D) Effect of tbf1-ΔN, tbf1-25, or tbf1-26 mutation on cell proliferation. tbf1Δ mutants carrying the URA3-marked YCpU-TBF1 plasmid were transformed with pRS314-TBF1-FLAG, pRS314-tbf1-ΔN-FLAG, pRS314-tbf1-25-FLAG, pRS314-tbf1-26-FLAG, or the control vector. Transformants were streaked and grown on plates containing medium with or without 5-FOA. The strain used here is derived from a TBF1/tbf1Δ diploid strain (KSC2505). (E) Effect of tbf1-25 or tbf1-26 mutation on protein–protein interaction. tbf1-d cells (KSC2756) were transformed with pRS316-TBF1-HA and pRS314-TBF1-FLAG, pRS316-tbf1-25-HA and pRS314-tbf1-25-FLAG, or pRS316-tbf1-26-HA and pRS314-tbf1-26-FLAG. Protein–protein interaction of Tbf1 was examined as in C. (F, G) Effect of Tbf1-25 or Tbf1-26 protein tethering on Mre11 and Tel1 association with adjacent TG81 ends. LacO8-TG81-HO tbf1-d cells expressing Mre11-HA (KSC2865) or Tel1-HA (KSC2866) were transformed with the GAL-HO plasmid and placI-TBF1, placI-tbf1-25, placI-tbf1-26, or the empty placI plasmid. Transformed cells were treated as in C and subjected to ChIP assay as described in Figure 1B to monitor the association of Mre11 (F) or Tel1 (G) with DNA ends.
Tbf1 shows significant conservation with fungal Tbf1 proteins in the N-terminal region (Cockell et al., 2009; Figure 6A). As shown, Tbf1 inhibits MRX localization independently of sequence-specific DNA binding. We therefore tested the hypothesis that the N-terminus of Tbf1 mediates inhibition of MRX localization to DNA ends. We first investigated the effect of deletion of the N-terminus on protein–protein interaction of Tbf1. To exclusively monitor the protein–protein interaction of mutant proteins, we performed coimmunoprecipitation experiments in the tbf1-d mutants at 37°C after depletion of functional Tbf1 protein. Extracts from cells expressing HA- or FLAG-tagged N-terminal truncated Tbf1 protein (Tbf1-ΔN) were subjected to immunoprecipitation and subsequent immunoblotting as described. Protein–protein interaction between Tbf1-ΔN proteins was undetectable (Figure 6C), indicating that the conserved N-terminal domain is required for efficient protein–protein interaction. Because the TBF1 gene is essential for cell proliferation, we examined whether the N-terminal truncation affects Tbf1 function. Both FLAG-tagged and untagged versions of the tbf1-ΔN mutation failed to rescue the lethality associated with the tbf1Δ mutataion at 25, 30, or 37°C (Figure 6D; data not shown).
The foregoing result raises a possibility that Tbf1 function requires protein–protein interaction. However, it is equally possible that some Tbf1 functions operate independently of protein–protein interaction. We generated several substitutions by replacing conserved residues with alanine in the N-terminal conserved region and determined whether loss of function is coupled to defective protein–protein interaction. Similar to the tbf1-ΔN mutation, two substitution mutations, tbf1-25 and tbf1-26, regardless of whether they were tagged or not, each failed to rescue proliferation defects associated with the tbf1Δ mutation at 25, 30, or 37°C (Figure 6D, data not shown). Because the conserved region is required for protein–protein interaction, we determined whether these tbf1 mutations disrupt complex formation. As described, tbf1-d cells were transformed with plasmids carrying HA- or FLAG-tagged wild-type TBF1 or mutant tbf1 constructs. After depletion of Tbf1-degron protein, cells were subjected to coimmunoprecipitation experiments. Neither tbf1-25 nor tbf1-26 mutation affected complex formation (Figure 6E).
We then addressed whether the tbf1-25 or tbf1-26 mutation confers defects in inhibiting MRX localization to DNA ends. To examine the function of the Tbf1-mutant protein, we tested the effect of LacI-Tbf1-25 or LacI-Tbf1-26 expression on MRX or Tel1 localization to LacO8-TG81 ends in the tbf1-d background after depletion of functional Tbf1 protein (Figure 6, F and G). Cells expressing Mre11-myc or Tel1-HA were transformed with the GAL-HO plasmid and placI-TBF1, placI-tbf1-25, placI-tbf1-26 plasmid or the control vector (placI). Transformants were grown in sucrose at 30°C and then incubated in galactose at 37°C to induce HO expression, as well as Tbf1 degradation. Cells were analyzed by ChIP assay as described, to monitor Mre11 or Tel1 binding at DNA ends. Both tbf1-25 and tbf1-26 mutations conferred defects in inhibiting MRX localization; Mre11 or Tel1 binding was increased in cells expressing a LacI-Tbf1-25 or LacI-Tbf1-26 fusion compared with those expressing the wild-type LacI-Tbf1 fusion. Neither the tbf1-25 nor the tbf1-26 mutation altered the expression level of LacI-Tbf1 fusion protein (data not shown). These results suggest that the conserved N-terminal region of Tbf1 mediates inhibition of MRX localization to telomeric DNA ends.
Effect of Tbf1 depletion on checkpoint activation at endogenous telomeres
As shown here, Tbf1 and Rap1 inhibit MRX localization to DNA ends independently of Rif1 or Rif2. In parallel, Rap1 collaborates with Rif1 and Rif2 in inhibiting Tel1 localization. We previously showed that the 162–base pair TG162 sequence inhibits MRX and Tel1 localization in a Rif1- and Rif2-dependent manner, whereas the 81–base pair TG81 sequence does not (Hirano et al., 2009). We therefore tested the hypothesis that Tbf1 is required to maintain genome stability of chromosomal ends containing short telomeric sequences.
