

Abstract
Mitochondrial complex I (CI) is a large assembly of 45 different subunits, and defects in its biogenesis are the most frequent cause of mitochondrial disorders. In vitro evidence suggests a stepwise assembly process involving pre-assembled modules. However, whether these modules also exist in vivo is as yet unresolved. To answer this question, we here applied submitochondrial fluorescence recovery after photobleaching to HEK293 cells expressing 6 GFP-tagged subunits selected on the basis of current CI assembly models. We established that each subunit was partially present in a virtually immobile fraction, possibly representing the holo-enzyme. Four subunits (NDUFV1, NDUFV2, NDUFA2, and NDUFA12) were also present as highly mobile matrix-soluble monomers, whereas, in sharp contrast, the other two subunits (NDUFB6 and NDUFS3) were additionally present in a slowly mobile fraction. In the case of the integral membrane protein NDUFB6, this fraction most likely represented one or more membrane-bound subassemblies, whereas biochemical evidence suggested that for the NDUFS3 protein this fraction most probably corresponded to a matrix-soluble subassembly. Our results provide first time evidence for the existence of CI subassemblies in mitochondria of living cells.
The vast majority of cellular ATP is produced by mitochondrial oxidative phosphorylation (OXPHOS).2 The OXPHOS system consists of five multisubunit complexes of which NADH:ubiquinone oxidoreductase or complex I (CI) is the largest (∼1 MDa) and least understood (1). CI liberates electrons from NADH, channels them to ubiquinone, and uses part of their energy to expel protons from the mitochondrial matrix into the intermembrane space. The complex consists of 45 subunits, seven of which are encoded by the mtDNA and the remainder by the nuclear genome (2). Its catalytic core comprises 14 evolutionary conserved subunits (3), which, in humans, are encoded by the nuclear NDUFV1, NDUFV2, NDUFS1, NDUFS2, NDUFS3, NDUFS7, and NDUFS8 genes and the mitochondrial ND1-ND6 and ND4L genes. Once assembled, the complex has an l-shaped structure with one arm protruding into the matrix and the other arm embedded in the inner membrane. The matrix-protruding, or peripheral, arm is involved in electron channeling and contains the flavo-mononucleotide group and eight iron-sulfur clusters. The membrane arm includes all of the mtDNA-encoded subunits and is believed to play a role in proton translocation (3).
At present, it is still unclear how the different subunits are exactly pieced together during CI biogenesis. Understanding this process is also clinically relevant because mutations in CI subunits prevent proper CI assembly, causing a variety of mitochondrial disorders (reviewed in Ref. 4). The current hypothesis is that CI assembly occurs by a stepwise mechanism during which prefabricated modules, or assembly intermediates, are combined (5–9). In agreement with this idea, we recently demonstrated that inhibition of mitochondrial translation caused an accumulation of the NDUFS3 subunit in two distinct subassemblies (7). Apart from this, recent in vitro import and assembly studies revealed a mechanism involving the replacement of earlier incorporated subunits by newly formed ones (9). It is conceivable that such a mechanism might serve to maintain the functional integrity of CI.
Up to now, most insights into the mechanism of CI assembly were obtained from studies using blue native polyacrylamide gel electrophoresis (BN-PAGE) of mitochondrial cell fractions. Here, we extended our investigations into the CI assembly process to living cells. To allow doing so, we generated a collection of human cell lines that expressed AcGFP1-tagged CI subunits at a low level. In these cell lines, the mobility of the tagged subunits was investigated using fluorescence recovery after photobleaching (FRAP) (10–17).
By comparison with matrix-soluble and mitochondrial inner membrane (MIM)-bound versions of AcGFP1 of different size, we could establish the presence of these CI subunits in mobile (matrix-soluble) and/or immobile (membrane-bound) subassemblies. Our findings provide the first evidence that CI assembly occurs in a modular manner in vivo.
EXPERIMENTAL PROCEDURES
Generation of Inducible Cell Lines—cDNA fragments of the following genes were generated by Gateway-adapted PCR procedures according to the manufacturer's instructions (Invitrogen): the cox8 leader sequence (first 210 base-pairs of sequence NM_00004074), NDUFS3 (NM_004551), NDUFV1 (NM_007103), NDUFA2 (NM_002488), NDUFA12 (NM_018838), NDUFB6 (NM_002493; transcript variant 1). Entry clones were generated from the resulting PCR products by recombination with pDONR201 (Invitrogen) using Gateway Clonase II Enzyme Mix (Invitrogen). Entry clones containing NDUFV2 (BC_001632; Clone HsCD00041552) and human ANT1 (BC_008664; Clone HsCD00040509) without stop codons were ordered from The Plasmid Information data base (18). A destination vector was generated by subcloning the BamHI/NotI restriction fragment of pAcGFP1-N1 (Clontech, Westburg, Leusden, The Netherlands) in-frame behind Gateway Reading Frame Cassette B (Invitrogen) in pcDNA5/FRT/TO (Invitrogen). To obtain an inducible vector containing mitochondrial-targeted AcGFP1 and C-terminally AcGFP1-tagged proteins, the entry vectors were recombined with the AcGFP1-Destination vector by using Gateway LR Clonase II Enzyme Mix (Invitrogen). In the same manner, a tandem mitochondrial AcGFP1 expression vector was created by first making an entry vector containing the cox8 leader sequence linked to the N terminus of AcGFP1 (without the stop codon) and then recombining this entry clone with the AcGFP1-Destination vector. Flp-In T-Rex293 cells (Invitrogen) were stably transfected using Superfect Transfection Reagent (Qiagen, Venlo, The Netherlands) according to the manufacturer's procedures and cultured for selection in the presence of 200 μg/ml hygromycin (Calbiochem, Brunschwig, Amsterdam, The Netherlands) in Dulbecco's modified Eagle's medium (Biowhittaker, Walkersville, MD) supplemented with 10% fetal calf serum (v/v), 1% penicillin/streptomycin (Invitrogen, Breda, The Netherlands) and 50 μg/ml blasticin (Invitrogen). To induce expression of the fusion proteins, 1 μg/ml of doxycycline (Sigma) was added to the medium, followed by incubation for 24 h.
