

Abstract
There is compelling evidence that G protein-coupled receptors exist as homo- and heterodimers, but the way these assemblies function at the molecular level remains unclear. We used here the purified leukotriene B4 receptor BLT1 stabilized in its dimeric state to analyze how a receptor dimer activates G proteins. For this, we produced heterodimers between the wild-type BLT1 and a BLT1/ALXR chimera. The latter is no longer activated by leukotriene B4 but is still activated by ALXR agonists. In this heterodimer, agonist binding to either one of the two protomers induced asymmetric conformational changes within the receptor dimer. Of importance, no G protein activation was observed when using a dimer where the ligand-loaded protomer was not able to trigger GDP/GTP exchange due to specific mutations in its third intracellular loop, establishing that the conformation of the agonist-free protomer is not competent for G protein activation. Taken together, these data indicate that although ligand binding to one protomer in the heterodimer is associated with cross-conformational changes, a trans-activation mechanism where the ligand-free subunit would trigger GDP/GTP exchange cannot be considered in this case for G protein activation. This observation sheds light into the way GPCR dimers, in particular heterodimers, could activate their cognate G proteins.
GPCRs2 are versatile biological sensors that are responsible for the majority of cellular responses to hormones and neuro-transmitters as well as for the senses of sight, smell, and taste (1, 2). Signal transduction is associated with a set of changes in the tertiary structure of the receptor that are recognized by the associated intracellular partners, in particular the G proteins (3).
It has been recently shown that receptor monomers can efficiently activate their G protein partners (4-7). However, both homo- and heterodimers have been described for many GPCRs in transfected cells, although more work is needed to extend these observations to native tissues (8). The exact molecular mechanisms governing the functioning of GPCR dimers/oligomers are not clear so far. There is increasing evidence that the two protomers in a dimer are not totally equivalent. In the case of the LTB4 receptor BLT1 homodimer, we have evidence that only one of the protomers is activated at one time (9). In the same way, only about half of the rhodopsin present in lipid nanodiscs containing two rhodopsins is available to interact with transducin, suggesting an asymmetric functioning in this case also (10). A similar explanation has been provided for the differences in G protein activation between the neurotensin receptor monomer and dimer (7). Finally, for class C receptors, several data also support a model where the receptor dimer functions in an asymmetrical way, a single heptahelical domain being activated at a time (11, 12). In the same way, only one subunit (GB2) is able to activate G proteins in the heterodimeric GABA(B) receptor (13, 14).
The existence of GPCR dimers has led to the concept that interreceptor communication within receptor dimers/oligomers by cross-conformational changes is a conceivable mechanism whereby functional properties of receptor hetero-complexes might be regulated. We investigated here the cross-conformational changes within the dimeric BLT1 receptor and their relationship to G protein activation. For this, we used a previously described chimera between BLT1 and the LXA4 receptor ALXR (15). This chimeric receptor is no longer activated by LTB4 but still by ALXR agonists. Reconstituting a dimer between a chimeric and a wild-type receptor provided us with a heterodimer where each of the subunits was selectively activated by a structurally different ligand. Our data indicate that agonist-induced activation of one of the protomers in this heterodimeric assembly, although leading to a cross-conformational change of the other subunit, is not associated with a trans-activation mechanism where the agonist-free protomer would activate G proteins.
EXPERIMENTAL PROCEDURES
Materials—LTB4 was purchased from BIOMOL Laboratories, and the WKYMVm peptide was purchased from PerkinElmer Life Sciences. 5HW was from Sigma. Asolectin was purchased from Fluka. All detergents were from Anatrace.
Protein Production—The BLT1/ALXR chimera (BLT1chim) was produced as described by Chiang et al. (15) using BLT1-W41L/W83L/W142L/W161L (16) as a template. The i3-1 mutation was introduced in BLT1 using the QuikChange multisite-directed mutagenesis kit (Stratagene) and, as a template, BLT1-W41L/W83L/W142L/W161L. All mutations were confirmed by nucleotide sequencing. BLT1, BLT1chim, and BLT1i3-1 were all expressed and purified as described by Damian et al. (9). For producing the different heterodimers used throughout this work, BLT1 or BLT1i-3 and BLT1chim were expressed as fusion proteins with an S-tag and a Strep-tag sequence after the thrombin cleavage site, respectively. The heterodimers were then refolded and purified as described by Damian et al. (9). 5HW was introduced in the receptors by biosynthetic labeling using the method described by Mesnier and Banères (17).
Ligand Binding Assays—LTB4 binding was assayed as previously described (16, 17). 125I-Labeled WKYMVm binding was measured by equilibrium dialysis. Dialysis cells from Dianorm were used with two 800-μl cavities separated by a 12-14-kDa molecular mass cut-off dialysis membrane (Spectra/Por). A series of experiments in the same conditions but in the absence of protein were carried out in parallel to measure nonspecific binding. Competition experiments were carried out in the same conditions, in the presence of 1 nm WKYMVm and increasing concentrations of the competitor compound. All protein concentrations were calculated from UV absorptivity values (Cary 400 spectrophotometer; Varian) using the extinction coefficient calculated by the method of Gill and von Hippel (18). G protein concentrations in the 10 μm range were used to ensure an efficient coupling between the receptor and the G protein, based on the Kd value measured for the receptor-G protein interaction.3 The binding profiles are presented as the degree of binding X as a function of ligand concentration. The degree of binding X is defined as mol of ligand bound/mol of receptor dimer (for a definition of the degree of binding, see Ref. 19). The titration data were analyzed using the PRISM software version 4.0 (Graphpad Inc.) by considering a set of usual models for describing ligand-receptor interactions.