MRX plays a key role in activating the Mec1-dependent checkpoint pathway, as well as the Tel1 pathway (Nakada et al., 2003a, 2004). We first examined the effect of Tbf1 depletion on checkpoint activation at normal-length telomeres. Checkpoint activation is controlled in a cell cycle–dependent manner (Ira et al., 2004; Vodenicharov and Wellinger, 2006). We therefore monitored phosphorylation of Rad53 in G2/M-arrested cells (Figure 7A). No Rad53 phosphorylation was detected after Tbf1 depletion, whereas MMS treatment induced Rad53 phosphorylation in Tbf1-depleted cells. G2/M synchronization with nocodazole did not alter the expression level of Tbf1 protein (Supplemental Figure S1). The Cdc13-Stn1-Ten1 complex acts as a telomere cap to protect telomeres from DNA degradation (Petreaca et al., 2007; Xu et al., 2009). It is possible that Tbf1 functions as a backup for the Cdc13-Stn1-Ten1 complex; therefore, the effect of Tbf1 depletion might be detectable only at uncapped telomeres. We next examined the effect of Tbf1 depletion on checkpoint activation in cells carrying the temperature-sensitive stn1-13 mutation (Grandin et al., 2001; Figure 7B). Rad53 was phosphorylated in stn1-13 mutants after shifting to 37°C, but Tbf1 depletion did not affect the phosphorylation status. It should be noted that the stn1-13 mutation does not cause telomere shortening (Grandin et al., 2001). Thus Tbf1 is dispensable for telomere protection in cells containing normal-length or elongated telomeres.
FIGURE 7:

Effect of Tbf1 depletion on checkpoint response to endogenous telomeres. (A) Effect of Tbf1 depletion on checkpoint activation in cells carrying normal-length telomeres. Wild-type (KSC1602) or tbf1-d (KSC2756) cells were cultured as in Figure 6C to degrade Tbf1 protein. Cells were further treated with 0.1% methyl methanesulfonate (MMS) for 1 h at 37°C. Aliquots of cells were collected and subjected to immunoblotting analysis with anti-Rad53 antibodies. Tubulin was detected as a loading control. (B) Effect of Tbf1 depletion on checkpoint activation in stn1-13 mutant cells. tbf1-d (KSC2756) or tbf1-d stn1-13 (KS2891) cells carrying YCpT-TBF1 or the control vector were grown and treated with nocodazole at 25°C. After synchronization, the culture was incubated with galactose and concomitantly shifted to 37°C. Aliquots of cells were collected at the indicated times after incubation with galactose at 37°C. As a control, aliquots of cells were treated with 0.1% MMS for 1 h at 37°C (labeled as M). Cells were subjected to immunoblotting analysis as in A.
Effect of Tbf1 binding on checkpoint activation at DNA ends with the 22–base pair TG sequence
Checkpoint activation occurs at DNA ends adjacent to the 22–base pair TG (TG22) sequence although telomeres are added at TG22 ends (Hirano and Sugimoto, 2007). We next addressed whether Tbf1 attenuates checkpoint activation at DNA ends with the TG22 sequence (TG22 ends) nearby (Figure 8). The TG22-HO-CA cassette was used or further modified to monitor checkpoint activation at a single DNA end after DSB induction (Hirano and Sugimoto, 2007; Figure 1A). To test the effect of Tbf1 binding, we fused the XIIIR sequence centromere-proximal to the TG22-HO-CA sequence, generating the XIIIR-TG22-HO-CA cassette (Figure 1A). We also replaced the TG22 sequence of the TG22-HO-CA cassette, constructing the XIIIR-HO-CA cassette (Figure 1A). The TG22 sequence contains a single Rap1-binding motif; ChIP assays confirmed Rap1 binding at the TG22 sequence (Supplemental Figure S2). TG22-HO-CA, XIIIR-TG22-HO-CA, or XIIIR-HO-CA cells carrying the GAL-HO plasmid were first grown in sucrose and then incubated with galactose to induce HO expression (Figure 8A). Checkpoint activation was examined by monitoring the phosphorylation status of Rad53. Rad53 was phosphorylated in TG22-HO-CA cells after HO expression. Rad53 phosphorylation was detected in XIIIR-HO-CA cells as well. However, no apparent phosphorylation was observed in XIIIR-TG22-HO-CA cells. We also examined the effect of the XIIIR sequence on accumulation of Mre11, Tel1, or Mec1 at DNA ends (Figure 8, B– D). Accumulation of Mre11, Tel1, or Mec1 was significantly decreased at XIIIR-TG22 ends compared with that at TG22 or XIIIR ends. Thus the combination of the XIIIR and the TG22 sequence inhibits checkpoint activation at nearby DNA ends.
FIGURE 8:

Effect of Tbf1 binding on checkpoint activation at adjacent DNA ends with the 22–base pair TG22 sequence. (A) Effect of the XIIIR and 22–base pair TG22 sequence on Rad53 phosphorylation after HO expression. TG22-HO-CA (KSC2230), XIIIR-HO-CA (KSC2894), or XIIIR-TG22-HO-CA (KSC2834) cells were transformed with the GAL-HO plasmid. Transformed cells were grown in sucrose and synchronized at G2/M with nocodazole. After arrest, the culture was incubated with galactose to induce HO expression. Aliquots of cells were collected at the indicated times after HO expression, and cell extracts were analyzed by immunoblotting analysis with anti-Rad53 antibodies. (B) Effect of the XIIIR and 22–base pair TG22 sequence on Mre11 accumulation at DNA ends. XIIIR-TG22-HO-CA MRE11-myc (KSC2892), TG22-HO-CA MRE11-myc (KSC2901), or XIIIR-HO-CA MRE11-myc (KSC2905) cells carrying the GAL-HO plasmid were analyzed by ChIP assay as in Figure 1B. (C) Effect of the XIIIR and 22–base pair TG22 sequence on Tel1 accumulation at DNA ends. XIIIR-TG22-HO-CA TEL1-HA (KSC2896), TG22-HO-CA TEL1-HA (KSC2895), or XIIIR-HO-CA TEL1-HA (KSC2897) cells carrying the GAL-HO plasmid were analyzed by ChIP assay as B. (D) Effect of the XIIIR and 22–base pair TG22 sequence on Mec1 accumulation at DNA ends. XIIIR-TG22-HO-CA MEC1-HA (KSC2899), TG22-HO-CA MEC1-HA (KSC2898), or XIIIR-HO-CA MEC1-HA (KSC2900) cells carrying the GAL-HO plasmid were analyzed by ChIP assay as in B. (E) Effect of Tbf1 depletion on checkpoint signaling from XIIIR-TG22 ends. TG22-HO-CA (KSC2230), XIIIR-TG22-HO-CA (KSC2839), or XIIIR-TG22-HO-CA tbf1-d (KSC2836) cells were transformed with the GAL-HO plasmid. Transformants were treated as in Figure 6C to induce HO expression and Tbf1 degradation. Cells were subjected to immunoblotting analysis as in A.
