

Abstract
Abnormalities of chromosome copy number are called aneuploidies and make up a large health load on the human population. Many aneuploidies are lethal because the resulting abnormal gene dosage is highly deleterious. Nevertheless, some whole chromosome aneuploidies can lead to live births. Alterations in the copy number of sections of chromosomes, which are also known as segmental aneuploidies, are also associated with deleterious effects. Here we examine how aneuploidy of whole chromosomes and segmental aneuploidy of chromosomal regions are modeled in the mouse. These models provide a whole animal system in which we aim to investigate the complex phenotype-genotype interactions that arise from alteration in the copy number of genes. Although our understanding of this subject is still in its infancy, already research in mouse models is highlighting possible therapies that might help alleviate the cognitive effects associated with changes in gene number. Thus, creating and studying mouse models of aneuploidy and copy number variation is important for understanding what it is to be human, in both the normal and genomically altered states.
1. Introduction
Traditionally, aneuploidy was defined as a deletion or duplication of a whole chromosome. This genomic abnormality is thought to occur in at least 5% of all clinically recognized pregnancies, usually resulting in spontaneous abortion [1]. Aneuploidy is thought to be usually highly deleterious because many genes are “dosage-sensitive” in that their expression is affected by their copy number in the genome, and changes in gene expression levels may result in altered phenotypes that can be lethal [2]. As well as whole chromosome aneuploidy, deletion of a few kilobases or megabases of DNA (microdeletion) or similarly a duplicated region (microduplication) within a chromosome can also result in changes in gene copy number. Recent advances in genomic technologies have revealed the association of many of these segmental aneuploidies (microdeletions and duplications) with specific genetic syndromes and diseases [3–5].
The most frequently occurring full autosomal aneuploidy is that of trisomy of human chromosome 21 (Hsa21), which causes Down syndrome (DS). DS is the most common cause of genetic intellectual disability, occurring in ~1 in 750 live births in all populations. People with DS have an increased risk of developing cardiac defects, certain leukemias, and early onset Alzheimer's disease as well as many other phenotypes [6]. Trisomies of chromosomes 18 (Hsa18) (Edwards syndrome) and 13 (Hsa13) (Patau syndrome) occur at lower frequency than DS (1 in 4300 and 1 in 7100 live births, respectively), and infants with these conditions have a very short life expectancy, typically less than 1 year and less than 5 years, respectively [7]. Aneuploidy of the sex chromosomes can occur with multiple copies of the X or Y chromosome, or loss of the X or Y chromosome. Relatively common sex chromosome aneuploidies include Klinefelter syndrome (KS) (47 XXY, males with an additional copy of the X chromosome), ~1 in 500–1000 males [8], and Turner syndrome (TS) (45,X, females with monosomy of the X chromosome), 1 in ~4000 live births [9].
Segmental aneuploidies, otherwise known as partial aneuploidies or segmental aneusomies may be more compatible with life than whole chromosomal aneuploidies, and result in a large number of well-defined syndromes (Table 1). Many of these conditions are associated with neurodevelopmental and growth problems that result in epilepsy, intellectual disability, and autism.
Table 1.
Examples of mouse models of segmental aneuploidies.
| Human syndrome | Associated genetic change | Aneuploid mouse models |
|---|---|---|
| Angelman syndrome | deletion of maternal 15q11–13 | PatDp [37] |
| MatDf(Ube3a-Gabrb3) [38] | ||
| Prader-Willi syndrome | deletion of paternal 15q11–13 | MatDp [37] |
| Autism risk factor | Duplication 15q11–13 | matDp; pat Dp [57] |
| Smith-Magenis syndrome | deletion of 17p11/17p11.2 | Df(11)17 [58] |
| Df(11)17-1; Df(11)17-2; Df(11)17-3 [59] | ||
| Potocki-Lupski syndrome | duplication of 17p11/17p11.2 | Dp(11)17 [58] |
| DiGeorge syndrome | deletion of 22q11.2 | Df1 [44] |
| Idd-Ctp [45] | ||
| Idd-Arvcf [46] | ||
| Df2; Df3; Df4; Df5 [60] | ||
| Williams-Beuren | deletion of 7q11 | PD and DD [53] |
| — | deletion/duplication of 17q21 | Df11[ 1] and Dp11[ 1] [35] |
The challenge facing scientists and clinicians from the aneuploidy syndromes is how to unravel the interaction between abnormal gene dosage and abnormal gene expression that leads to the specific phenotypes of each syndrome, and then to find therapies for intervention for these phenotypes.