GTPγS Binding Assays—GTPγS binding assays were carried out as described by Glass and Northup (20). The G proteins were produced as previously described (21). The assays were all carried out at 5 nm receptor concentration, 100 nm Gα, and 200 nm Gβγ. We systematically checked by FRET with labeled receptors (17) that no dissociation of the receptor dimer occurs at this protein concentration. G protein-binding activity was also systematically measured in the absence of agonist. The assays were carried out at 30 °C in a buffer of 10 mm MOPS, pH 7.5, 2 mm MgCl2, 1 mm EDTA, 100 mm NaCl, 0.5% (w/v) BSA, 4 μm GDP, and [35S]GTPγS (2-5 × 105 cpm). [35S]GTPγS was added to the receptor preparations, and the reaction was incubated for 10 min. Assays were stopped by the addition of 400 μl of a 0.5% cholate solution as described in Ref. 22.
Fluorescence Measurements—Fluorescence emission spectra were recorded at 20 °C on a Cary Eclipse spectrofluorimeter (Varian) with an excitation wavelength of 315 nm (band-width 2 nm). Emission was recorded 15 min after adding the ligand. We systematically checked that the fluorescence changes were not sensitive to fluid phase conditions at the detergent/lipid ratios used. For the time-dependent measurements, the ligand was added, and fluorescence was recorded at 337 nm each minute for 175 min. Receptor concentrations in the 10-8 m range were used. As in the ligand-binding measurements, a G protein concentration in the micromolar range was used to ensure an efficient coupling between the receptor and the G protein. Buffer contributions were subtracted under the same experimental conditions. The normalized fluorescence change is defined as the ratio of the 5HW emission intensity at 337 nm in the presence of agonist to that of the ligand-free receptor.
RESULTS
Production of a Chimeric BLT1 Receptor—We previously used heterodimeric complexes between the wild-type BLT1 receptor and a mutant that displayed a reduced affinity for LTB4 to analyze the ligand-induced conformational changes in a receptor dimer (9, 17). However, as we previously noted, the major limitation of this system was that the mutant still bound LTB4. To obtain a complex where we could specifically load either of the protomers with an agonist with no possibility of cross-binding between the two protomers, we produced here a heterodimer where one subunit was BLT1 and the other one a chimera between the LTB4 and the LXA4 receptors. LXA4 is an endogenous lipid that regulates leukocyte trafficking by inter-acting with a specific G protein-coupled receptor, ALXR (23). It had been shown that a chimeric BLT1 receptor encompassing the third extracellular loop and the seventh transmembrane domain of ALXR displayed specific binding of ALXR ligands with an affinity comparable with that for wild-type ALXR but failed to bind LTB4 (15). The expression level and signaling properties of this chimera were similar to those of wild-type receptors, indicating that the substitution did not affect the main three-dimensional fold of the receptor (15).
We produced here the same BLT1/ALXR chimeric receptor (referred to throughout as BLT1chim). To simplify the analysis of the fluorescence profiles used to monitor receptor activation (see below), the changes were introduced in a BLT1 receptor where all of the tryptophan residues besides Trp234 were replaced by leucines (9). Trp234 is located in TM6 (transmembrane 6) and is the only Trp residue in BLT1 whose fluorescence properties are sensitive to the activation state of the receptor (16). Mutating all of the Trp residues to Leu except for Trp234 affects neither the ligand binding nor the structural properties of BLT1 (16). BLT1chim was expressed, refolded, and purified under similar conditions as the wild-type BLT1, indicating that both receptors are likely to display similar physico-chemical properties. No significant differences were observed between both receptors when comparing their fluorescence and far-UV circular dichroism spectra (not shown), indicating that there was no major difference in their structural organization, at least in a way these methods can detect.
We next assessed the ligand-binding and agonist-induced activation properties of the chimeric receptor. Ligand binding experiments were all carried out in the presence of saturating concentration of purified Gαi2βγ proteins (see “Experimental Procedures”) to stabilize the high affinity state of the receptor. As shown in Fig. 1, BLT1chim specifically bound the ALXR agonist, WKYMVm, with high affinity (Kd = 1.26 ± 0.32 nm; n = 3). LXA4 efficiently competed with WKYMVm for binding to the purified chimeric receptor (Fig. 1B). In contrast, no specific binding of LTB4 was observed (Fig. 1B), in agreement with the data of Chiang et al. (15).
FIGURE 1.

Ligand binding to BLT1chim. Shown are direct binding of the ALXR 125I-labeled WKYMVm agonist (A) and 125I-labeled WKYMVm displacement by LXA4 (closed circles) or LTB4 (open circles) (B). Ligand-binding experiments were carried out by equilibrium dialysis as described under “Experimental Procedures.” The direct binding data are presented as a plot of the binding degree X as a function of the ligand concentration. The binding degree is defined by bound mol of ligand/mol of receptor. The experiments illustrated here are representative of three independent trials, each performed in duplicate.
Agonist-induced conformational changes were then monitored by intrinsic fluorescence, as previously described for the wild-type receptor (17). As stated above, BLT1chim contains a single Trp residue, Trp234 in TM6, whose emission spectrum is sensitive to the activation state of the receptor (16, 21). LXA4 was used as the agonist in all of the fluorescence experiments to prevent fluorescence effects arising from the peptide ligand itself. As shown in Fig. 2, LXA4 binding to BLT1chim induced an increase in the fluorescence emission intensity similar to that obtained for BLT1 in the presence of LTB4. This indicates that these two receptors are likely to be stabilized in a similar active conformation upon binding their respective agonists.
FIGURE 2.