BN-PAGE Analysis—Native mitochondrial proteins were isolated, and BN 5–15% gradient gels were loaded with 40–80 μg of protein as described before (19). After electrophoresis, in-gel fluorescence was determined using an imaging analyzer FLA5000 (Fujifilm, Tilburg, The Netherlands) (473-nm excitation laser, FITC filter >510 nm). After scanning, gels were further processed for in-gel activity assays and Western blotting, as described previously (19).
SDS-PAGE Analysis—Mitochondrial lysates were separated on a 10% SDS-PAGE gel as described previously (7) and scanned for in-gel fluorescence.
Mitochondrial Subfractionation—NDUFS3 cells were resuspended in isotonic buffer (0.25 m sucrose, 0.2 mm EDTA, 5 mm Tris-HCl, pH 7.4) and mechanically disrupted with a 5-ml glass/Teflon Potter-Elvehjem homogenizer (clearance 0.025 mm; 8 strokes at 1800 rpm; 0 °C). After adding 1 ml of isotonic buffer the homogenate was centrifuged (10 min; 600 × g; 4 °C). The supernatant was centrifuged again (25 min; 10,000 × g; 4 °C). The pellet was resuspended in ACBT (1.5 m aminocaproic acid, 75 mm Bis-Tris) and sonicated for 10 s on ice. Unbroken mitochondria were pelleted by centrifugation (30 min; 10,000 × g; 4 °C). The supernatant was divided over ultracentrifuge tubes and centrifuged (60 min; 100,000 × g; 4 °C). Each pellet was resuspended in 50 μl of ACBT, and dodecyl maltoside (DDM) was added to a final concentration of 0, 0.5, and 2%. The samples were incubated on ice for 10 min and centrifuged (60 min; 100,000 × g, 4 °C). The supernatant was used for BN-PAGE analysis.
Antibodies and ECL Detection—Immunodetection of OXPHOS proteins was performed using primary antibodies against NDUFA9 (Invitrogen), the CII 70-kDa subunit (SDHA; Invitrogen), and EGFP (a kind gift of Dr. F. J. van Kuppeveld, Dept. of Medical Microbiology, RUNMC, Nijmegen). Secondary detection was performed using peroxidase-conjugated anti-mouse or anti-rabbit IgGs (Invitrogen). The signal was generated using ECL (Pierce).
FRAP Analysis—For FRAP analysis, HEK293 cells were seeded on a Wilco dish (Intracel Ltd., Royston, UK) and grown to ∼70% confluence. During FRAP measurements cells were maintained in a colorless HEPES-Tris (HT) solution (132 mm NaCl, 4.2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 5.5 mm d-glucose, and 10 mm HEPES, pH 7.4). As a reference, a cox8-AcGFP1-expressing cell line was included in the analysis on each day of experiments. FRAP measurements were carried out at 20 °C to minimize mitochondrial movement (20) using a ZEISS LSM510 Meta confocal microscope (Carl Zeiss B.V., Sliedrecht, The Netherlands). Images were acquired at a rate of 10 Hz using a ×63 oil immersion objective (N.A. 1.4; Carl Zeiss). Routinely, a zoom factor of 4 was used, and pinhole settings were chosen to achieve an optical section thickness of <2 μm. Only single mitochondria that were fully within the focal plane were used for analysis. This was confirmed by an axial (Z) scan through the filament. A FRAP region of 10 × 10 pixels (1.4 × 1.4 μm) was used. After acquiring the base level of fluorescence, the AcGFP1 in the FRAP region was photobleached during 500 ms using 488-nm light delivered by an argon-ion laser set at 100% transmission. Importantly, intense illumination of AcGFP1 is not phototoxic (21). On resuming image acquisition, AcGFP1 fluorescence intensity was reduced by 60–70% (Table 1, F0). Only mitochondria in which FRAP was paralleled by fluorescence loss in photobleaching (FLIP) in a part distal to the FRAP region were considered to possess a continuous mitochondrial matrix and included in the analysis. Following bleaching, fluorescence recovery in the FRAP region was monitored using normal 488-nm excitation (0.1% transmission). Individual FRAP curves (F(t)) were calculated as described previously (14) using Equation 1.
