

Abstract
The trp RNA-binding attenuation protein (TRAP) is a paradigmatic allosteric protein that regulates the tryptophan biosynthetic genes associated with the trp operon in Bacilli. The ring-shaped 11-mer TRAP is activated for recognition of a specific trp-mRNA target by binding up to 11 tryptophan molecules. To characterize the mechanisms of tryptophan-induced TRAP activation we have performed methyl relaxation dispersion (MRD) nuclear magnetic resonance (NMR) experiments that probe the time-dependent structure of TRAP in the microsecond to millisecond “chemical exchange” time window. We find significant side chain flexibility localized to the RNA and tryptophan binding sites of the apo protein, and that these dynamics are dramatically reduced upon ligand binding. Analysis of the MRD NMR data provides insights into the structural nature of transiently populated conformations sampled in solution by apo TRAP. The MRD data are inconsistent with global two-state exchange, indicating that conformational sampling in apo TRAP is asynchronous. These findings imply a temporally heterogeneous population of structures that are incompatible with RNA binding, and substantiate the study of TRAP as a paradigm for probing and understanding essential dynamics in allosteric, regulatory proteins.
Keywords: TRAP, tryptophan, trp, CPMG, methyl relaxation dispersion
Introduction
Allosteric effects are pervasive in cellular mechanisms for regulating gene expression. The undecameric (11-mer) trp RNA-binding attenuation protein (TRAP) is a paradigmatic allosteric protein that regulates production of enzymes involved in biosynthesis of the amino acid tryptophan (Trp) in bacilli1 via transcription attenuation and translation inhibition2. When the cellular concentration of Trp is low, TRAP remains in its apo state, and an anti-terminator hairpin is formed in nascent trp-mRNA transcripts that enables transcription and translation of the encoded enzymes, and hence production of Trp (Fig. S1). As cellular concentration of Trp increases, the amino acid binds to TRAP, thus activating it for high-affinity binding to trp-mRNA. The interaction of holo (Trp-bound) TRAP with nascent RNA transcripts leads to formation of a transcriptional terminator, thus precluding downstream production of Trp. In some bacilli, binding of Trp-activated TRAP to RNA can be prevented through prior interaction with the Anti-TRAP protein, thus favoring Trp production3.
Qualitative solution NMR studies of this 91 kDa oligomer4, and quantitative thermodynamic studies5 suggest that TRAP activation involves changes in both structure and dynamics, such that the flexibility exclusive to the apo protein masks its RNA-binding surface. In turn, this localized disorder hinders detailed structural insights from crystallographic studies of apo TRAP6, whereas the structure of the Trp-bound state has been defined7. In the holo TRAP structure, the Trp ligand is completely buried and secluded from solvent; thus, in order for bound Trp to be released, thereby deactivating TRAP, holo TRAP must also be somewhat flexible (albeit in a manner distinct from apo TRAP, such that RNA and Anti-TRAP can bind). Moreover, the structure of holo TRAP appears nearly unchanged when bound to RNA8-10, or to Anti-TRAP3, underscoring the importance of Trp binding to the mechanism of TRAP activation.
Severe line broadening in previous 15N-edited NMR studies of TRAP5 provided limited quantitative insights into the mechanism of TRAP activation. Here, we have used quantitative methyl-directed NMR-based chemical shift and relaxation dispersion methods to study the solution structure and dynamics of TRAP in its inactive apo and Trp-activated holo states. Experiments that focus on the sidechain methyl groups of Ile, Leu, and Val11, 12 reveal both structural differences between apo and holo TRAP, and a high degree of flexibility only in the apo protein. NMR experiments probing single- and multiple-quantum coherences, both acquired at two magnetic field strengths, enable both kinetic and structural insight into the conformational exchange processes exhibited by apo TRAP. Importantly, asynchrony in the detected dynamic processes reveals an underlying temporally heterogeneous population of structures that differ from the RNA binding, Trp-activated conformation, and underscores the role of protein dynamics in gene regulation by TRAP.
Results
Tryptophan binding alters the structure of TRAP: NMR CSPs (ΔδH, ΔδC)
Bacillus stearothermophilus TRAP is a 74-residue protein that adopts a beta sandwich fold with seven strands (A-G), and assembles into an axially-symmetric undecameric ring, with binding sites for Trp between adjacent protomers8, 13. To study the structure and dynamics of TRAP sidechains, we prepared samples in which the methyl groups of Ile, Leu, and Val were isotopically labeled with 13C and 1H, while other carbons were unlabeled and their attached hydrogens were deuterated. In addition, we substituted Ala 28 with Ile in order to probe the Trp-binding loop between strands B and C. (This point mutant binds both Trp and RNA with near-WT affinity, and exhibits similar structure and dynamics, (Figs. S2 and S3), and is hereafter referred to as “TRAP.” Residue numbers correspond to those of B. subtilis TRAP, matching the scheme used in the crystal structure of B. stearothermophilus TRAP, 1C9S.)