Agonist-induced receptor activation. Shown are fluorescence emission spectra of 5HW-labeled wild-type (A) or chimeric (B) receptors in the absence of ligand (free) or in the presence of LTB4 or LXA4. Fluorescence emission spectra were recorded as described under “Experimental Procedures.”
Finally, we compared the extent of GTPγS binding at the level of Gαi2 induced by BLT1 and BLT1chim in the presence of saturating concentrations in LTB4 and LXA4, respectively. As shown in Fig. 3, very similar GTPγS binding was observed in both cases. This again indicates that the mutations introduced in BLT1chim do not affect the G protein activation properties of the receptor.
FIGURE 3.

Receptor-catalyzed [35S]GTPγS binding to the G protein. GDP/GTP exchange on Gαi catalyzed by BLT1 or BLT1chim in the presence of saturating concentrations in LTB4 or LXA4, respectively. Data are expressed as the percentage of maximal binding. In all cases, data represent the mean ± S.E. from three independent experiments.
The BLT1-BLT1chim Heterodimer—We then associated BLT1chim and the wild-type BLT1 in the same dimeric complex. BLT1 in this dimer was also devoid of its tryptophan residues besides Trp234 to simplify the fluorescence emission spectra analyses. The BLT1-BLT1chim heterodimer was produced as previously described for the dimer with a mutated protomer (17). Similar amounts of heterodimer were obtained as compared with what we described previously for the homodimer, indicating that BLT1chim is likely to behave as the wild-type receptor as far as the dimerization properties are considered. In agreement with this assumption, the heterodimeric assembly was stable as a dimer for a period of time compatible with the experiments described below (fluorescence resonance energy transfer and size exclusion chromatography evidence not shown).
We then analyzed the ligand-binding properties of the BLT1-BLT1chim dimer. All of the ligand-binding experiments reported here were carried out in the presence of purified Gαi2βγ proteins and in the absence of GTP to stabilize the high affinity state of the receptor (21). As shown in Fig. 4, the BLT1-BLT1chim heterodimeric assembly bound one LTB4 and one WKYMVm molecule, in agreement with the subunit composition of this complex. The affinity measured for each of these molecules (Kd = 0.91 ± 0.21 and 1.55 ± 0.38 nm, respectively) were similar to those reported for the isolated BLT1 and BLT1chim, indicating that associating these receptors in the same dimer does not affect the ligand-binding properties of the two protomers compared with what is observed in the homodimers.
FIGURE 4.

Ligand binding to the BLT1-BLT1chim heterodimer. Ligand-binding experiments were carried out by equilibrium dialysis as described under “Experimental Procedures” using 3H-labeled LTB (A) or 125I-labeled WKYMVm (B). The binding experiments were carried out in the presence (open symbols) or absence (closed symbols) of 500 μm WKYMVm (A) or LTB4 (B). The binding data are presented as a plot of the binding degree X as a function of the ligand concentration. The binding degree is defined by bound mol of ligand/mol of receptor. The experiments illustrated here are representative of three independent trials, each performed in duplicate.
We also measured the affinity of each of the protomers in the dimer in the presence of saturating concentrations of the agonist for the other protomer (Fig. 4). Interestingly, in this case, a strong decrease was observed in the affinity for the ligand (Kd = 107 ± 9 and 169 ± 11 nm for LTB4 and WKYMVm, respectively), indicating that occupation of one of the two ligand-binding sites in the heterodimer leads to a significant decrease in the affinity of the second protomer for its own agonist. This observation is in line with previous data establishing negative cooperative effects in heterodimeric assemblies (24). The simplest explanation would be to consider, as we previously established in the case of the purified BLT1 homodimer (9), that this effect is due to the fact that a single G protein interacts with one subunit only of the GPCR dimer, therefore stabilizing a single subunit in the high affinity state (see “Discussion”).
Functional coupling between the BLT1-BLT1chim dimer and the G protein was finally assessed by examining the ability of the purified receptor to stimulate GDP/GTPγS exchange on the αi subunit. As shown in Fig. 5, full G protein activation was observed with either LTB4 or LXA4. This indicates that both protomers in the heterodimer are fully functional in terms of G protein activation. This result also confirms what we previously showed with the BLT1 homodimer (9) (i.e. that a receptor dimer with a single protomer loaded with an agonist can trigger G protein activation).
FIGURE 5.

BLT1-BLT1chim-catalyzed [35S]GTPγS binding to the G protein. GDP/GTP exchange on Gαi catalyzed by the BLT1-BLT1chim heterodimer in the absence of agonist, in the presence of saturating concentrations in LTB4 or in the presence of saturating concentrations in LXA4. Data are expressed as the percentage of maximal binding. In all cases, data represent the mean S.E. from three independent experiments.
Agonist-induced Conformational Changes in the BLT1-BLT1chim Dimer—We next analyzed agonist-induced conformational changes in the BLT1-BLT1chim dimer using intrinsic fluorescence with dimers where one of the two protomers was labeled with 5HW (9). 5HW has a significant shoulder in its absorption spectrum at 315 nm that is absent from that of tryptophan, so that excitation at 315 nm in a mixture of 5HW-labeled and unlabeled proteins produces a fluorescence signal centered at 337 nm that is exclusively from the 5HW label (25).
Either BLT1 or BLT1chim in the BLT1-BLT1chim heterodimer was therefore labeled with 5HW, and the conformational changes of the labeled subunit were selectively monitored by fluorescence. As shown in Fig. 6, binding of the agonist to either of the subunits led to a full conformational change of the ligand-loaded protomer as well as to a partial change in the conformation of the ligand-free one, in agreement with our previous data with the BLT1 homodimer (9).