2. Mouse Models of Aneuploidy
The use of mouse models of aneuploidy allows scientists to study the direct effects of abnormal gene dosage on specific syndromes, at the molecular, cellular, physiological, and behavioural level. Many technologies exist to manipulate the mouse genome to mutate, overexpress, and knock-out specific genes of interest, and help to define which dosage sensitive genes are causative for any given phenotype (reviewed in [10]). Such technologies now include chromosome engineering whereby large regions of the mouse genome can be deleted or duplicated corresponding to the partial aneuploidies found in humans (reviewed in [11, 12]). However, one confounding factor is that each human chromosome has syntenic regions to two or more mouse chromosomes. An alternative approach has been to transfer an entire human chromosome into a mouse, to overcome this problem [13]. Here, we discuss the contribution of mouse models of whole chromosome and segmental aneuploidy to our biological understanding and highlight possible future models and how they may further our knowledge.
3. Mouse Models of Whole Chromosome Aneuploidies
3.1. Down Syndrome
A number of mouse models have been developed to study the most frequently occurring autosomal aneuploidy, Down syndrome (Figure 1). The Tc1 transchromosomic mouse model contains a freely segregating maternally inherited copy of Hsa21 and is trisomic for approximately 75% of Hsa21 genes [13]. This mouse has altered learning and memory, synaptic plasticity, a reduced cerebellar neuronal number, heart anomalies, reduced solid tumor development, and defects in angiogenesis and megakaryopoiesis [13–18]. Other mouse models of DS contain an additional copy of regions of mouse chromosomes 16, 17, and 10, which are syntenic with Hsa21. The Ts65Dn mouse model is the most widely used; it contains an extra copy of a segment of mouse chromosome 16 (Mmu16) and is trisomic for about 50% of the genes found on Hsa21 [19]. This model shows impaired learning and motor deficits [19], neuronal degeneration similar to that observed in people with Alzheimer's disease (which is part of the DS phenotype) and heart and angiogenesis defects [20–22]. Another commonly used model is the Ts1Cje mouse which contains a smaller segmental trisomy of Mmu16 including approximately 68 genes; it also exhibits learning and behavioral deficits, but does not exhibit neuronal degeneration [23]. The newest model of DS, developed by Yu and colleagues, contains three copies of all Hsa21 homologs on mouse chromosomes 16, 17, and 10 and shows learning and memory deficits that may be similar to some of the cognitive problems that people with DS experience [24, 25].
Figure 1.
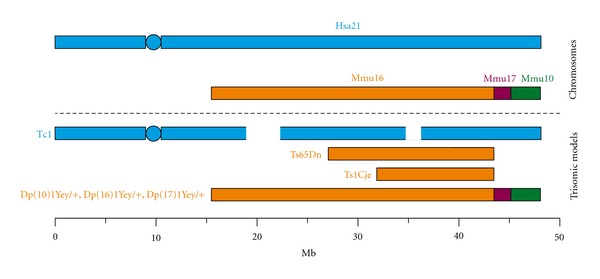
Mouse models of Down syndrome. Hsa21 (in blue) and the syntenic mouse chromosomes (Mmu 16, orange, Mmu 17, purple, Mmu10, green). The trisomic regions of several of the well-established mouse models of DS, the Tc1 mouse, Ts65Dn, Ts1Cje, and Dp(10)1Yey/+, Dp(16)1Yey/+, Dp(17)1Yey/+ are aligned to the corresponding parts of the human and mouse genome.
To determine the identity of trisomic genes that cause specific phenotypes, aneuploid mouse models of DS can be crossed with mouse models of segmental Hsa21 monosomy (Ms1Yah and Ms4Yah) [26–29] or to gene knockouts to alter dosage of individual genes within a region of trisomy. These techniques have been recently used to identify the genes responsible for trisomy-21-related protection against the tumour formation [30], furthering our understanding of the biology that underlies these important processes. Mouse models of Hsa21 trisomy have been used also for demonstrating the potential for cognitive enhancement therapies for people who have DS [21, 31, 32]. A number of the drugs tested in studies of DS mouse models for their effects on learning and memory are currently in small-scale clinical trials, demonstrating the utility of these mice to combat the deleterious effects of DS.
