

Abstract
It is of pivotal importance for genome stability that repair DNA polymerases (Pols), such as Pols λ and β, which all exhibit considerably reduced fidelity when replicating undamaged DNA, are tightly regulated, because their misregulation could lead to mutagenesis. Recently, we found that the correct repair of the abundant and highly miscoding oxidative DNA lesion 7,8-dihydro-8-oxo-2′-deoxyguanine (8-oxo-G) is performed by an accurate repair pathway that is coordinated by the MutY glycosylase homologue (MutYH) and Pol λ in vitro and in vivo. Pol λ is phosphorylated by Cdk2/cyclinA in late S and G2 phases of the cell cycle, promoting Pol λ stability by preventing it from being targeted for proteasomal degradation by ubiquitination. However, it has remained a mystery how the levels of Pol λ are controlled, how phosphorylation promotes its stability, and how the engagement of Pol λ in active repair complexes is coordinated. Here, we show that the E3 ligase Mule mediates the degradation of Pol λ and that the control of Pol λ levels by Mule has functional consequences for the ability of mammalian cells to deal with 8-oxo-G lesions. Furthermore, we demonstrate that phosphorylation of Pol λ by Cdk2/cyclinA counteracts its Mule-mediated degradation by promoting recruitment of Pol λ to chromatin into active 8-oxo-G repair complexes through an increase in Pol λ’s affinity to chromatin-bound MutYH. Finally, MutYH appears to promote the stability of Pol λ by binding it to chromatin. In contrast, Pol λ not engaged in active repair on chromatin is subject for proteasomal degradation.
Keywords: base excision repair, Mule E3 ubiquitin ligase
Genetic stability is of crucial importance for any form of life and if not properly maintained can result in many human diseases (1). Reactive oxygen species (ROS) are among the many insults that can affect the stability of DNA by causing damage to the highly reactive DNA bases, such as guanine. Because of its prevalence and high mutagenic potential, 8-oxo-2′-deoxyguanine (8-oxo-G) is recognized as one of the most abundant mutagenic oxidative DNA lesions arising from such insults (reviewed in ref. 2). The cardinal problem with 8-oxo-G is that the majority of polymerases (Pols), including the three replicative Pols α, δ, and ϵ, bypass 8-oxo-G in an inaccurate manner by frequently incorporating the “wrong” adenine (A) opposite 8-oxo-G. This error can lead to the formation of GC → TA transversion mutations, which in turn can give rise to diseases such as cancer (3). In sharp contrast to the other Pols, mammalian Pol λ, a member of the X family Pols (4), is the main Pol capable of correctly handling an oxidatively damaged DNA strand with very high fidelity in collaboration with the auxiliary factors proliferating cell nuclear antigen (PCNA) and replication protein A (RP-A), and incorporates over 1000-fold more efficiently the correct cytosine (C) opposite 8-oxo-G than the incorrect A (5, 6) in vitro. Furthermore, we have shown the existence of an accurate repair pathway for 8-oxo-G that is coordinated by the MutY glycosylase homologue (MutYH) and Pol λ in vitro and in vivo (7). These findings suggest that Pol λ is the most likely candidate among the fifteen mammalian Pols to play an important role in the accurate repair of oxidative DNA lesions and that this task is achieved by correctly using the damaged (oxidized) DNA strand as a template (7, 8).
Components of DNA repair complexes and especially of base excision repair (BER) need to be tightly regulated in order to guarantee that they are active only when needed [(9) and as discussed in ref. 10]. This regulation is of special importance for the DNA repair Pols, which show a much lower fidelity in polymerization of long stretches of DNA than the replicative Pols, and therefore could introduce many point mutations when replicating undamaged DNA. This hypothesis is supported by increasing evidence that deregulation of Pol λ and also other translesion synthesis Pols including Pol β can lead to diseases in general (11) and cancer in particular (12). Also, it has been shown that repair Pols are overexpressed in many tumors, a feature that may contribute to disease manifestation (13) further strengthening the idea that a tight control of repair Pols is pivotal. Nevertheless, the regulation of DNA repair enzymes, and Pols in particular, is so far poorly understood. Posttranslational modifications (PTMs) constitute a fascinating means of regulation to ensure proper temporal and spatial organization of repair components in the cell. Data from the Dianov lab have shown that BER components, such as Pol β, a close relative to Pol λ, undergo ubiquitination by the ubiquitin E3 ligase carboxy terminus of Hsc70 interacting protein (CHIP), which is an E3 ubiquitin ligase containing a C-terminal U box domain providing interaction with an E2 enzyme and an N-terminal tetratricopeptide mediating its interaction with heat shock proteins (14). CHIP plays an important role in the heat shock response (15, 16) and has been shown to be involved in regulating cellular levels of proteins like p53 (17). CHIP-mediated ubiquitination of Pol β leads to its degradation by the proteasome under normal circumstances (9, 18). However, upon DNA damage, those BER proteins are stabilized and recruited to chromatin to fulfill their roles in the maintenance of genomic integrity in vivo.