To determine whether Tbf1 indeed contributes to the checkpoint inhibition, we examined the effect of Tbf1 depletion on checkpoint activation at XIIIR-TG22 ends (Figure 8E). XIIIR-TG22-HO-CA TBF1 or tbf1-d cells carrying the GAL-HO plasmid were initially grown in sucrose and incubated with galactose at 37°C to induce HO expression and degrade Tbf1-d protein. After 3 h of incubation, phosphorylation of Rad53 protein was monitored by immunoblotting analysis. The tbf1-d mutation restored Rad53 phosphorylation in XIIIR-TG22-HO-CA cells after HO expression. These results support the idea that Tbf1 binding inhibits checkpoint activation at DNA ends with short telomeric repeats.
Effect of Tbf1 depletion on checkpoint activation at short telomeres
We next asked whether Tbf1 depletion triggers checkpoint activation at short telomeres (Figure 9A). To shorten telomeres, we introduced a deletion mutation of EST1, which encodes a regulatory protein for telomerase, into tbf1-d mutants. Cells carrying an est1Δ mutation shorten telomeres gradually and eventually suffer senescence because of the end replication problem (Nugent and Lundblad, 1998). Growth of the est1Δ strains was maintained by wild-type EST1 on a URA3 plasmid. Cells were transformed with YCpT-TBF1 or the control vector. To select for loss of the complementing EST1 plasmid, transformants were grown on plates containing 5-fluoroorotic acid (5-FOA). The resulting single colonies were inoculated into liquid culture. The cultures were diluted fourfold and grown for 7 h for successive serial dilutions. Cells from each culture were arrested with nocodazole to block further telomere attrition. Southern blotting analysis confirmed that telomeres in est1Δ mutants became shorter after successive dilutions compared with those in wild-type cells (Figure 9A). After nocodazole block, cells were incubated with galactose at 37°C to degrade Tbf1-d protein. After 3 h of incubation, phosphorylation of Rad53 protein was monitored by immunoblotting analysis. Rad53 phosphorylation was detected after three successive dilutions in tbf1-d mutant cells but not in cells expressing wild-type Tbf1 protein.
FIGURE 9:

Effect of Tbf1 depletion on checkpoint response to short telomeres. (A) Effect of Tbf1 depletion on checkpoint response in telomerase-deficient cells. Telomerase-deficient est1Δ tbf1-d (KSC2867) cells carrying YCpT-TBF1 or the control vector were obtained from the strain carrying the URA3-marked EST1 plasmid. Colonies arisen on plates containing 5-FOA were grown to the late log phase. The culture was diluted fourfold to allow cells to undergo cell division twice for 7 h at 30°C. This cycle was repeated four times. Cells from each serial dilution culture were synchronized at G2/M with nocodazole. Aliquots of cells were harvested to monitor the telomere length by Southern blots (top). Cells were incubated with galactose at 37°C to degrade Tbf1-d protein. After 3 h of incubation cells were harvested to monitor Rad53 phosphorylation by immunoblotting with anti-Rad53 antibodies (bottom). Tubulin was detected as a loading control. (B) Effect of Tbf1 depletion on checkpoint response in telomerase-deficient cells. Telomerase-deficient est1Δ tbf1-d (KSC2867) cells were obtained after streaking the strain carrying the URA3-marked EST1 plasmid. Colonies arisen on plates containing 5-FOA were grown to the late log phase. Cells were continuously grown after dilution and then synchronized with nocodazole at 30°C as in A. Aliquots of cells were harvested to monitor the telomere length by Southern blots (top). After synchronization, half of the rest was incubated with galactose at 37°C to degrade Tbf1 protein. The other half was incubated with glucose at 30°C to retain Tbf1 expression. After 3 h of incubation, cells were harvested to monitor Rad53 phosphorylation as in A.
It remained possible that telomere attrition occurs slightly faster in tbf1-d mutants than with wild-type TBF1 cells; therefore, checkpoint is activated earlier in tbf1-d mutants than in TBF1 cells. To exclude this possibility, tbf1-d est1Δ cells were grown in sucrose for several generations as described. After nocodazole block, half of the culture was incubated with galactose at 37°C and the other half was grown with glucose at 30°C (Figure 9B). Rad53 phosphorylation was detected after three successive dilutions in Tbf1-depleted cells but not in nondepleted cells. Incubation of cells with galactose at 37°C does not elicit Rad53 phosphorylation (Figure 7; Vodenicharov et al., 2010). Thus Tbf1 depletion accelerated triggering of checkpoint responses in cells containing short telomeres but not normal-length telomeres. Together, our results support the model in which Tbf1 protein plays a pivotal role in maintaining genome stability of short telomeres.