To monitor Trp-induced conformational changes in TRAP, 1H-13C two-dimensional correlation spectra of the protein were acquired in its apo and Trp-bound holo states. These are of high quality (Fig. 1a), reflecting the eleven-fold symmetry of the molecule, and enable resolution and stereospecific assignment of all 29 labeled methyl groups in apo and holo TRAP. Upon binding Trp, the chemical shift perturbations (CSPs) for 1H and 13C, ΔδH and ΔδC, provide a site-specific metric for the structural changes coincident with activation14 (Figs. 1a, S5); the magnitude of the CSPs are mapped to the structure of TRAP as observed in ternary complex with Trp and RNA (1C9S8). The largest CSPs map to the methyl probes nearest the Trp-binding site, including those in the inter-protomer interface, the BC loop and the RNA-binding surface (Fig. 2a). The core of the beta sandwich fold and base of the oligomeric ring exhibit no significant CSPs and thus appear structurally insensitive to Trp. These observations are consistent with the allosteric function of TRAP: Trp binding induces structural changes that propagate to the RNA-binding sites of the protein.
Fig. 1.

Chemical shift perturbations and methyl relaxation dispersion demonstrate site-specific changes in the structure and μs-ms dynamics of TRAP upon binding Trp. (a) 1H-13C correlation spectra of apo TRAP (blue) and holo TRAP (red) at 25°C. Lines connect the apo and holo signals for each of 29 methyl probes. The signal for *Ile 28δ1 at δC = 13.5 ppm in holo TRAP suggests this methyl group samples a minor state, which may reflect flexibility at the supra-ms timescale. (Inset in a) A single protomer from the 11-mer is shown in blue, with 11 bound Trp molecules in gold. (b) Medium to large dispersions exclusively in apo TRAP, but not holo TRAP, for Ile 22δ1 and Leu 24δ1 from 13C-SQ MRD at 25°C. Solid lines are fits to the site-specific two-state exchange model and dashed lines are fits to the no-exchange model. Uncertainties in the R Eff2 values (see methods) were generally on the order of 1-3 Hz and smaller than symbols shown. For Leu 24δ1, the double-headed arrow illustrates that the value of Rex corresponds to the height of the dispersion curve.
Fig. 2.
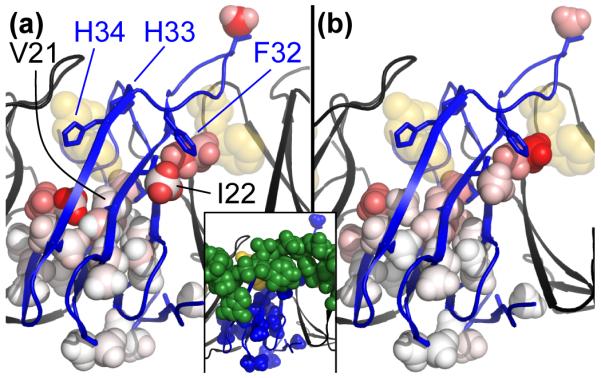
Chemical shift perturbations and Rex values mapped to 29 methyl probes in a model based on the crystal structure of holo TRAP. One TRAP protomer is shown in blue, neighboring protomers in black, bound Trp molecules in gold, and RNA shown in green spheres only in the inset figure. (a) 1H and 13C CSPs upon Trp binding, ΔδH and ΔδC, indicate the largest structural differences between apo and holo TRAP are at the Trp-binding loop, the inter-protomer interface and regions of the core near the Trp pocket and RNA surface. The color scale ranges from white (0 ppm) to red (0.37 ppm for 1H and 1.3 ppm for 13C). (b) Exchange broadening, Rex, is largest in the Trp binding loop, the inter-protomer interface and regions of the core beneath the Trp pocket and the RNA surface. In contrast, methyls at the base report Rex near zero. The color scale ranges from white (0 /s) to red (73 /s). (Inset) The RNA-binding surface coincides with regions of large apo-holo CSPs and apo Rex.
Tryptophan binding eliminates the flexibility of apo TRAP: MRD exchange-broadening (Rex)
To examine the hypothesis that protein dynamics is important for the function of TRAP, we investigated the dynamics of apo and holo TRAP in the μs-ms time window by performing 13C single quantum methyl relaxation dispersion NMR experiments (13C-SQ MRD15). The resulting “relaxation dispersion curves,” , encapsulate effects from dynamic processes that enable the protein to transiently populate alternative conformations with different chemical shifts. Fitting each curve reliably yields the dynamic metric Rex, the contribution of exchange to the transverse relaxation rate (Fig. 1b)16. We found apo TRAP to be remarkably flexible in the μs-ms time window, with 23 of 29 methyl probes yielding non-zero Rex (Figs. 2b, S4, and S5). These values are largest in the Trp-binding loop, the inter-protomer interface, and regions of the core beneath the Trp pocket and the RNA surface, whereas Rex vanishes near the base (Fig. 2b). This localized flexibility may both permit Trp-binding and prevent RNA-binding, thus enabling the functions of apo TRAP without disrupting the integrity of its oligomeric structure. Upon binding Trp, Rex is reduced significantly from an average of 14±18 /s for 29 apo signals to 1±2 /s for 30 holo signals (Figs. 1b, S4, and S5). It is worth noting that methyl groups experiencing conformational exchange may nevertheless exhibit zero Rex if the processes are too fast or slow and/or involve alternative states that are not sufficiently populated or distinguished via chemical shift16. However, the multitude of NMR probes provides confidence in the conclusion that μs-ms dynamics present in apo TRAP are almost entirely eliminated in holo TRAP, in response to Trp binding and activation.
