

Abstract
Uracil-DNA glycosylase (UDG) is an important repair enzyme in all organisms to remove uracil bases from DNA. Recent biochemical studies have revealed that human nuclear UDG (UNG2) forms a multiprotein complex in replication foci and initiates the base excision repair pathway by interacting with proliferating cell nuclear antigen (PCNA). Here, we show the physical and functional interactions between UDG and PCNA from the hyperthermophilic euryarchaeon, Pyrococcus furiosus. The physical interaction between the two proteins was identified by a surface plasmon resonance analysis. Furthermore, the uracil glycosylase activity of P. furiosus UDG is stimulated by P. furiosus PCNA (PfuPCNA) in vitro. This stimulatory effect was observed only when wild type PfuPCNA, but not a monomeric PCNA mutant, was present in the reaction. Mutational analyses revealed that our predicted PCNA-binding region (AKTLF) in P. furiosus UDG is actually important for the interaction with PfuPCNA. This is the first report describing the functional interaction between archaeal UDG and PCNA.
Hydrolytic deamination of cytosine to uracil, which is a common type of mutagenic base lesion, leads to the formation of U:G base pairs in DNA (1). The uracil base is also generated by the misincorporation of dUMP during DNA replication, which results in the formation of U:A base pairs. Damaged bases are removed and restored by the base excision repair (BER)2 pathway (reviewed in Refs. 2 and 3). Uracil-DNA glycosylase (UDG) initiates the BER pathway by catalyzing the hydrolysis of the N-glycosyl bonds linking the uracil base to the sugar-phosphate backbone in DNA. The repair of the abasic site thus produced is achieved by other enzymes, including AP endonuclease, which produces an incision or nick in duplex DNA immediately 5′ to the AP site. Currently, the UDGs are classified into five families, based on their substrate specificity and amino acid sequence motifs in the active site (referred to as motifs A and B), although the UDGs form a single protein superfamily with a common structural fold (reviewed in Refs. 4 and 5). The family 1 UDGs, including the Ung proteins from Escherichia coli, herpes simplex virus type 1, and humans, have been most extensively studied. The Ung enzymes remove the uracil base from both single- and double-stranded DNA. In human cells, two types of enzymes are produced by alternative splicing and are distributed to mitochondria (UNG1) and the nucleus (UNG2). The family 2 enzymes can catalyze the removal of uracil and thymine that is mispaired with guanine in double-stranded DNA. Mismatch-specific uracil-DNA glycosylase and thymine-DNA glycosylase belong to this family. The family 3 UDGs exhibit unique substrate specificity for single-stranded DNA and therefore have been named single strand-selective monofunctional uracil-DNA glycosylase. This enzyme family actually has broader specificity than the Ungs, and double-stranded DNA is a substrate as well as single-stranded DNA. The family 4 UDGs are known as the thermostable UDGs found in some thermophilic bacteria and Archaea. Biochemical studies of Pyrobaculum aerophilum UDG and the crystallographic analysis of Thermus thermophilus UDG (TthUDG) revealed that the most characteristic feature of the family 4 UDGs is the presence of structural iron-sulfur (Fe-S) centers (6, 7). Recently, several reports have demonstrated the existence of Fe-S-containing enzymes that are involved in DNA transactions (8, 9). The family 5 UDGs were identified as a novel type of thermostable UDG based on the second UDG activity (UDGb) in P. aerophilum (10) and T. thermophilus (11). Although the family 4 and family 5 UDGs share sequence similarity, their active site forms are different. The active site of the family 5 UDGs lacks a polar residue corresponding to a catalytic residue, glutamate, in the family 4 UDGs. In addition, the family 5 UDG from P. aerophilum can reportedly excise hypoxanthine (deaminated adenine) in DNA (10).
Several biochemical studies using human cell extracts have provided evidence for the existence of a UNG2-associated repair complex at replication foci (12-14). The complex contains a DNA sliding clamp called proliferating cell nuclear antigen (PCNA), which acts as a processivity factor for replicative DNA polymerases (reviewed in Ref. 15). Consistent with these observations, physical and functional interactions between human UNG2 and PCNA in vitro were confirmed with the purified proteins (16). Besides UNG2, several DNA glycosylases reportedly interact with PCNA (17-19). It is widely known that many other replicative and repair enzymes interact with PCNA, via a consensus sequence called the PCNA-interacting protein (PIP) box (15). The PIP box consists of the sequence QXX(L/I/M)XX(F/Y)(F/Y). In many cases, the hydrophobicity of the aromatic side chain of the PCNA is important for the interaction with PCNA-binding proteins (20-22). It was previously suggested that human UNG2 interacts with PCNA via a typical PCNA-binding motif, 4QKTLYSFF11, at the N terminus of the protein (12).
In Archaea, the third domain of life, physical interactions between PCNA and the family 4 UDGs from P. aerophilum and Sulfolobus solfataricus have been reported (23, 24). These previous studies revealed that the two successive hydrophobic amino acid residues within the C-terminal PCNA-binding motif are important for the binding to their cognate PCNA (193PITLDNFL200 for S. solfataricus UDG and 185GGG-LDRFL192 for P. aerophilum UDG). Although the direct interactions between PCNA and the UDGs from the two crenarchaeal organisms described above were well documented, the functional meaning of the interaction is not fully understood. In addition, it is interesting that the unusual PCNA-binding motifs found in these crenarchaeal UDGs are not conserved in Archaea, and especially, several euryarchaeal UDGs lack the C-terminal region containing the PCNA-binding motif of the crenarchaeal UDGs (23). Thus, it is unclear whether all of the archaeal UDGs can form complexes with PCNA. Moreover, there has been no report describing the purification of a BER complex from archaeal cell extracts. Thus, little is currently known about the interactions of archaeal UDGs with other related protein factors. Interestingly, the complex formation between archaeal family B DNA polymerase and UDG was described previously (25). The family B DNA polymerases from the hyperthermophilic Archaea specifically recognize a uracil base in the template strand and stall DNA polymerization in vitro. This property of the archaeal DNA polymerases has been implicated as an intrinsic ability for the removal of the uracil base (25-27). The rate of cytosine deamination to uracil is accelerated at high temperature (28). These findings raised the possibility that a unique mechanism for uracil excision repair may exist in hyperthermophilic Archaea.