To date, very little is known about the regulation of Pol λ during the cell cycle. In previous work, we have shown that Pol λ interacts with cyclin-dependent kinase 2 (Cdk2) and is phosphorylated in vitro by the Cdk2/cyclinA complex (19). Phosphorylation per se does not affect the polymerization activity of Pol λ, but phosphorylation is decreased when Pol λ interacts with PCNA. Furthermore, the phosphorylation pattern of Pol λ coincides with the presence of Cdk2/cyclinA during the cell cycle. In follow-up work we demonstrated that phosphorylation prevents Pol λ from being degraded by the ubiquitin-proteasome pathway in vivo (20).
In the present study, we were interested in elucidating how the levels of Pol λ are controlled, how phosphorylation promotes its stability, and how the engagement of Pol λ in active repair complexes is coordinated. We found that Pol λ can be ubiquitinated by the E3 ligase Mule in vitro and in vivo and that this interaction is functionally connected to the phosphorylation-dependent stabilization of Pol λ by Cdk2/cyclinA. Importantly, the control of Pol λ levels by Mule has functional consequences for the ability of mammalian cells to deal with 8-oxo-G lesions in vitro. Furthermore, we demonstrate that phosphorylation of Pol λ by Cdk2/cyclinA counteracts its Mule-mediated degradation by promoting recruitment of Pol λ to chromatin into active 8-oxo-G repair complexes through an increase in Pol λ’s affinity to chromatin-bound MutYH in vitro and in vivo. Finally, MutYH appears to promote the stability of Pol λ by binding it to chromatin. In contrast, Pol λ not engaged in active repair on chromatin is subject for proteasomal degradation. Our data elucidate how precisely and tightly PTMs can control Pol λ’s activity status along with its overall cellular levels by orchestrating its subcellular localization and stability.
Results
Identification of Mule as E3 Ligase for DNA Polymerase λ.
In order to shed more light on the regulation of Pol λ in vivo, we set out to identify the E3 ubiquitin ligase responsible for the ubiquitination of Pol λ. To this end, we tested fractions originating from an assay established by the Dianov lab (9), where Pol λ ubiquitination activity was purified from HeLa whole cell extracts in a series of biochemical fractionations via chromatographic columns combined with an in vitro ubiquitination assay (Fig. S1A). The final Mono Q fractions D14–D10 clearly displayed Pol λ mono- and polyubiquitination activity of Pol λ (Fig. 1A), and they were sent for mass spectrometric analysis to identify the E3 ligase present in the fractions. The MS/MS data clearly identified Mule to be the major ubiquitin E3 ligase present in these fractions (18). In the same fractions, the E3 ligase CHIP was also identified and has been shown to ubiquitinate Pol λ in vitro (21). Mule is a 482 kDa protein in which the catalytically active homologous to the E6-AP carboxyl terminus (HECT) domain has been mapped to the C-terminal 370 amino acids (22) and has previously been shown to play a role in the regulation of another member of the Pol X family, Pol β (18). We thus used the recombinant truncated catalytically active HECT domain of Mule and confirmed that this protein mono- and polyubiquitinates Pol λ in a concentration-dependent manner in vitro (Fig. 1B). The ubiquitination reaction could be efficiently supported by any of the three E2 conjugating enzymes H5b, H5c, or H7 (Fig. S1B). When the band of in vitro ubiquitinated Pol λ was excised from a Coomassie stained SDS-PAGE gel (Fig. S1C) and subjected to MS/MS analysis, two lysines (K27 and K273) were identified to be ubiquitinated (Fig. S1D). In vitro ubiquitination assays were performed by using Pol λ with mutated K27, K273, or both residues using a mutant ubiquitin, not capable of forming polyubiquitin chains, in order to better quantitatively visualize the total ubiquitination. Although the single K273R mutant did not display any significantly reduced in vitro ubiquitination by Mule, both the single K27R and the double (K27R/K273R; named K2R) ubiquitination-deficient mutants of Pol λ showed 10 and 5 times reduced in vitro ubiquitination by Mule, respectively (Fig. S1E), suggesting that K27 is the major site of ubiquitination. The residual ubiquitination of the 2KR mutant Pol λ that was observed most probably stems from other K residues in the vicinity of K27 and K273 that are minor ubiquitination sites and can be ubiquitinated more strongly after the loss of the two main ubiquitination sites identified in the MS/MS approach.