Describing apo TRAP dynamics: evidence for independent motions
The observed values of Rex suggest important μs-ms dynamics that are unique to the inactive apo state of TRAP (Figs. 1b, 2b, S4, S5). To understand how these dynamics affect the function of apo TRAP, we sought to address the extent to which these motions reflect a “global” dynamic process involving all methyl probes moving in synchrony, versus set of “site-specific” motions wherein different parts of the protein move independently of one another16. Hence, additional MRD data were acquired and analyzed using a site-specific two-state exchange model A↔B, which comprises the kinetics (kex = kA + kB), thermodynamics (PA = kB / kex) and structural changes (|ΔωH|, |ΔωC|), for each methyl probe in apo TRAP. In this way, the presence of global motions can be evidenced by the uniformity of site-specific kex and PA values.
A rigorous fitting procedure applied to signal intensities from the 29 methyl probes reveals that 18 of 29 exhibit exchange (Rex > 0 and therefore kex > 0), 13 of 18 yield reliable kex, and 5 of 13 also yield reliable PA, |ΔωH|, and |ΔωC|, in part because they are not in the fast-exchange regime (Table S2). These fitted parameters suggest that some TRAP side chains undergo independent motions, as evidenced by non-uniform kex values (Fig. 3), which range from ~1000 /s at Leu 24δ2 at the inter-protomer interface to ~2400 /s at Ile 28δ1 in the Trp-binding loop (average 1629±366 /s; N = 13). The PA values corroborate this result by a similarly wide distribution (Fig. 4a), from 87.3±2.5% at Leu 24δ2 to 98.6±0.62% at Ile 22δ1 at the RNA surface (average 94.8±4.4%; N = 5). Moreover, some probes which appear dynamically similar by values of kex, such as Ile 22δ1, Leu 24δ1, and Leu 24δ2, are shown to be different by values of PA, which quantifies the energetic preference of their respective A states.
Fig. 3.

Site-specific exchange rate constants, kex, suggest dynamics in apo TRAP is asynchronous, not a “global” exchange process. (a) MRD data for Leu 24δ2 and Ile 28δ1 illustrate the difference between the slowest and fastest exchanging probes, at 1047±104 /s and 2391±238 /s, respectively. (b) Of the 29 methyl probes, 11 exhibit zero Rex (black rectangles at kex = 0), 13 of 29 exhibit exchange from which site-specific kex can be determined (black squares, with error bars obtained from standard deviations from Monte Carlo error analysis), and 5 of 29 exhibit exchange with ill-defined kex, due to poor signal to noise (magenta circles at kex = average(kex) with error bars spanning min(kex) to max(kex)). Because some pairs of probes correspond to the same residue number, the second probe is offset by 0.5 residue units (e.g., Val 10γ1 and Val 10γ2 are shown at Residue = 10 and 10.5, respectively). (c) kex values mapped to the structure of holo TRAP, with a color scale ranging from white (0 /s) to red (2400 /s); magenta designates kex > 0, but undefined.
Fig. 4.

Exchange kinetics and chemical shift differences obtained from relaxation dispersion data for methyl probes in apo TRAP. (a) For the 5 methyl probes that are not in the fast-exchange regime, the fits to RD data yield the population fraction, PA. The spread in these values quantitatively illustrates the extent to which these probes independently sample their alternate local conformations. In addition to site-specific fits (black), to consider the effect of a hypothetical concerted motion in TRAP, a global fit was performed that imposes a uniform kex and PA for all probes, each fixed to the average value (red circle). (b) Site-specific chemical shift differences from MRD, |ΔωC|, compared to the Trp-induced CSP |ΔδC| (Figs. 1a and S5) for methyls analyzed site-specifically (black square), or globally (red circles). The observation that most data lie above the diagonal (i.e., |ΔωC| > |ΔδC|) suggests that apo TRAP samples conformations that exceed the structural differences between the apo and holo forms of the protein. All error bars are standard deviations from Monte Carlo error analysis, except for the “Global fit” errors in (a), which correspond to standard deviations from site-specific kex and PA, and for the ΔδC errors in (b), which correspond to the line width of each NMR signal. The curved dotted line indicates the difference in |ΔωC| values between the global fit and site-specific fit, for both Ile 22δ1 and Ile 28δ1.
Considering the sparse data and the simplifying advantages to detailed analysis by assuming global two-state exchange, we examined whether the data could be reasonably explained by a global dynamic process by two fitting procedures. First, a global fit which fixes kex and PA to their average values, while optimizing site-specific |ΔωH|, |ΔωC|, and , is unable to adequately fit several signals whose site-specific fits are far from this average, as evidenced by non-random residuals (Fig. S8). Second, a global fit of the fastest-exchanging probe, Ile 28δ1, with either of the two slowest-exchanging probes, Leu 24δ1 or Leu 24δ2, also appears inadequate, even when the shared kex and PA values are variable fitting parameters (Fig. S9). Finally, although these data could be fit using more complex models of exchange such as global three-state or two-state and a local jump, the additional model parameters would increase uncertainty in the fitted parameters with only modest improvements in the residuals. Hence, these simple two-state analyses are consistent with the interpretation that μs-ms dynamics in apo TRAP is asynchronous, at least in part, with different methyl groups experiencing different exchange processes. By comparison, holo TRAP exhibits more conformational coherence in this time window, as per the small values of Rex (Figs. S4, S5).