In this study, we describe the physical and functional interactions between the PCNA and UDG proteins from the hyperthermophilic euryarchaeon, Pyrococcus furiosus. The physical interaction between P. furiosus UDG (PfuUDG) and P. furiosus PCNA (PfuPCNA) was detected by a surface plasmon resonance (SPR) analysis. The stimulatory effect of PfuPCNA on the uracil-N-glycosylase activity of PfuUDG in vitro was also observed. Based on our knowledge, the PCNA-interacting site was predicted in the PfuUDG sequence, and the pentapeptide sequence 152AKTLF156 in PfuUDG was demonstrated to be directly involved in binding to PfuPCNA. Interestingly, the PCNA-binding motif found in PfuUDG was not located at the extreme C terminus, as in the crenarchaeal UDGs, but toward the middle of the peptide chain.
EXPERIMENTAL PROCEDURES
Cloning the Genes Encoding PfuUDG and Its Mutant Proteins—The uracil DNA glycosylase gene (udg) was amplified by PCR directly from P. furiosus genomic DNA, using the oligonucleotides 5′-GCGCCATATGTCAAAGCATGAGCTAATGAA-3′ and 5′-GCGCGGATCCCTAAATCCCTAGTTTTTCTA-3′ as the forward and reverse primers, respectively. The amplified gene was cloned into the pGEM-T Easy vector (Promega), and its nucleotide sequence was confirmed. The cloned gene was digested by NdeI-BamHI and inserted into the corresponding sites of the expression vector, pET-21a (Novagen). The resultant plasmid was designated pET-udg. Amino acid substitutions were introduced into the udg gene on the pET-udg plasmid by a PCR-mediated mutagenesis (QuikChange site-directed mutagenesis kit; Stratagene) using the appropriate primers. Their sequences are available upon request.
Protein Purification—To obtain recombinant PfuUDG, E. coli Rosetta(DE3)pLysS cells (Novagen) carrying pET-udg were grown in 1 liter of LB medium, containing 50 μg/ml ampicillin and 34 μg/ml chloramphenicol, at 37 °C. The cells were cultured to A600 = 0.60, and expression of the udg gene was induced by adding isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mm and continuing the culture for 3 h at 37 °C. After cultivation, the cells were harvested and disrupted by sonication in buffer A (50 mm Tris-HCl, pH 8.0, 0.5 mm DTT, and 10% glycerol). The soluble cell extract, obtained by centrifugation at 12,000 × g for 20 min, was heated at 80 °C for 20 min. The heat-resistant fraction obtained by centrifugation was treated with 0.15% polyethyleneimine to remove the nucleic acids. The soluble proteins were precipitated by 80% saturated ammonium sulfate. The precipitate was resuspended in buffer B (50 mm Tris-HCl, pH 8.0, 1 m (NH4)2SO4, 0.5 mm dithiothreitol, and 10% glycerol) and was subjected to chromatography on a Hitrap phenyl column (GE Healthcare). The proteins containing the UDG activity were eluted at 0 m ammonium sulfate, and the eluted proteins were dialyzed against buffer C (50 mm Tris-HCl, pH 8.0, 0.1 m NaCl, 0.5 mm dithiothreitol, and 10% glycerol). The dialysate was loaded onto a Hitrap Q column (GE Healthcare), and the proteins were eluted in the flow-through fraction. The eluted proteins were subjected to chromatography on a Hitrap SP column (GE Healthcare). The proteins containing UDG activity were eluted at 0.25 m sodium chloride, pooled, and stored at 4 °C. The mutated PfuUDG proteins prepared in this study were purified by the same procedures. The purification of PfuPCNA and its mutants was performed as described previously (29). P. furiosus Pol BI (PfuPolBI) was prepared as described previously (30). The purity of each protein used in this study was evaluated by SDS-PAGE. No extra band was detected by Coomassie Brilliant Blue staining of the gel containing 2 μg of each purified protein. The protein concentrations were calculated by measuring the absorbance at 280 nm. The theoretical molecular absorption coefficient of each molecule was calculated based on its tryptophan and tyrosine content, as described (31). The calculated molar extinction coefficients of PfuUDG, PfuPCNA, and PfuPolBI are 24,410, 7450, and 129,040, respectively.
Uracil DNA Glycosylase Assay—The 49-mer deoxynucleotide (5′-AGCTATGACCATGATTACGAATTGUTTAATTCGTGCAGGCATGGTAGCT-3′) labeled with 32P at the 5′ terminus was annealed to either the 49-mer deoxynucleotide, 5′-AGCTACCATGCCTGCACGAATTAAGCAATTCGTAATCATGGTCATAGCT-3′, or 5′-AGCTACCATGCCTGCACGAATTAAACAATTCGTAATCATGGTCATAGCT-3′ in TAM buffer (40 mm Tris acetate, pH 7.8, and 0.5 mm magnesium acetate) to produce a double-stranded substrate with a G:U base pair or a A:U base pair at the center of the 49-mer. The purified PfuUDG proteins (at different concentrations for each experiment, as described in the figure legends) were incubated with 5 nm DNA substrate, prepared as described above, in 20 μl of assay buffer (50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1 mm dithiothreitol, and 0.1 μg/ml bovine serum albumin) at 60 °C for 15 min. The reactions were then subjected to hot alkali treatment by the addition of 2 μl of 200 mm NaOH and an incubation for 10 min at 90 °C. After the treatment, the same volume of 200 mm HCl was added to neutralize the samples, and then a 6-μl aliquot of loading dye (98% formamide, 10 mm EDTA, 0.1% bromphenol blue, and 0.1% xylene cyanol) was added. Samples were heated at 98 °C for 5 min and chilled rapidly on ice prior to loading onto a denaturing 12% polyacrylamide gel containing 7 m urea. After electrophoresis, the gels were dried and were scanned with a FLA5000 imager to detect the 32P-labeled DNA. Three independent experiments were carried out in succession for each condition required in this study, and the S.E. values are shown as vertical lines on the plots in each graph.
Measurements of UV-visible Spectra—The purified wild-type PfuUDG, C17S, C20S, and C17/20S proteins were concentrated to 180 μm with Ultrafree-0.5 centrifugal filter devices (Millipore). The UV-visible absorption spectra of the proteins were measured with an Ultrospec 3100 Pro (GE Healthcare).
Surface Plasmon Resonance Analysis—A Biacore system was used to study the physical interaction between PfuUDG and PfuPCNA. Highly purified recombinant PfuUDG or PfuPCNA was bound to a CM5 sensor chip (research grade; Biacore) according to the manufacturer's recommendations. To measure the kinetic parameters, various concentrations of PfuUDG (0.4, 0.6, 0.8, 1.0, and 1.2 μm) were applied to the immobilized PCNA. The physical interaction between PfuUDG and PfuPolBI was evaluated in the same manner. All measurements were conducted at 25 °C, in buffer containing 10 mm HEPES (pH 7.4), 150 mm NaCl, and 0.005% Tween 20. At the end of each cycle, the bound protein was removed by washing with 2 m NaCl. The equilibrium constant (KD) for PfuUDG binding to PfuPCNA was determined from the association and dissociation curves of the sensorgrams, using the BIAevaluation program (Biacore).