3.2. Edwards Syndrome and Patau Syndrome
A mouse model of Edwards syndrome has yet to be developed. Hsa18 is 78 Mb in length and has conserved synteny with 5 principal regions encoded on three mouse chromosome (Mmu 1, 17, and 18). Similarly, no animal model of Patau syndrome has been reported; Hsa13 has conserved synteny with six mouse chromosome segments. Thus, although technically challenging, it would be possible to generate models of these syndromes by duplication of the mouse syntenic regions. These models could be used to further our understanding of the biology of this devastating conditions.
3.3. Turner Syndrome and Klinefelter Syndrome
Mouse models with both paternal and maternally inherited 45,X karyotypes exhibit behavioural changes including reduced attention, growth retardation, and hearing defects, which resemble aspects of human Turner syndrome (TS) (reviewed by [33]). These models have been useful for understanding the X-parent-of-origin-effect on TS-associated phenotypes. However, 39,X mice do not manifest some TS-associated phenotypes, such as motor deficits; this may reflect differences in X-inactivation between mouse and humans. Mouse models of Klinefelter syndrome (XXY male) develop hypogonadism and cognitive problems and have impaired fertility (reviewed by [34]), phenotypes that resemble aspects of KS. Molecular studies undertaken in XXY mouse models have shed light on the possible chemical alterations in the brain that cause cognitive problems observed in KS [34], and this knowledge may lead to the development of therapeutic strategies.
4. Mouse Models of Segmental Aneuploidies
Mouse models of segmental aneuploidy are invaluable for our understanding of which dosage-sensitive genes result in the deleterious phenotypes that are associated with these genomic changes in humans. Moreover, mouse models of segmental aneuploidy, not associated with a specific human syndrome, can also be used to understand the relationship between gene and phenotype. For example, mouse models with 0.8 Mb reciprocal chromosomal deletions and duplications have been used to identify the role of Stat5 in immune-hypersensitivity and metabolic syndrome [35].
A number of models of Prader-Willi syndrome (PWS) (deletion of paternal 15q11–13) and Angelman syndrome (AS) (deletion of maternal 15q11–13) have been reported [36–38]. PWS is also associated with chromosome 15 maternal disomy and AS with paternal chromosome 15 disomy. Mouse models of these genetic changes have been reported and both exhibit reduced viability and neonatal growth retardation [36, 37]. Deficits in learning and memory have also been observed in a mouse model with a maternally inherited segmental deletions (Ube3a-Gabrb3) corresponding to part of the region lost in AS [38]. Mouse models deficient in the Ube3a and Gabrb3 PWS/AS candidate genes exhibit neurodevelopment and behavior changes, highlighting the key role these genes play in the syndromes [39–41].
Interestingly, maternal duplications of the PWS/AS associated region, 15q11–13, are associated with autism [42]. A mouse model of the duplication of the mouse syntenic region of chromosome 7 exhibits some features that resemble autism, but only when the duplication is paternally inherited in contrast to the inheritance pattern observed in humans [43]. These models will help give insight into the genetic and biochemical abnormalities causing autism.
Mouse models of DiGeorge syndrome (deletion of 1.5–3 Mb at 22q11) have been crucial to the molecular understanding of this condition. A series of complementary mouse models with full or partial deletions of the region syntenic with 22q11 identified the key deleted gene, Tbx1, responsible for the syndrome's deleterious phenotypes [44–46]. The 22q11 deletion is also the largest known genetic risk factors for schizophrenia [47, 48], and the DiGeorge mouse models may also be useful to further understanding of this condition [49]. Similarly, mouse models of the complete 3.7 Mb deletion and duplication associated with Smith-Magenis (SMS) and Potocki-Lupski (PTLS) syndromes have helped identify one of the key dosage sensitive genes, Rai1 [50]. Moreover, these models have also been used to investigate the relative effect of genomic rearrangement versus gene copy number change on gene expression [51]. Work in this field has also highlighted the very complex interactions of genes both within the copy number altered region and those elsewhere in the genome [52], as the penetrance of some PTLS-like features in the mouse models vary with the size of the region disrupted and the genetic background of the model. The effect of genetic background on the penetrance of aneuploidy-associated phenotypes has also been highlighted in the Tc1 mouse model in which DS-like heart defects appear with a greater penetrance on a C57BL/6 mouse inbred line background [15].