Apo TRAP samples divergent structures: MRD chemical shifts (Δω)
The magnitude of chemical shift differences between exchanging A and B states, |ΔωH| and |ΔωC|, were obtained from fits to the MRD data for the five methyl probes. Here, we focus on |ΔωC| because it is a more robust indicator of sidechain conformational change than its 1H counterpart, which is strongly influenced by through-space effects14, 17. Although the magnitude of a chemical shift change is not a perfect metric for the magnitude of structural change, the finding that |ΔωC| for these methyl probes exceeds the magnitude of the shift perturbations induced by Trp binding, |ΔδC|, suggests that these methyl probes in apo TRAP exhibit structural excursions in excess of the differences between the apo and Trp-bound states (Fig. 4b). For example, Ile 22δ1 exhibits a mere |ΔδC| = 0.1 ppm shift upon Trp-binding, but in the apo state, the sampled orientations differ by |ΔωC| = 1.5 ppm! This structural interpretation holds even if exchange kinetics are considered globally uniform, wherein |Δω| values can be extracted for more than five probes (Figs. 3 and 4a). Hence, the inferred high degree of conformational sampling, which is exclusive to the apo state, is consistent with the dynamics of apo TRAP both masking the RNA binding site and making the otherwise-secluded Trp-pocket accessible for ligand entry.
Discussion
Solution structure of apo TRAP
Trp-induced activation of TRAP likely requires changes in both structure and dynamics4,5. In order to understand the unique structural features of apo TRAP that preclude RNA binding, we interpret the CSPs (Table S1) as perturbations to the holo TRAP crystal structure (1C9S8). In general, the 1H shift δH is strongly dependent on through-space effects from electron-rich functional groups, whereas the 13C shift δC encapsulates through-bond effects from side chain structure14, 17. In other words, δC reports methyl orientation relative to its side chain, whereas δH reports methyl orientation relative to its local environment, and therefore, reports the conformations of adjacent residues. Although we note that the magnitude of chemical shift changes need not necessarily be linearly correlated to a unique conformational coordinate, this analysis (detailed in Table S1) forms the basis for the following structural hypotheses describing the inactive apo state of TRAP.
The observation that CSPs are localized to the vicinity of the Trp-binding pocket and RNA-binding site (Fig 2a) suggest that RNA binding to apo TRAP is precluded by only a sparse set of highly localized uniquely-apo side chain conformations; this implies that the structural component of the TRAP activation requires only a few key intramolecular interactions.
The small ΔδH and ΔδC from Val 21γ1, which is proximal to bound Trp (Fig. 2a), might be explained by a conformational change that allows either His 33 or His 3413 to occupy the empty pocket in apo TRAP. Such a conformational change could conceivably replace the effects on δH from the Trp ring current with those from the His sidechain, yet without facilitating other intramolecular contacts that stabilize the activated conformation of TRAP. Notably, mutation of each of these conserved histidines to Ala results in binding defects to both Trp and RNA18. (In the crystallographic model of apo TRAP, the sidechain of Arg 31 is modeled in the Trp-binding pocket, albeit with weak electron density6.)
Methyl CSPs suggest that the conserved Phe 3213, which contacts both RNA8, 18 and Anti-TRAP3 in their respective complexes, adopts a different orientation in apo TRAP. This hypothesis is primarily evidenced by the adjacent Ile 22δ1, which reports only a small change in sidechain structure upon Trp binding, via ΔδC, but a large change in local magnetic field, via ΔδH (Fig 2a). This pattern implies that Phe 32 has reoriented upon Trp binding because it is the only proximal functional group that could magnetically shield the structurally-undisturbed Ile 22. Additional support for this hypothesis arises via large ΔδH in Leu 24δ1 and Leu 24δ2, but is confounded by their proximity to His 34 and the Trp ligand, both of which may contribute to the observed ΔδH.
Dynamic conformational sampling by apo TRAP
In order to synthesize our knowledge regarding the role of dynamics in the functions of apo TRAP, we interpret the two-state exchange parameters kex, PA, |ΔωH|, and |ΔωC| from the MRD experiments (Tables S1 and S2). The observation of pervasive μs-ms dynamics via kex (Fig. 3c) indicates that the side chains of apo TRAP are nearly uniformly flexible, except near the base of the ring, and suggests that TRAP activation involves a relatively global restriction in protein dynamics (as opposed to the sparse set of structural changes induced by Trp binding, as reported by CSPs, Fig. 2).
