

Abstract
Parkinson disease (PD) is the most common neurodegenerative movement disorder. An increase in the amount of α-synuclein protein could constitute a cause of PD. α-Synuclein is degraded at least partly by chaperone-mediated autophagy (CMA). The I93M mutation in ubiquitin C-terminal hydrolase L1 (UCH-L1) is associated with familial PD. However, the relationship between α-synuclein and UCH-L1 in the pathogenesis of PD has remained largely unclear. In this study, we found that UCH-L1 physically interacts with LAMP-2A, the lysosomal receptor for CMA, and Hsc70 and Hsp90, which can function as components of the CMA pathway. These interactions were abnormally enhanced by the I93M mutation and were independent of the monoubiquitin binding of UCH-L1. In a cell-free system, UCH-L1 directly interacted with the cytosolic region of LAMP-2A. Expression of I93M UCH-L1 in cells induced the CMA inhibition-associated increase in the amount of α-synuclein. Our findings may provide novel insights into the molecular links betweenα-synuclein and UCH-L1 and suggest that aberrant interaction of mutant UCH-L1 with CMA machinery, at least partly, underlies the pathogenesis of PD associated with I93M UCH-L1.
Parkinson disease (PD)4 is the most common neurodegenerative movement disorder characterized by progressive degeneration confined mostly to dopaminergic neurons in the substantia nigra pars compacta. Although the majority of PD cases occur sporadically, nine genes have been reported to be associated with familial forms of PD. Several missense mutations in the α-synuclein gene are linked to dominantly inherited PD (1–3). Duplication and triplication of the α-synuclein gene were also shown to cause familial PD or parkinsonism (4–6), indicating that increases in the levels of α-synuclein could constitute a cause of PD. α-Synuclein is a major component of cytoplasmic inclusions called Lewy bodies in the brains of patients with sporadic PD (7, 8). These findings raised the idea that α-synuclein plays a central role in the pathogenesis of PD. Therefore, elucidating the molecular relationships between α-synuclein and other familial PD-associated proteins is important for understanding the mechanisms that underlie the pathology of PD.
A missense mutation in the ubiquitin C-terminal hydrolase L1 (UCH-L1) gene, leading to an I93M substitution at the protein level, has been reported in two affected siblings of a German family with dominantly inherited PD (9). In this family, four of seven family members were affected with PD. However, the family members, except the two siblings, were not geno-typed. There was an unaffected presumed carrier of the I93M mutation in the family. Therefore, the link between the I93M mutation and the development of PD has been questioned (10, 11). To clarify the link between the mutation and PD, we have generated UCH-L1I93M transgenic mice and reported that these mice exhibit progressive dopaminergic cell loss (12). In addition, we have shown that, compared with UCH-L1WT, UCH-L1I93M exhibits increased insolubility and levels of interactions with other proteins in mammalian cells, features that are characteristic of several neurodegenerative disease-linked mutants (13). These findings suggest that the I93M mutation in UCH-L1 contributes to the pathogenesis of PD. UCH-L1 has also been identified as a component of several inclusion bodies characteristic of neurodegenerative diseases including Lewy bodies (14). A polymorphism in the UCH-L1 gene, resulting in an S18Y substitution at the amino acid residue level, has been reported to be associated with decreased risk of PD in certain populations but not in other populations (15, 16). We have also reported that UCH-L1I93M and carbonyl-modified UCH-L1, which is associated with sporadic PD (17), display shared aberrant properties (13), suggesting that carbonyl-modified UCH-L1 constitutes one of the causes of sporadic PD.
UCH-L1 is one of the most abundant proteins in the brain (1–5% of total soluble protein) (18) and is thought to hydrolyze ubiquitin conjugates into monoubiquitin (19). UCH-L1 was also reported to function as a ubiquitin ligase for monoubiquitinated α-synuclein in a cell-free system (20). Other than these enzymatic activities, we have reported that UCH-L1 stabilizes monoubiquitin by binding to monoubiquitin in neurons (21). Although the hydrolase activity of UCH-L1I93M and the binding of UCH-L1I93M to monoubiquitin are decreased compared with those of UCH-L1WT (9, 13, 22), we have shown that mice deficient in UCH-L1 do not display obvious dopaminergic cell loss (21, 23). These observations indicate that the main cause of UCH-L1I93M-associated PD may not be a loss of UCH-L1 function but an acquired toxicity of UCH-L1I93M. Our previous studies also suggest that aberrantly enhanced physical interactions between UCH-L1I93M and multiple proteins, including tubulin, underlie the toxic functions of UCH-L1I93M (13).
However, the molecular relationship between α-synuclein and UCH-L1 in the pathogenesis of PD has remained largely unclear. α-Synuclein is known to be degraded at least partly by chaperone-mediated autophagy (CMA) (24), in which substrate proteins are selectively transported to and degraded in lysosomes (25). In this study, we sought to identify novel UCH-L1-interacting proteins. We found that UCH-L1 physically interacts with lysosome-associated membrane protein type 2A (LAMP-2A), heat shock cognate protein 70 (Hsc70), and heat shock protein 90 (Hsp90), all of which are components of the CMA pathway (26). These interactions were enhanced by the I93M mutation in UCH-L1 and were independent of the interaction between monoubiquitin and UCH-L1. We also provide the data suggesting that the aberrant interaction of UCH-L1 with CMA machinery results in the accumulation of α-synuclein.
EXPERIMENTAL PROCEDURES
Plasmids—pCI-neo-hUCH-L1 plasmids containing human WT UCH-L1 and UCH-L1 variants with or without a FLAG tag were prepared as described previously (13). The regulatory expression plasmids pTRE-Tight-hUCH-L1 containing WT and I93M UCH-L1 with a FLAG tag at the C terminus of UCH-L1 were constructed by ligating the cDNA encoding UCH-L1 into the pTRE-Tight (Clontech) vector. The expression plasmid pCI-neo-hα-synuclein was constructed using the pCI-neo mammalian expression vector (Promega), and the expression plasmid pCI-neo-ΔDQ α-synuclein was generated using a QuikChange site-directed mutagenesis kit (Stratagene).