FIGURE 6.

Receptor activation in the BLT1-BLT1chim heterodimer. A, relative change in 5HW fluorescence in the BLT1-BLT1chim dimer where either of the protomers is labeled with 5HW in the absence of agonist or in the presence of either LTB4 or LXA4. The species analyzed are schematically depicted in each case, where the open box represents the wild-type protomer, the gray box represents the BLT1chim protomer, and the star represents 5HW labeling. B, relative change in 5HW fluorescence in the BLT1-BLT1chim dimer, where the labeled protomer is the chimeric one in the absence of ligand, after the addition of LTB4, or after the addition of LTB4 and then LXA4. In all of the cases, the error bar corresponds to the S.D. value calculated from three independent experiments.
To assess if the asymmetrical behavior we previously reported for the wild-type homodimer also occurred in the BLT1-BLT1chim heterodimer, we monitored the fluorescence changes in the chimeric subunit after the addition of LTB4 and, in a second stage, LXA4. Under these conditions, we should visualize the conformational changes induced by the binding of an agonist to the second protomer of a dimer where the first ligand-binding site is already occupied by its specific agonist. As clearly shown in Fig. 6B, first adding LTB4 led to a cross-conformational change in the ligand-free BLT1chim protomer. The subsequent addition of LXA4 did not affect the fluorescence emission properties of this protomer, even if binding occurred (see the binding profile in Fig. 4). As expected, the opposite behavior was observed when monitoring changes in the conformation of the wild-type protomer with a heterodimer, where the 5HW-labeled subunit was the wild-type one (not shown).
If the absence of full activation of the chimeric protomer is due to the fact that the wild-type protomer is already occupied by its own agonist, one should observe a time-dependent reversal of these effects upon displacement of LTB4 from the high affinity sites at high LXA4/LTB4 concentration ratios. To determine if this is really the case, we first bound LTB4 to the wild-type protomer, added LXA4 in excess, and monitored the changes in the emission properties of either 5HW-labeled R or Rchim as a function of time. As shown in Fig. 7, we observed under these conditions a very slow increase in the emission intensity of the Rchim protomer that was accompanied by a parallel very slow decrease in the emission intensity of the R subunit. We previously postulated that the asymmetry in the conformational features of receptor dimer was due to the G protein (9). A possibility for the very slow interconversion between the two conformations would be to consider that no dissociation of the G protein from the LTB4-activated receptor occurred in the absence of GTP. To assess if this is indeed the case, we carried out the same kind of experiment in the presence of GTP. As shown in Fig. 7, we observed in this case a significantly faster parallel change in the emission properties of R and Rchim. This indicates that asymmetry in the conformational features of the two protomers in our BLT1 heterodimer is indeed certainly due to an asymmetric coupling of the G protein to the receptor. Our observation of G protein-dependent asymmetric conformational changes is probably related to the observation that the negative cooperative effects in ligand binding by the chemokine homo- and heterodimers appear to be also promoted by the G protein (26).
FIGURE 7.

Cross-conformational changes in the R-Rchim heterodimer. Fluorescence emission changes of 5HW-labeled R (profiles 1 and 3) or Rchim (profiles 2 and 4) as a function of time. LTB4 was first bound to the R protomer in the R-Rchim dimer, a large excess in LXA4 was added (LXA4/LTB4 ratio 1000:1), and the changes in the emission properties were recorded as a function of time. Measurements were carried out in the absence (profiles 1 and 2) or the presence (profiles 3 and 4) of 0.1 mm GTP.
G Protein Activation—We next assessed if the conformation adopted by the ligand-free subunit upon binding of the agonist to the neighboring protomer was a competent one in terms of G protein activation. For this, we introduced in the wild type receptor a four-residue deletion in its third intracellular loop (BLT1Δ203-206 or BLT1i3-1). The BLT1i3-1 mutant was characterized when expressed in HEK-293 and COS cells (27). This deletion was shown to impair Gαi activation (27). We introduced the same four-residue deletion in the i3 loop of BLT1. The resulting protein was expressed and refolded under similar conditions as the wild-type receptor. No difference between the overall structure of BLT1 and BLT1i3-1 was detected by CD or fluorescence spectroscopy (data not shown), indicating that the mutation did not affect the major structural features of BLT1.
We next assessed if the i3-1 mutation indeed abrogated Gαi activation in vitro as reported under in vivo conditions. As shown in Fig. 8A, no significant GTPγS binding was observed with BLT1i3-1 even in the presence of saturating concentrations in LTB4. This indicates that BLT1i3-1 lacks the ability to trigger GDP/GTP exchange at the level of Gαi in vitro as it does in vivo. We then associated BLT1i3-1 with BLT1chim in a dimeric complex and analyzed the G protein activation properties of this BLT1i3-1-BLT1chim complex. As shown in Fig. 8B, no significant GTPγS binding on Gαi was induced by this heterodimer in the presence of saturating concentrations in LTB4.NoGprotein activation was observed whatever the LTB4 concentration was, indicating that the absence of G protein activation was not dependent on the agonist concentration (supplemental Fig. 1). Such an absence of G protein activation was not due to the fact that the two ligands were required for an efficient G protein activation, since a significant increase in GTPγS binding was observed when using the wild-type BLT1 instead of BLT1i3-1 under the same conditions (Fig. 5). Moreover, full G protein activation was obtained in the presence of LXA4, indicating that the BLT1chim protomer is fully competent for G protein activation, provided it is loaded with its agonist.
FIGURE 8.