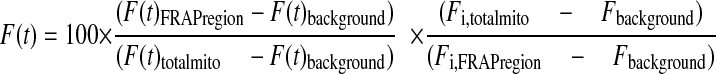 |
(Eq. 1) |
Here the fluorescence intensity in the bleached mitochondrial region (F(t)FRAPregion) and for the total mitochondrion (F(t)totalmito) is background-corrected (F(t)background) at each time point. Next, the corrected fluorescence signal in the bleached region is divided by the corrected intensity of the total mitochondrion to correct for the loss of fluorescence during the bleach. This is important because in our experiments the bleach area is relatively large compared with the size of the mitochondrion. The data are normalized to the background-corrected pre-bleach intensity (Fi,totalmito and Fi,FRAPregion) and multiplied by 100 to yield a percentage of initial fluorescence. Mean recovery curves were calculated by averaging individual FRAP curves and fitted by a two-component exponential association model (17) using Equation 2,
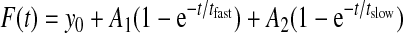 |
(Eq. 2) |
with tfast and tslow being the time constant of the fast and slow recovery phase, respectively and A1 + A2 + y0 being the fluorescence intensity (F∞) at t = t∞. Importantly, for all FRAP experiments the two-component model was more appropriate than a single-component exponential model (i.e. a model without the 3rd term in Equation 2) (22). The mobile fraction (Fm) was calculated from the averaged recovery curves using Equation 3 (14, 16, 17),
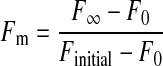 |
(Eq. 3) |
with F∞ as the fluorescence intensity at t = t∞ (as calculated from the fit), F0 as the starting fluorescence level of fluorescence recovery (Table 1), and Finitial as 100%.
TABLE 1.
FRAP curve fitting results of HEK293 cells expressing AcGFP1-tagged mitochondrial control proteins and complex I subunits
| Cell line | MWa | Nb | R2c | tfastd | tslowe | F0f | Fmg |
|---|---|---|---|---|---|---|---|
| AcGFP1-tagged control proteins | |||||||
| GFP | 27.9 | 171 | 0.997 | 0.216 ± 0.009h | 1.12 ± 0.034 | 39 ± 1.1 | 1.01 |
| GFP2 | 55.8 | 58 | 0.997 | 0.442 ± 0.024 | 2.42 ± 0.087 | 27 ± 1.2 | 0.99 |
|
ANT
|
62.8
|
55
|
0.969
|
n.d.
|
8.04 ± 0.644
|
29 ± 1.7
|
0.63
|
| AcGFP1-tagged complex I subunits | |||||||
| NDUFV1 | 77.5 | 22 | 0.882 | 0.401 ± 0.084 | 5.04 ± 1.20 | 36 ± 3.5 | 0.58 |
| NDUFV2 | 52.6 | 39 | 0.981 | 0.326 ± 0.031 | 3.26 ± 0.218 | 37 ± 1.5 | 0.63 |
| NDUFS3 | 55.3 | 43 | 0.928 | 0.364 ± 0.061 | 5.41 ± 0.896 | 42 ± 2.3 | 0.52 |
| NDUFA2 | 39.7 | 20 | 0.920 | 0.230 ± 0.057 | 1.58 ± 0.213 | 39 ± 3.3 | 0.72 |
| NDUFA12 | 46.0 | 32 | 0.971 | 0.176 ± 0.028 | 2.30 ± 0.125 | 36 ± 1.6 | 0.66 |
| NDUFB6 | 44.3 | 57 | 0.960 | n.d. | 8.39 ± 0.90 | 30 ± 1.9 | 0.46 |
Predicted MW (in kDa), including size of linker (2 kDa) and AcGFP1 (27.9 kDa)
Total number of mitochondria analyzed on at least 2 different days
Coefficient of determination, being the proportion of variability in the average FRAP curve that is accounted for by the fit model (calculated using Eq. 2)
Time constant of fast FRAP recovery phase in seconds (larger means slower). n.d., not determined
Time constant of slow FRAP recovery phase in seconds (larger means slower)
Starting level of fluorescence recovery (% of pre-bleach level)
Mobile fraction (calculated using Eq. 3)
Statistics: errors indicate S.E.
Image Processing, Curve Fitting, and Statistics—Image processing and analysis were performed using Image Pro Plus 5.1 (Media Cybernetics, Bethesda, MD). Non-linear curve fitting (using the Levenberg-Marquardt algorithm) and statistical analysis were performed using Origin Pro 7.5 (Originlabs, Northampton, MA). Values from multiple experiments are expressed as mean ± S.E.
RESULTS
AcGFP1-tagged CI Subunits Are Mitochondrially Localized and Incorporated in CI—For live cell analysis of CI assembly, we used HEK293 cell lines that expressed a monomeric variant of GFP, AcGFP1, fused to the C terminus of a nDNA-encoded CI subunit of interest. Subunits were selected on the basis of current CI assembly models (Fig. 1A and Ref. 8). A total of six cell lines were generated (schematically depicted in Fig. 1B) for inducible expression of subunits of either the dehydrogenase (green, NDUFV1 and NDUFV2), hydrogenase (purple, NDUFS3), or membrane (yellow, NDUFB6) module or accessory subunits (pink, NDUFA2 and NDUFA12). Confocal microscopy showed that all AcGFP1-tagged subunits were readily expressed and mitochondrially localized (supplemental Fig. S1).
FIGURE 1.