Cell Culture and Transfection—COS-7 cells were maintained in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum (JRH Biosciences, Lenexa, KS). IMR-90 cells, which have been used to study CMA (27), were cultured as described in the literature (27). NIH-3T3 cells stably expressing human UCH-L1 with a FLAG-hemagglutinin double tag at the N terminus were cultured as described previously (13). Transient transfection of COS-7 and IMR-90 cells with each vector was performed using Lipofectamine reagent (Invitrogen) and Lipofectamine LTX reagent (Invitrogen), respectively. There was no notable difference in the transfection efficiency among the culture dishes (wells) in our experimental conditions (data not shown).
Immunoblotting and Immunoprecipitation—Preparation of the detergent (1% Triton X-100)-soluble fraction was performed as described previously (28). The cytosolic fraction that does not contain LAMP-2, a marker of lysosomes, and the crude lysosomal fraction containing LAMP-2 (supplemental Fig. S1A) were prepared according to the method described by Pertoft et al. (29). SDS-PAGE was performed under reducing conditions. Immunoblotting was performed according to standard procedures as described previously (30). For some experiments, Can Get Signal Immunoreaction Enhancer Solution (Toyobo, Osaka, Japan) was used. The signal intensity was quantified by densitometry using FluorChem software (Alpha Innotech, San Leandro, CA). Immunoprecipitation was performed using anti-FLAG M2 affinity gel (Sigma) or 10 μg/ml antibodies (unless otherwise mentioned) with protein G-Sepharose (GE Healthcare), as described previously (13). The antibodies used were as follows. Antibodies against UCH-L1, Cu,Zn-superoxide dismutase 1, Hsc70, and Hsp90 were purchased from Ultra-Clone, Stressgen Bioreagents (Victoria, Canada), Affinity BioReagents (Golden, CO), and BD Transduction Laboratories (Franklin Lakes, NJ), respectively. Anti-β-actin, Mcl-1, and FLAG antibodies were from Sigma. Antibodies against α-synuclein and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were from Chemicon (Temecula, CA). Anti-p53 and Bcl-xL antibodies were from Cell Signaling. Anti-Bcl-2, Ubc9, NF-κB p65, and LAMP-2 antibodies were from Santa Cruz Biotechnology. The rabbit polyclonal anti-LAMP-2A antibody was raised in rabbit against a synthetic peptide (CYFIGLKHHHAGYEQF) containing an amino acid sequence corresponding to the cytosolic region of human LAMP-2A. The specificity of the anti-LAMP-2A antibody was confirmed as shown in supplemental Fig. S1, B and C.
Pulldown Assay—Recombinant human UCH-L1 proteins without a tag were prepared as described previously (13). A pulldown assay was performed as described previously (13) with slight modifications. Streptavidin-Sepharose (GE Healthcare) was blocked with 3% bovine serum albumin for 15 h to prevent nonspecific binding of UCH-L1 to the beads and washed three times with phosphate-buffered saline containing 0.05% Triton X-100. Ten μg of UCH-L1 (wild-type or I93M) and 2 nmol of synthetic peptides conjugated to biotin (control or LAMP-2A peptide, Invitrogen) were mixed and incubated for 15 h in phosphate-buffered saline containing 0.05% Triton X-100. Twenty μl of streptavidin beads blocked with bovine serum albumin was then added, and incubation was continued for 1 h. After beads were washed three times with phosphate-buffered saline containing 0.05% Triton X-100, proteins were eluted with SDS sample buffer and subjected to SDS-PAGE.
UCH-L1 Degradation Assay—COS-7 cells were cotransfected with pTet-Off and pTRE-Tight-hUCH-L1. Twenty-four h after transfection, transcription of UCH-L1-FLAG gene was suppressed by adding 100 ng/ml doxycycline and incubating for 4 h. Then, cells were harvested at the 0-, 24-, and 48-h time points after the suppression of the gene and analyzed by immunoblotting. Pulse-chase analyses were performed as described previously (21) with some modifications. COS-7 cells transfected with pCI-neo-hUCH-L1-FLAG were washed and incubated with methionine-, cysteine-, and cystine-free medium for 1 h. The cells were pulsed with 0.1 mCi/ml [35S]Met and [35S]Cys (Expre35S35S protein labeling mixture, PerkinElmer Life Sciences) for 1 h and then washed and chased with 3 mm methionine and cysteine for 48 h. At the 0-, 24-, and 48-h time points, the cells were harvested for immunoprecipitation with anti-FLAG M2 affinity gel. Following SDS-PAGE on a 15% gel, radioactive bands were detected and analyzed by using a BAS-5000 imaging analyzer (Fujifilm, Tokyo, Japan).
Statistical Analysis—For comparison of two groups, the statistical significance of differences was determined by the Student's t test.