Furthermore, based on site-specific differences in kex and/or PA (Figs. 3b, 4a), there appear to be multiple independent modes of motion in apo TRAP, which are reported on by methyl side chains at the Trp binding loop, at the inter-protomer interface, and at the core of each protomer (n.b., this independence is amplified in the context of the TRAP 11-mer, which is relevant for RNA binding, if each protomer is independently flexible). Interestingly, the Leu 24 methyl groups, δ1 and δ2, report different dynamic experiences (Fig 4a), presumably because they probe distinct structural locations of TRAP. Specifically, Leu 24δ1 is pointed toward the protomer core in holo TRAP, and reports a large |ΔωH| of 0.1±0.02 ppm in apo TRAP, which likely results from the aromatic sidechain of Phe 32. In contrast, Leu 24δ2 is pointed toward the inter-protomer interface in holo TRAP, and reports a near-zero |ΔωH| of 0±0.02 ppm. This result makes it unlikely that the dynamics of apo TRAP involve coherent sampling of a B state structure with a particular function (e.g., a holo-like state). Instead, we suggest that dynamic incoherence in apo TRAP ensures structural heterogeneity between adjacent protomers, thereby preventing population of conformations of the TRAP ring that are active for binding to RNA or Anti-TRAP; the dynamics likely also function locally to permit binding and dissociation of Trp.
Finally, the magnitude of conformational sampling in apo TRAP is relatively large, as indicated by the site-specific values of |ΔωC| (Fig 4b). This result underscores the importance of the observed dynamic excursions (i.e., they should not be ignored in a mechanistic description of TRAP), and clarifies the magnitude of conformational restriction that is coincident with Trp-binding and subsequent activation.
Dynamics of holo TRAP
Holo TRAP exhibits only minor flexibility in the μs-ms time window, most apparent under the RNA-binding surface and inter-protomer interface via Ile 22δ1 and Ile 45δ1 (Figs. 1b and S4). Exchange at these sites is slower and of smaller magnitude than its apo counterpart, consistent with activation via altered dynamics. Moreover, slower dynamics in the BC loop may allow sampling a conformation that permits release of bound Trp (Fig. 1a).
Mechanism for allosteric activation of TRAP
Protein dynamics are becoming broadly understood as necessary for enabling the allosteric behavior of macromolecules19-21, whether allostery occurs through induced fit22 or conformational sampling23 mechanisms. In the case of TRAP, the data suggest the undecameric ring exhibits both types of behaviors. Asynchronous conformational sampling at the protomer level makes it unlikely that multiple adjacent protomers will adopt a holo-like conformation, though such conformations can be stabilized in apo TRAP at reduced temperatures6. The conformational sampling likely also involves conformations that enable binding of Trp to an otherwise solvent-secluded site. Upon binding of Trp to one or multiple binding sites, the energetics of the conformational landscape are likely dramatically shifted in an induced-fit manner24 that favors structural, if not dynamic, synchrony between adjacent protomers, thereby favoring the activated state of TRAP.
In addition to supporting this general premise for the role of dynamics in allostery, the findings reported here move beyond this broader perspective to provide site-specific insight into the manner by which protein dynamics enable the function of apo TRAP. Analysis of methyl chemical shift perturbations and relaxation dispersion suggests that apo TRAP is characterized by incoherent μs-ms dynamic motions (Figs. 3 and 4a) that may both enable Trp binding and prevent RNA binding in the absence of adequate amounts of Trp. In contrast, Trp-activated holo TRAP is characterized by a discontinuous set of structural adjustments (Fig. 2a), and broader attenuation of μs-ms dynamics (Figs 1b and 5) that accommodate binding to RNA. Moreover, this extensive conformational restriction has been corroborated by a large negative heat capacity change ΔCp upon Trp binding5. Thus, allosteric activation of TRAP occurs via (1) a sparse set of localized, structural changes, and (2) a global change in incoherent protein dynamics. This finding underscores the importance of dynamics in TRAP function and suggests that attenuation of TRAP dynamics may be sufficient to enable RNA binding, whether induced by Trp binding, mutagenesis25, 26 or reduced temperatures6. This study substantiates the role of TRAP as a paradigm for probing and understanding essential dynamics in allosteric, regulatory proteins.
Fig. 5.

Tryptophan binding alters the conformational landscape of TRAP, which permits distinct functions of the apo and holo states. Apo TRAP dynamics permit binding to Trp, yet not to trp-mRNA. Hypothetically, this requires flexibility in the Trp loop and the core, which links the Trp pocket and RNA surface. As these motions are incoherent, the inactive protein is characterized by a “rugged” distribution of conformations in the μs-ms time window. In contrast, holo TRAP functions to bind trp-mRNA, and to release bound Trp. Activation by Trp attenuates the μs-ms flexibility, thus focusing the distribution of conformations and imparting a large negative heat capacity change ΔCp5, which may introduce the RNA scaffold. The μs-ms dynamics are quantified via Rex from 13C-SQ MRD at 25°C, and are color-mapped to 29 sites in each of 11 protomers of the apo and holo donuts. The color scales range from white (0 /sec, more rigid) to red (73 /sec, more flexible).