A mouse model of Williams-Beuren syndrome (WBS) has been developed recently that exhibits a large number of informative neurodevelopmental and behavioural abnormalities [53]. This model is likely to be crucial to further understanding of WBS.
5. Future of Aneuploid Mouse Models
Complete and partial mouse models of aneuploidy have significantly contributed to our understanding of the complex relationship between dosage of individual genes and the resulting phenotypes that arise in individual aneuploidy syndromes. Unfortunately, even for the most widely studied aneuploidy disorders such as DS, we are a long way from understanding much of the molecular basis of the pathology. However, the rate of progress in understanding the effects of gene copy number and expression levels is increasing, and we now know that there is a considerable variation of small genomic regions, copy number variation (CNV), across the entire human genome in normal individuals. These regions can be up to a megabase in size and affect much of normal human phenotypic variation, including susceptibility or resistance to common disorders (e.g., see [54–56]). New mouse models of CNVs will be beneficial to study not only the effects of gene dosage but also to dissect the effects of altering copy number for the regulatory elements found in these regions of the genome.
Advances in our understanding of the human genome will present new opportunities for the development of novel mouse aneuploid models. Equally, findings from existing mouse models will continue to influence human genetic studies. Thus, complementary human and mouse genetic studies are key to unraveling the links between gene copy number and phenotype.
Acknowledgments
The authors thank Ray Young for assistance with preparation of the figure. F. K. Wiseman, O. Sheppard, A. Ruparelia, and E. M. C. Fisher are funded by the UK Medical Research Council, the Wellcome Trust, the AnEUploidy Grant from Framework Programme 6 of the European Union Commission, and the Alzheimer's Research Trust, the Brain Research Trust. V. L. J. Tybulewicz is funded by the UK Medical Research Council, the AnEUploidy grant from Framework Programme 6 of the European Union Commission, and the Wellcome Trust.
Authors' Contribution
O. Sheppard and F. K. Wiseman contributed equally to this publication.
References
- 1.Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nature Reviews Genetics. 2001;2(4):280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 2.Dierssen M, Herault Y, Estivill X. Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiological Reviews. 2009;89(3):887–920. doi: 10.1152/physrev.00032.2007. [DOI] [PubMed] [Google Scholar]
- 3.Morrow EM. Genomic copy number variation in disorders of cognitive development. Journal of the American Academy of Child and Adolescent Psychiatry. 2010;49(11):1091–1104. doi: 10.1016/j.jaac.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annual Review of Medicine. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 5.Shaffer LG, Ledbetter DH, Lupski JR. Molecular cytogenetics of contiguous gene syndromes: mechanisms and consequences of gene dosage imbalance. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, Childs B, editors. The Metabolic and Molecular Bases of Inherited Diseases. New York, NY, USA: McGraw–Hill; 2001. pp. 1291–1326. [Google Scholar]
- 6.Wiseman FK, Alford KA, Tybulewicz VLJ, Fisher EMC. Down syndrome—recent progress and future prospects. Human Molecular Genetics. 2009;18(1):R75–R83. doi: 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savva GM, Walker K, Morris JK. The maternal age-specific live birth prevalence of trisomies 13 and 18 compared to trisomy 21 (Down syndrome) Prenatal Diagnosis. 2010;30(1):57–64. doi: 10.1002/pd.2403. [DOI] [PubMed] [Google Scholar]
- 8.Giltay JC, Maiburg MC. Klinefelter syndrome: clinical and molecular aspects. Expert Review of Molecular Diagnostics. 2010;10(6):765–776. doi: 10.1586/erm.10.63. [DOI] [PubMed] [Google Scholar]
- 9.Davenport ML. Approach to the patient with Turner syndrome. Journal of Clinical Endocrinology and Metabolism. 2010;95(4):1487–1495. doi: 10.1210/jc.2009-0926. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen D, Tian X. The expanding role of mouse genetics for understanding human biology and disease. Disease Models and Mechanisms. 2008;1(1):56–66. doi: 10.1242/dmm.000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tybulewicz VLJ, Fisher EMC. New techniques to understand chromosome dosage: mouse models of aneuploidy. Human Molecular Genetics. 2006;15(2):R103–R109. doi: 10.1093/hmg/ddl179. [DOI] [PubMed] [Google Scholar]