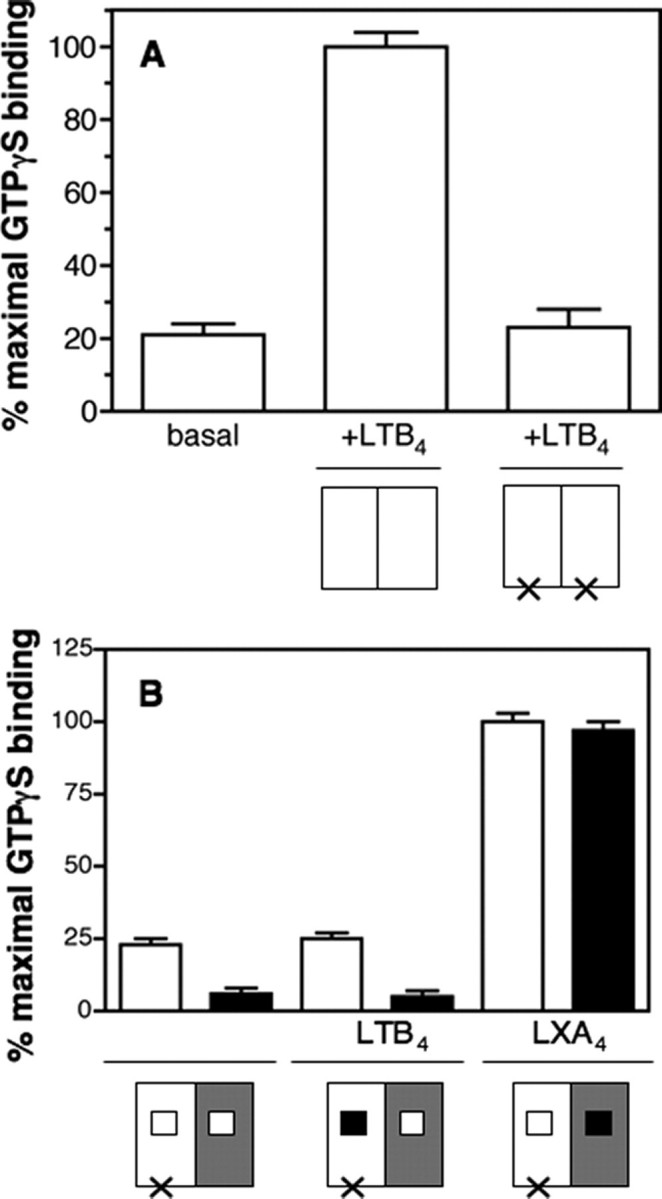
BLT1i3-1-BLT1chim-catalyzed [35S]GTPγS binding to the G protein. A, GDP/GTP exchange on Gαi catalyzed by BLT1 or BLT1i3-1 in the presence of saturating concentrations in LTB4. Data are expressed as the percentage of maximal binding. The species analyzed are schematically depicted in each case, where the open box represents the wild-type protomer and the cross represents the i3-1 mutation. In all cases, data represent the mean S.E. from three independent experiments. B, GDP/GTP exchange on Gαi (open boxes) or Gαo (closed boxes) catalyzed by the BLT1i3-1-BLT1chim heterodimer in the absence of agonist, in the presence of saturating concentrations in LTB4 or in the presence of saturating concentrations in LXA4. Data are expressed as the percentage of maximal binding. The species analyzed are schematically depicted in each case where the open box represents the wild-type protomer, the gray box represents the BLT1chim protomer, the cross represents the i3-1 mutation, and the black box represents the ligand. In all cases, data represent the mean S.E. from three independent experiments.
BLT1 has been shown to couple to different G protein sub-types, including Gαo and Gαi1, besides Gαi2 (28). To assess that the absence of trans-activation was not due to a particular interaction with Gαi2, we carried out the same kind of experiment using Gαo instead of Gαi2 in the αβγ complex. As shown in Fig. 8, no GTPγS binding was observed in this case either. The same effect was also observed when using Gαi1 (not shown). This clearly indicates that the absence of trans-activation reported here is not correlated to the G protein subtype used. All of these data indicate that the intermediate conformation induced at the level of the ligand-free protomer upon agonist binding to the other subunit in the dimer is probably not an active one in terms of G protein activation.
DISCUSSION
We analyzed here the molecular mechanisms whereby a GPCR dimer activates its cognate G protein. For this, we produced a heterodimer between the wild-type BLT1 and a BLT1/ALXR chimera. The latter retains the structural features and G protein-activation properties of BLT1 but can only be activated by ALXR agonists. This allows a specific activation of BLT1 or BLT1chim with LTB4 or LXA4, respectively. Using this model heterodimer, we clearly established that agonist binding to one of the subunits, although leading to a cross-conformational change, does not trigger full activation of the ligand-free subunit. This observation argues against a trans-activation mechanism where the signal would be transmitted from the subunit where the ligand binds to the neighboring subunit in the receptor dimer.
Although it clearly appears that receptor monomers can activate G proteins (4-7), compelling results from different receptors indicate that GPCRs exist as dimers and/or higher order oligomers at the cell surface and that dimerization can in some cases modulate the functional properties of the receptors. It has been shown with the neurotensin receptor that the kinetics of G protein activation are different depending on whether one considers the receptor monomer or dimer, indicating that the two protomers in the dimer are not equivalent in terms of G protein activation (7). Recent work with rhodopsin (10) inserted in lipid nanodiscs has also suggested that the functioning of the dimer involves some asymmetry. The two protomers in a receptor dimer are probably not equivalent for binding the ligand either, as suggested by the observation of cooperative effects for different receptor homo- and heterodimers (24, 26, 29-35).