Model Building of PfuUDG·PfuPCNA·DNA—The molecular model was constructed from the crystal structures of T. thermophilus UDG (Protein Data Bank code 2ddg) and P. furiosus PCNA (Protein Data Bank code 1isq) by using two in-house programs, BIOMOL and SEARCHCMP, and MOE (Ryoka Systems Inc.). BIOMOL can calculate the biological quaternary structure from the asymmetric unit of a crystal structure (32). SEARCHCMP assembles two molecular structures by searching for and superposing the same or similar molecules in two Protein Data Bank files. The trimeric form of PfuPCNA was made with BIOMOL, and then the UDG·PCNA complex (complex-1) was assembled by superposing the corresponding new PCNA interaction site in TthUDG and the bound PIP-box peptide in the PfuPCNA trimer. The homology model of PfuUDG was constructed with MOE from TthUDG and was used to replace the TthUDG in the complex-1 model. The position of the dsDNA was based on that in the co-crystal structure of TthUDG-DNA, which was recently published (33). Since the bound dsDNA substrate co-crystallized with TthUDG was not long enough to penetrate PCNA, a B-type dsDNA model was also assembled into complex-1 to build the ternary complex (complex-2). Finally, the assembled model was energy-minimized with MOE to obtain the final model of the PfuUDG·PfuPCNA·DNA complex (complex-3).
RESULTS
Biochemical Properties of PfuUDG—Sequence homology searches revealed the presence of a family 4 UDG homolog in the P. furiosus genome. An amino acid sequence alignment between the P. furiosus UDG candidate and the T. thermophilus UDG reviewed that the active site motifs are well conserved between them (Fig. 1). Furthermore, four cysteine residues (Cys17, Cys20, Cys88, and Cys104), which would be important for the formation of the Fe-S cluster, are completely conserved between the two sequences (Fig. 1). Therefore, we cloned the gene (open reading frame PF1385), expressed it in E. coli cells, and purified the recombinant protein to investigate its biochemical properties. The purified protein had a brown color, suggesting the existence of an Fe-S cluster, as shown previously for other family 4 UDGs (7, 34). The uracil excision activity of the protein was detected by using a synthetic oligonucleotide containing a uracil base at the center as the substrate, and we designated the gene product as PfuUDG, a member of the family 4 UDGs. As shown in Fig. 2A, PfuUDG can remove uracil from both single- and double-stranded DNAs. Like the other family 4 UDGs, divalent cations were not required for the activity. In addition, the maximum enzyme activity of PfuUDG was observed at 80 °C (data not shown).
FIGURE 1.

Amino acid sequence comparison of PfuUDG and TthUDG. The amino acid sequences of the two family 4 UDGs were aligned, using the program ClustalW. *, identical residues between the two UDGs; colons and periods, residues with conserved and semiconserved substitutions, respectively. The dashes indicate gaps. The secondary structure of TthUDG is shown above the corresponding residues, based on the crystal structure (7). Amino acid sequences of the active site motifs A and B are boxed, and the conserved cysteine residues for the Fe-S cluster formation are shown in black boxes. The PCNA-binding motif in PfuUDG, proposed in this study, is underlined.
FIGURE 2.
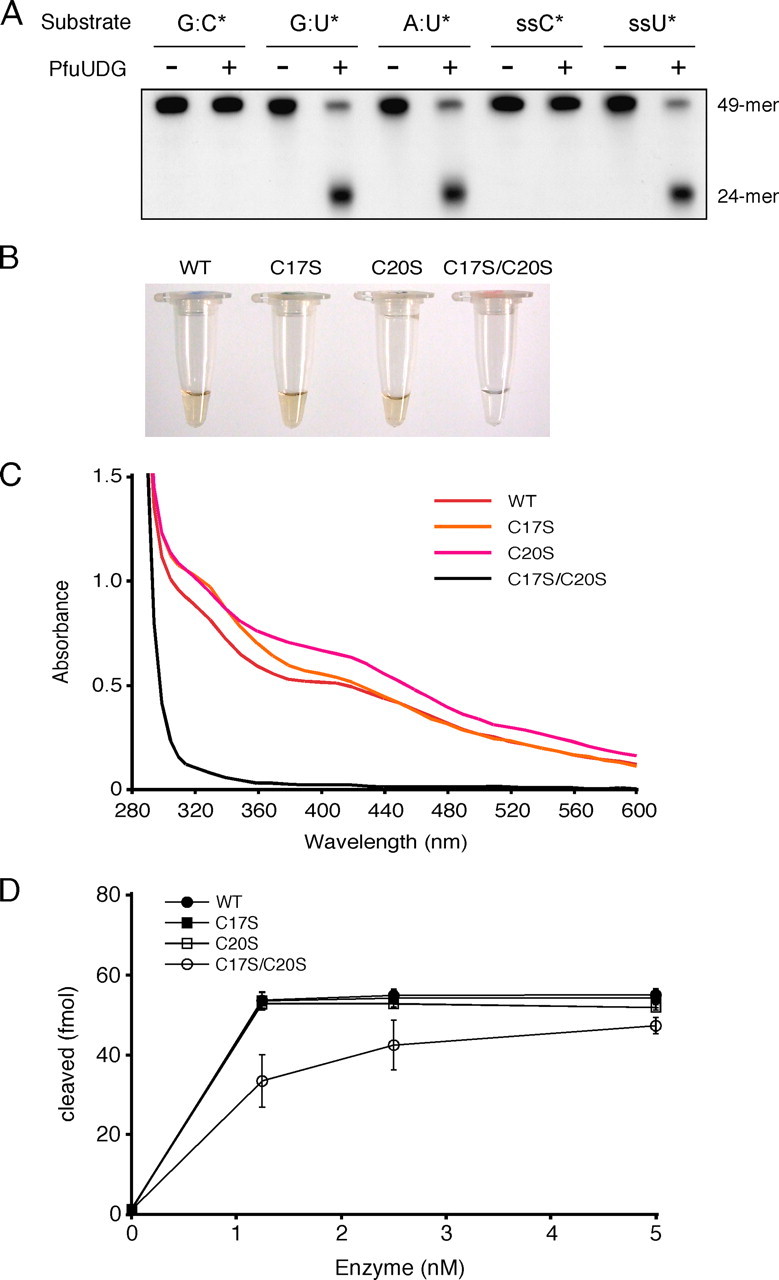
Biochemical properties of PfuUDG. A, the substrate specificity of PfuUDG was examined by using a synthetic 49-mer oligonucleotide containing a uracil base at the center. Wild-type PfuUDG (20 fmol) was incubated with 100 fmol of the double-stranded DNA substrates containing G:C*, G:U*, and A:U* base pairs (the asterisk indicates the 32P-labeled oligonucleotide) in the reaction, as described under “Experimental Procedures.” A single-stranded (ss) DNA was also tested in the same manner as the double-stranded DNA substrates. B, the colors of the purified wild-type (WT) and mutant (C17S, C20S, and C17S/C20S) PfuUDG protein solutions at a concentration of 180 μm are shown. C, the UV-visible absorption spectra (280-600 nm) of the PfuUDG proteins at a concentration of 180 μm are shown. D, comparison of the enzyme activities of the wild-type and mutant (C17S, C20S, and C17S/C20S) PfuUDGs. Various amounts of the PfuUDG proteins, as indicated, were incubated with 100 fmol of the uracil-containing DNA substrate (G:U*) in the reaction, as described under “Experimental Procedures.” The uracil excision efficiency is plotted as a function of the enzyme concentration.