AcGFP1-tagging of CI subunits. A, current CI assembly model. Biogenesis of the hydrogenase module (containing NDUFS3) is thought to form the starting point of assembly (steps 1, 2, and 3). This module is coupled to a membrane module (at step 4), which contains at least ND1. Later in assembly, this membrane module is completed by other hydrophobic subunits (i.e. NDUFB6). Subunits of the NADH dehydrogenase module (containing NDUFV1, NDUFV2) are presumably added after formation of subcomplex 6. Accessory subunits (NDUFA2, NDUFA12) are also thought to enter in a late stage. B, schematic representation of the AcGFP1-tagged CI subunits (fusion proteins) expressed by inducible HEK293 cell lines. Colors correspond to the functional modules in A. C, BN-PAGE fluorograms (f) and anti-EGFP antibody immunodetection (a) of different cell lines expressing low levels of the AcGFP1-tagged CI subunits. The position of holo-CI is marked with closed arrowheads. Open arrowheads (NDUFS3 cell line) indicate CI assembly intermediates (1–6) or monomeric (AcGFP1-NDUFS3) subunit (m).
Five of the six cell lines (NDUFV1, NDUFV2, NDUFS3, NDUFA2, and NDUFA12) expressed the tagged subunit already under non-induced conditions (leakage). This gave us the opportunity to minimize the risk of overexpression artifacts. An additional advantage is that under non-induced conditions incorporation of tagged subunit is at steady state (7). Fluorescence scanning of native gels of mitochondrial fractions (Fig. 1C, lanes indicated with f) of these five cell lines showed a fluorescent band at the height of holo-CI (closed arrowheads). Their incorporation in CI was confirmed by immunodetection with an anti-EGFP antibody (Fig. 1C, lanes indicated with a). Distinct bands of lower MW, previously identified as CI assembly intermediates (7), were found in NDUFS3 cells (Fig. 1C, open arrowheads 1–6). Other (faint) bands of lower MW, not seen before, were observed in NDUFV1, NDUFV2, and NDUFA2 cells. Both detection methods showed for low-level induced NDUFB6 cells a dim smear at and below the height of the holo-complex, which was in line with the assembly pattern of endogenous NDUFB6 (7). Further induction of the tagged subunits increased their incorporation into CI without altering the CI amount and in-gel activity (supplemental Fig. S2). This indicates that the AcGFP1 tag did not interfere with proper assembly and functioning of the holo-complex.
Submitochondrial FRAP Discriminates between Matrix-soluble and MIM-associated Proteins—Before FRAP analysis of the AcGFP1-tagged CI subunits was performed, we first established that submitochondrial FRAP analysis was able to distinguish matrix-soluble from MIM-associated proteins.
Given the fact that the mobility of matrix-soluble proteins might depend on their size (23), we first assessed the relationship between protein size and mobility in the matrix. To this end, we generated two inducible HEK293 cell lines expressing either an N-terminal fusion from AcGFP1 to the mitochondrial leader sequence of cox8 or a mitochondria-targeted tandem fusion of AcGFP1 (Fig. 2A, cell lines designated GFP and GFP2, respectively). Fluorescence scanning (Fig. 2B, lanes indicated f) and anti-EGFP immunostaining of Western blots (Fig. 2B, lanes indicated a) revealed a specific band of the expected size on SDS-PAGE gels. Fig. 2C depicts a typical FRAP experiment with GFP cells. Following the bleach pulse, the fluorescence signal in the FRAP region (square) rapidly recovered to prebleach values. Recovery kinetics was appropriately fitted by a two-component model. Averaged FRAP curves are shown in Fig. 2D. Calculation of the mobile fraction (Fm) revealed values of 1.01 and 0.99 for GFP and GFP2 cells, respectively (Table 1), demonstrating that both unconjugated matrix proteins were fully mobile. In GFP cells, fluorescence recovered with time constants of 0.216 s (tfast) and 1.12 s (tslow). For GFP2 cells, both time constants (0.442 s and 2.42 s for tfast and tslow, respectively) were 2-fold slower, indicating that the mobility of matrix-soluble GFP decreases with size.
FIGURE 2.

FRAP analysis of matrix-soluble and MIM-bound AcGFP1 in living cells. A, schematic representation of control proteins expressed in inducible HEK293 cell lines: matrix-targeted AcGFP1 (GFP), matrix-targeted tandem version of AcGFP1 (GFP2), and ANT1-linked AcGFP1 (ANT). B, SDS-PAGE fluorograms (f) and anti-EGFP antibody immunodetection (a) of GFP, GFP2, and ANT cells. Filled arrowheads indicate fluorescent products of the expected MW. C, typical example of images acquired during a FRAP experiment in GFP cells. Following bleaching, AcGFP1 fluorescence rapidly recovered in the FRAP region (square). This was paralleled by fluorescence redistribution across the mitochondrial filament. D, average fluorescence recovery curve (symbols, mean ± S.E.) and the fitted two-component model (lines) for GFP, GFP2, and ANT cells (see also Table 1).
Next, we verified whether submitochondrial FRAP could distinguish between GFP, GFP2, and AcGFP1 fused to an integral protein of the MIM. To this end, an inducible HEK293 cell line was generated that expressed an N-terminal fusion from AcGFP1 to mitochondrial adenine nucleotide translocase 1 (Fig. 2A, cell line designated ANT). Fluorescence scanning (Fig. 2B, lanes indicated f) and anti-EGFP immunostaining of Western blots (Fig. 2A, lanes indicated a) confirmed the presence of a specific product of the correct size after SDS-PAGE. The averaged ANT recovery curve is shown in Fig. 2D. Calculation of Fm revealed a value of 0.63 for ANT cells (Table 1). In contrast to GFP and GFP2, which were fully mobile, 37% of AcGFP1-tagged ANT was virtually immobile. The remaining fraction of AcGFP1-tagged ANT displayed a much slower mobility than GFP and GFP2 (Fig. 2D). This is reflected by tslow, which was much larger for ANT cells (8.04 s) than for GFP (1.12 s) and GFP2 (2.42 s) cells. Proper calculation of tfast was not feasible for ANT cells given the limited number of data points constituting the fast phase of the recovery curve (Fig. 2D). Taken together, these results demonstrate that matrix-soluble and MIM-associated proteins display different submitochondrial FRAP kinetics. This property can be used to study CI assembly, which is thought to occur in the mitochondrial matrix and MIM (Fig. 1A).