Materials and Methods
Sample preparation
TRAP mutagenesis, expression, and purification protocols were described previously4, with minor alterations. Mutagenesis was performed using the Stratagene QuikChange method using the WT B. stearothermophilus gene as the template and oligonucleotide primers designed to change Ala 28 to Ile. Candidate clones were sequenced by the Roswell Park Biopolymers sequencing facility and a clone with the desired substitutions was transformed into E. coli BL21(DE3). Three isotope labeling schemes were used: (1) U-[2H, 13C, 15N], ILV-[13CH3]27 for total correlation spectroscopy (TOCSY) and out-and-back spectra, (2) 10%-[13C] for stereo-assignments28, and (3) U-[2H, 15N], ILV-[13CH3] for relaxation measurements29. Samples for resonance assignments were 600-900 μM TRAP (11-mer) in H2O NMR buffer (100 mM NaCl, 50 mM NaPO4, pH 8.0 at 25°C, 0.02% NaN3), while MRD spectra were recorded with 130-145 μM TRAP 11-mer in D2O NMR buffer.
Molecular visualization
Pymol (http://pymol.sourceforge.net) was used to generate figures and introduce the A28I in silico mutation to 1C9S8.
NMR spectroscopy
Apo and holo WT TRAP methyl signals were assigned using several experiments, NMR samples (1) and (2), and backbone assignments4, all at 50-55°C. Assignments were transferred from WT to A28I TRAP by spectral comparison (Fig. S2). Apo TRAP was assigned from six NMR spectra: (1) 3D C(CO)NH-TOCSY, (2) 3D H(CCO)NH-TOCSY, (3, 4, 5) 2D methyl “out and back” experiments27 correlating (Cm−x, Hm) where x = {0, 1, 2} is the number of C-C magnetization-transfers from methyl carbon towards backbone, and (6) a 2D 13C-edited 1H-1H NOESY. Holo TRAP was assigned from five NMR spectra: (1) 3D C(CO)NH-TOCSY, (2) 3D H(CCO)NH-TOCSY and the methyl “out and back” experiments. Stereo-specific assignments for apo and holo TRAP were obtained from a 2D non-decoupled 1H, 13C-HSQC28.
Dispersion curves for each of 4 MRD conditions were obtained by recording 12 2D spectra: one reference spectrum with T = νCPMG = 0, and 11 CPMG spectra with T = 20 ms acquired in random order using νCPMG = 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1000 Hz. The 4 MRD conditions consisted of 1H,13C-MQ30, acquired twice with different 13C carrier frequencies30, or 13C-SQ15: (1) 25°C, 800 MHz, MQ, (2) 25°C, 800 MHz, SQ (apo and holo), (3) 25°C, 600 MHz, MQ, and (4) 25°C, 600 MHz, SQ. Processing, assignment, and analysis were conducted with NMRPipe31 and NMRViewJ32. Temperatures were calibrated with ethylene glycol, and chemical shifts were referenced with DSS.
MRD analysis
Dispersions were calculated via , where I(νCPMG) is the signal intensity in the spectrum with refocusing duration TCPMG at frequency νCPMG, and I0 is the intensity in the spectrum with no refocusing block. Intensities were extracted using NMRPipe31, with errors via standard deviation from repeat experiments. Each dispersion curve was fit with a custom software package, GUARDD (Kleckner, et al., in press; http://code.google.com/p/guardd/), programmed in-house using MATLAB (The Mathworks, Inc.). Dispersion curves were fit to Carver-Richards-style formulae that describe two-site exchange A↔B for MQ or SQ dispersions30, 33. Each curve requires specification of 5 types of parameters: (1, 2) |ΔωH| and |ΔωC|, the ppm magnitude of 1H and 13C chemical shift differences between states A and B (note: |ΔωH| is fixed to zero only for 13C-SQ curves), (3) PA, the equilibrium population fraction of state A, (4) kex = kA + kB,, the rate of exchange between states A and B. kA and kB are the rates of exchange from A→B and B→A, respectively, with kA = (1-PA)kex and kB = PAkex. (5) , the transverse relaxation rate in the absence of exchange assuming 33, 34. When fitting multiple dispersion curves, these 5 parameters were shared by way of three different models. (A) The site-specific model aggregates up to all four curves from a single NMR probe into a group (e.g., Ile 28δ1), and optimizes shared values of |ΔωH|, |ΔωC|, P, and kex for all curves, as well as one for each curve. (B) The group-optimized model aggregates curves from multiple NMR probes into a group (e.g., two probes: Ile 28δ1 + Leu 24δ1), with variable PA and kex used for all curves in the group, and variable |ΔωH| and |ΔωC| for each set of curves from the same NMR probe, and one variable for each curve. (C) The group-fixed model is the same as (B) except that PA and kex are fixed for all curves in the group, instead of being variable fitted parameters (because for large groups, varying PA and kex in addition to site-specific |Δω| makes it difficult to find the global best fit). In general, RD data can be challenging to interpret because of their multivariate nature35, 36, and because PA and Δω are unknown if the NMR probe is in the fast-exchange regime (kex >> Δω, or α→2.0, where α = d(ln(Rex )) d(ln(Δω)) 37). Hence, to assess the sensitivity of the final fit to the initial conditions, the data in each group for models (A) and (B) were fit multiple times using a grid search over 4D parameter space which varied |ΔωH| from [0.01, 0.1] ppm in 2 steps, |ΔωC| from [0.1, 2.0] ppm in 3 steps, PA from [0.8, 0.999] in 4 steps and kex from [500, 3,500] /s in 4 steps. Each grid point specified initial conditions for the fit optimization, which minimized the sum of fitted residuals, χ2, via MATLAB’s fmincon function. For each group, a χ2 map was produced and visually inspected to either (a) select the best fit, or (b) conclude that the data cannot be fit accurately (e.g., due to multiple apparently-reasonable fitting solutions). If a single reasonable fit was obtained, 100 iterations of Monte Carlo error estimation was used to assess the sensitivity of the final fit to noise in the data38. For many probes, fast-exchange precluded knowledge of PA, |ΔωH|, and |ΔωC|, as evidenced by a wide range in parameter values that yield an equivalent quality of fit, assessed via χ2, as well as the calculated value of α which approached 2.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01GM077234 to M.P.F. and P.G. The authors thank L. Kay and P. Neudecker (Univ. of Toronto), C. Yuan and J. Sachleben (Ohio State Univ.) for aid in NMR experiments. The authors are grateful for paradigmatic discussions with Foster Lab members, and to A. Simmons for manuscript review.