To test the importance of the cysteine residues for the formation of the Fe-S cluster, we constructed the serine-substituted mutants C17S, C20S, and C17S/C20S. All of the serine-substituted PfuUDGs were purified by the same procedure as used for the wild-type PfuUDG, and the yields of purified proteins (3 mg from 1 liter of culture) were not different from that of wild type PfuUDG. The purified C17S and C20S proteins had a brown color similar to that of the wild type protein, whereas the C17S/C20S mutant was purified as a colorless fraction (Fig. 2B). As shown in Fig. 2C, the UV-visible absorption spectra of the wild-type, C17S, and C20S protein had a broad shoulder at a wavelength of 400 nm, which is specific for Fe-S proteins (34). In contrast, a decrease in the absorbance at 400 nm was observed in the spectrum of the C17S/C20S mutant (Fig. 2C). The colorless fractionation of the C17S/C20S protein indicates the loss of the Fe-S cluster by these substitutions. Due to color fading after the protein purification, the spectra of the colored proteins (wild-type, C17S, and C20S) were inconsistent. A similar observation about the fading was reported previously (9). Since the amount of purified C17S/C20S mutant protein was the same as that of wild type from the same procedure, including the heat treatment (80 °C for 20 min), as that of the wild type protein, the apparent thermostability of the mutant protein must not be very different from that of the wild-type enzyme. In order to examine the role of the Fe-S cluster in the enzymatic reaction, the specific activities of the mutant proteins were compared. Decreased activity was observed for the C17S/C20S mutant protein, but the mutant enzyme still possesses substantial activity, indicating that the Fe-S cluster does not participate in the glycosylase activity directly (Fig. 2D).
Physical Interaction between PfuUDG and PCNA—It was previously reported that two crenarchaeal family 4 UDGs (from P. aerophilum and S. solfataricus) physically interact with PCNA (23, 24). However, the C-terminal PCNA-binding motifs found in these UDGs were not completely conserved in the archaeal UDGs. Especially, several euryarchaeal UDGs, including PfuUDG, have slightly shorter sequences as compared with the above crenarchaeal UDGs and lack the PCNA-binding motifs (23). We therefore performed SPR experiments to investigate the physical interaction between PfuUDG and PfuPCNA. Purified PfuPCNA was immobilized on the Biacore CM5 sensor chip, and wild-type PfuUDG was injected at different concentrations. A clear sensorgram indicating the physical interaction between PfuUDG and the immobilized PfuPCNA was obtained, as shown in Fig. 3. The calculated equilibrium constant (KD) for wild-type PfuUDG from the sensorgrams obtained with five different concentrations was 2.2 × 10-7 m, which was comparable with that for the P. furiosus DNA ligase (PfuLig)-PfuPCNA interaction (1.1 × 10-7 m) and the PfuPolB-PfuPCNA interaction (9.9 × 10-8 m) determined by our SPR analysis (35, 36). These observations suggested that PfuUDG can interact with PfuPCNA via an unidentified binding motif.
FIGURE 3.

Physical interaction between PfuUDG and PfuPCNA. The SPR analysis was performed using a Biacore system to detect the physical interaction between PfuUDG and PfuPCNA. Purified PfuPCNA was immobilized on a Biacore sensor chip, and various concentrations of purified PfuUDG (0.4-1.2 μm) were injected for 120 s. The equilibrium constant (KD) was calculated from the obtained sensorgrams.
Enhancement of Uracil Glycosylase Activity by the PCNA Trimer—Some PCNA-interacting enzymes are reportedly stimulated by PCNA in vitro. Therefore, we performed the enzyme assay of PfuUDG in the presence of 0-450 nm PCNA (as a monomer concentration) to investigate the effect of PfuPCNA on the PfuUDG activity. As shown in Fig. 4, the uracil-cleaving activity of PfuUDG was increased in a PCNA concentration-dependent manner. The PfuUDG activity was stimulated 2.3-fold by PfuPCNA at a concentration of 300 nm. It is not clear why the stimulation effect of PCNA on the activity is relatively low at this stage; however, the effect is the same as the case of human UNG2 and PCNA, as reported earlier (16). In our previous study of PfuLig-PfuPCNA interaction, stimulation effect of PCNA on the ligation reaction was especially distinct at high salt reaction conditions, which are more similar to the physiological condition in the P. furiosus cells (35). In the case of PfuUDG, however, the glycosylase activity was drastically decreased at high salt concentrations (over 0.3 m KCl or potassium glutamate) both in the presence and absence of PfuPCNA (data not shown).
FIGURE 4.

Stimulatory effect of the PCNA trimer on the uracil excision ability of PfuUDG. A, PfuUDG (20 fmol) was incubated with 100 fmol of uracil-containing DNA substrate (G:U*) and 0-450 nm of wild-type (WT) or a monomeric mutant (D143A/D147A) PfuPCNA, in a reaction containing 0.1 m KCl, as described under “Experimental Procedures.” B, the uracil excision efficiencies are plotted as a function of the PCNA concentration as a monomer.
In addition, a vast excess of PCNA is required for the functional interaction in vitro, as shown in our previous study (35). This discrepancy could be explained by the difficulty of loading PCNA onto the DNA fragment in the assay mixture. The PCNA trimer loads by diffusion onto the double-stranded DNA substrate from the ends, without a clamp loader (replication factor C) in this case. This is probably a limited process, and efficient loading requires an excess amount of PCNA, as discussed previously for the human DNA ligase-PCNA interaction (37). Since no stimulation effect was observed in the case of the monomeric mutant PCNA, D143A/D147A, the toroidal structure of PCNA is required for the functional interaction. This experiment also excludes the possibility that the increasing activity of PfuUDG is due to a stabilization effect by higher protein concentrations in the reaction mixtures.