Fig. 1.

Identification of Mule as an E3 ubiquitin ligase regulating cellular protein levels of DNA polymerase λ. (A) E3 ubiquitin ligase activity of the final Mono Q fractions from HeLa cell extracts (D14–D10) against Pol λ protein. (B) Ubiquitination of Pol λ by the purified recombinant HECT domain of Mule. (C) Effect of siRNA-mediated Mule knockdown on Pol λ levels in HEK 293T cells, analyzed by Western blotting. (D and F) Quantification of protein levels shown in C and E (three independent experiments each) showing mean + SD and p values obtained from one-sample t tests performed on the data. The Pol λ signal was normalized to tubulin. (E) Effect of siRNA-mediated ARF knockdown on Pol λ levels in HEK 293T cells, analyzed by Western blotting.
Next, we addressed the question of whether Mule also regulates the protein levels of Pol λ in vivo. To test this, we knocked down Mule in HEK 293T cells by siRNA and found that this knockdown resulted in a significant increase in Pol λ protein levels (p = 0.006) (Fig. 1 C and D). The increase in Pol λ levels upon knockdown of Mule was less pronounced, but still significant (p = 0.006), in HeLa cells (Fig. S2 A and B), which are known to overexpress Mule protein (23). At the same time, the mRNA level of Pol λ remained constant (Fig. S2E), which suggests that transcription of the POLL gene was not affected by the knockdown of Mule. The effect of the Lipofectamine control was similar to controls using either nonspecific siRNA or siRNA against luciferase (Fig. S3 A and B).
Mule directly binds and ubiquitinates targets like p53 (22) and, thus, represses p53-mediated tumor suppressor functions leading to cell cycle arrest or aging (24). In response to DNA damage, the alternative reading frame of the INK4a/ARF locus (ARF) protein is induced, inhibits the activity of Mule, and thus leads to a stabilization of Mule targets as p53 (22) and Pol β (18). Importantly, ARF has not been shown to influence the levels of Mule but merely to inhibit its ubiquitin ligase activity (18, 22). To test whether this inhibitory effect of ARF on Mule affects Pol λ levels as well, we next knocked down ARF by siRNA. As expected, protein levels of Pol λ decreased upon knockdown of ARF in either HEK 293T (Fig. 1E and F) in HeLa cells (Fig. S2 C and D) and reflected the effect of increased Mule activity, even though the decrease was too small to prove significant (p = 0.204 for HEK and p = 0.166 for HeLa, respectively). At the same time, the mRNA level of Pol λ remained constant (Fig. S2E), which further supports the evidence that Mule regulates Pol λ protein levels by ubiquitination-dependent proteasomal degradation.
The Correct 8-oxo-G Bypass by DNA Polymerase λ Is Regulated Through Mule.
We have previously shown that Pol λ is the Pol that bypasses 8-oxo-G containing DNA most accurately and incorporates the correct C opposite the lesion in vitro and that the specificity of correct nucleotide incorporation is provided by RP-A, PCNA, and MutYH in vitro (5–7). Therefore, we set out to determine whether modulation of Pol λ levels by Mule affects the amount of bypass of 8-oxo-G lesions. To assess this question, single-nucleotide incorporation assays using crude cell extracts with a primer-template combination that allows the quantitative monitoring of the incorporation of incorrect dATP or correct dCTP opposite a template containing 8-oxo-G (Fig. 2A) were performed as previously described (5). Crude cell extracts from HEK 293T cells treated with either siRNA against Mule [showing elevated levels of Pol λ (Fig. 1 C and D)] or Lipofectamine as a control were prepared, and their single-nucleotide incorporation activity opposite 8-oxo-G was analyzed. The extracts from cells with a Mule knockdown showed higher levels of correct C incorporation opposite 8-oxo-G than the control extracts, consistent with a role for Pol λ, or another Pol that is regulated by Mule, in the bypass of 8-oxo-G in vivo (Fig. 2 B and C and Fig. S4). When the relative differences of dCTP to dATP incorporation were analyzed, the extracts treated with siRNA against Mule showed a higher relative difference of dCTP to dATP incorporation (Fig. 2C) compared to the control-treated extracts. This difference could be further stimulated by the addition of PCNA and RP-A to the reaction, which is in line with our previous findings that PCNA and RP-A stimulate the correct bypass of 8-oxo-G by Pol λ (5).
Fig. 2.