DISCUSSION
Rap1 binds to double-stranded telomeric sequences and recruits Rif1 and Rif2 proteins via its C-terminal domain (Conrad et al., 1990; Lustig et al., 1990; Hardy et al., 1992; Wotton and Shore, 1997). Both Rif1 and Rif2 inhibit the localization of Tel1, but not the Mre11-Rad50-Xrs2 complex, to adjacent DNA ends (Hirano et al., 2009). Rap1 collaborates with Rif1 and Rif2 and inhibits MRX accumulation at DNA ends (Hirano et al., 2009). The budding yeast Tbf1 protein binds to TTAGGG sequences (Brigati et al., 1993; Koering et al., 2000; Lavoie et al., 2010), which are found at the telomere junction of subtelomeres (Louis, 1995). In this study we show that Tbf1 and Rap1 collaborate to inhibit the localization of MRX to DNA ends independently of Rif1 or Rif2 function. Tbf1 depletion triggers checkpoint activation from short telomeres but not from normal-length telomeres. These observations support a model in which Tbf1 collaborates with Rap1 and circumvents genomic instability at short telomeres. Our results also provide an explanation of why TTAGGG repeats, if placed centromere-proximal to TG repeats at telomeres, are counted as a telomere sequence (Berthiau et al., 2006; Bah et al., 2011).
Tbf1 and Rap1 collaborate to inhibit MRX and Tel1 localization to DNA ends with short TG repeats nearby. Localization of MRX complex to DNA ends leads to accumulation of ssDNA tracts at DNA ends, thereby activating the Mec1-checkpoint pathway as well (Nakada et al., 2003a, 2004). Tbf1 depletion enhances checkpoint activation in cells containing short telomeres but does not stimulate checkpoint activation in cells containing normal-length telomeres. Moreover, Tbf1 is essential for inhibition of checkpoint activation at DNA ends where a subtelomeric sequence is placed together with a short TG repeat sequence. These observations support the model in which Tbf1 plays a critical role in maintaining telomere homeostasis at short telomeres. The presence of the Tbf1-binding TTAGGG sequence at the telomere junctions could reduce the length of the adjacent telomeric TG sequence (Berthiau et al., 2006; Bah et al., 2011). As telomeres shorten, the TG sequence disappears earlier than the centromere-proximal TTAGGG sequence. Binding of Rap1 and Rif2 decreases in proportion as telomeres become short (McGee et al., 2010). Rif1 might remain associated with short telomeres, but Rif1 by itself does not inhibit MRX localization. Both Rif1 and Rif2 require Rap1 to inhibit MRX localization (Hirano et al., 2009). Moreover, Rif1 function is partially dependent on Rif2 function when Rif1 binding near DNA ends is limited (Hirano et al., 2009). Thus Rif1 and Rif2 functions become attenuated at short telomeres. Of note, Tbf1 and Rap1 act independently of Rif1 or Rif2. Not all subtelomeres possess multiple Tbf1-binding consensus motifs at the subtelomere–telomere junctions. A single unrepairable DNA break, however, induces the damage checkpoint response (Sandell and Zakian, 1993; Lee et al., 1998). These observations are consistent with the idea that Tbf1 plays a critical role in DNA-end protection at short telomeres adjacent to subtelomeric TTAGGG repeats. Inhibition of MRX and Tel1 recruitment could prevent degradation of DNA ends but might also decrease telomerase recruitment. Previous studies showed, however, that the N-terminus of Tbf1 fused to the Gal4-DNA–binding domain (Gbd-Tbf1N) promotes telomere addition near the Gal4 binding sequence in tel1Δ mutants when telomeres are short (Arneric and Lingner, 2007). Tbf1 thus may stimulate telomerase independent of the MRX-Tel1 pathway at short telomeres. Several lines of evidence established that the Cdc13-Stn1-Ten1 complex plays a key role in preventing checkpoint activation at telomeres (Weinert et al., 1994; Garvik et al., 1995; Grandin et al., 2001; Vodenicharov and Wellinger, 2006; Vodenicharov et al., 2010; Petreaca et al., 2007; Xu et al., 2009). Cdc13 can bind to single-strand TTAGGG DNA; however, the affinity to TTAGGG repeat DNA is much weaker than that to TG repeat DNA (Lin and Zakian, 1996). Tbf1 and Rap1 thus appear to function as the last bastion to resist nucleolytic activities that intrude into non-TG sequence.
Rap1 and Tbf1 both contain Myb-related DNA-binding domains, but sequence-specific DNA binding is dispensable for the inhibition of MRX localization. Substitution mutations of TBF1 at the N-terminal region confer defects in inhibiting MRX localization without affecting protein–protein interaction. These results suggest that the N-terminus of Tbf1 constitutes a domain that mediates inhibition of MRX localization. TBF1 is required for cell proliferation (Brigati et al., 1993). The substitution mutations disrupt the essential function of Tbf1 as well. Other studies showed that another N-terminal region of Tbf1 regulates telomere length independently of the Tel1 pathway (Berthiau et al., 2006; Arneric and Lingner, 2007). Thus the N-terminus mediates multiple functions of Tbf1 protein. Recent studies indicate that like Rap1, Tbf1 controls the transcriptional activation of genes required for ribosome biosynthesis (Lavoie et al., 2010; Preti et al., 2010). However, it seems less likely that transcriptional activation leads to inhibition of MRX recruitment. Although lacI-Gal4 functions as a transcriptional activator similar to lacI-Tbf1, no inhibition of MRX localization was observed with the combination of lacI-Gal4 and TetR-Rap1 (data not shown). Further studies will be needed to understand the mechanism of how Rap1 and Tbf1 attenuate MRX binding at DNA ends. At this moment, it is not precisely determined how the MRX complex recognizes and associates with DNA ends.
Budding yeast tolerate vertebrate-sequence telomeres containing only TTAGGG repeats (Alexander and Zakian, 2003). Neither Rap1 nor Rif2 binding was detected at TTAGGG telomeres, indicating that Tbf1 contributes to telomere maintenance independently of Rap1. Tbf1 therefore could protect DNA ends similarly to Rap1. Consistent with the idea, while this manuscript was under revision, Tbf1 was shown to inhibit MRX localization at a DNA end flanked by a 230–base pair TTAGGG repeat (Ribaud et al., 2011). Rap1 collaborates with Rif1 and Rif2 at wild-type TG telomeres (Hardy et al., 1992; Wotton and Shore, 1997), whereas Tbf1 appears to act with other telomere-binding proteins at TTAGGG telomeres. Whereas yeast strains with wild-type TG telomeres undergo cell division to senescence after telomerase loss, telomerase is essential for cell proliferation of strains with TTAGGG telomeres (Bah et al., 2011). Of note, telomere-capping Cdc13 protein was much more abundant at TTAGGG telomeres than at wild-type TG telomeres (Alexander and Zakian, 2003). Thus Rap1 and Tbf1 appear to exert related but different functions at distal telomeric regions.