Identification of a PCNA-binding Motif in PfuUDG—The typical PIP box sequence is not present within the PfuUDG sequence. To determine the region responsible for PCNA binding in PfuUDG, we constructed a series of truncated PfuUDG genes. However, not all of the truncated genes could be expressed at levels sufficient for characterization. Previous studies of P. aerophilum UDG and S. solfataricus UDG revealed that the successive hydrophobic amino acid residues, Phe-Leu, are important for the physical interaction with PCNA (23, 24). We previously demonstrated that the hydrophobicity of the aromatic residues (103QKSFF107) is critical for the physical and functional interactions between PfuLig and PfuPCNA (35). Moreover, the cluster of basic amino acids adjacent to the binding motif seemed to be important for the archaeal DNA ligase-PCNA interactions (35). Based on these findings, we visually scanned the sequence to predict the PCNA-binding site in PfuUDG and found the amino acid sequence (152AKTLF156), which is similar to the PCNA-binding motif of PfuLig (Fig. 5A). The putative PCNA-binding motif found in PfuUDG may be a shorter version of the PIP box sequence within the human UDG2, as in the case of DNA ligases (Fig. 5B).
FIGURE 5.

Putative PCNA-binding motif in PfuUDG. A, the PCNA-binding motifs found in PfuPolB (36) and PfuLig (35) are shown. Based on the PfuLigPCNA interaction, a PCNA-binding motif 152AKTLF156 was predicted in PfuUDG. The basic residue and the glutamine (Q), serine/threonine (S/T) and phenylalanine (F) residues are highlighted in blue, magenta, light blue, and yellow, respectively. The locations of the PCNA-binding motifs in PfuPolB, PfuLig, and PfuUDG are shown in red boxes. B, comparison of the amino acid sequences of the PCNA-binding motifs among the Ligs and UDGs from eukaryotes and Archaea. Identical and similar amino acid residues are connected by solid and broken lines, respectively.
To examine whether the putative binding motif of PfuUDG is actually involved in PCNA binding, site-specific mutational analyses were performed. At the first, we substituted the last two hydrophobic residues with alanine (L155A/F156A). The SPR analysis showed that the PCNA binding affinity of the L155A/F156A mutant protein was obviously decreased, but it still bound to PCNA (Fig. 6). In addition, the uracil glycosylase activity of the L155A/F156A mutant was stimulated by PCNA in vitro (1.9-fold as compared with 2.3-fold for wild type UDG as described above). These observations indicated that other amino acid residues also function in PCNA binding. Therefore, we constructed and purified two more mutant UDG proteins, K153A and T154A. As a result, the decreased binding affinity to PCNA was also observed with the T154A mutant, whereas the mutant K153A interacted with immobilized PCNA with slightly higher affinity, as compared with wild-type PfuUDG (Fig. 6). The KD value for the K153A mutant-PCNA interaction was calculated to be 1.7 × 10-7 m (Fig. S1). These observations suggested that the basic residue Lys153 does not play an important role in PCNA binding. We next prepared the triple alanine-substituted mutant T154A/L155A/F156A (hereafter designated AKAAA) to examine the importance of both the hydroxyl group and hydrophobic side chains in the putative PCNA-binding motif. As a result, the sensorgram showed that the AKAAA mutant had almost no binding affinity to PfuUDG (Fig. 6). Consistently, no stimulatory effect of PfuPCNA on the AKAAA mutant was observed in the uracil glycosylase assay (Fig. 7). The sensorgram of AKAAA indicated that the mutant may still have very weak binding ability, suggesting the possibility that a physical interaction via Ala152 cannot be ruled out, because the detailed role of the Ala152 was not evaluated by an alanine-substituted mutant analysis. The specific activity of the AKAAA mutant was confirmed to be the same as that of wild-type PfuUDG (Fig. S2). Hence, the decreased response of the AKAAA mutant to PCNA is not due to the lower enzyme activity. Taken together, we suggest that the short amino acid sequence, AKTLF, which resembles the PCNA-binding motif found in PfuLig, plays a critical role in the physical and functional interactions between PfuUDG and PfuPCNA.
FIGURE 6.

Physical interaction between PfuUDG mutants and PCNA. Using the PCNA-immobilized Biacore sensor chip, as described in the legend to Fig. 3, wild-type (WT) and mutant (K153A, T154A, L155A/F156A, and T154A/L155A/F156A) PfuUDG proteins (1.0 μm) were assayed to investigate their physical interactions with PCNA.
FIGURE 7.

The functional interaction between PfuUDG and PCNA. A, wild-type (WT) and mutant (T154A/L155A/F156A) PfuUDGs (20 fmol each) were incubated with 100 fmol of uracil-containing DNA substrate (G:U*) and 0-450 nm wild-type PfuPCNA in a reaction mixture containing 0.1 m KCl, as described under “Experimental Procedures.” B, the uracil excision efficiencies are plotted as a function of the PCNA concentration as a trimer. These reactions were performed three times independently for each combination of UDG and PCNA, including the cases presented in Fig. 4, at the same time, and therefore, the plot with the error bar for PCNA WT is exactly the same as that shown in Fig. 4.
DISCUSSION
We have shown that the open reading frame PF1385, with a family 4 UDG-like sequence, is actually a uracil DNA glycosylase in P. furiosus cells. Our biochemical analyses revealed that the PfuUDG is a typical family 4 UDG containing an Fe-S cluster. Recent studies have demonstrated that the Fe-S cluster is important for the archaeal XPD helicase and primase activities (8, 9). The functional roles of the Fe-S cluster have been discussed in the studies of family 4 UDGs from P. aerophilum (35) and T. thermophilus (7). These reports suggest that the Fe-S cluster in the UDGs is not involved directly in catalysis, because the Fe-S cluster is far from the active site in the ternary structure of the UDG proteins. Our site-directed mutagenesis analyses of PfuUDG support the idea, and the Fe-S cluster probably contributes to stabilization of the local loop structure of the UDG proteins, as suggested previously. Further biochemical and biophysical studies will be required to understand the detailed role of the Fe-S cluster in the family 4 UDGs.