Evidence for Coexistence of Mobile and Immobile CI Assemblies within the Mitochondrion—After having established the FRAP characteristics of matrix-soluble and MIM-bound proteins, we next applied this technique to the cell lines expressing the tagged CI subunits (Fig. 3, A–F, red curve). Again, recovery kinetics was adequately described with a two-component model. Calculation of Fm revealed that in all six cell lines significant amounts of the AcGFP1-tagged subunit resided in the immobile fraction (Table 1).
FIGURE 3.

FRAP recovery kinetics and immobile fraction of AcGFP1-tagged CI subunits in living cells. Average FRAP curves (symbols) and two-component model fit (red line) in non-induced (leakage) cells for: NDUFV1 (A), NDUFV2 (B), NDUFS3 (C), NDUFA2 (D), NDUFA12 (E), and induced NDUFB6 (F) cells (see also Table 1). Smooth curves represent the two-component model fits to the average GFP (green) and ANT (blue) FRAP curves depicted in Fig. 2.
Concerning the mobilities of GFP-tagged subunits, the results obtained for GFP and GFP2 cells show that these can only be compared if the size of the subunits is taken into account. Thus, a plot of tfast versus MW shows that the values for fully mobile GFP and GFP2 lie on a line through the origin, whereas those for the AcGFP1-tagged subunits are below this line (Fig. 4A). This suggests that our cell lines contained AcGFP1-tagged product(s) that move faster than predicted on the basis of the MW of the intact fusion protein. NDUFA2 and NDUFA12 cells displayed similar tfast values as observed in GFP cells, demonstrating the presence of fully mobile AcGFP1 in these two cell lines. To enquire the presence of fluorescent breakdown products, we analyzed mitochondrial enriched fractions by SDS-PAGE. The fluorograms (Fig. 4B, lanes indicated f) show that mast cell lines displayed, in addition to a clear band of correct size (closed arrowheads), bands of lower MW. The relative intensity of these latter bands was low in NDUFS3 and NDUFA2 cells, but high in NDUFA12 cells. Because GFP fluorescence is extremely sensitive to truncations (24), the lower MW breakdown products most probably consist of full-length AcGFP1 alone or fused to partially truncated subunit. Together, these results support the conclusion that the fast component of the FRAP signal reflects the movement of lower MW breakdown products demonstrated to be present in these cell lines. This is substantiated by the fact that the fast component of the FRAP signal was virtually absent in ANT and NDUFB6 cells, which do not contain lower MW breakdown products (see Figs. 2B and 4B).
FIGURE 4.

Relationship between FRAP recovery kinetics and molecular weight of the AcGFP1-tagged CI subunits in living cells. The kinetics of the fast (tfast) and slow (tslow) recovery phase of the FRAP curve were quantified by fitting a two-component model. A, plot of average tfast (mean ± S.E.) of fusion proteins versus their MW. The values for GFP and GFP2 are on a line through the origin (p < 0.05). B, SDS-PAGE fluorograms (f) and anti-EGFP antibody immunodetections (a) of cells expressing different AcGFP1-tagged subunits. Filled arrowheads indicate fluorescent products of the expected MW. C, plot of average tslow(mean ± S.E.) of subunits versus their MW. The values for GFP and GFP2 are on a line through the origin (p < 0.05). Blue lines indicate 95% confidence limits of the fit lines.
Also in a plot of tslow versus MW, the values for GFP and GFP2 lie on a line through the origin (Fig. 4C). In this case, however, the values for the AcGFP1-tagged CI subunits are either on this line (NDUFA2 and NDUFA12 cells), above this line but within the 95% confidence interval (NDUFV2 and NDUFV1 cells) or well above the 95% upper confidence limit (NDUFS3 and NDUFB6 cells), indicating that these subunits moved either as predicted (NDUFA2 and NDUFA12 cells), possibly slower than predicted (NDUFV2 and NDUFV1 cells) or significantly slower than predicted (NDUFS3 and NDUFB6 cells) on the basis of their MW. The tslow value for NDUFS3 cells is clearly above the line through the origin but below the tslow value for ANT cells, suggesting that AcGFP1-tagged NDUFS3 moves faster than expected when bound to the MIM. In contrast, the tslow value for NDUFB6 cells is above the ANT line, suggesting that this AcGFP1-tagged subunit moves considerably slower than expected when bound to the MIM. The most simple explanation for these results is that in NDUFS3 cells the slow component of the FRAP signal reflects the movement of a matrix-soluble assembly of the AcGFP1-tagged subunit and several other proteins, whereas in NDUFB6 cells, it reflects a membrane-bound assembly of this AcGFP1-tagged subunit and several other proteins.