Mammalian TRF1 and TRF2 and fission yeast Taz1 proteins form homodimers through a homodimerization domain at the N-terminus and bind to telomeric TTAGGG repeats through a Myb-related domain at the C-terminus (Palm and de Lange, 2008; Moser and Nakamura, 2009). Similarly, Tbf1 proteins interact with each other and bind to DNA through a Myb-related domain at the C-terminus. Although the N-terminus of Tbf1 is not structurally related to that of TRF1, TRF2, or Taz1, it is required for protein–protein interaction of Tbf1. Tbf1 does not bind to telomeres; it is concentrated at subtelomeric and intergenic regions (Hogues et al., 2008; Lavoie et al., 2010). Of interest, recent evidence indicates that mammalian TRF2 localizes to TTAGGG-rich extratelomeric regions as well (Martinez et al., 2010; Simonet et al., 2011; Yang et al., 2011). TRF1, TRF2, and Taz1 are involved in telomere protection (Miller and Cooper, 2003; Denchi and de Lange, 2007; Palm and de Lange, 2008; Martinez et al., 2009). TRF2 and Taz1 act in complex with RAP1 and Rap1, respectively (Palm and de Lange, 2008; Moser and Nakamura, 2009). We have shown that budding yeast Tbf1 collaborates with Rap1 to protect DNA ends of short telomeres. Further characterization of Tbf1 should contribute to better understanding of telomere maintenance and telomere evolution.
MATERIALS AND METHODS
Plasmids
The pXIIIR-TG81-HO and pXIIIR-HO plasmid were generated as follows. Complementary oligonucleotides containing the chromosome XIII-R subtelomeric sequence (KS2438 and KS2439) were annealed and digested with BamHI and EcoRI, generating the BamHI-EcoRI XIIIR fragment. Two DNA fragments, containing both the TG81 sequence and the HO cleavage site or the HO cleavage site alone, were obtained by PCR with oligonucleotides (KS1679 and KSX007) using the pTG81-HO plasmid (Hirano and Sugimoto, 2007) as a template. A DNA fragment containing both the TG81 sequence and the HO cleavage site was digested with MfeI and NotI and cloned together with the BamHI-EcoRI XIIIR fragment into BamHI/NotI–treated pTG81-HO, resulting in pXIIIR-TG81-HO. Another DNA fragment containing the HO cleavage site alone was digested with EcoRI and NotI and cloned together with the BamHI-EcoRI XIIIR fragment into BamHI/NotI–treated pTG81-HO, resulting in pXIIIR-HO. The pTAG2-TG81-HO, pTAG4-TG81-HO, pTAG10-TG81-HO, or pCTA10-TG81-HO plasmid was constructed as follows. First, DNA fragments containing 2xTTAGGG, 4xTTAGGG, 10xTTAGGG, or 10xCCCTAA were generated by PCR using pTG81-HO as a template. The PCR primers are KS1255 and KS2324 for 2x TTAGGG, KS2325 for 4xTTAGGG, KS2110 for 10xTTAGGG, or KS2327 for 10xCCCTAA. The resulting PCR fragments were treated with NcoI and EcoRI and cloned together with the MfeI-NotI DNA fragment containing both the TG81 sequence and the HO cleavage site (see earlier discussion) into NcoI/NotI–treated pTG81-HO. The placO8-TG81-HO plasmid was generated as follows. First, the placO8-HO plasmid containing 8xlacO was generated. The 4xlacO sequence was fused to the KanMX cassette by PCR with the primer pair (KS1255 and KS1746) using pTG81-HO as a template, and the resulting PCR fragment was digested with NcoI and EcoRI and cloned into EcoRI/NcoI–treated pTG81-HO, generating pLacO4-HO. To duplicate 4xlacO, an MfeI-NotI fragment containing 4xlacO was cloned into EcoRI/NotI–treated placO4-HO plasmid. Finally, the MfeI-NotI fragment containing the TG81 sequence and HO cleavage site (see earlier discussion) was cloned into EcoRI/NotI–treated placO8-HO. Similarly, an MfeI-NotI fragment containing the 4xtetO sequence and the HO site was prepared from ptetO4-HO (Hirano et al., 2009) and cloned into EcoRI/NotI-treated placO8, generating the placO8-tetO4-HO plasmid. The pXIIIR-TG22-HO-CA plasmid was constructed as follows. First, the TG22 sequence and the XIIIR sequence were fused by PCR with the KSX006-KS2577 primer set using the pTG22-HO-CA plasmid (Hirano and Sugimoto, 2007) as a template. The PCR product was treated with EcoRI and SalI and cloned into EcoRI/SalI–digested pTG22-HO-CA. The BamHI-EcoRI XIIIR fragment (see earlier discussion) was introduced into BamHI/EcoRI–treated pTG22-HO-CA, generating pXIIIR-HO-CA. The HO-CA construct was described previously (Hirano and Sugimoto, 2007). All the foregoing plasmids were cleaved with NotI and SalI to direct integration into the ADH4 locus. The XIIIR and 10XTTAGGG sequence fused to the TG81 sequence were counted as a telomere sequence after the TG81 ends were converted to telomeres (Supplemental Figure 3), consistent with the previous findings that TTAGGG repeats, if placed centromere-proximal to TG repeats, behave as a telomere sequence (Berthiau et al., 2006; Bah et al., 2011).