We presented here the physical and functional interaction between PfuUDG and PfuPCNA in vitro. The PCNA ring loaded on the DNA strand probably contributes to decreasing the dissociation of bound UDG from DNA. It is also possible that PCNA-UDG complex formation serves to increase the affinity of UDG for DNA strand. We identified the PCNA-binding site in the UDG protein. To investigate whether the identified PCNA-binding site is actually functional in the BER complex, we attempted to build a three-dimensional structure model of the PfuUDG·PfuPCNA·DNA complex, using the available structural data. The molecular model was constructed from the co-crystal structures of T. thermophilus UDG·dsDNA (Fig. S3a) and P. furiosus PCNA (Fig. S3b). The trimer of PfuPCNA was formed (Fig. S3c), and then the UDG·PCNA complex (supplemental Fig. S3d) was assembled by superposing the corresponding region in TthUDG on the newly identified PCNA-binding site of PfuUDG. The homology model of PfuUDG was constructed from TthUDG (Fig. S3e) and then was used in place of the TthUDG in the complex-1 model. The position of the dsDNA was based on that in the co-crystal structure of TthUDG-DNA. However, the TthUDG-bound dsDNA was not long enough to penetrate PCNA, and therefore, a B-type dsDNA model (Fig. S3f) was also assembled into complex-1, to build complex 2 (Fig. S3g). The assembled model was energy-minimized with MOE to obtain the final PfuUDG·PfuPCNA·DNA complex (complex-3; supplemental Fig. S3h). The structure model is shown in Fig. 8A. This ternary structure model revealed that the dsDNA in complex with PfuUDG would pierce the hole of the PfuPCNA ring, when the newly found PCNA-binding site (colored red) was properly placed at the PIP-binding site of PfuPCNA. The bound dsDNA displayed a 15° inclination from the 3-fold axis (normal to the PCNA ring) of PfuPCNA. The PCNA-binding motif sequence was nearby, as shown in Fig. 8B (blue in the stick model). The Leu-155 and Phe-156 residues in PfuUDG were placed in the conserved, hydrophobic pocket of PfuPCNA, including Leu-48 and Leu-242. This hydrophobic interaction mode is clearly conserved in the PfuPCNA-RFCL interaction, in which Leu-473 and Phe-474 correspond to the two hydrophobic residues. The positively charged Lys-153 in PfuUDG might interact with Glu-224 in PfuPCNA, although the mutation analysis showed that the interaction is not critical for PfuUDG·PfuPCNA binding, as described above.
FIGURE 8.
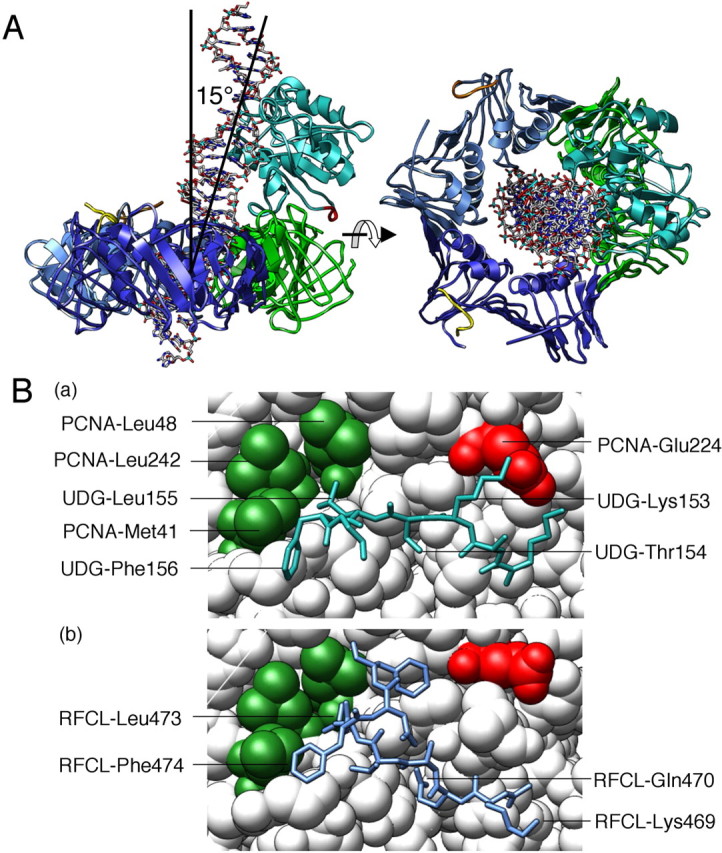
Molecular model of the PfuUDG·PCNA·DNA complex. A, the assembled model revealed that the dsDNA (stick model) complexed with PfuUDG (colored mint) pierces the hole of the PfuPCNA trimer (subunits are colored blue, dark blue, and green, respectively) when the newly identified PCNA-binding site (colored red) was properly placed at the PIP-binding site of PfuPCNA. B, close-up view of the PfuUDG PIP-box peptide (blue in the stick model) bound to PfuPCNA (a). The hydrophobic Leu-155 and Phe-156 of PfuUDG are placed in the conserved hydrophobic pocket of Leu-48 and Leu-242 in PfuPCNA. This interaction mode is clearly conserved in the interaction of PfuPCNA-RFCL as shown in (b) drawn from our previous study (43).
Direct evidence to show the functional interaction between PfuUDG and PfuPCNA in the P. furiosus cells should be obtained. However, our biochemical and structural investigations presented here strongly support that PfuUDG and PfuPCNA work together in the replisome, and therefore, PfuUDG may be a functional counterpart of hUNG2 in the replication-associated repair pathway at replication fork, as proposed previously for the cus (24) UDGs. Recently, a multiprotein complex, including UNG2, APE1 (AP endonuclease), XRCC1, Polα, Polβ, Polδ, Polε, DNA ligase 1, and DNA-dependent protein kinase, was isolated from the nuclei of human cycling cells (14). It should be investigated whether the same kind of protein complex can be isolated from the chromatin fraction of P. furiosus cells. We already cloned and expressed the gene for an AP endonuclease from P. furiosus.3 We are presently trying to analyze the interaction between PfuAP and PfuPCNA as well as that between PfuUDG and PfuAP.
It is well known that the archaeal family B DNA polymerases have uracil recognition ability to stop the DNA strand synthesis before the uracil site in the template in vitro, although it is not clear whether the uracil recognition actually functions in the cells. One possibility is that DNA polymerase stalls at a uracil site, and then it recruits UDG to initiate a uracil excision repair pathway (25, 38). If PolBI and UDG interact with each other to form a complex in the replisome, then it would be an efficient replication/repair unit for the uracil removal. In this case, the PolBI·PCNA complex would have to synthesize DNA through the template containing an abasic site. We investigated whether PolBI and PCNA directly interact with each other by an SPR analysis. Purified PfuUDG was immobilized on the Biacore CM5 sensor chip, and wild-type PfuPCNA was injected as a control. The physical interaction between PCNA and immobilized PfuUDG was identified by the SPR sensorgram. On the other hand, we could not detect a direct interaction between PfuPolBI and PfuUDG (data not shown). This result suggests that PfuUDG cannot form a stable complex with PfuPolBI, at least by itself. We demonstrated previously that the processivity of PolBI is enhanced by PfuPCNA (36, 39, 40) and identified the PCNA-binding site in Pol BI (36). It is possible that PfuUDG is recruited onto the stalled uracil site by PCNA, which binds PolBI, and then the AP endonuclease is recruited to reorganize the replisome.