To substantiate this conclusion, we determined the submitochondrial localization of AcGFP1-tagged subunit in induced NDUFS3 cells. Mitochondria were isolated, sonicated and sequentially centrifuged at 10,000 × g (30 min) and 100,000 × g (60 min) to obtain a 100,000 × g soluble and membrane fraction (25). To assess the strength of membrane binding, aliquots of the membrane fraction were treated with either 0.5% or 2.0% lauryl maltoside for 10 min at 4 °C and centrifuged at 100,000 × g (60 min). Fluorescence scanning (Fig. 5A) and Western blotting (Fig. 5B) of native gels of the initial soluble fraction and the supernatants of the lauryl maltoside-treated membrane fractions revealed that subassembly 1 was both soluble and membrane-bound, whereas subassemblies 2 and 3 were relatively loosely membrane-bound and subassemblies 4, 5, and 6 were relatively tightly membrane-bound. These results support the idea that the slow component of the FRAP signal reflects the movement of a matrix-soluble assembly of AcGFP1-tagged NDUFS3 and other proteins. Possibly this matrix-soluble assembly is represented by the soluble part of subassembly 1. The immobile FRAP fraction would then be composed of the membrane-bound part of subassembly 1 and the membrane-bound subassemblies 2, 3, 4, 5, and 6, as well as the holo-complex. Together, our results indicate that CI assembly is localized in both the matrix and the mitochondrial inner membrane.
FIGURE 5.

Topology and membrane anchorage of subcomplexes in induced NDUFS3 cells. A, BN-PAGE fluorograms of membrane-solubilized proteins. Left lane represents the soluble (matrix-containing) fraction. Proteins were solubilized by treatment with DDM (concentrations are given in % (w/v)). The height of holo-CI is marked with closed arrowheads. Open arrowheads indicate NDUFS3 assembly intermediates (1–6) or monomeric subunit (m). The asterisk indicates an a-specific signal, also present in wild-type cells after contrast optimization (not shown). B, duplicate experiment as depicted in A was used for Western blotting and immunodetection with antibodies against EGFP. In this figure, the signal marked by a question mark most likely reflects sonication-induced breakdown of the peripheral arm of holo CI.
DISCUSSION
The biogenesis of CI is complicated by its large size and its regulation by two genomes, with seven subunits encoded by the mtDNA and the remainder by nuclear genes. Knowledge of this process might provide important insights into the pathogenesis of many hitherto unexplained OXPHOS disorders. Previous in vitro studies strongly support the idea that the 45 subunits assemble via different routes to form the functional enzyme. Proper understanding of how macromolecular assemblies such as CI are built up requires investigation of protein interactions in the dynamic environment of the living cell. Here, we reasoned that if CI assembly occurs through pre-assembled modules it should be possible to demonstrate their existence by submitochondrial FRAP in living cells. Indeed, FRAP analysis revealed that the NDUFS3 subunit, proposed to be incorporated at an early stage of the assembly process, displayed mobilities that were consistent with its presence in both slowly mobile and virtually immobile assemblies. Using differential extraction, we confirmed the presence of this subunit in a matrix-soluble assembly, probably representing the slowly mobile fraction, and several membrane-bound assemblies including the holo-enzyme, probably representing the immobile fraction. In contrast to the NDUFS3 subunit, the NDUFV1, NDUFV2, NDUFA2, and NDUFA12 subunits, proposed to be incorporated at a late stage during the assembly of CI, were either immobile or present as highly mobile matrix-soluble monomers. Consistent with its possession of a transmembrane domain, NDUFB6 was either immobile or part of one or more slowly diffusing membrane-bound subassemblies. Together, these results show for the first time that CI subassemblies exist in vivo and that they might be present in both the matrix and the MIM.
To allow proper interpretation of the FRAP data, we first determined the mobilities of a matrix-targeted version of AcGFP1 (GFP), a matrix-targeted tandem version of AcGFP1 (GFP2), and an MIM-targeted version of AcGFP1 (ANT). The FRAP mobility data obtained with GFP and GFP2 demonstrate that a small change in size of a freely diffusing and non-interacting protein influences its mobility in the mitochondrial matrix. GFP and GFP2 were fully mobile and displayed a biphasic FRAP curve. Importantly, both the fast and slow time-constant of this curve (tfast and tslow) showed a linear relationship with the MW. These findings are compatible with the idea that mitochondrial structures (e.g. cristae) can temporarily trap freely moving and non-interacting proteins (26). In this context, evidence has been presented in the literature indicating that mitochondrial cristae are connected to the periphery of the MIM by narrow tubular junctions (cristae junctions, (27)), which might be responsible for the observed molecular sieving effect. Additionally, we showed that binding of AcGFP1 to the MIM through its fusion with ANT1 slowed down the recovery kinetics by a factor 4 compared with matrix-soluble GFP2. This result agrees with previous data on ANT diffusion (10). About 37% of AcGFP1-tagged ANT1 was immobile. Most probably this represents the fraction of molecules bound to the mitochondrial permeability transition pore complex (28).
All AcGFP1-tagged subunits displayed an immobile fraction. Previous estimates of holo-CI diffusion in mitoplasts revealed a diffusion constant of 0.0046 μm2/s (29, 30). Assuming that this value is similar to the mobility of holo-CI in living cells and comparing it to the diffusion constant of matrix-soluble EYFP in living cells (25 μm2/s; (22)), holo-CI can be expected to move ∼5100 times slower than unconjugated AcGFP1. Based on these considerations, it is reasonable to conclude that the virtually immobile fraction observed with all subunits tested consists of holo-CI. This conclusion is supported by earlier work of Bush et al. (31) who observed a speckled distribution of GFP- and RFP-tagged CI during ongoing mitochondrial fusion and fission.