Abbreviations
- TRAP
trp RNA-binding attenuation protein
- Trp
tryptophan
- NMR
nuclear magnetic resonance
- CSP
chemical shift perturbation
- MRD
methyl relaxation dispersion
- SQ
single quantum
- MQ
multiple quantum
- Rex
NMR exchange broadening
- sec
second
- s
second
- ms
millisecond (10−3 s)
- μs
microsecond (10−6 s)
- Δδ
chemical shift difference upon binding tryptophan
- Δω
chemical shift difference upon exchange from A state to B state
- PA
population of A state
- kex
total exchange rate between A and B states
- kA
exchange rate from A to B
- kB
exchange rate from B to A
- α
scaling parameter for timescale of chemical exchange
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
Author contributions: I.R.K., P.G., and M.P.F. designed research, I.R.K. and P.G. performed research, I.R.K. and MPF analyzed data, and I.R.K. and M.P.F. wrote the paper.
Glossary
(Not applicable)
Accession numbers
Data deposition: The NMR chemical shifts of apo and holo B. stearothermophilus A26I TRAP methyl groups have been deposited to the BioMagResBank (www.bmrb.wisc.edu), with accession numbers 17400 and 17413, respectively.
References
- 1.Gollnick P, Babitzke P, Antson A, Yanofsky C. Complexity in regulation of tryptophan biosynthesis in bacillus subtilis. Annu. Rev. Genet. 2005;39:47–68. doi: 10.1146/annurev.genet.39.073003.093745. [DOI] [PubMed] [Google Scholar]
- 2.Du H, Tarpey R, Babitzke P. The trp RNA-binding attenuation protein regulates trpg synthesis by binding to the trpg ribosome binding site of bacillus subtilis. J. Bacteriol. 1997;179:2582–2586. doi: 10.1128/jb.179.8.2582-2586.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watanabe M, Heddle JG, Kikuchi K, Unzai S, Akashi S, Park S, Tame JRH. The nature of the TRAP-anti-TRAP complex. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2176–2181. doi: 10.1073/pnas.0801032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McElroy C, Manfredo A, Wendt A, Gollnick P, Foster M. Trosy-NMR studies of the 91kda TRAP protein reveal allosteric control of a gene regulatory protein by ligand-altered flexibility. J. Mol. Biol. 2002;323:463–473. doi: 10.1016/s0022-2836(02)00940-3. [DOI] [PubMed] [Google Scholar]
- 5.McElroy CA, Manfredo A, Gollnick P, Foster MP. Thermodynamics of tryptophan-mediated activation of the trp RNA-binding attenuation protein. Biochemistry. 2006;45:7844–7853. doi: 10.1021/bi0526074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malay AD, Watanabe M, Heddle JG, Tame JRH. Crystal structure of unliganded TRAP: implications for dynamic allostery. Biochem. J. 2011;434:427–434. doi: 10.1042/BJ20101813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antson AA, Brzozowski AM, Dodson EJ, Dauter Z, Wilson KS, Kurecki T, Otridge J, Gollnick P. 11-fold symmetry of the trp RNA-binding attenuation protein (TRAP) from bacillus subtilis determined by x-ray analysis. J. Mol. Biol. 1994;244:1–5. doi: 10.1006/jmbi.1994.1698. [DOI] [PubMed] [Google Scholar]
- 8.Antson AA, Dodson EJ, Dodson G, Greaves RB, Chen X, Gollnick P. Structure of the trp RNA-binding attenuation protein, TRAP, bound to RNA. Nature. 1999;401:235–242. doi: 10.1038/45730. [DOI] [PubMed] [Google Scholar]
- 9.Hopcroft NH, Manfredo A, Wendt AL, Brzozowski AM, Gollnick P, Antson AA. The interaction of RNA with TRAP: the role of triplet repeats and separating spacer nucleotides. J. Mol. Biol. 2004;338:43–53. doi: 10.1016/j.jmb.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 10.Hopcroft NH, Wendt AL, Gollnick P, Antson AA. Specificity of TRAP-RNA interactions: crystal structures of two complexes with different RNA sequences. Acta Crystallogr. D Biol. Crystallogr. 2002;58:615–621. doi: 10.1107/s0907444902003189. [DOI] [PubMed] [Google Scholar]
- 11.Foster MP, McElroy CA, Amero CD. Solution NMR of large molecules and assemblies. Biochemistry. 2007;46:331–340. doi: 10.1021/bi0621314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruschak AM, Kay LE. Methyl groups as probes of supra-molecular structure, dynamics and function. J. Biomol. NMR. 2010;46:75–87. doi: 10.1007/s10858-009-9376-1. [DOI] [PubMed] [Google Scholar]
- 13.Chen XP, Antson AA, Yang M, Li P, Baumann C, Dodson EJ, Dodson GG, Gollnick P. Regulatory features of the trp operon and the crystal structure of the trp RNA-binding attenuation protein from bacillus stearothermophilus. J. Mol. Biol. 1999;289:1003–1016. doi: 10.1006/jmbi.1999.2834. [DOI] [PubMed] [Google Scholar]