To construct YCp plasmids carrying TBF1, the coding region of TBF1 together with the 5′- and 3′-noncoding region was amplified from genomic DNA by PCR with the KS2330-KS2331 primer set and digested with BamHI and SalI. The resulting fragment was cloned into YCplac22 or YCplac33 (Gietz and Sugino, 1988), generating YCpT-TBF1 or YCpU-TBF1, respectively. To construct HA- or FLAG-tagged TBF1, two copies of an HA or FLAG epitope were fused to the N-terminal coding sequence by PCR using the KS2337-2338 or KS2335-2336 primer set, respectively (Wakayama et al., 2001). The tagged constructs were cloned into YCplac22, YCplac33, pRS314, or pRS316 (Sikorski and Hieter, 1989). The N-terminal deletion or amino acid substitution mutations of TBF1 were introduced by PCR-based methods (Hirano et al., 2009). The oligonucleotides for PCR were KS2446 and KS2492 (tbf1-ΔN), KS2497 and KS2498 (tbf1-25; L177A, Q180A), or KS2503 and KS2504 (tbf1-26; L187A, Q190A). The resulting DNA fragments containing tbf1 mutations were cloned into pRS314 or pRS316. The plasmids containing LacI or LacI-Tbf1 fusion genes were constructed as follows. A DNA fragment containing the lacI coding sequence and the SV40 nuclear localization signal were amplified by PCR with the KS1747-KS1756 primer set from pAFS78 (Straight et al., 1996) and digested with SacI and SalI. The resulting DNA fragment was cloned into YCplac33 or YCplac111 (Gietz and Sugino, 1988) carrying a truncated version of the ADH1 promoter, resulting in YCpU-lacI or YCpL-lacI, respectively. The coding sequences of TBF1 wild-type or mutation genes were amplified by PCR with the KS2091-2092 primer set, digested with SacI and SacII, and cloned into YCpU-lacI or YCpL-lacI. The LacI-Tbf1, FLAG-TBF1, or HA-TBF1 construct fully rescues the lethality of tbf1Δ mutants (data not shown). The YCpU-EST1 plasmid was constructed by transferring the PstI-XbaI fragment of the EST1 gene on pVL499 (obtained from V. Lundblad, Salk Institute) to YCplac33. The plasmid ptetR-RAP1 was previously described (Hirano et al., 2009). The ADE1- or TRP1 marked GAL-HO plasmid was described elsewhere (Hirano et al., 2009).
Strains
The heat-inducible TBF1 degron (tbf1-d) was constructed as described previously (Kanemaki et al., 2003). As a result the tbf1-d construct is fused to a myc-epitope. A his3 version of the KSC006 strain (ade1, his2, leu2, trp1, ura3) (Wakayama et al., 2001) was constructed as follows. First, a HIS2 fragment (obtained from C. Wittenberg, Scripps Research Institute) was introduced into its own locus to create a His+ strain. Next the HIS3 gene was disrupted by a PCR strategy using the hisG-URA3-hisG construct (Alani et al., 1987). Finally, Ura− cells were selected out on plates containing 5-FOA (Boeke et al., 1987). To delete the KanMX marker from the degron construction, KanMX was replaced with URA3 by using the plasmid M4758 (Voth et al., 2003). Cells carrying an ura3 version of the degron construct were obtained by selecting on plates containing 5-FOA. Epitope tagging and integration or deletion construction were performed by PCR-based methods (Wach et al., 1994; Reid et al., 2002; Janke et al., 2004). The MEC1-HA, MRE11-myc, RAP1-HA, and TEL1-HA constructs were as described previously (Wakayama et al., 2001; Nakada et al., 2003a; Hirano and Sugimoto, 2007; Hirano et al., 2009). The stn1-13 mutation was obtained from M. Charbonneau (Universite de Tours, France; Grandin et al., 2001). Strains used in this study are listed in Supplemental Table S1.
Telomere shortening by loss of the EST1 gene
First, tbf1-d est1Δ cells carrying YCpU-EST1 were streaked on plates containing 5-FOA to obtain Ura− colonies that had lost the EST1 plasmid. Cells from a single colony were grown in sucrose medium overnight. The culture was then diluted fourfold and grown for 7 h. Similar dilution and culture were repeated. During the fifth serial dilution culture, cells showed decreased growth rate (IJpma and Greider, 2003). Cells from the first, second, third, or fourth serial dilution cultures were grown in sucrose medium for 2 h after diluting to a density of ∼5 × 107/ml. Cells were then treated with nocodazole (15 μg/ml) at 30°C for 2 h to block further cell division. The cultures were then shifted to 37°C after addition of galactose (to a final concentration of 2%) to deplete Tbf1 protein or maintained at 30°C after addition of glucose to retain Tbf1 expression.
Other methods
Southern blotting was performed as described, except for using 32P-labeled hybridization DNA probes (Hirano et al., 2009). The ChIP assay was described previously (Hirano et al., 2009; Fukunaga et al., 2011). The PCR primers were KS1744 and KS1745 for the HO site and KS2037 and KS2038 for the SMC2 locus. HO cleavage for each cassette was analyzed by performing real-time PCR with input samples used for the ChIP assay. HO cutting efficiency represents the ratio of the signal from the HO site using the cutting-site primer pair (KS1311 and KS1312) after HO induction to the signal before HO induction. Signals from the HO site were normalized to signals from the control SMC2 locus. Immunoprecipitation and immunoblotting were performed as described (Fukunaga et al., 2011). Rad53 was detected with anti-Rad53 antibody (gift from John Diffley, Cancer Research UK London Research Institute). Tubulin was detected with the monoclonal TAT-1 antibody (Woods et al., 1989). DNA flow cytometry analysis was performed as described previously (Burke et al., 2000; Nakada et al., 2003b). Oligonucleotides used in this study are listed in Supplemental Table S2.
Supplementary Material
Acknowledgments
We thank C. Newlon for critical reading; Y. Kwon, Y. Ishino, and T. Shirai for helpful discussion; E. Henry for technical assistance; and H. Araki, M. Charbonneau, J. Diffley, V. Lundblad, D. Stillman, and C. Wittenberg for sending materials. This work was supported by National Institutes of Health Grants GM073876 and CA148939 (K.S.).