RESULTS
UCH-L1 Interacts with LAMP-2A, Hsc70, and Hsp90—We have previously shown that soluble UCH-L1 interacts with multiple proteins in mammalian cells and that one of the UCH-L1-interacting proteins is α/β-tubulin (13). In this study, we further screened for UCH-L1-interacting proteins using a coimmunoprecipitation assay (Fig. 1A). We identified LAMP-2 as a novel UCH-L1-interacting protein (Fig. 1A). To confirm this interaction in vivo, a coimmunoprecipitation assay was performed using mouse whole brain lysate. Interaction between endogenous UCH-L1 and endogenous LAMP-2 was observed (Fig. 1B). LAMP-2 exists in three different isoforms, LAMP-2A, LAMP-2B, and LAMP-2C, which are produced by the alternative splicing of the LAMP-2 pre-mRNA (31). LAMP-2A forms a complex with chaperones such as Hsc70 and Hsp90 and functions as a receptor for CMA at the lysosomal membrane (26). Because α-synuclein has been reported to interact with LAMP-2A (24), we tested for interactions between UCH-L1 and LAMP-2A, Hsc70, and Hsp90. The UCH-L1 immunoprecipitant included LAMP-2A as well as Hsc70 and Hsp90 (Fig. 1D). These results indicate that UCH-L1 interacts with LAMP-2A, Hsc70, and Hsp90 in mammalian cells.
FIGURE 1.

Physical interactions of UCH-L1 with LAMP-2A, Hsc70, and Hsp90. A, lysates of NIH-3T3 cells stably expressing FLAG-hemagglutinin (HA)-tagged UCH-L1 were immunoprecipitated (IP) with antibodies against cell death- or protein degradation-related proteins and analyzed by immunoblotting using anti-UCH-L1 antibody. A representative blot including immunoprecipitant with anti-LAMP-2 antibody is shown. B, mouse (C57BL/6J) whole brain lysates were immunoprecipitated with control IgG, anti-LAMP-2, or anti-UCH-L1 antibody and immunoblotted with anti-UCH-L1 and LAMP-2 antibodies. C, lysates of COS-7 cells transfected with the indicated constructs were immunoprecipitated with 5μg/ml control IgG or anti-LAMP-2 antibody and analyzed by immunoblotting using anti-UCH-L1 antibody. D, lysates of COS-7 cells transfected with the indicated constructs (–, empty vector) were immunoprecipitated with anti-FLAG beads and immunoblotted using anti-LAMP2, LAMP-2A, Hsc70, Hsp90, and UCH-L1 antibodies.
UCH-L1 Can Be Degraded by Macroautophagy—Although UCH-L1 physically interacts with LAMP-2A, UCH-L1 is not a presumable substrate for CMA because UCH-L1 does not contain a KFERQ-like motif, which is required for substrate proteins to be degraded by CMA (32). Therefore, we speculated that UCH-L1 is degraded by other degradation pathways in mammalian cells. We used a regulatory protein expression system to switch off the expression of UCH-L1 by adding doxycycline, to follow UCH-L1 degradation. Degradation of UCH-L1 was observed 24 or 48 h after expression was switched off, compared with the time point at which expression was switched off (Fig. 2A). The half-life of UCH-L1 was >48 h (Fig. 2A). Long-lived proteins are known to be mainly degraded by macroautophagy (33). We therefore investigated whether UCH-L1 was degraded by macroautophagy using 3-MA, an inhibitor of macroautophagy (24, 28, 34). The 3-MA treatment significantly inhibited the degradation of UCH-L1 (Fig. 2A). Similar results were obtained when we used UCH-L1I93M (Fig. 2B). Pulse-chase experiments also showed that the degradations of UCH-L1WT and UCH-L1I93M were significantly inhibited by 3-MA treatment (Fig. 2, C and D). These results suggest that macroautophagy is one of the major pathways that degrade UCH-L1 in our cell model.
FIGURE 2.
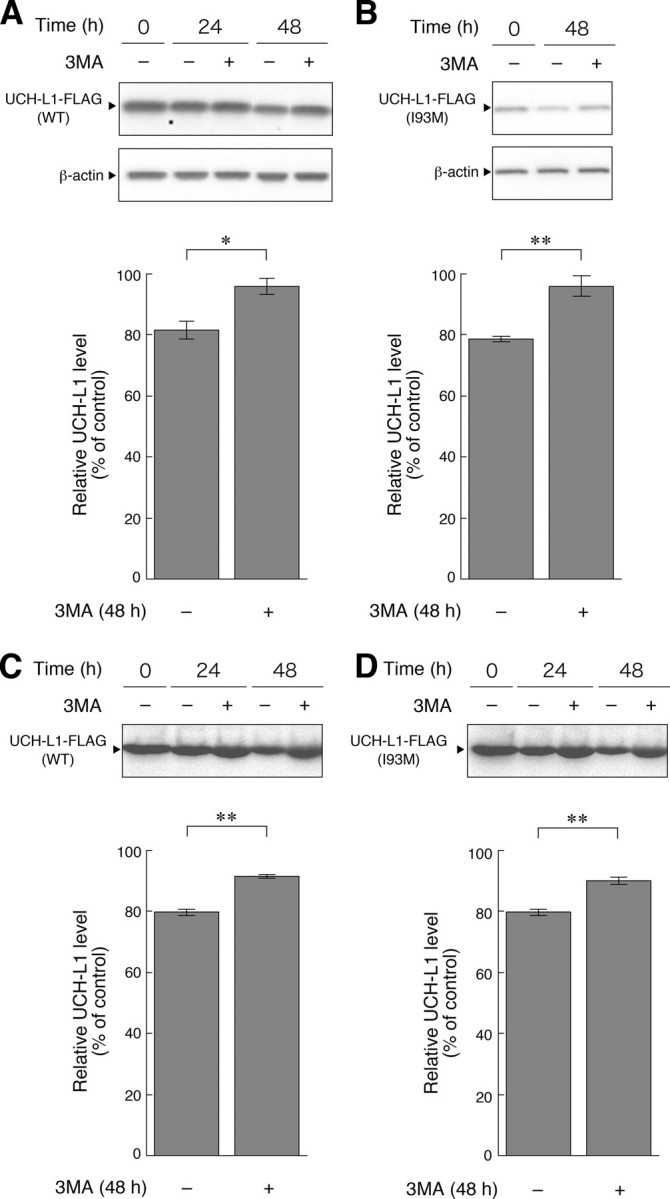
Degradation of UCH-L1 by macroautophagy. A and B, COS-7 cells were transfected with pTet-Off and pTRE-Tight-hUCH-L1WT (A) or pTRE-Tight-hUCH-L1I93M (B). Twenty-four h after transfection, transcription of UCH-L1-FLAG gene was suppressed by adding 100 ng/ml doxycycline and incubating for 4 h. Then, 3-MA (+) or vehicle (–) was added, and cells were harvested at the indicated times after the suppression of the gene and analyzed by immunoblotting (upper panels). The relative levels of UCH-L1-FLAG at 48 h after the suppression (% of 0-h control) were quantified by densitometry. Mean values are shown with S.E. (A, n = 4; B, n = 3). *, p < 0.05; **, p < 0.01. C and D, COS-7 cells were transfected with pCI-neo-hUCH-L1WT-FLAG (C) or pCI-neo-hUCH-L1I93M-FLAG (D). Twenty-four h after transfection, cells were labeled with [35S]Met and [35S]Cys. Autoradiograms of anti-FLAG immunoprecipitates pulse-chased at the indicated times in the absence or presence of 3-MA are shown (upper panels). Relative band intensities at 48 h (% of 0-h control) are quantified. Mean values are shown with S.E. (n = 3). **, p < 0.01.