We previously reported an asymmetry in the purified BLT1 homodimer with both protomers in a different conformation even in the presence of saturating concentrations in the LTB4 agonist. We further investigated this behavior with a heterodimeric complex where each of the subunits was selectively activated by a structurally different ligand. Using this dimer, we again established that agonist binding to one of the receptor subunits leads to conformational changes of the other subunit, thus reinforcing the model of a cross-conformational change transmitted between the two protomers within a receptor dimer.
As previously reported, the subsequent addition of the second agonist did not led to a subsequent change in the conformation of the second protomer, indicating that this subunit could not reach its fully activated state even if its agonist is present. It was only by both adding a large excess of the second agonist and allowing the rearrangement of the receptor-G protein complex in the presence of GTP that an interconversion between the two conformational states was observed. This again favors the asymmetric model we proposed, where only one protomer in a dimeric assembly is able to reach a fully activated state (9, 11, 12). Such a model could explain different observations reported for GPCR heterodimers, for instance the recent observation with μ-opioid receptor-α2A adrenergic receptor dimers where morphine binding to μ-opioid receptor induced an inhibition in the agonist-induced conformational changes of the α2A adrenergic protomer (36).
It is tempting to relate the G protein-dependent asymmetry in receptor conformation reported here to the G protein-promoted cooperative ligand-binding properties previously reported for different receptors. For instance, there is evidence in the neurotensin receptor indicating that G protein and agonist sites in the receptor dimer are linked in a negative cooperative manner (7). Negative cooperative effects have also been reported for chemokine homo- and heterodimers that are promoted by the G protein (26). If one considers a model where a single G protein interacts with the receptor heterodimer (37), the interaction of a G protein heterotrimer with a receptor dimer is likely to be asymmetrical, since there are two opposite ways for the G protein to contact the dimer. If this is the case, the major component of the asymmetry of the receptor dimer within the receptor-G protein complex might be the position of the Gα subunit relative to the agonist-occupied monomer. A possible model would be that activation of one of the protomers favors an oriented interaction of the receptor dimer with the G protein in such a way that the agonist-occupied protomer directly interacts with the G protein and is therefore stabilized in a fully active, high affinity conformation, whereas the other protomer remains in an uncoupled, low affinity conformation. Such a model would be consistent with the full activation of G protein by receptor monomers (4-7).
The question that then arises is whether cross-conformational changes within the dimeric complex necessarily imply trans-activation (i.e. whether the unliganded protomer can nevertheless activate G proteins). This is of crucial importance for our understanding of signaling through receptor dimers, in particular in the case of heterodimers. We observed here that occupancy of a single ligand-binding site in the BLT1 dimer by an agonist triggers G protein activation. However, this does not answer the question as to whether the signal is transmitted directly from the agonist-loaded protomer to the G protein or if the agonist-free protomer stabilized in its intermediate conformation can also activate the G protein. On this aspect, the situation is not clear.
On the one hand, there are lines of evidence suggesting that subunits of GPCR oligomers function independently in the process of G protein activation. For instance, co-expression of ligand binding-defective and G protein coupling-defective mutant α-factor receptors did not significantly improve signaling, suggesting that the signal of agonist binding was not transferred from the G protein coupling-defective mutant to the agonist-binding defective mutant (38). Co-expression of different mutants of the angiotensin II receptor could not restore signaling (39), indicating that complete trans-complementation did not occur in this case either.
On the other hand, it was shown that in cells cotransfected with chimeras between the adrenergic and muscarinic receptors, signaling occurred upon stimulation with a muscarinic agonist (40). However, this observation does not necessarily imply trans-activation, since one may speculate that the formation of a functional protomer involving the intermolecular exchange of N- and C-terminal receptor domains underlies this phenomenon. Finally, data with mutant GPCR-G protein fusions where either the G protein or the receptor had been specifically deactivated through specific mutations also suggested that GPCRs could operate through trans-activation (41). However, as noted by the authors, the possibility cannot be excluded in this case that the amino acid linker of the fusion protein is long enough to allow the Gα subunit some degree of motion, resulting in Gα subunit interaction by the nonfused receptor (42). Trans-activation has also been reported for GPCRs with a large, extracellular, ligand-binding domain, such as the glycoprotein hormone receptors (30, 43, 44) and class C receptors (45, 46). This is particularly striking for the GB1-GB2 heterodimeric GABA(B) receptor where only GB1 binds the ligand, whereas G protein activation is mediated by GB2 (13). However, the situation for this class of receptors is likely to be more complex, since one has to consider also the relative movements of the two extracellular domains combined with those of the transmembrane portion for receptor and G protein activation.
Our data with a receptor dimer where the protomer that binds the ligand is no more able to activate its G protein partner due to a specific mutation in the third intracellular loop clearly indicate that no trans-activation occurs in this complex. Indeed, no receptor-catalyzed GTPγS binding was observed in the presence of LTB4 (i.e. under conditions where only the i3 mutated wild-type protomer is loaded with the agonist). This is not due to the fact that both protomers need to be loaded with an agonist for GTPγS binding to occur, since Gαi full activation occurred in the presence of LXA4 (i.e. under conditions where only the BLT1chim protomer is loaded with an agonist). This observation therefore implies that in our model system, signaling through the cognate G protein essentially occurs through the protomer where the agonist binds (see the model in Fig. 9). This observation would explain why a single receptor monomer fully activates G proteins (4-7). Caution needs to be exerted in the sense that our data only consider signal transduction within an isolated receptor-G protein purified complex. Our results directly establish that the conformation of the ligand-unloaded protomer is not per se competent for G protein activation. The possibility cannot be excluded, however, that other components in a cellular environment could facilitate a coupling of the ligand-free protomer with other partners and/or G proteins.