Abbreviations used:
- ATM
ataxia-telangiectasia mutated
- ChIP
chromatin immunoprecipitation
- DSB
double-strand break
- 5-FOA
5-fluoroorotic acid
- MMS
methyl methanesulfonate
- MRX
Mre11-Rad50-Xrs2
- RPA
replication protein A
- STR
subtelomeric repeat element
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-06-0568) on November 30, 2011.
REFERENCES
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MK, Zakian VA. Rap1p telomere association is not required for mitotic stability of a C(3)TA(2) telomere in yeast. EMBO J. 2003;22:1688–1696. doi: 10.1093/emboj/cdg154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneric M, Lingner J. Tel1 kinase and subtelomere-bound Tbf1 mediate preferential elongation of short telomeres by telomerase in yeast. EMBO Rep. 2007;8:1080–1085. doi: 10.1038/sj.embor.7401082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bah A, Gilson E, Wellinger RJ. Telomerase is required to protect chromosomes with vertebrate-type T2AG3 3′ ends in Saccharomyces cerevisiae. J Biol Chem. 2011;286:27132–27138. doi: 10.1074/jbc.M111.220186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthiau AS, Yankulov K, Bah A, Revardel E, Luciano P, Wellinger RJ, Geli V, Gilson E. Subtelomeric proteins negatively regulate telomere elongation in budding yeast. EMBO J. 2006;25:846–856. doi: 10.1038/sj.emboj.7600975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Shore D. Increased association of telomerase with short telomeres in yeast. Genes Dev. 2007;21:1726–1730. doi: 10.1101/gad.438907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:164–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- Brevet V, Berthiau AS, Civitelli L, Donini P, Schramke V, Geli V, Ascenzioni F, Gilson E. The number of vertebrate repeats can be regulated at yeast telomeres by Rap1-independent mechanisms. EMBO J. 2003;22:1697–1706. doi: 10.1093/emboj/cdg155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigati C, Kurtz S, Balderes D, Vidali G, Shore D. An essential yeast gene encoding a TTAGGG repeat-binding protein. Mol Cell Biol. 1993;13:1306–1314. doi: 10.1128/mcb.13.2.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Dawson D, Stearns T. Methods in Yeast Genetics. NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Chang M, Arneric M, Lingner J. Telomerase repeat addition processivity is increased at critically short telomeres in a Tel1-dependent manner in Saccharomyces cerevisiae. Genes Dev. 2007;21:2485–2494. doi: 10.1101/gad.1588807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockell MM, Lo Presti L, Cerutti L, Cano Del Rosario E, Hauser PM, Simanis V. Functional differentiation of tbf1 orthologues in fission and budding yeasts. Eukaryot Cell. 2009;8:207–216. doi: 10.1128/EC.00174-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad MN, Wright JH, Wolf AJ, Zakian VA. RAP1 protein interacts with yeast telomeres in vivo: overproduction alters telomere structure and decreases chromosome stability. Cell. 1990;63:739–750. doi: 10.1016/0092-8674(90)90140-a. [DOI] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- Diede SJ, Gottschling DE. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell. 1999;99:723–733. doi: 10.1016/s0092-8674(00)81670-0. [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Fukunaga K, Kwon Y, Sung P, Sugimoto K. Activation of protein kinase Tel1 through recognition of protein-bound DNA ends. Mol Cell Biol. 2011;31:1959–1971. doi: 10.1128/MCB.05157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Grandin N, Damon C, Charbonneau M. Ten1 functions in telomere end protection and length regulation in association with Stn1 and Cdc13. EMBO J. 2001;20:1173–1183. doi: 10.1093/emboj/20.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Sussel L, Shore D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992;6:801–814. doi: 10.1101/gad.6.5.801. [DOI] [PubMed] [Google Scholar]
- Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- Hector RE, Shtofman RL, Ray A, Chen BR, Nyun T, Berkner KL, Runge KW. Tel1p preferentially associates with short telomeres to stimulate their elongation. Mol Cell. 2007;27:851–858. doi: 10.1016/j.molcel.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Fukunaga K, Sugimoto K. Rif1 and Rif2 inhibit localization of Tel1 to DNA ends. Mol Cell. 2009;33:312–322. doi: 10.1016/j.molcel.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Sugimoto K. Cdc13 telomere capping decreases Mec1 association but does not affect Tel1 association with DNA ends. Mol Biol Cell. 2007;18:2026–2036. doi: 10.1091/mbc.E06-12-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogues H, Lavoie H, Sellam A, Mangos M, Roemer T, Purisima E, Nantel A, Whiteway M. Transcription factor substitution during the evolution of fungal ribosome regulation. Mol Cell. 2008;29:552–562. doi: 10.1016/j.molcel.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJpma A, Greider CW. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Kanemaki M, Sanchez-Diaz A, Gambus A, Labib K. Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo. Nature. 2003;423:720–724. doi: 10.1038/nature01692. [DOI] [PubMed] [Google Scholar]
- Koering CE, Fourel G, Binet-Brasselet E, Laroche T, Klein F, Gilson E. Identification of high affinity Tbf1p-binding sites within the budding yeast genome. Nucleic Acids Res. 2000;28:2519–2526. doi: 10.1093/nar/28.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol. 2010;8:e1000329. doi: 10.1371/journal.pbio.1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Zakian VA. The Saccharomyces CDC13 protein is a single-strand TG1-3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc Natl Acad Sci USA. 1996;93:13760–13765. doi: 10.1073/pnas.93.24.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008;22:125–140. doi: 10.1101/gad.1626908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis EJ. The chromosome ends of Saccharomyces cerevisiae. Yeast. 1995;11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- Louis EJ, Vershinin AV. Chromosome ends: different sequences may provide conserved functions. Bioessays. 2005;27:685–697. doi: 10.1002/bies.20259. [DOI] [PubMed] [Google Scholar]