The Interactions of UCH-L1 with LAMP-2A, Hsc70, and Hsp90 Are Enhanced by the I93M Mutation in UCH-L1 and Are Independent of the Interaction between Monoubiquitin and UCH-L1—We have previously shown that the amount of each protein interacting with UCH-L1I93M is mostly higher than the amount interacting with UCH-L1WT (13). Consistent with this observation, we found that the amount of LAMP-2A, Hsc70, and Hsp90 interacting with UCH-L1I93M is higher than the amount interacting with UCH-L1WT (∼1.8-, 1.3-, and 1.3-fold increases, respectively) (Fig. 1, C and D, and supplemental Fig. S2A). The interactions of LAMP-2, Hsc70, or Hsp90 with UCH-L1S18Y, UCH-L1D30K, which lacks hydrolase activity and binding affinity for ubiquitin (21), and UCH-L1C90S, which lacks hydrolase activity but maintains binding affinity for ubiquitin (21), were not notably changed compared with those of UCH-L1WT (Fig. 1C, supplemental Fig. S2A, and data not shown). These results suggest that the interaction between UCH-L1 and CMA machinery is independent of both UCH-L1-binding affinity for ubiquitin and the hydrolase activity of UCH-L1. To further show that these interactions are independent of monoubiquitin binding to UCH-L1, and to elucidate the amino acid residues of UCH-L1 involved in the interaction with LAMP-2A, Hsc70, and Hsp90, we performed coimmunoprecipitation assays using a series of alanine substitutions (13) of basic and acidic residues located on the surface of UCH-L1 (Fig. 3A). The R63A mutant displayed increased levels of interactions with LAMP-2, Hsc70, and Hsp90, whereas other mutations had no notable effect on the interactions (Fig. 3A). We further performed alanine-scanning mutagenesis experiments and found that E174A, D176A, and H185A mutants also displayed increased levels of interactions with LAMP-2, Hsc70, and Hsp90 (Fig. 3B and data not shown). Glu174, Asp176, and His185 are located near Arg63 (Fig. 3C). The surface region containing Arg63 and His185 possesses features that are characteristic of a protein-protein interacting site (35). These observations suggest that this surface region, which is distinct from the ubiquitin-binding region (13, 35), is involved in the interactions with LAMP-2, Hsc70, and Hsp90. The R63A, E174A, D176A, or H185A mutation possibly causes partial misfolding, resulting in increased interactions.
FIGURE 3.

Alanine-scanning mutagenesis of UCH-L1. A and B, lysates of COS-7 cells transfected with the indicated constructs were immunoprecipitated (IP) with anti-FLAG antibody and analyzed by immunoblotting. C, a structural model for human UCH-L1 is shown. Arg63, Glu174, Asp176, and His185 are shown in blue, green, magenta, and red, respectively, using Cn3D software (version 4.1) and NCBI structural model mmdbId:38174 (35).
UCH-L1 Directly Interacts with the Cytoplasmic Region of LAMP-2A—LAMP-2 is a type 1 membrane protein, consisting of a short cytoplasmic tail (12 amino acids), one transmembrane domain, and a glycosylated luminal domain (31). To test whether UCH-L1 directly interacts with the cytosolic region of LAMP-2A, we prepared purified recombinant wild-type and I93M UCH-L1 proteins, a peptide containing an amino acid sequence corresponding to the C-terminal cytoplasmic tail of LAMP-2A, and a control peptide (Fig. 4A). Purified UCH-L1 proteins and the peptides were mixed, and pulldown assays were performed. A direct interaction between wild-type UCH-L1 and the cytosolic region of LAMP-2A was observed (Fig. 4, B and C). Consistent with the results of the coimmunoprecipitation assay, UCH-L1I93M exhibited an abnormally increased level of interaction with the cytosolic region of LAMP-2A compared with wild-type UCH-L1 (Fig. 4D). Because chaperones, including Hsc70, are considered to be required for the interaction of the CMA substrates with LAMP-2A (36), our results may indicate that UCH-L1 interacts with LAMP-2A in a manner different from the interaction between CMA substrates and LAMP-2A.
FIGURE 4.