There are several DNA glycosylases in the cell, and they probably share the base excision repair functions for each specific aberrant base. The family 5 UDGs are now recognized as functioning counteract the mutagenic threats of cytosine and adenine deaminations, because this family of enzymes catalyzes the removal of hypoxanthine as well as uracil. The family 5 UDGs have so far been found only in hyperthermophilic Archaea and eubacteria and are predicted to be important for habitation at elevated temperatures. Actually, one family 4 and one family 5 UDG are present in P. aerophilum and T. thermophilus. There is no PCNA-binding motif-like sequence in the P. aerophilum UDGb (family 5), and therefore, a PCNA-independent BER pathway, in addition to the PCNA-dependent pathway with UDGa (family 4), probably functions at least in these archaeal cells. In the genome sequence of P. furiosus, however, there is only one family 4 UDG-like sequence as characterized in this study and no family 5 UDG. It would be interesting to search for other DNA glycosylases with a new family sequence in P. furiosus cells.
Furthermore, several BER pathways are known, and two types of repair synthesis/ligation, long patch repair and short patch repair, have been well studied. However, there are no sequence homologs corresponding to Polβ and LigIII, which purportedly function in the short patch repair process, in the P. furiosus genome. It would also be interesting to investigate the process of BER after UDG/AP endonuclease reactions in P. furiosus.
As shown in Fig. 9A, the PCNA-binding motif found in PfuUDG is conserved in Thermococcales (Pyrococcus and Thermococcus species), although the basic residue at the second position has diversified (Lys for P. furiosus, Arg for P. abyssi and P. horikoshii, and His for Thermococcus kodakarensis). Our SPR analyses showed that the K153A mutant interacts with immobilized PfuPCNA with slightly stronger affinity. It is not clear at this stage why the Lys to Ala substitution provides PfuUDG with more affinity to PfuPCNA, but the result implies, at least, that the basic residue found in this type of PCNA-binding motif is not important for the interaction. In general, a glutamine residue is highly conserved in the typical PIP box motif; however, the pentapeptide motif found in this study lacks a glutamine residue. Our prediction of the PCNA-binding motifs in archaeal DNA ligases revealed that not all of the putative sequences have a glutamine residue (35, 41). In addition, the experimentally confirmed PCNA-binding motifs in crenarchaeal UDG lack a glutamine residue (23, 24). Thus, a PCNA-binding motif without a glutamine residue may commonly exist in archaeal PCNA-binding proteins, especially in the short binding motifs.
FIGURE 9.

Conservation of the PCNA-binding motif found in PfuUDG. A, amino acid sequence alignment of the C-terminal regions of the archaeal family 4 UDGs. To investigate the conservation of the PCNA-binding motif identified in this study, a BLAST search was performed (available on the World Wide Web). Some crenar-chaeal species were intentionally omitted from the results to focus the discussion on the conservation of PCNA-binding motifs mainly in euryarchaeal UDGs. Experimentally confirmed and putative PCNA-binding motifs are shown in black boxes and gray boxes, respectively. B, alignment of the amino acid sequences of the N-terminal regions of the archaeal family 4 UDG.
Interestingly, the PCNA-binding motif discovered in PfuUDG is clearly deleted in the UDG sequences deduced from the genomes of the uncultured euryarchaea, Alv-FOS1 and Alv-FOS4 (Fig. 9A). Instead, a PCNA-binding motif-like sequence, 2QTSLF6, is present at the N-terminal region in the Alv-FOS UDG sequences (Fig. 9B). In Methanoculleus marisnigri UDG, no recognizable PCNA-binding motif sequence is present in either the N- or C-terminal region. Since the three amino acid residues, 154TLF156, in PfuUDG are important for the PCNA binding, the short amino acid sequence 171LLF173 in M. marisnigri UDG may play an important role in PCNA binding (Fig. 9A). It has been proposed that a family 4 UDG from the euryarchaeon Archaeoglobus fulgidus has a PCNA-binding motif at the extreme C-terminal region, like the other crenar-chaeal UDGs (42), suggesting that the locations of the PCNA-binding sites in euryarchaeal UDGs may be diversified. It would be evolutionally very interesting to determine why the positions of the PCNA-binding sites are so diverse in the amino acid chains of UDG if these motif sequences actually interact with PCNA. Three-dimensional structural analyses of the UDG-PCNA-DNA complexes will shed light on the puzzle to elucidate the biological meaning of the variety of PCNA binding positions among these enzymes with the same function.
Supplementary Material
Acknowledgments
We thank Takehiro Yoshimochi for help with a figure.
This work was supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to Y. I.). This work was also supported by the Hou-ansha Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S3.
Footnotes
The abbreviations used are: BER, base excision repair; UDG, uracil-DNA glycosylase; TthUDG, T. thermophilus UDG; PCNA, proliferating cell nuclear antigen; PIP, PCNA-interacting protein; SPR, surface plasmon resonance; PfuUDG, P. furiosus UDG; PfuPCNA, P. furiosus PCNA; PfuPolBI, P. furiosus Pol BI; dsDNA, double-stranded DNA; AP, apurinic/apyrimidinic; AP endonuclease, apurinic/apyrimidinic endonuclease.