Although we used low expression levels of the AcGFP1-tagged subunits, inspection of the tfast values revealed that all five cell lines displayed a fast component with a mobility that corresponded to or was faster than expected on the basis of the MW of the AcGFP1-tagged subunit. Fluorograms of SDS-PAGE gels confirmed that these faster mobilities were caused by the presence of lower MW fluorescent breakdown products. The fact that these breakdown products remained fluorescent suggests that CI subunits, if not incorporated, are readily prone to proteolytic breakdown.
The tslow values of the cells lines expressing AcGFP1-tagged NDUFA2, NDUFA12, NDUFV2, or NDUFV1 were in good agreement with the predicted values. Thus, these four subunits were present either as part of the virtually immobile holo-complex (see above) or in their free form, i.e. not bound to any other larger-sized complex. This demonstrates that these four subunits are incorporated at a relatively late stage during in vivo CI assembly. In a recent study we described a patient with a mutation in the NDUFA2 subunit of CI (32). Interestingly, this patient accumulated a CI subassembly of ∼830 kDa, indicating that the NDUFA2 subunit is indeed a late subunit. A similar ∼830-kDa subassembly was observed in a patient with a mutation in the NDUFV1 subunit (33). Experimental evidence for this subunit being a late subunit comes from metabolic labeling studies showing that the top of matrix-protruding part of the holo-enzyme, which contains at least the NDUFV1 and NDUFV2 subunits, is added at the very end of the CI assembly process (9). It has been suggested that the NDUFA12 subunit, similar to the NDUFV1 and NDUFV2 subunits, is added after the formation of the ∼830-kDa subassembly (34). Our results provide first in vivo evidence that the NDUFA12 subunit indeed is a late subunit.
NDUFS3-expressing cells displayed a slow component with a mobility that was slower than expected on the basis of the MW of the AcGFP1-tagged subunit, but faster than expected when linked to a membrane-bound protein. This result is indicative for the presence of a matrix-soluble subassembly. Evidence in support of this idea was provided by a differential extraction experiment, revealing not only a matrix-soluble NDUFS3-AcGFP1 containing subassembly (subassembly 1) but also two loosely membrane-associated subassemblies (subassemblies 2 and 3) and four strongly membrane-bound assemblies (subassemblies 4 to 6 and holo-CI) containing this fusion protein. We previously demonstrated that subassemblies 4 to 6 and holo-CI contain the MIM spanning ND1 subunit (7). In this respect, the membrane-associated characteristics of subassemblies 2 and 3 are unexpected because, so far, only soluble proteins have been identified in these subcomplexes (6, 7, 9). In contrast to the current assembly models, these results support the conclusion that subassemblies 2 and 3 are assembled at the MIM. It is tempting to speculate that linkage of these two subassemblies to the MIM requires a, currently unknown, membrane-anchoring protein. Further analysis of the composition of these subassemblies is required to provide clarity on this issue.
In summary, this study provides first time evidence that CI subassemblies exist in vivo in the matrix and the inner membrane. This indicates that a modular CI assembly pathway is operational in different mitochondrial compartments in the living cell.
Supplementary Material
Acknowledgments
We thank E. Leijen for optimization of the fractionation protocol.
This work was supported by a grant from the Radboud University Nijmegen Medical Centre (RUNMC, to W. J. H. K. and L. G. J. N.) and equipment grants of ZON (Netherlands Organization for Health Research and Development, 903-46-176) and NWO (Netherlands Organization for Scientific Research, No: 911-02-008). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: OXPHOS, oxidative phosphorylation; CI, complex I; DDM, dodecyl maltoside; FRAP, fluorescence recovery after photobleaching; MIM, mitochondrial inner membrane; NDUF, NADH dehydrogenase ubiquinone flavoprotein; GFP, green fluorescent protein; BN-PAGE, blue native polyacrylamide gel electrophoresis.