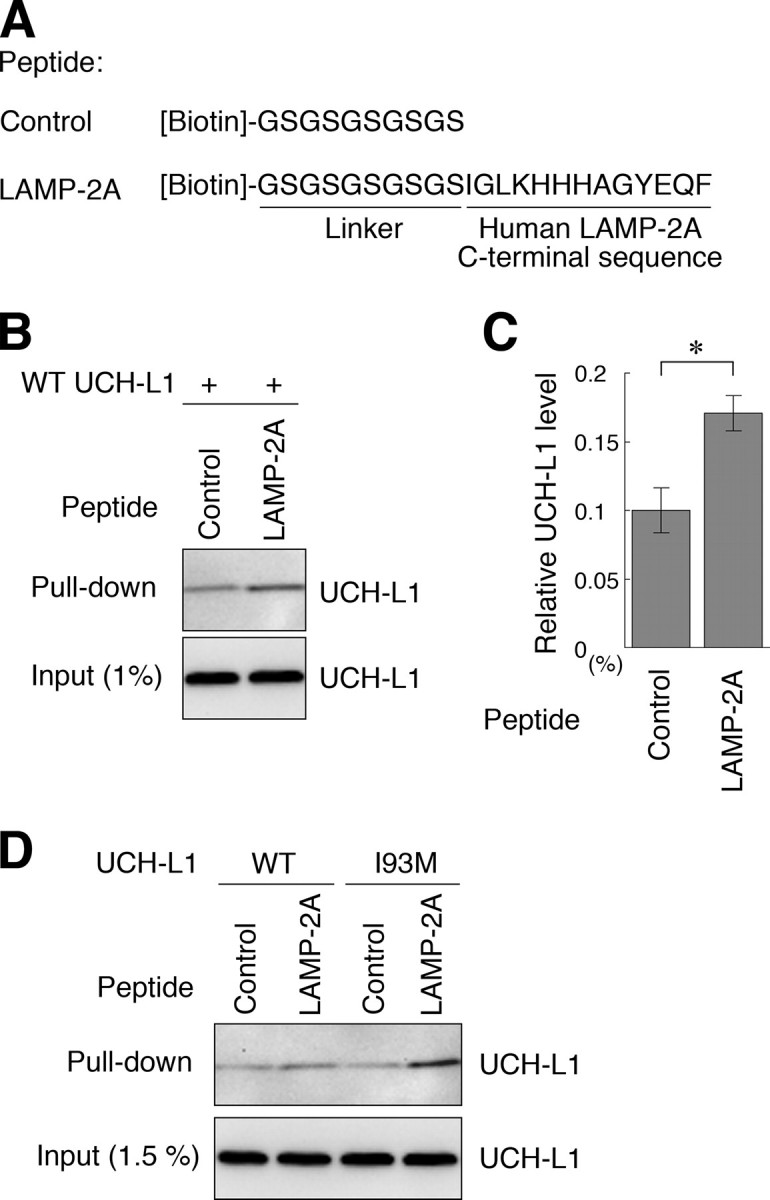
Direct interaction between UCH-L1 and the cytosolic region of LAMP-2A. A, an amino acid sequence of biotin-conjugated peptides is shown. B and C, 10μg of recombinant UCH-L1 and 2 nmol of peptides (control or LAMP-2A peptide) were mixed, and a pulldown assay was performed using streptavidin beads. Precipitants were analyzed by immunoblotting (B). The levels of UCH-L1 relative to input were quantified by densitometry. Mean values are shown with S.E. (n = 3). *, p < 0.05. D, 10 μg of UCH-L1 (wild-type or I93M) and 2 nmol of peptides (control or LAMP-2A peptide) were mixed, and a pulldown assay was performed.
UCH-L1I93M Causes Accumulation of α-Synuclein—It has been reported that α-synucleinWT is a CMA substrate, but pathogenic mutants A30P and A53T α-synuclein inhibit CMA by tight binding to LAMP-2A (24). Thus, UCH-L1I93M, which exhibits elevated interactions with LAMP-2A, Hsc70, and Hsp90, may also inhibit CMA. To examine this possibility in mammalian cells, we assessed the effects of UCH-L1I93M on the protein level of GAPDH, an established substrate of CMA (24), in the lysosomal fraction and whole-cell lysate. The GAPDH level in whole-cell lysate was increased in cells expressing UCH-L1I93M compared with that in cells expressing UCH-L1WT (an ∼1.5-fold increase) (Fig. 5A), whereas the GAPDH level in the lysosomal fraction was decreased in cells expressing UCH-L1I93M (an ∼2.1-fold decrease) (Fig. 5B), supporting the idea that the aberrant interaction of UCH-L1I93M with CMA machinery inhibits CMA. The inhibition of CMA also results in the accumulation of other CMA substrates, including α-synuclein (24). We found that the amount of α-synucleinWT was increased in cells expressing UCH-L1I93M compared with cells expressing UCH-L1WT (∼1.7 and 1.4-fold increases, respectively) (Fig. 5, C and D) or control mock cells (data not shown). The physical interaction between UCH-L1 and α-synuclein was not detected under these experimental conditions (data not shown). These results suggest that the accumulation of α-synuclein in cells expressing UCH-L1I93M is due to the inhibition of CMA-dependent degradation of α-synuclein. α-Synuclein contains a CMA recognition motif, 95VKKDQ99, and mutant α-synucleinΔDQ, in which 98DQ99 is replaced by Ala-Ala, is not degraded by CMA (24). To confirm that the accumulation of α-synuclein in cells expressing UCH-L1I93M is associated with CMA-dependent degradation of α-synuclein, we used mutant α-synucleinΔDQ and found that the I93M mutation does not affect the α-synucleinΔDQ level (∼1.0 and 1.0-fold increases, respectively) (Fig. 5, E and F).
FIGURE 5.