FIGURE 9.

A model for BLT1-BLT1chim-induced G protein activation. The white box corresponds to BLT1, and the gray box corresponds to BLT1chim. The G protein is represented by the box marked G. The different shapes correspond to the different conformations adopted by the receptor. Binding of the agonist to one of the protomers (e.g. BLT1) induces a conformational change of the ligand-loaded subunit and a cross-conformational change of the ligand-free protomer. However, only the agonist-loaded protomer can trigger G protein activation.
Our observation does not rule out either that the intermediate conformation of the ligand-free protomer could activate alternative pathways involving other G proteins and/or other signaling partners, such as arrestins, although in this latter case, there is evidence also that receptor trafficking is determined by the activated dimeric partner (47-49). An interesting view would be to consider that the intermediate conformation of the ligand-free protomer is responsible for triggering activation of pathways other than those induced by the fully activated conformation of the receptor loaded with the agonist, and this could explain the differential signaling pathways observed in some cases when comparing heterodimers and homodimers.
Supplementary Material
Acknowledgments
We thank Joseph Parello for a critical reading of the manuscript.
This work was supported by CNRS and by French Ministry of Research Grants ACI BCMS 328 and ANR BLAN06-3_135092. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are: GPCR, G protein-coupled receptor; 5HW, 5-hydroxytryptophan; i3, third intracellular loop; LTB4, leukotriene B4; LXA4, lipoxin A4; MOPS, 4-morpholinepropanesulfonic acid; GTPγS, guanosine 5′-O-(thiotriphosphate).
M. Damian, J.-P. Pin, and J.-L. Banères, unpublished observations.
References
- 1.Bockaert, J., and Pin, J. P. (1999) EMBO J. 18 1723-1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bockaert, J., Claeysen, S., Becamel, C., Pinloche, S., and Dumuis, A. (2002) Int. Rev. Cytol. 212 63-132 [DOI] [PubMed] [Google Scholar]
- 3.Kobilka, B. K. (2007) Biochim. Biophys. Acta 1768 794-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whorton, M. R., Bokoch, M. P., Rasmussen, S. G., Huang, B., Zare, R. N., Kobilka, B., and Sunahara, R. K. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 7682-7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whorton, M. R., Jastrzebska, B., Park, P. S., Fotiadis, D., Engel, A., Palczewski, K., and Sunahara, R. K. (2007) J. Biol. Chem. 283 4387-4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ernst, O. P., Gramse, V., Kolbe, M., Hofmann, K. P., and Heck, M. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 10859-10864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White, J. F., Grodnitzky, J., Louis, J. M., Trinh, L. B., Shiloach, J., Gutierrez, J., Northup, J. K., and Grisshammer, R. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 12199-12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pin, J. P., Neubig, R., Bouvier, M., Devi, L., Filizola, M., Javitch, J. A., Lohse, M. J., Milligan, G., Palczewski, K., Parmentier, M., and Spedding, M. (2007) Pharmacol. Rev. 59 5-13 [DOI] [PubMed] [Google Scholar]
- 9.Damian, M., Martin, A., Mesnier, D., Pin, J. P., and Banères, J. L. (2006) EMBO J. 25 5693-5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bayburt, T. H., Leitz, A. J., Xie, G., Oprian, D. D., and Sligar, S. G. (2007) J. Biol. Chem. 282 14875-14881 [DOI] [PubMed] [Google Scholar]
- 11.Goudet, C., Kniazeff, J., Hlavackova, V., Malhaire, F., Maurel, D., Acher, F., Blahos, J., Prezeau, L., and Pin, J. P. (2005) J. Biol. Chem. 280 24380-24385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hlavackova, V., Goudet, C., Kniazeff, J., Zikova, A., Maurel, D., Vol, C., Trojanova, J., Prezeau, L., Pin, J. P., and Blahos, J. (2005) EMBO J. 24 499-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galvez, T., Duthey, B., Kniazeff, J., Blahos, J., Rovelli, G., Bettler, B., Prezeau, L., and Pin, J. P. (2001) EMBO J. 20 2152-2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duthey, B., Caudron, S., Perroy, J., Bettler, B., Fagni, L., Pin, J. P., and Prezeau, L. (2002) J. Biol. Chem. 277 3236-3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiang, N., Fierro, I. M., Gronert, K., and Serhan, C. N. (2000) J. Exp. Med. 191 1197-1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banères, J. L., Martin, A., Hullot, P., Girard, J. P., Rossi, J. C., and Parello, J. (2003) J. Mol. Biol. 329 801-814 [DOI] [PubMed] [Google Scholar]
- 17.Mesnier, D., and Banères, J. L. (2004) J. Biol. Chem. 279 49664-49670 [DOI] [PubMed] [Google Scholar]
- 18.Gill, S. C., and von Hippel, P. H. (1989) Anal. Biochem. 182 319-326 [DOI] [PubMed] [Google Scholar]