The extent of error-free bypass of 8-oxo-G by DNA polymerase λ in human and mouse cell extracts is dependent on Mule. (A) 5′ labeled DNA primer/template pair used for the single-nucleotide incorporation assays. The first incorporation event is opposite 8-oxo-G. The incorporation of dATP yields a 40-mer product, whereas incorporation of dCTP gives rise to a 40-mer and a 41-mer. (B) Single-nucleotide incorporation by crude extracts from HEK 293T cells treated with siRNA against Mule. Experiments were performed with 10 μg of extracts, 10 μM dATP or dCTP, and +/- RP-A (40 nM) and PCNA (100 nM), respectively. (C) Quantification for B, mean of three independent experiments ± SD. (D) siRNA-mediated knockdown of Mule in Pol λ +/+ or -/- MEFs. (E) Single-nucleotide incorporation by extracts shown in D, performed with 10 μg of crude extracts and 0.5 μM dATP or dCTP, respectively. (F) Quantification for B, mean of four independent experiments ± SD.
To show that the amount of error-free bypass of 8-oxo-G lesions specifically depends on Pol λ and not on another Pol, and to confirm the functional effect of modulation of Pol λ levels by Mule in another cell line, we knocked down Mule by siRNA in Pol λ + /+ and Pol λ - /- mouse embryonic fibroblasts (MEFs) (Fig. 2D). We found that in Pol λ + /+ MEFs the cellular protein levels of Pol λ were increased upon Mule knockdown (Fig. 2D). Importantly, only in cell extracts generated from MEFs containing Pol λ (Pol λ + /+ MEFs) was the incorporation of dCTP higher in extracts when Mule was knocked down in comparison to the Lipofectamine-only–treated cells (Fig. 2 E and F). This result corroborated the fact that the increase in 8-oxo-G lesion bypass observed after Mule knockdown specifically depends on Pol λ.
The Phosphorylation Status of DNA Polymerase λ Regulates Its Subcellular Localization and thus Orchestrates Its Degradation Mediated by Mule.
In order to investigate the functional link between the stabilizing phosphorylation of Pol λ and its degradation by Mule, we analyzed HEK 293T cells stably transfected with either myc-Pol λ WT or myc-Pol λ 4A (a phosphorylation-deficient mutant of Pol λ lacking all four phosphorylation sites S167A, S177A, S230A, and T553A), because we previously observed that the lack of phosphorylation in this mutant increases its ubiquitination and thus leads to decreased cellular protein levels (20). As expected, examination of total cellular protein levels confirmed a reduction of 75% of the levels of Pol λ 4A compared to the WT protein (Fig. 3A). Furthermore, the majority of the Pol λ WT protein was present in a phosphorylated form (P-Pol λ), whereas the 4A mutant clearly showed a faster mobility following SDS-PAGE analysis. Treatment of these cells with siRNA against Mule dramatically increased the levels of Pol λ 4A by 3.9-fold compared to an increase of 1.4-fold in the WT protein (Fig. 3 B and C), which hints toward an inhibitory effect of phosphorylation of Pol λ on its degradation by Mule. In order to examine the mechanism of this cross-talk between phosphorylation and ubiquitination more closely, we next fractionated the stable HEK 293T Pol λ WT and 4A cells into cytoplasmic, nuclear, and chromatin-bound protein fractions. Analysis of these extracts revealed a substantial decrease in chromatin-bound Pol λ 4A mutant compared to the WT protein (Fig. 3 D and E). A different fractionation protocol of these cells into soluble and chromatin fractions under harsher conditions further corroborated the difference in chromatin association between the WT Pol λ protein and its 4A mutant (Fig. S5). Taken together, these findings indicated that phosphorylation of Pol λ establishes or enhances its interaction either directly with chromatin or with a protein tightly bound to chromatin and that this binding to chromatin prevents Pol λ from being degraded by Mule-mediated ubiquitination.
Fig. 3.

Phosphorylation of DNA polymerase λ inhibits its ubiquitination by Mule and promotes its binding to chromatin. (A) Total cellular Pol λ protein levels in HEK cells stably transfected with myc-Pol λ WT or myc-Pol λ 4A constructs. Relative Pol λ levels were normalized to tubulin and are indicated below the respective column. P-Pol λ, phosphorylated form of Pol λ. (B) siRNA mediated knockdown of Mule or Lipofectamine control treatment in HEK cells stably transfected with myc-Pol λ WT or myc-Pol λ 4A constructs.(C) Quantification for B, mean of three independent experiments +SD. The Pol λ signal was normalized to tubulin. The relative Pol λ levels of siMule treated fractions to the respective Lipofectamine-treated controls are indicated. (D) Cell fractionation of HEK cells stably transfected with myc-Pol λ WT or myc-Pol λ 4A constructs. Cyto = cytoplasmic, Nuc = nuclear, and CHR = chromatin-bound fraction. (E) Quantification for D, mean of two independent experiments ± SD. The Pol λ signal was normalized to tubulin (for the cytoplasmatic fraction), to fibrillarin (for the nuclear fraction), or to histone H1 (for the chromatin-bound fraction). 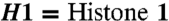 .