Effects of the I93M mutation of UCH-L1 on CMA and α-synuclein levels. A and B, COS-7 cells were transfected with the indicated constructs. Forty-eight h after transfection, whole-cell lysates (A) and a lysosomal fraction (B) were prepared and analyzed by immunoblotting. C–G, COS-7 cells (C, E, and G) or IMR-90 cells (D and F) were transfected with the indicated constructs. Cell lysates were prepared and analyzed by immunoblotting. Accumulation of α-synucleinWT in cells transfected with UCH-L1I93M was observed in COS-7 (C), IMR-90 (D), and SH-SY5Y cells (data not shown). The assays were performed at least three times; representative results are shown. SOD1, Cu,Zn-superoxide dismutase 1.
G93A Cu,Zn-superoxide dismutase 1 and WT Cu,Zn-superoxide dismutase 1 are not presumable substrates for CMA because Cu,Zn-superoxide dismutase 1 does not contain a KFERQ-like motif, but they can be degraded by the proteasome and macroautophagy (28). Protein levels of G93A Cu,Zn-superoxide dismutase 1 and WT Cu,Zn-superoxide dismutase 1 in cells transfected with UCH-L1I93M were not increased compared with those in cells expressing UCH-L1WT (an ∼1.0-fold increase) (Fig. 5G and data not shown), suggesting that the I93M mutation does not considerably affect the degradation of proteins by macroautophagy and the proteasome under these experimental conditions.
Contrary to UCH-L1I93M, UCH-L1D30K and UCH-L1C90S did not increase the amount of α-synuclein in cells (supplemental Fig. S2B), indicating that the accumulation of α-synuclein in cells expressing UCH-L1I93M is independent of the hydrolase activity of UCH-L1 and the interaction between monoubiquitin and UCH-L1. These observations are consistent with the results showing that the interaction between UCH-L1 and LAMP-2A, Hsc70, or Hsp90 is independent of the enzymatic activity of UCH-L1 and the interaction between monoubiquitin and UCH-L1 (Figs. 1C and 3) and also with the idea that the main cause of UCH-L1I93M-associated PD is not a loss of UCH-L1 function but an acquired toxicity of UCH-L1I93M.
DISCUSSION
An increase in the amount of α-synuclein protein could constitute a pathogenic factor underlying sporadic PD because the heterozygous duplication of the α-synuclein gene causes familial PD (4, 5), and the deposition of α-synuclein protein is associated with sporadic PD (7, 8, 37). α-SynucleinWT is a CMA substrate, but mutant A30P and A53T α-synuclein inhibit CMA by aberrant tight binding to LAMP-2A (24). Thus, inhibition of CMA by mutant α-synuclein might result in an increase in the amount of α-synuclein protein, leading to the neurodegeneration in familial PD associated with mutant α-synuclein. To date, the relationships between α-synuclein and other familial PD-associated mutant proteins in the pathogenesis of PD have remained largely unclear. Although it was reported that UCH-L1 polyubiquitinates monoubiquitinated α-synuclein in a cell-free system (20), the relationship between UCH-L1 and non-ubiquitinated α-synuclein in the pathogenesis of PD has also remained unknown.
In the present study, we have shown that familial PD-associated UCH-L1I93M abnormally interacts with LAMP-2A, Hsc70, and Hsp90 and causes an increase in the amounts of α-synuclein and GAPDH, which are CMA substrates, in cultured cells. The increase can be explained by an inhibition of CMA via an aberrant interaction between UCH-L1I93M and CMA machinery because the GAPDH level in the lysosomal fraction was decreased in cells expressing UCH-L1I93M (Fig. 5B), and the I93M mutation in UCH-L1 does not affect the levels of α-synucleinΔDQ, which is not degraded by CMA (Fig. 5, E and F). These findings suggest that an increase in the amount of α-synuclein protein by inhibition of CMA via the interaction between UCH-L1I93M and CMA machinery underlies one of the causes of familial PD associated with mutant UCH-L1. It is also possible that increases in the amount of other CMA substrates, such as GAPDH, are involved in the pathogenesis of PD. Taken together with a report that pathogenic mutant A30P and A53T α-synuclein exhibit an enhanced interaction with LAMP-2A compared with α-synucleinWT (24), our results indicate that UCH-L1I93M and mutant A30P and A53T α-synuclein share aberrant biochemical properties with respect to their interactions with LAMP-2A. These observations further support the idea that the I93M mutation in UCH-L1 contributes to the pathogenesis of PD.
We revealed that the R63A, E174A, D176A, or H185A substitution in UCH-L1 increases the levels of interactions of UCH-L1 with LAMP-2, Hsc70, and Hsp90 (Fig. 3), suggesting that the surface region containing Arg63 and His185 in UCH-L1 (35) is involved in its interaction with LAMP-2, Hsc70, and Hsp90. We have previously reported that the R63A or H185A substitution in UCH-L1 enhances the interaction of UCH-L1 with tubulin (13). These results suggest that tubulin, LAMP-2A, Hsc70, and Hsp90 interact with the same region in UCH-L1. Arg63 and His185 are distinct from Asp30, which is one of the ubiquitin-binding sites (21, 38), and from Cys90 (13), which is a catalytic center cysteine residue. We have shown that D30K or C90S mutation in UCH-L1 does not alter its interactions with tubulin (13) and LAMP-2 (Fig. 1C). Thus, the interactions of UCH-L1 with tubulin and LAMP-2 are independent of the monoubiquitin-binding and hydrolase activity of UCH-L1.