References
- 1.Shoubridge, E. A. (2001) Hum. Mol. Genet. 10 2277-2284 [DOI] [PubMed] [Google Scholar]
- 2.Carroll, J., Fearnley, I. M., Skehel, J. M., Shannon, R. J., Hirst, J., and Walker, J. E. (2006) J. Biol. Chem. 281 32724-32727 [DOI] [PubMed] [Google Scholar]
- 3.Brandt, U. (2006) Annu. Rev. Biochem. 75 69-92 [DOI] [PubMed] [Google Scholar]
- 4.Janssen, R. J., Nijtmans, L. G., van den Heuvel, L. P., and Smeitink, J. A. (2006) J. Inherit. Metab Dis. 29 499-515 [DOI] [PubMed] [Google Scholar]
- 5.Antonicka, H., Ogilvie, I., Taivassalo, T., Anitori, R. P., Haller, R. G., Vissing, J., Kennaway, N. G., and Shoubridge, E. A. (2003) J. Biol. Chem. 278 43081-43088 [DOI] [PubMed] [Google Scholar]
- 6.Ugalde, C., Vogel, R., Huijbens, R., Van Den, H. B., Smeitink, J., and Nijtmans, L. (2004) Hum. Mol. Genet. 13 2461-2472 [DOI] [PubMed] [Google Scholar]
- 7.Vogel, R. O., Dieteren, C. E., van den Heuvel, L. P., Willems, P. H., Smeitink, J. A., Koopman, W. J., and Nijtmans, L. G. (2007) J. Biol. Chem. 282 7582-7590 [DOI] [PubMed] [Google Scholar]
- 8.Vogel, R. O., Smeitink, J. A., and Nijtmans, L. G. (2007) Biochim. Biophys. Acta. 1767 1215-1227 [DOI] [PubMed] [Google Scholar]
- 9.Lazarou, M., McKenzie, M., Ohtake, A., Thorburn, D. R., and Ryan, M. T. (2007) Mol. Cell. Biol. 27 4228-4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupte, S. S., Chazotte, B., Leesnitzer, M. A., and Hackenbrock, C. R. (1991) Biochim. Biophys. Acta 1069 131-138 [DOI] [PubMed] [Google Scholar]
- 11.Seksek, O., Biwersi, J., and Verkman, A. S. (1997) J. Cell Biol. 138 131-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haggie, P. M., and Verkman, A. S. (2002) J. Biol. Chem. 277 40782-40788 [DOI] [PubMed] [Google Scholar]
- 13.Lippincott-Schwartz, J., tan-Bonnet, N., and Patterson, G. H. (2003) Nat. Cell Biol. (suppl.), S7-S14 [PubMed]
- 14.Goodwin, J. S., and Kenworthy, A. K. (2005) Methods 37 154-164 [DOI] [PubMed] [Google Scholar]
- 15.Roy, S., Plowman, S., Rotblat, B., Prior, I. A., Muncke, C., Grainger, S., Parton, R. G., Henis, Y. I., Kloog, Y., and Hancock, J. F. (2005) Mol. Cell. Biol. 25 6722-6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen, Y., Lagerholm, B. C., Yang, B., and Jacobson, K. (2006) Methods 39 147-153 [DOI] [PubMed] [Google Scholar]
- 17.Picard, D., Suslova, E., and Briand, P. A. (2006) Exp. Cell Res. 312 3949-3958 [DOI] [PubMed] [Google Scholar]
- 18.Zuo, D., Mohr, S. E., Hu, Y., Taycher, E., Rolfs, A., Kramer, J., Williamson, J., and LaBaer, J. (2007) Nucleic Acids Res. 35 D680-D684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nijtmans, L. G., Henderson, N. S., and Holt, I. J. (2002) Methods 26 327-334 [DOI] [PubMed] [Google Scholar]
- 20.Koopman, W. J., Visch, H. J., Smeitink, J. A., and Willems, P. H. (2006) Cytometry A 69 1-12 [DOI] [PubMed] [Google Scholar]
- 21.Bulina, M. E., Chudakov, D. M., Britanova, O. V., Yanushevich, Y. G., Staroverov, D. B., Chepurnykh, T. V., Merzlyak, E. M., Shkrob, M. A., Lukyanov, S., and Lukyanov, K. A. (2006) Nat. Biotechnol. 24 95-99 [DOI] [PubMed] [Google Scholar]
- 22.Koopman, W. J., Hink, M. A., Verkaart, S., Visch, H. J., Smeitink, J. A., and Willems, P. H. (2007) Biochim. Biophys. Acta. 1767 940-947 [DOI] [PubMed] [Google Scholar]
- 23.Sprague, B. L., and McNally, J. G. (2005) Trends Cell Biol. 15 84-91 [DOI] [PubMed] [Google Scholar]
- 24.Li, X., Zhang, G., Ngo, N., Zhao, X., Kain, S. R., and Huang, C. C. (1997) J. Biol. Chem. 272 28545-28549 [DOI] [PubMed] [Google Scholar]
- 25.Cardol, P., Boutaffala, L., Memmi, S., Devreese, B., Matagne, R. F., and Remacle, C. (2008) Biochim. Biophys. Acta. 1777 388-396 [DOI] [PubMed] [Google Scholar]
- 26.Olveczky, B. P., and Verkman, A. S. (1998) Biophys. J. 74 2722-2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mannella, C. A. (2006) Biochim. Biophys. Acta. 1763 542-548 [DOI] [PubMed] [Google Scholar]
- 28.Kroemer, G., Galluzzi, L., and Brenner, C. (2007) Physiol. Rev. 87 99-163 [DOI] [PubMed] [Google Scholar]
- 29.Chazotte, B., and Hackenbrock, C. R. (1991) J. Biol. Chem. 266 5973-5979 [PubMed] [Google Scholar]
- 30.Lenaz, G., and Genova, M. L. (2007) Am. J. Physiol. Cell Physiol. 292 C1221-C1239 [DOI] [PubMed] [Google Scholar]
- 31.Busch, K. B., Bereiter-Hahn, J., Wittig, I., Schagger, H., and Jendrach, M. (2006) Mol. Membr. Biol. 23 509-520 [DOI] [PubMed] [Google Scholar]
- 32.Hoefs, S. J., Dieteren, C. E., Distelmaier, F., Janssen, R. J., Epplen, A., Swarts, H. G., Forkink, M., Rodenburg, R. J., Nijtmans, L. G., Willems, P. H., Smeitink, J. A., and van den Heuvel, L. P. (2008) Am. J. Hum. Genet. 82 1306-1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel, R. O., van den Brand, M. A., Rodenburg, R. J., van den Heuvel, L. P., Tsuneoka, M., Smeitink, J. A., and Nijtmans, L. G. (2007) Mol. Genet. Metab. 91 176-182 [DOI] [PubMed] [Google Scholar]
- 34.Lazarou, M., Thorburn, D. R., Ryan, M. T., and McKenzie, M. (2008) Biochim. Biophys. Acta , in press [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.