.
Phosphorylation of DNA Polymerase λ Enhances Its Interaction with MutYH on Chromatin and thus Regulates Its Activity State in the Cell.
The next question to be addressed was how and why phosphorylation orchestrates the subcellular localization of Pol λ and, more precisely, its binding to chromatin. We hypothesized that phosphorylation might enable and/or strengthen the interaction of Pol λ with a binding partner on chromatin. MutYH, a known interactor of Pol λ (7), recognizes 8-oxo-G:A mismatches and catalyzes the excision of the wrong A (reviewed in ref. 2). This step is followed by incision of the apurinic/apyrimidinic (AP) site by the action of apurinic/apyrimidinic endonuclease 1 (Ape1) to generate the substrate for Pol that performs the subsequent gap filling reaction (25). Thus MutYH is a protein that, according to our model of Pol λ’s involvement in correct incorporation of C opposite 8-oxo-G (7), precedes the action of Pol λ in repair of 8-oxo-G lesions. To test whether MutYH influences the subcellular localization of Pol λ, we treated HEK 293T cells with siRNA against MutYH. Fractionation of siRNA- and Lipofectamine-only–treated cells revealed that both the total cellular levels as well as the chromatin-bound fraction of Pol λ were markedly reduced upon treatment with siRNA against MutYH (Fig. 4 A and B). This finding suggested that the interaction of Pol λ with MutYH stabilizes, and possibly also recruits, Pol λ to chromatin into active repair complexes and that this interaction is dependent on, or can be enhanced by, phosphorylation of Pol λ. Thus, we next asked whether the phosphorylation status of Pol λ has any effect on its binding to MutYH. To investigate this question, recombinant purified His-tagged Pol λ was phosphorylated by Cdk2/cyclinA in an in vitro phosphorylation assay and GST-pull-down experiments were carried out by using recombinant GST-MutYH and different amounts of His-Pol λ (Fig. 4C). The results of this interaction study clearly show that phosphorylation of Pol λ strongly enhances its interaction with MutYH.
Fig. 4.

Chromatin-binding of DNA polymerase λ is mediated by MutYH, enhanced by phosphorylation of DNA polymerase λ by Cdk2/cyclinA, and inducible by oxidative DNA damage. (A) Cell fractionation of HEK 293T cells treated with siRNA against MutYH. Cyto = cytoplasmic, Nuc = nuclear, and CHR = chromatin-bound fraction. (B) Quantification for A, mean of two independent experiments ± SD. The Pol λ signal was normalized to tubulin (for the cytoplasmic fraction), to fibrillarin (for the nuclear fraction), or to histone H1 (for the chromatin-bound fraction). 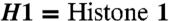 . (C) GST-pull-down of different amounts of recombinant purified His-Pol λ by recombinant purified GST-MutYH after in vitro phosphorylation of Pol λ WT by Cdk2/cyclinA. Non-P-Pol, λ = non-phosphorylated control reactions for Pol λ, were carried out as the phosphorylation reaction but without the addition of Cdk2/cyclinA. Negative control for unspecific binding was carried out by adding GST instead of GST-MutYH to the reactions. Immunoblot: GST = GST-MutYH. (D) Analysis of chromatin-bound Pol λ levels in T24 cells upon induction of oxidative DNA damage in the early S phase by H2O2 treatment. The Pol λ signal was normalized to Histone H3 and the 0h control cell time point. The cell cycle progression was monitored by analysis of cyclin E and cyclin B. (E) Quantification of D, mean of two independent experiments ± SD.
. (C) GST-pull-down of different amounts of recombinant purified His-Pol λ by recombinant purified GST-MutYH after in vitro phosphorylation of Pol λ WT by Cdk2/cyclinA. Non-P-Pol, λ = non-phosphorylated control reactions for Pol λ, were carried out as the phosphorylation reaction but without the addition of Cdk2/cyclinA. Negative control for unspecific binding was carried out by adding GST instead of GST-MutYH to the reactions. Immunoblot: GST = GST-MutYH. (D) Analysis of chromatin-bound Pol λ levels in T24 cells upon induction of oxidative DNA damage in the early S phase by H2O2 treatment. The Pol λ signal was normalized to Histone H3 and the 0h control cell time point. The cell cycle progression was monitored by analysis of cyclin E and cyclin B. (E) Quantification of D, mean of two independent experiments ± SD.