It is known that the majority of PD cases occur sporadically and that oxidative/carbonyl stresses are elevated in PD brains (17, 39). In the brains of sporadic PD patients, UCH-L1 is a major target of carbonyl formation (17). We previously reported that carbonyl-modified UCH-L1 and UCH-L1I93M share biochemical properties: both of these UCH-L1 variants display increased insolubility, elevated interactions with multiple proteins including tubulin, and decreased interaction with monoubiquitin compared with UCH-L1WT (13). We have also shown that both carbonyl-modified UCH-L1 and UCH-L1I93M abnormally promote tubulin polymerization (13). Our previous studies using circular dichroism suggest that both of these UCH-L1 variants display decreased α-helix and increased β-sheet content (13, 22, 40). Thus, both carbonyl modification and the I93M mutation in UCH-L1 may alter its conformation, resulting in changes in the biochemical and functional properties of UCH-L1. It is an interesting issue whether carbonyl-modified UCH-L1 can also inhibit CMA. Other than tubulin, LAMP-2A, Hsc70, and Hsp90, UCH-L1 interacts with multiple proteins (13). These other interactors may also be involved in the mechanism of UCH-L1-mediated PD and are currently under investigation. It is also possible that the interaction of Hsc70 or Hsp90 with UCH-L1 plays roles other than in the CMA pathway.
α-Synuclein and UCH-L1 have been reported to be expressed abundantly in dopaminergic neurons in the human brain (41). Thus, UCH-L1I93M is possibly overproduced in dopaminergic neurons in familial PD, leading to an accumulation of α-synuclein and the selective loss of dopaminergic neurons. In conclusion, familial PD-associated mutant UCH-L1I93M physically interacts with LAMP-2A, Hsc70, and Hsp90 and causes an increase in the amount of α-synuclein in cells. We propose that aberrant interaction of mutant UCH-L1 with CMA machinery, at least in part, underlies the pathogenesis of familial PD associated with UCH-L1I93M.
Supplementary Material
Acknowledgments
We thank Dr. Yasuyuki Suzuki (National Institute of Neuroscience) and Dr. Rieko Setsuie (National Institute of Neuroscience) for scientific comments and Takeshi Mitsui (National Institute of Neuroscience) for technical assistance.
This work was supported by grants-in-aid for scientific research from the Japan Society for the Promotion of Science; a research grant in priority area research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; grants-in-aid for scientific research from the Ministry of Health, Labor, and Welfare, Japan; and the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation and the New Energy and Industrial Technology Development Organization, Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures,” Figs. S1 and S2, and an additional reference.
Footnotes
The abbreviations used are: PD, Parkinson disease; UCH-L1, ubiquitin C-terminal hydrolase L1; WT, wild-type; CMA, chaperone-mediated autophagy; LAMP-2, lysosome-associated membrane protein type 2; Hsc70, heat shock cognate protein 70; Hsp90, heat shock protein 90; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
References
- 1.Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997) Science 2762045 –2047 [DOI] [PubMed] [Google Scholar]
- 2.Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J. T., Schols, L., and Riess, O. (1998) Nat. Genet. 18106 –108 [DOI] [PubMed] [Google Scholar]
- 3.Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L., Hoenicka, J., Rodriguez, O., Atares, B., Llorens, V., Gomez Tortosa, E., del Ser, T., Munoz, D. G., and de Yebenes, J. G. (2004) Ann. Neurol. 55164 –173 [DOI] [PubMed] [Google Scholar]
- 4.Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., Levecque, C., Larvor, L., Andrieux, J., Hulihan, M., Waucquier, N., Defebvre, L., Amouyel, P., Farrer, M., and Destee, A. (2004) Lancet 3641167 –1169 [DOI] [PubMed] [Google Scholar]
- 5.Ibanez, P., Bonnet, A. M., Debarges, B., Lohmann, E., Tison, F., Pollak, P., Agid, Y., Durr, A., and Brice, A. (2004) Lancet 3641169 –1171 [DOI] [PubMed] [Google Scholar]
- 6.Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M. R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J., and Gwinn-Hardy, K. (2003) Science 302 841. [DOI] [PubMed] [Google Scholar]
- 7.Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997) Nature 388839 –840 [DOI] [PubMed] [Google Scholar]
- 8.Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998) Proc. Natl. Acad. Sci. U. S. A. 956469 –6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leroy, E., Boyer, R., Auburger, G., Leube, B., Ulm, G., Mezey, E., Harta, G., Brownstein, M. J., Jonnalagada, S., Chernova, T., Dehejia, A., Lavedan, C., Gasser, T., Steinbach, P. J., Wilkinson, K. D., and Polymeropoulos, M. H. (1998) Nature 395451 –452 [DOI] [PubMed] [Google Scholar]
- 10.Setsuie, R., and Wada, K. (2007) Neurochem. Int. 51105 –111 [DOI] [PubMed] [Google Scholar]
- 11.Healy, D. G., Abou-Sleiman, P. M., and Wood, N. W. (2004) Cell Tissue Res. 318189 –194 [DOI] [PubMed] [Google Scholar]