- 19.Wyman, J., and Gill, S. J. (1990). Binding and Linkage: Functional chemistry of biological macromolecules (Kelly, A., ed) pp. 33-61, University Science Books, Mill Valley, CA
- 20.Glass, M., and Northup, J. K. (1999) Mol. Pharmacol. 56 1362-1369 [DOI] [PubMed] [Google Scholar]
- 21.Banères, J. L., and Parello, J. (2003) J. Mol. Biol. 329 815-829 [DOI] [PubMed] [Google Scholar]
- 22.Kurose, H., Regan, J. W., Caron, M. G., and Lefkowitz, R. J. (1991) Bio-chemistry 30 3335-3341 [DOI] [PubMed] [Google Scholar]
- 23.Chiang, N., Serhan, C. N., Dahlen, S. E., Drazen, J. M., Hay, D. W., Rovati, G. E., Shimizu, T., Yokomizo, T., and Brink, C. (2006) Pharmacol. Rev. 58 463-487 [DOI] [PubMed] [Google Scholar]
- 24.Springael, J. Y., Urizar, E., Costagliola, S., Vassart, G., and Parmentier, M. (2007) Pharmacol. Ther. 115 410-418 [DOI] [PubMed] [Google Scholar]
- 25.Ross, J. B. A., Senear, D. F., Waxman, E., Kombo, B. B., Rusinova, E., Huang, Y. T., Laws, W. R., and Hasselbacher, C. A. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 12023-12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Springael, J. Y., Le Minh, P. N., Urizar, E., Costagliola, S., Vassart, G., and Parmentier, M. (2006) Mol. Pharmacol. 69 1652-1661 [DOI] [PubMed] [Google Scholar]
- 27.Kuniyeda, K., Okuno, T., Terawaki, K., Miyano, M., Yokomizo, T., and Shimizu, T. (2007) J. Biol. Chem. 282 3998-4006 [DOI] [PubMed] [Google Scholar]
- 28.Masuda, K., Sakihama, T., Akiyama, C., Takahashi, K., Yokomizo, T., Shimizu, T., Kodama, T., and Hamakubo, T. (2003) J. Biol. Chem. 278 24552-24562 [DOI] [PubMed] [Google Scholar]
- 29.Durroux, T. (2005) Trends Pharmacol. Sci. 26 376-384 [DOI] [PubMed] [Google Scholar]
- 30.Urizar, E., Montanelli, L., Loy, T., Bonomi, M., Swillens, S., Gales, C., Bouvier, M., Smits, G., Vassart, G., and Costagliola, S. (2005) EMBO J. 24 1954-1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Springael, J. Y., Urizar, E., and Parmentier, M. (2005) Cytokine Growth Factor Rev. 16 611-623 [DOI] [PubMed] [Google Scholar]
- 32.El-Asmar, L., Springael, J. Y., Ballet, S., Andrieu, E. U., Vassart, G., and Parmentier, M. (2005) Mol. Pharmacol. 67 460-469 [DOI] [PubMed] [Google Scholar]
- 33.Albizu, L., Balestre, M. N., Breton, C., Pin, J. P., Manning, M., Mouillac, B., Barberis, C., and Durroux, T. (2006) Mol. Pharmacol. 70 1783-1791 [DOI] [PubMed] [Google Scholar]
- 34.Maggio, R., Vogel, Z., and Wess, J. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 3103-3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sohy, D., Parmentier, M., and Springael, J. Y. (2007) J. Biol. Chem. 282 30062-30069 [DOI] [PubMed] [Google Scholar]
- 36.Vilardaga, J. P., Nikolaev, V. O., Lorenz, K., Ferrandon, S., Zhuang, Z., and Lohse, M. J. (2008) Nat. Chem. Biol. 4 126-131 [DOI] [PubMed] [Google Scholar]
- 37.Filipek, S., Krzysko, K. A., Fotiadis, D., Liang, Y., Saperstein, D. A., Engel, A., and Palczewski, K. (2004) Photochem. Photobiol. Sci. 3 628-638 [DOI] [PubMed] [Google Scholar]
- 38.Chinault, S. L., Overton, M. C., and Blumer, K. J. (2004) J. Biol. Chem. 279 16091-16100 [DOI] [PubMed] [Google Scholar]
- 39.Monnot, C., Bihoreau, C., Conchon, S., Curnow, K. M., Corvol, P., and Clauser, E. (1996) J. Biol. Chem. 271 1507-1513 [DOI] [PubMed] [Google Scholar]
- 40.Maggio, R., Vogel, Z., and Wess, J. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 3103-3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrillo, J. J., Pediani, J., and Milligan, G. (2003) J. Biol. Chem. 278 42578-42587 [DOI] [PubMed] [Google Scholar]
- 42.Snook, L. A., Milligan, G., Kieffer, B. L., and Massotte, D. (2006) J. Phar-macol. Exp. Ther. 318 683-690 [DOI] [PubMed] [Google Scholar]
- 43.Osuga, Y., Hayashi, M., Kudo, M., Conti, M., Kobilka, B., and Hsueh, A. J. (1997) J. Biol. Chem. 272 25006-25012 [DOI] [PubMed] [Google Scholar]
- 44.Ji, I., Lee, C., Song, Y., Conn, P. M., and Ji, T. H. (2002) Mol. Endocrinol. 16 1299-1308 [DOI] [PubMed] [Google Scholar]
- 45.Kniazeff, J., Bessis, A. S., Maurel, D., Ansanay, H., Prezeau, L., and Pin, J. P. (2004) Nat. Struct. Mol. Biol. 11 706-713 [DOI] [PubMed] [Google Scholar]
- 46.Brock, C., Oueslati, N., Soler, S., Boudier, L., Rondard, P., and Pin, J. P. (2007) J. Biol. Chem. 282 33000-33008 [DOI] [PubMed] [Google Scholar]
- 47.Stanasila, L., Perez, J. B., Vogel, H., and Cotecchia, S. (2003) J. Biol. Chem. 278 40239-40251 [DOI] [PubMed] [Google Scholar]
- 48.Terrillon, S., Barberis, C., and Bouvier, M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 1548-1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson, S. J., Dowling, J. K., Zhao, L., Carnish, E., and Smyth, E. M. (2007) Arterioscler. Thromb. Vasc. Biol. 27 290-296 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.