S. Kiyonari, S. Tahara, and Y. Ishino, unpublished results.
References
- 1.Lindahl, T. (1993) Nature 362709 -715 [DOI] [PubMed] [Google Scholar]
- 2.Barnes, D. E., and Lindahl, T. (2004) Annu. Rev. Genet. 38445 -476 [DOI] [PubMed] [Google Scholar]
- 3.Berti, P. J., and McCann, J. A. (2006) Chem. Rev. 106506 -555 [DOI] [PubMed] [Google Scholar]
- 4.Aravind, L., and Koonin, E. V. (2000) Genome Biol. 20000007.1 -0007.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearl, L. H. (2000) Mutat. Res. 460165 -181 [DOI] [PubMed] [Google Scholar]
- 6.Sartori, A. A., Schär, P., Fitz-Gibbon, S., Miller, J. H., and Jiricny, J. (2001) J. Biol. Chem. 27629979 -29986 [DOI] [PubMed] [Google Scholar]
- 7.Hoseki, J., Okamoto, A., Masui, R., Shibata, T., Inoue, Y., Yokoyama, S., and Kuramitsu, S. (2003) J. Mol. Biol. 333515 -526 [DOI] [PubMed] [Google Scholar]
- 8.Rudolf, J., Makrantoni, V., Ingledew, W. J., Stark, M. J., and White, M. F. (2006) Mol. Cell 23 801-880 [DOI] [PubMed] [Google Scholar]
- 9.Klinge, S., Hirst, J., Maman, J. D., Krude, T., and Pellegrini, L. (2007) Nat. Struct. Mol. Biol. 14 875-877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sartori, A. A., Fitz-Gibbon, S., Yang, H., Miller, J. H., and Jiricny, J. (2002) EMBO J. 213182 -3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Starkuviene, V., and Fritz, H. J. (2002) Nucleic Acids Res. 302097 -2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otterlei, M., Warbrick, E., Nagelhus, T. A., Haug, T., Slupphaug, G., Akbari, M., Aas, P. A., Steinsbekk, K., Bakke, O., and Krokan, H. E. (1999) EMBO J. 183834 -3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akbari, M., Otterlei, M., Peña-Diaz, J., Aas, P. A., Kavli, B., Liabakk, N. B., Hagen, L., Imai, K., Durandy, A., Slupphaug, G., and Krokan, H. E. (2004) Nucleic Acids Res. 325486 -5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parlanti, E., Locatelli, G., Maga, G., and Dogliotti, E. (2007) Nucleic Acids Res. 351569 -1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moldovan, G. L., Pfander, B., and Jentsch, S. (2007) Cell 129665 -679 [DOI] [PubMed] [Google Scholar]
- 16.Ko, R., and Bennett, S. E. (2005) DNA Repair 41421 -1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oyama, M., Wakasugi, M., Hama, T., Hashidume, H., Iwakami, Y., Imai, R., Hoshino, S., Morioka, H., Ishigaki, Y., Nikaido, O., and Matsunaga, T. (2004) Biochem. Biophys. Res. Commun. 321183 -191 [DOI] [PubMed] [Google Scholar]
- 18.Xia, L., Zheng, L., Lee, H. W., Bates, S. E., Federico, L., Shen, B., and O'Connor, T. R. (2005) J. Mol. Biol. 3461259 -1274 [DOI] [PubMed] [Google Scholar]
- 19.Dou, H., Theriot, C. A., Das, A., Hegde, M. L., Matsumoto, Y., Boldogh, I., Hazra, T. K., Bhakat, K. K., and Mitra, S. (2008) J. Biol. Chem. 2833130 -3140 [DOI] [PubMed] [Google Scholar]
- 20.Haracska, L., Johnson, R. E., Unk, I., Phillips, B., Hurwitz, J., Prakash, L., and Prakash, S. (2001) Mol. Cell. Biol. 217199 -7206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomes, X. V., and Burgers, P. M. (2000) EMBO J. 193811 -3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin, D. S., McKenna, A. E., Motycka, T. A., Matsumoto, Y., and Tomkinson, A. E. (2000) Curr. Biol. 10 919-922 [DOI] [PubMed] [Google Scholar]
- 23.Yang, H., Chiang, J. H., Fitz-Gibbon, S., Lebel, M., Sartori, A. A., Jiricny, J., Slupska, M. M., and Miller, J. H. (2002) J. Biol. Chem. 27722271 -22278 [DOI] [PubMed] [Google Scholar]
- 24.Dionne, I., and Bell, S. D. (2005) Biochem. J. 387859 -863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connolly, B. A., Fogg, M. J., Shuttleworth, G., and Wilson, B. T. (2003) Biochem. Soc. Trans. 31 699-702 [DOI] [PubMed] [Google Scholar]
- 26.Greagg, M. A., Fogg, M. J., Panayotou, G., Evans, S. J., Connolly, B. A., and Pearl, L. H. (1999) Proc. Natl. Acad. Sci. U. S. A. 969045 -9050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fogg, M. J., Pearl, L. H., and Connolly, B. A. (2002) Nat. Struct. Biol. 9922 -927 [DOI] [PubMed] [Google Scholar]
- 28.Lindahl, T., and Nyberg, B. (1974) Biochemistry 133405 -3410 [DOI] [PubMed] [Google Scholar]
- 29.Matsumiya, S., Ishino, S., Ishino, Y., and Morikawa, K. (2003) Protein Sci. 12 823-831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komori, K., and Ishino, Y. (2000) Protin Eng. 1341 -47 [DOI] [PubMed] [Google Scholar]
- 31.Jarvis, T. C., Paul, L. S., and von Hippel, P. H. (1989) J. Biol. Chem. 26412709 -12716 [PubMed] [Google Scholar]
- 32.Shirai, T., Hung, V. S., Morinaka, K., Kobayashi, T., and Ito, S. (2008) Proteins, in press [DOI] [PubMed]
- 33.Kosaka, H., Hoseki, Jun, Nakagawa, N., Kuramitsu, S., and Masui, R. (2007) J. Mol. Biol. 373839 -850 [DOI] [PubMed] [Google Scholar]
- 34.Hinks, J. A., Evans, M. C., De Miguel, Y., Sartori, A. A., Jiricny, J., and Pearl, L. H. (2002) J. Biol. Chem. 27716936 -16940 [DOI] [PubMed] [Google Scholar]
- 35.Kiyonari, S., Takayama, K., Nishida, H., and Ishino, Y. (2006) J. Biol. Chem. 28128023 -28032 [DOI] [PubMed] [Google Scholar]
- 36.Tori, K., Kimizu, M., Ishino, S., and Ishino, Y. (2007) J. Bacteriol. 1895652 -5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tom, S., Henricksen, L. A., Park, M. S., and Bambara, R. A. (2001) J. Biol. Chem. 27624817 -24825 [DOI] [PubMed] [Google Scholar]
- 38.Grogan, D. W. (1998) Mol. Microbiol. 281043 -1049 [DOI] [PubMed] [Google Scholar]
- 39.Cann, I. K., Ishino, S., Hayashi, I., Komori, K., Toh, H., Morikawa, K., and Ishino, Y. (1999) J. Bacteriol. 1816591 -6599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishino, Y., Tsurimoto, T., Ishino, S., and Cann, I. K. (2001) Genes Cells 6 699-706 [DOI] [PubMed] [Google Scholar]
- 41.Kiyonari, S., Kamigochi, T., and Ishino, Y. (2007) Extremophiles 11675 -684 [DOI] [PubMed] [Google Scholar]
- 42.Sandigursky, M., and Franklin, W. A. (2000) J. Biol. Chem. 27519146 -19149 [DOI] [PubMed] [Google Scholar]
- 43.Matsumiya, S., Ishino, S., Ishino, Y., and Morikawa, K. (2002) Genes Cells 7 911-922 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.