- Lustig AJ, Kurtz S, Shore D. Involvement of the silencer and UAS binding protein RAP1 in regulation of telomere length. Science. 1990;250:549–553. doi: 10.1126/science.2237406. [DOI] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Carlos AR, Gomez-Lopez G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12:768–780. doi: 10.1038/ncb2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee JS, Phillips JA, Chan A, Sabourin M, Paeschke K, Zakian VA. Reduced Rif2 and lack of Mec1 target short telomeres for elongation rather than double-strand break repair. Nat Struct Mol Biol. 2010;17:1438–1445. doi: 10.1038/nsmb.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Cooper JP. The telomere protein Taz1 is required to prevent and repair genomic DNA breaks. Mol Cell. 2003;11:303–313. doi: 10.1016/s1097-2765(03)00041-8. [DOI] [PubMed] [Google Scholar]
- Moser BA, Nakamura TM. Protection and replication of telomeres in fission yeast. Biochem Cell Biol. 2009;87:747–758. doi: 10.1139/o09-037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Hirano Y, Sugimoto K. Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol Cell Biol. 2004;24:10016–10025. doi: 10.1128/MCB.24.22.10016-10025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Hirano Y, Tanaka Y, Sugimoto K. Role of the C terminus of mec1 checkpoint kinase in its localization to sites of DNA damage. Mol Biol Cell. 2005;16:5227–5235. doi: 10.1091/mbc.E05-05-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003a;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Shimomura T, Matsumoto K, Sugimoto K. The ATM-related Tel1 protein of Saccharomyces cerevisiae controls a checkpoint response following phleomycin treatment. Nucleic Acids Res. 2003b;31:1715–1724. doi: 10.1093/nar/gkg252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Ribaud V, Bianchi A, Shore D. DNA breaks are masked by multiple Rap1 binding in yeast: implications for telomere capping and telomerase regulation. Genes Dev. 2007;21:292–302. doi: 10.1101/gad.400907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent CI, Hughes TR, Lue NF, Lundblad V. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 1996;274:249–252. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- Nugent CI, Lundblad V. The telomerase reverse transcriptase: components and regulation. Genes Dev. 1998;12:1073–1085. doi: 10.1101/gad.12.8.1073. [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Jeggo PA. The role of double-strand break repair—insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Pennock E, Buckley K, Lundblad V. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell. 2001;104:387–396. doi: 10.1016/s0092-8674(01)00226-4. [DOI] [PubMed] [Google Scholar]
- Petreaca RC, Chiu HC, Nugent CI. The role of Stn1p in Saccharomyces cerevisiae telomere capping can be separated from its interaction with Cdc13p. Genetics. 2007;177:1459–1474. doi: 10.1534/genetics.107.078840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Stark JM, Araujo FD, Moynahan ME, Berwick M, Jasin M. Double-strand breaks and tumorigenesis. Trends Cell Biol. 2001;11:S52–59. doi: 10.1016/s0962-8924(01)02149-3. [DOI] [PubMed] [Google Scholar]
- Preti M, Ribeyre C, Pascali C, Bosio MC, Cortelazzi B, Rougemont J, Guarnera E, Naef F, Shore D, Dieci G. The telomere-binding protein Tbf1 demarcates snoRNA gene promoters in Saccharomyces cerevisiae. Mol Cell. 2010;38:614–620. doi: 10.1016/j.molcel.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Reid RJ, Lisby M, Rothstein R. Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol. 2002;350:258–277. doi: 10.1016/s0076-6879(02)50968-x. [DOI] [PubMed] [Google Scholar]
- Ribaud V, Ribeyre C, Damay P, Shore D. DNA-end capping by the budding yeast transcription factor and subtelomeric binding protein Tbf1. EMBO J. 2011;31:138–149. doi: 10.1038/emboj.2011.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol Cell. 2002;9:857–869. doi: 10.1016/s1097-2765(02)00507-5. [DOI] [PubMed] [Google Scholar]
- Sabourin M, Tuzon CT, Zakian VA. Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell. 2007;27:550–561. doi: 10.1016/j.molcel.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Zakian VA. Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell. 2002;9:1055–1065. doi: 10.1016/s1097-2765(02)00532-4. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet T, et al. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res. 2011;21:1028–1038. doi: 10.1038/cr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- Straight AF, Belmont AS, Robinett CC, Murray AW. GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol. 1996;6:1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol. 2005;15:1364–1375. doi: 10.1016/j.cub.2005.06.063. [DOI] [PubMed] [Google Scholar]
- Vega LR, Mateyak MK, Zakian VA. Getting to the end: telomerase access in yeast and humans. Nat Rev Mol Cell Biol. 2003;4:948–959. doi: 10.1038/nrm1256. [DOI] [PubMed] [Google Scholar]
- Viscardi V, Bonetti D, Cartagena-Lirola H, Lucchini G, Longhese MP. MRX-dependent DNA damage response to short telomeres. Mol Biol Cell. 2007;18:3047–3058. doi: 10.1091/mbc.E07-03-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodenicharov MD, Laterreur N, Wellinger RJ. Telomere capping in non-dividing yeast cells requires Yku and Rap1. EMBO J. 2010;29:3007–3019. doi: 10.1038/emboj.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodenicharov MD, Wellinger RJ. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol Cell. 2006;24:127–137. doi: 10.1016/j.molcel.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Voth WP, Jiang YW, Stillman DJ. New “marker swap” plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast. 2003;20:985–993. doi: 10.1002/yea.1018. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;13:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wakayama T, Kondo T, Ando S, Matsumoto K, Sugimoto K. Pie1, a protein interacting with Mec1, controls cell growth and checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:755–764. doi: 10.1128/MCB.21.3.755-764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J Cell Sci. 1989;93:491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- Wotton D, Shore D. A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev. 1997;11:748–760. doi: 10.1101/gad.11.6.748. [DOI] [PubMed] [Google Scholar]
- Xu L, Petreaca RC, Gasparyan HJ, Vu S, Nugent CI. TEN1 is essential for CDC13-mediated telomere capping. Genetics. 2009;183:793–810. doi: 10.1534/genetics.109.108894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, Songyang Z. Human telomeric proteins occupy selective interstitial sites. Cell Res. 2011;21:1013–1027. doi: 10.1038/cr.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B-BS, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.