Chromatin-association of DNA Polymerase λ Is Inducible by Oxidative Stress.
The ultimate question to be answered was whether this fine-tuning of Pol λ levels had a physiological relevance under conditions of increased cellular stress due to oxidative DNA damage. To address this, cell fractionation experiments were performed by using T24 cells. T24 cells are human bladder carcinoma cells with the particular property that they are arrested in the G0 phase of the cell cycle by contact inhibition upon reaching 100% confluency. Importantly, these cells synchronously reenter the cycling phase upon seeding and, thus, enable analysis of synchronized cells without the need to use DNA damaging and cellular stress-inducing synchronization regimes, which could interfere with cell-cycle-dependent analysis of DNA repair pathways. G0-arrested T24 cells were seeded, either treated with 500 μM H2O2 for 45 min or mock-treated in the early S phase and fractionated at 2-h intervals upon release into normal medium. Analysis of chromatin fractions of H2O2-treated cells revealed an increase in chromatin-bound Pol λ levels that peaked to 1.7-fold 4 h after release compared to the control fractions, which did not show alterations in Pol λ levels (Fig. 4 D and E). Although the control cells clearly progressed through S to the G2/M phase, the H2O2-treated cells arrested in the S phase and failed to enter G2/M for the duration of the experiment (Fig. 4D). These results are consistent with an inducible chromatin recruitment of Pol λ dependent on oxidative DNA damage during the S phase.
Discussion
The data presented in this paper shed more light on the intricate control mechanisms that are in place to regulate protein levels of Pol λ, its subcellular localization, and its engagement into active repair complexes on chromatin upon induction of oxidative DNA damage. We now identify Mule as an E3 ligase responsible for ubiquitination of Pol λ, leading to degradation of Pol λ via the ubiquitin-proteasome pathway. Although we found that Mule mainly monoubiquitinates Pol λ, the formation of di- or polyubiquitin chains can also be observed. It is still unclear whether it is Mule alone that is responsible for the degradation of Pol λ or if, like in the case of Pol β (18), monoubiquitination by Mule stimulates polyubiquitination by another E3 ligase. The possible role of the E3 ligase CHIP, also found to ubiquitinate Pol λ in vitro (21), in the regulation of Pol λ levels in vivo still remains to be assessed. In line with our findings concerning the regulation of Pol λ levels by Mule, we demonstrate that this regulation influences the capacity of HEK 293T cells to perform correct 8-oxo-G repair. Furthermore, we show that this repair is mainly carried out by Pol λ, as shown with single-nucleotide incorporation experiments using Pol λ + /+ or Pol λ - /- MEF cell extracts depleted of Mule. Experiments assessing the phosphorylation of Pol λ, which has previously been found to stabilize Pol λ by decreasing its ubiquitination (20), reveal an decrease in chromatin association of nonphosphorylated Pol λ, as determined by fractionation experiments comparing HEK 293T cells stably transfected with Pol λ WT or Pol λ 4A phosphomutant. The phosphorylation-dependent chromatin binding protects and stabilizes Pol λ levels, as it prevents Pol λ from being shuttled to the cytoplasm, where it is subsequently ubiquitinated by Mule (a cytoplasmic protein) and degraded by the proteasome. Therefore, levels of Pol λ are controlled by means of changes in subcellular localization, which is dependent on the protein’s phosphorylation status. MutYH is the DNA glycosylase that catalyzes the excision of an incorrect A opposite 8-oxo-G, followed by the action of Ape1, and thus precedes the role of Pol λ in incorporating a correct C opposite 8-oxo-G. We show that phosphorylation of Pol λ enhances its binding to MutYH and that depletion of MutYH in HEK 293T cells leads to a decrease in total cellular levels of Pol λ, as well as a decrease in the chromatin-associated Pol λ fraction. Phosphorylation of Pol λ by Cdk2/cyclinA has been shown to take place in the late S and G2 phases of the cell cycle. Considering that frequent misincorporation of A opposite 8-oxo-G is performed by the replicative Pols δ and ϵ, high levels of A:8-oxo-G mispairs are expected to be present immediately after DNA synthesis in the S phase. It is pivotal that those mispairs are corrected before mitosis proceeds, because otherwise GC to TA transversion mutations can manifest themselves. Under these circumstances, the Cdk2/cyclinA phosphorylation-dependent recruitment of Pol λ to chromatin makes a lot of sense, because Pol λ is so far the most likely candidate to work together with MutYH to achieve a correct repair of A:8-oxo-G lesions. This model is further substantiated by the finding that the chromatin-bound fraction of Pol λ can be increased 1.7-fold upon induction of oxidative stress (Fig. 4 D and E) in the early S phase and is in line with previous findings implicating the involvement of Pol λ in repair of oxidative DNA damage in vivo (7).