- 12.Ramírez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378(6558):720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- 13.O’Doherty A, Ruf S, Mulligan C, et al. Genetics: an aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alford KA, Slender A, Vanes L, et al. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010;115(14):2928–2937. doi: 10.1182/blood-2009-06-227629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunlevy L, Bennett M, Slender A, et al. Down’s syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovascular Research. 2010;88(2):287–295. doi: 10.1093/cvr/cvq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galante M, Jani H, Vanes L, et al. Impairments in motor coordination without major changes in cerebellar plasticity in the Tc1 mouse model of Down syndrome. Human Molecular Genetics. 2009;18(8):1449–1463. doi: 10.1093/hmg/ddp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morice E, Andreae LC, Cooke SF, et al. Preservation of long-term memory and synaptic plasticity despite short-term impairments in the Tcl mouse model of down syndrome. Learning and Memory. 2008;15(7):492–500. doi: 10.1101/lm.969608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynolds LE, Watson AR, Baker M, et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Downs syndrome. Nature. 2010;465(7299):813–817. doi: 10.1038/nature09106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reeves RH, Irving NG, Moran TH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics. 1995;11(2):177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 20.Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Salehi A, Faizi M, Colas D, et al. Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Science translational medicine. 2009;1(7):7–ra17. doi: 10.1126/scitranslmed.3000258. [DOI] [PubMed] [Google Scholar]
- 22.Cooper JD, Salehi A, Delcroix JD, et al. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sago H, Carlson EJ, Smith DJ, et al. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu T, Liu C, Belichenko P, et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Research. 2010;1366:162–171. doi: 10.1016/j.brainres.2010.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu T, Li Z, Jia Z, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Human Molecular Genetics. 2010;19(14):2780–2791. doi: 10.1093/hmg/ddq179. Article ID ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Besson V, Brault V, Duchon A, et al. Modeling the monosomy for the telomeric part of human chromosome 21 reveals haploinsufficient genes modulating the inflammatory and airway responses. Human Molecular Genetics. 2007;16(17):2040–2052. doi: 10.1093/hmg/ddm152. [DOI] [PubMed] [Google Scholar]
- 27.Duchon A, Pothion S, Brault V, et al. The telomeric part of the human chromosome 21 from Cstb to Prmt2 is not necessary for the locomotor and short-term memory deficits observed in the Tc1 mouse model of Down syndrome. Behavioural Brain Research. 2011;217(2):271–281. doi: 10.1016/j.bbr.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson LE, Richtsmeier JT, Leszl J, Reeves RH. A chromosome 21 critical region does not cause specific down syndrome phenotypes. Science. 2004;306(5696):687–690. doi: 10.1126/science.1098992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu T, Clapcote SJ, Li Z, et al. Deficiencies in the region syntenic to human 21q22.3 cause cognitive deficits in mice. Mammalian Genome. 2010;21(5-6):258–267. doi: 10.1007/s00335-010-9262-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses ApcMin-mediated tumours in mouse models of Down’s syndrome. Nature. 2008;451(7174):73–75. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- 31.Costa ACS, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33(7):1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience. 2007;10(4):411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 33.Lynn PMY, Davies W. The 39,XO mouse as a model for the neurobiology of Turner syndrome and sex-biased neuropsychiatric disorders. Behavioural Brain Research. 2007;179(2):173–182. doi: 10.1016/j.bbr.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Wistuba J. Animal models for Klinefelter’s syndrome and their relevance for the clinic. Molecular Human Reproduction. 2010;16(6):375–385. doi: 10.1093/molehr/gaq024. Article ID gaq024. [DOI] [PubMed] [Google Scholar]
- 35.Ermakova O, Piszczek L, Luciani L, et al. Sensitized phenotypic screening identifies gene dosage sensitive region on chromosome 11 that predisposes to disease in mice. EMBO Molecular Medicine. 2011;3(1):50–66. doi: 10.1002/emmm.201000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cattanach BM, Barr JA, Beechey CV, Martin J, Noebels J, Jones J. A candidate model for Angelman syndrome in the mouse. Mammalian Genome. 1997;8(7):472–478. doi: 10.1007/s003359900479. [DOI] [PubMed] [Google Scholar]
- 37.Cattanach BM, Barr JA, Evans EP, et al. A candidate mouse model for Prader-Willi syndrome which shows an absence of Snrpn expression. Nature Genetics. 1992;2(4):270–274. doi: 10.1038/ng1292-270. [DOI] [PubMed] [Google Scholar]