- 12.Setsuie, R., Wang, Y. L., Mochizuki, H., Osaka, H., Hayakawa, H., Ichihara, N., Li, H., Furuta, A., Sano, Y., Sun, Y. J., Kwon, J., Kabuta, T., Yoshimi, K., Aoki, S., Mizuno, Y., Noda, M., and Wada, K. (2007) Neurochem. Int. 50119 –129 [DOI] [PubMed] [Google Scholar]
- 13.Kabuta, T., Setsuie, R., Mitsui, T., Kinugawa, A., Sakurai, M., Aoki, S., Uchida, K., and Wada, K. (2008) Hum. Mol. Genet. 171482 –1496 [DOI] [PubMed] [Google Scholar]
- 14.Lowe, J., McDermott, H., Landon, M., Mayer, R. J., and Wilkinson, K. D. (1990) J. Pathol. 161153 –160 [DOI] [PubMed] [Google Scholar]
- 15.Maraganore, D. M., Lesnick, T. G., Elbaz, A., Chartier-Harlin, M. C., Gasser, T., Kruger, R., Hattori, N., Mellick, G. D., Quattrone, A., Satoh, J., Toda, T., Wang, J., Ioannidis, J. P., de Andrade, M., and Rocca, W. A. (2004) Ann. Neurol. 55512 –521 [DOI] [PubMed] [Google Scholar]
- 16.Healy, D. G., Abou-Sleiman, P. M., Casas, J. P., Ahmadi, K. R., Lynch, T., Gandhi, S., Muqit, M. M., Foltynie, T., Barker, R., Bhatia, K. P., Quinn, N. P., Lees, A. J., Gibson, J. M., Holton, J. L., Revesz, T., Goldstein, D. B., and Wood, N. W. (2006) Ann. Neurol. 59627 –633 [DOI] [PubMed] [Google Scholar]
- 17.Choi, J., Levey, A. I., Weintraub, S. T., Rees, H. D., Gearing, M., Chin, L. S., and Li, L. (2004) J. Biol. Chem. 27913256 –13264 [DOI] [PubMed] [Google Scholar]
- 18.Wilkinson, K. D., Lee, K. M., Deshpande, S., Duerksen-Hughes, P., Boss, J. M., and Pohl, J. (1989) Science 246670 –673 [DOI] [PubMed] [Google Scholar]
- 19.Larsen, C. N., Krantz, B. A., and Wilkinson, K. D. (1998) Biochemistry 373358 –3368 [DOI] [PubMed] [Google Scholar]
- 20.Liu, Y., Fallon, L., Lashuel, H. A., Liu, Z., and Lansbury, P. T., Jr. (2002) Cell 111209 –218 [DOI] [PubMed] [Google Scholar]
- 21.Osaka, H., Wang, Y. L., Takada, K., Takizawa, S., Setsuie, R., Li, H., Sato, Y., Nishikawa, K., Sun, Y. J., Sakurai, M., Harada, T., Hara, Y., Kimura, I., Chiba, S., Namikawa, K., Kiyama, H., Noda, M., Aoki, S., and Wada, K. (2003) Hum. Mol. Genet. 121945 –1958 [DOI] [PubMed] [Google Scholar]
- 22.Nishikawa, K., Li, H., Kawamura, R., Osaka, H., Wang, Y. L., Hara, Y., Hirokawa, T., Manago, Y., Amano, T., Noda, M., Aoki, S., and Wada, K. (2003) Biochem. Biophys. Res. Commun. 304176 –183 [DOI] [PubMed] [Google Scholar]
- 23.Saigoh, K., Wang, Y. L., Suh, J. G., Yamanishi, T., Sakai, Y., Kiyosawa, H., Harada, T., Ichihara, N., Wakana, S., Kikuchi, T., and Wada, K. (1999) Nat. Genet. 2347 –51 [DOI] [PubMed] [Google Scholar]
- 24.Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004) Science 3051292 –1295 [DOI] [PubMed] [Google Scholar]
- 25.Cuervo, A. M. (2004) Trends Cell Biol. 1470 –77 [DOI] [PubMed] [Google Scholar]
- 26.Agarraberes, F. A., and Dice, J. F. (2001) J. Cell Sci. 1142491 –2499 [DOI] [PubMed] [Google Scholar]
- 27.Finn, P. F., and Dice, J. F. (2005) J. Biol. Chem. 28025864 –25870 [DOI] [PubMed] [Google Scholar]
- 28.Kabuta, T., Suzuki, Y., and Wada, K. (2006) J. Biol. Chem. 28130524 –30533 [DOI] [PubMed] [Google Scholar]
- 29.Pertoft, H., Warmegard, B., and Hook, M. (1978) Biochem. J. 174309 –317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabuta, T., Hakuno, F., Asano, T., and Takahashi, S. (2002) J. Biol. Chem. 2776846 –6851 [DOI] [PubMed] [Google Scholar]
- 31.Eskelinen, E. L., Cuervo, A. M., Taylor, M. R., Nishino, I., Blum, J. S., Dice, J. F., Sandoval, I. V., Lippincott-Schwartz, J., August, J. T., and Saftig, P. (2005) Traffic 61058 –1061 [DOI] [PubMed] [Google Scholar]
- 32.Dice, J. F. (1990) Trends Biochem. Sci. 15305 –309 [DOI] [PubMed] [Google Scholar]
- 33.Levine, B., and Kroemer, G. (2008) Cell 13227 –42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webb, J. L., Ravikumar, B., Atkins, J., Skepper, J. N., and Rubinsztein, D. C. (2003) J. Biol. Chem. 27825009 –25013 [DOI] [PubMed] [Google Scholar]
- 35.Das, C., Hoang, Q. Q., Kreinbring, C. A., Luchansky, S. J., Meray, R. K., Ray, S. S., Lansbury, P. T., Ringe, D., and Petsko, G. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 1034675 –4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majeski, A. E., and Dice, J. F. (2004) Int. J. Biochem. Cell Biol. 362435 –2444 [DOI] [PubMed] [Google Scholar]
- 37.Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., Trojanowski, J. Q., and Iwatsubo, T. (1998) Am. J. Pathol. 152879 –884 [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston, S. C., Riddle, S. M., Cohen, R. E., and Hill, C. P. (1999) EMBO J. 183877 –3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ischiropoulos, H., and Beckman, J. S. (2003) J. Clin. Investig. 111163 –169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naito, S., Mochizuki, H., Yasuda, T., Mizuno, Y., Furusaka, M., Ikeda, S., Adachi, T., Shimizu, H. M., Suzuki, J., Fujiwara, S., Okada, T., Nishikawa, K., Aoki, S., and Wada, K. (2006) Biochem. Biophys. Res. Commun. 339717 –725 [DOI] [PubMed] [Google Scholar]
- 41.Solano, S. M., Miller, D. W., Augood, S. J., Young, A. B., and Penney, J. B., Jr. (2000) Ann. Neurol. 47201 –210 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.