Studies assessing the regulation of Pol β protein levels have recently shown that the levels of Pol β are regulated by Mule and CHIP in vivo (9, 18). Importantly, these studies showed that ubiquitin-mediated proteasomal degradation mainly regulates the protein levels of Pol β and not its activity. So far, we do not have any evidence indicating that ubiquitination of either Pol λ or β influences the choice of a specific Pol in the BER pathway directly. Rather, the results from this study point to the possibility that this Pol choice is brought about by other PTMs, as phosphorylation, enabling a subtle regulation of Pol’s subcellular localization, and thus contributes to the regulation of its degradation. It remains to be seen whether the polymerase activity of any of the repair Pols can be stimulated directly by means of PTMs, if PTMs control their association with other proteins to form entire repair complexes, or if their repair activity can be enhanced simply by changes in subcellular localization.
In the early Lindahl paper, a steady state of 100–500 8-oxo-G per cell was suggested (26). Subsequent work warned about artifactual DNA oxidation during isolation and suggested an amount of approximately 1,500 8-oxo-G per genome measured (see, e.g., ref. 27). Friedberg et al. indicated that around 1,000–2,000 8-oxo-G can be repaired per cell per day (28). Such a high steady-state level of DNA oxidation asks for a robust and tightly controlled repair system. With the data presented here, not only do we address the fine-tuning of Pol λ levels during physiological cellular conditions, we also provide evidence for what occurs upon induction of oxidative stress. ROS encountering a C:G base pair during any cell cycle phase devoid of DNA synthesis will lead to the formation of a C:8-oxo-G base pair, which is a substrate for Ogg1 (reviewed in ref. 2). Ogg1 will remove the damaged base 8-oxo-G, and subsequent BER will take care of the resulting AP site. Consequently, 8-oxo-G lesions caused by ROS inflicted on the cell in any of these nonreplicative phases will mainly necessitate the action of Ogg1. On the other hand, A:8-oxo-G mispairs are thought to mainly arise from inaccurate bypass of an 8-oxo-G lesion by replicative Pols during the S phase. For this reason, late S and G2 are the phases during which the removal of A opposite 8-oxo-G is needed. This notion is supported by the fact that MutYH levels reach their maximum during the S phase (29) and that the repair of A:8-oxo-G mismatches in vivo by MutYH is fourteenfold more efficient when the substrate is replication-proficient compared to a nonreplicating one (30). Those results are in accordance with a replication-associated activity of MutYH. Hence, because Pol λ is stabilized by phosphorylation by Cdk2/cyclinA in late S and G2 as well (20), we believe that the stabilization of an interaction between MutYH and Pol λ during exactly these phases of the cell cycle can promote repair of A:8-oxo-G mismatches.
Taken together, we have unveiled an important feature in the dynamics and control of Pol λ, a repair Pol pivotal for correct repair of oxidative DNA damage, which is crucial for the maintenance of genetic stability. Our results are consistent with a model in which Pol λ is recruited to and/or retained on chromatin in a phosphorylation-dependent manner into active repair complexes by MutYH in the late S and G2 phases of the cell cycle. Importantly, this recruitment is increased upon exposure of S-phase cells to oxidative DNA damaging agents. This phosphorylation-dependent chromatin recruitment further protects Pol λ from being sent to the cytoplasm, where it undergoes ubiquitination by Mule and is then sent for proteasomal degradation.
Materials and Methods
In Vitro Ubiquitination of Pol λ.
Purification of the ubiquitination activity for Pol λ from HeLa whole cell extracts and the in vitro ubiquitination assays were performed as described in ref. 18.
In Vitro Phosphorylation of Pol λ.
This experiment was performed as outlined in ref. 20.
GST Pull-down Assay of Pol λ with MutYH.
This interaction was done as recently described in ref. 7. Single-nucleotide incorporation assays were performed as described in ref. 5 with modifications as indicated in the figure legends.
RNAi Interference.
Cells were transfected by using the Lipofectamine RNAi max (Invitrogen) according to the manufacturer’s protocol and analyzed 72 h after transfection.
Statistical Analysis.
For all the statistical analysis, the program GraphPad Prism (www.graphpad.com) was used.
Full Materials and Methods can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments.