- 38.Jiang YH, Pan Y, Zhu L, et al. Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS One. 2010;5(8) doi: 10.1371/journal.pone.0012278. Article ID e12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeLorey TM, Handforth A, Homanics GE, Olsen RW. Mice lacking the gabrb3 gene have epilepsy and behavioral characteristics of Angelman syndrome. Brain Research. 1998;809:p. A29. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLorey TM, Handforth A, Asatourian A, et al. Mice lacking the GABA(A) receptor beta(3) subunit gene have some of the characteristics of Angelmann syndrome. Journal of Neurochemistry. 1997;69:p. S236. [Google Scholar]
- 41.Jiang YH, Armstrong D, Albrecht U, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21(4):799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 42.Cook EH, Lindgren V, Leventhal BL, et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. American Journal of Human Genetics. 1997;60(4):928–934. [PMC free article] [PubMed] [Google Scholar]
- 43.Takumi T. A humanoid mouse model for autism by a chromosome engineering. Neuroscience Research. 2009;65:p. S27. [Google Scholar]
- 44.Lindsay EA, Botta A, Jurecic V, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401(6751):379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 45.Kimber WL, Hsieh P, Hirotsune S, et al. Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Human Molecular Genetics. 1999;8(12):2229–2237. doi: 10.1093/hmg/8.12.2229. [DOI] [PubMed] [Google Scholar]
- 46.Puech A, Saint-Jore B, Merscher S, et al. Normal cardiovascular development in mice deficient for 16 genes in 550 kb of the velocardiofacial/DiGeorge syndrome region. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10090–10095. doi: 10.1073/pnas.97.18.10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464(7289):763–767. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nature Reviews Neuroscience. 2010;11(6):402–416. doi: 10.1038/nrn2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meechan DW, Tucker ES, Maynard TM, LaMantia AS. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(38):16434–16439. doi: 10.1073/pnas.0905696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17)(p11.2p11.2) Journal of Clinical Investigation. 2006;116(11):3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ricard G, Molina J, Chrast J, et al. Phenotypic consequences of copy number variation: insights from smith-magenis and Potocki-Lupski syndrome mouse models. PLoS Biology. 2010;8(11) doi: 10.1371/journal.pbio.1000543. Article ID e1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan J, Bi W, Lupski JR. Penetrance of craniofacial anomalies in mouse models of Smith-Magenis syndrome is modified by genomic sequence surrounding Rai1: not all null alleles are alike. American Journal of Human Genetics. 2007;80(3):518–525. doi: 10.1086/512043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li HH, Roy M, Kuscuoglu U, et al. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Molecular Medicine. 2009;1(1):50–65. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Human Molecular Genetics. 2010;19(R2):R176–187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Need AC, Goldstein DB. Whole genome association studies in complex diseases: where do we stand? Dialogues in Clinical Neuroscience. 2010;12(1):37–46. doi: 10.31887/DCNS.2010.12.1/aneed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee C, Scherer SW. The clinical context of copy number variation in the human genome. Expert Reviews in Molecular Medicine. 2010;12:p. e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 57.Nakatani J, Tamada K, Hatanaka F, et al. Abnormal behavior in a chromosome- engineered mouse model for human 15q11-13 duplication seen in Autism. Cell. 2009;137(7):1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walz K, Caratini-Rivera S, Bi W, et al. Modeling del(17)(p11.2p11.2) and dup(17)(p11.2p11.2) contiguous gene syndromes by chromosome engineering in mice: phenotypic consequences of gene dosage imbalance. Molecular and Cellular Biology. 2003;23(10):3646–3655. doi: 10.1128/MCB.23.10.3646-3655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan J, Keener VW, Bi W, et al. Reduced penetrance of craniofacial anomalies as a function of deletion size and genetic background in a chromosome engineered partial mouse model for Smith-Magenis syndrome. Human Molecular Genetics. 2004;13(21):2613–2624. doi: 10.1093/hmg/ddh288. [DOI] [PubMed] [Google Scholar]
- 60.Lindsay EA, Baldini A. Recovery from arterial growth delay reduces penetrance of cardiovascular defects in mice deleted for the DiGeorge syndrome region. Human Molecular Genetics. 2001;10(9):997–1002. doi: 10.1093/hmg/10.9.997. [DOI] [PubMed] [Google Scholar]