- 14.Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Meth. Enzymol. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
- 15.Lundström P, Vallurupalli P, Religa TL, Dahlquist FW, Kay LE. A single-quantum methyl 13c-relaxation dispersion experiment with improved sensitivity. J. Biomol. NMR. 2007;38:79–88. doi: 10.1007/s10858-007-9149-7. [DOI] [PubMed] [Google Scholar]
- 16.Kleckner IR, Foster MP. An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta. 2011;1814:942–968. doi: 10.1016/j.bbapap.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.London RE, Wingad BD, Mueller GA. Dependence of amino acid side chain 13c shifts on dihedral angle: application to conformational analysis. J. Am. Chem. Soc. 2008;130:11097–11105. doi: 10.1021/ja802729t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang M, Chen XP, Militello K, Hoffman R, Fernandez B, Baumann C, Gollnick P. Alanine-scanning mutagenesis of bacillus subtilis trp RNA-binding attenuation protein (TRAP) reveals residues involved in tryptophan binding and RNA binding. J. Mol. Biol. 1997;270:696–710. doi: 10.1006/jmbi.1997.1149. [DOI] [PubMed] [Google Scholar]
- 19.Kern D, Zuiderweg ERP. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Tzeng S, Kalodimos CG. Protein dynamics and allostery: an NMR view. Curr. Opin. Struct. Biol. 2010;21:62–67. doi: 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 22.Koshland DEJ, Némethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 23.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 24.Sullivan SM, Holyoak T. Enzymes with lid-gated active sites must operate by an induced fit mechanism instead of conformational selection. Proc. Natl. Acad. Sci. U.S.A. 2008;105:13829–13834. doi: 10.1073/pnas.0805364105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Payal V, Gollnick P. Substitutions of Thr30 provide mechanistic insight into tryptophan-mediated activation of TRAP binding to RNA. Nucleic Acids Res. 2006;34:2933–2942. doi: 10.1093/nar/gkl383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li PTX, Gollnick P. Characterization of a trp RNA-binding attenuation protein (TRAP) mutant with tryptophan independent RNA binding activity. J. Mol. Biol. 2004;335:707–722. doi: 10.1016/j.jmb.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase g using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- 28.Senn H, Werner B, Messerele BA, Weber C, Traber R, Wuthrich K. Stereospecific assignment of the methyl H-1-NMR lines of valine and leucine in polypeptides by nonrandom C-13 labeling. FEBS Lett. 1989;249:113–118. [Google Scholar]
- 29.Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- 30.Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE. Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: application to a 723-residue enzyme. J. Am. Chem. Soc. 2004;126:3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- 31.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on unix pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 32.Johnson BA, Blevins RA. NMR view - a computer-program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 33.Korzhnev DM, Kloiber K, Kay LE. Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: theory and application. J. Am. Chem. Soc. 2004;126:7320–7329. doi: 10.1021/ja049968b. [DOI] [PubMed] [Google Scholar]
- 34.Ishima R, Torchia DA. Accuracy of optimized chemical-exchange parameters derived by fitting CPMG R2 dispersion profiles when R2(0a) not = R2(0b) J. Biomol. NMR. 2006;34:209–219. doi: 10.1007/s10858-005-6226-7. [DOI] [PubMed] [Google Scholar]
- 35.Kovrigin EL, Kempf JG, Grey MJ, Loria JP. Faithful estimation of dynamics parameters from CPMG relaxation dispersion measurements. J. Magn. Reson. 2006;180:93–104. doi: 10.1016/j.jmr.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Ishima R, Torchia DA. Error estimation and global fitting in transverse-relaxation dispersion experiments to determine chemical-exchange parameters. J. Biomol. NMR. 2005;32:41–54. doi: 10.1007/s10858-005-3593-z. [DOI] [PubMed] [Google Scholar]
- 37.Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J. Am. Chem. Soc. 2000;122:2867–2877. [Google Scholar]
- 38.Motulsky H, Christopoulos A. In: Fitting models to biological data using linear and nonlinear regression. a practical guide to curve fitting. Graphpad Software Inc., editor. Graphpad Software Inc.; 2003. p. 104. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.