The authors thank K. Neelsen (Zürich) and R. Santoro (Zürich) for their valuable input and comments as outside experts. We also thank B. Kessler and M. Edelmann (Oxford) for expert sample preparation and proteomic analysis. E.M., B.v.L., and U.H. were supported by the Swiss National Science Foundation (SNF) and Oncosuisse and E.M. by the MD-PhD grant by the SNF and the “Forschungskredit” of the University of Zurich. J.L.P. and G.L.D. are supported by the Medical Research Council and Cancer Research United Kingdom.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110449109/-/DCSupplemental.
References
- 1.Friedberg EC, et al. DNA repair: From molecular mechanism to human disease. DNA Repair. 2006;5:986–996. doi: 10.1016/j.dnarep.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 2.van Loon B, Markkanen E, Hubscher U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair. 2010;9:604–616. doi: 10.1016/j.dnarep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hubscher U, Spadari S, Villani G, Maga G. DNA Polymerases: Discovery, Characterization and Functions in Cellular DNA Transactions. Teaneck, NJ: World Scientific; 2010. [Google Scholar]
- 5.Maga G, et al. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- 6.Maga G, et al. Replication protein A and proliferating cell nuclear antigen coordinate DNA polymerase selection in 8-oxo-guanine repair. Proc Natl Acad Sci USA. 2008;105:20689–20694. doi: 10.1073/pnas.0811241106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Loon B, Hubscher U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc Natl Acad Sci USA. 2009;106:18201–18206. doi: 10.1073/pnas.0907280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubscher U, Spadari S, Villani G, Maga G. DNA Polymerase: Discovery, Characterization and Functionsin Cellular DNA Transactions. Teaneck, NJ: World Scientific; 2010. p. 321. [Google Scholar]
- 9.Parsons JL, et al. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell. 2008;29:477–487. doi: 10.1016/j.molcel.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 10.Sobol RW. CHIPping away at base excision repair. Mol Cell. 2008;29:413–415. doi: 10.1016/j.molcel.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Ramadan K, Maga G, Hübscher U. DNA Polymerases and Diseases. Heidelberg: Springer; 2007. pp. 69–102. [Google Scholar]
- 12.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albertella MR, Lau A, O’Connor MJ. The overexpression of specialized DNA polymerases in cancer. DNA Repair. 2005;4:583–593. doi: 10.1016/j.dnarep.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Kaye FJ, et al. A family of ubiquitin-like proteins binds the ATPase domain of Hsp70-like Stch. FEBS Lett. 2000;467:348–355. doi: 10.1016/s0014-5793(00)01135-2. [DOI] [PubMed] [Google Scholar]
- 15.McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: Chaperones culling corrupt conformations. Nat Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- 16.McDonough H, Patterson C. CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esser C, Scheffner M, Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J Biol Chem. 2005;280:27443–27448. doi: 10.1074/jbc.M501574200. [DOI] [PubMed] [Google Scholar]
- 18.Parsons JL, et al. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009;28:3207–3215. doi: 10.1038/emboj.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frouin I, Toueille M, Ferrari E, Shevelev I, Hubscher U. Phosphorylation of human DNA polymerase lambda by the cyclin-dependent kinase Cdk2/cyclin A complex is modulated by its association with proliferating cell nuclear antigen. Nucleic Acids Res. 2005;33:5354–5361. doi: 10.1093/nar/gki845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wimmer U, Ferrari E, Hunziker P, Hubscher U. Control of DNA polymerase lambda stability by phosphorylation and ubiquitination during the cell cycle. EMBO Rep. 2008;9:1027–1033. doi: 10.1038/embor.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markkanen E, van Loon B, Ferrari E, Hubscher U. Ubiquitylation of DNA polymerase lambda. FEBS Lett. 2011;585:2826–2830. doi: 10.1016/j.febslet.2011.03.069. [DOI] [PubMed] [Google Scholar]
- 22.Chen D, et al. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 23.Adhikary S, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123:409–421. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 24.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 25.Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 27.Ravanat JL, et al. Cellular background level of 8-oxo-7,8-dihydro-2′-deoxyguanosine: An isotope based method to evaluate artefactual oxidation of DNA during its extraction and subsequent work-up. Carcinogenesis. 2002;23:1911–1918. doi: 10.1093/carcin/23.11.1911. [DOI] [PubMed] [Google Scholar]
- 28.Friedberg EC, Walker GC, Siede W. In: DNA Repair and Mutagenesis. 2nd Ed. Friedberg Errol C, et al., editors. Washington, DC: ASM Press; 2006. p. xxix. [Google Scholar]
- 29.Boldogh I, et al. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001;29:2802–2809. doi: 10.1093/nar/29.13.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayashi H, et al. Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr Biol. 2002;12:335–339. doi: 10.1016/s0960-9822(02)00686-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.