

Abstract
Receptor-mediated apoptosis proceeds via two pathways: one requiring only a cascade of initiator and effector caspases (type I behavior) and the second requiring an initiator–effector caspase cascade and mitochondrial outer membrane permeabilization (type II behavior). Here, we investigate factors controlling type I versus II phenotypes by performing Lyapunov exponent analysis of an ODE-based model of cell death. The resulting phase diagrams predict that the ratio of XIAP to pro-caspase-3 concentrations plays a key regulatory role: type I behavior predominates when the ratio is low and type II behavior when the ratio is high. Cell-to-cell variability in phenotype is observed when the ratio is close to the type I versus II boundary. By positioning multiple tumor cell lines on the phase diagram we confirm these predictions. We also extend phase space analysis to mutations affecting the rate of caspase-3 ubiquitylation by XIAP, predicting and showing that such mutations abolish all-or-none control over activation of effector caspases. Thus, phase diagrams derived from Lyapunov exponent analysis represent a means to study multi-factorial control over a complex biochemical pathway.
Keywords: apoptosis, caspases, dynamical systems analysis, kinetic modeling, XIAP
Introduction
In mammalian cells, once the commitment to apoptosis has been made, caspase-mediated protein degradation must proceed to completion lest cells survive with damaged genomes. At a biochemical level, this involves all-or-none activation of potent effector caspases (caspase-3 and caspase-7). Extrinsic apoptosis, induced by extracellular death ligands such as Fas ligand (FasL), tumor necrosis factor (TNF), or TRAIL (TNF-related apoptosis-inducing ligand), begins with assembly of death inducing signaling complexes (DISCs) on receptor intracellular tails. Initiator pro-caspases (caspase-8 and caspase-10) dimerize at the DISC, leading to activation and concomitant cleavage of inhibitory pro-domains. In type I cells, a cascade of activated initiator (caspase-8/10) and effector (caspase-3/7) caspases is sufficient to generate a lethal level of caspase activity and mitochondrial outer membrane permeabilization (MOMP) is not required. In cells that exhibit type II behavior, active caspase-8/10 still cleaves pro-caspase-3/7 but MOMP is required for death (Scaffidi et al, 1998). Type I and type II phenotypes are generally assumed to be mutually exclusive: most epithelial cells exhibit a type II phenotype but some immune cells and cells having a ‘mesenchymal’ expression profile exhibit a type I phenotype (Algeciras-Schimnich et al, 2003). Identifying factors that control type I versus II apoptosis is not only relevant to understanding regulation of extrinsic apoptosis in immune editing, tumor surveillance and cancer therapy but also exemplifies the more general challenge of determining how different cell types use distinct but overlapping pathways to control essential physiological processes.
MOMP, the event controlling apoptosis in type II cells, is triggered by caspase-8/10-mediated cleavage of Bid into its active form (tBid) followed by tBid-mediated changes in the conformation and activity of Bak and Bax (Letai, 2008). Active Bak/Bax molecules are able to self-assemble into pores in the mitochondrial outer membrane but assembly is inhibited by anti-apoptotic members of the Bcl2 protein family. When levels of active Bak/Bax exceed those of anti-apoptotic Bcl2 proteins, pores form suddenly, allowing Smac/DIABLO and cytochrome c to translocate into the cytosol (Letai, 2008). Smac-mediated inhibition of XIAP, a protein that negatively regulates active caspase-3/7, and apoptosome-mediated cleavage of pro-caspase-3/7 generates a burst of active effector caspase able to cleave essential cellular substrates and cause cell death. Experiments with membrane-bound and soluble FasL suggest that a key distinction between type I and type II cells is the rate of DISC assembly and the consequent efficiency of pro-caspase-8/10 activation (Algeciras-Schimnich et al, 2003). In type I cells, caspase-8/10 is activated sufficiently rapidly to cleave pro-caspase-3/7 and trigger death (Scaffidi et al, 1998; Barnhart et al, 2003). In type II cells, the generation of active caspase-8/10 is proposed to be less efficient and MOMP is therefore necessary to amplify a weak initiator caspase signal (Barnhart et al, 2003). However, other studies suggest an important role for XIAP in determining the balance between type I and type II death (Eissing et al, 2004; Jost et al, 2009).
Using a mass-action model developed in our laboratory to describe key biochemical steps in extrinsic apoptosis (EARM1.4; Box 1; Albeck et al, 2008a, 2008b; Spencer et al, 2009), we searched for factors influencing type I and type II behaviors. This involved identifying factors that determined whether or not MOMP was required for efficient effector caspase activation. Such an analysis can be performed in a straightforward manner using the method of direct finite-time Lyapunov exponent analysis (DLEs; Box 2; Aldridge et al, 2006b; Rateitschak and Wolkenhauer, 2010). DLEs measure the influence of changes in initial protein concentrations on the future state of a model; in the case of EARM1.4, we examined timescales determined experimentally to be relevant to caspase activation in TRAIL-treated cells (∼8 h). When DLE analysis was used to compute a six-dimensional phase diagram of type I or II phenotypes, a distinct boundary (a separatrix) was observed to cut across multiple dimensions in concentration phase space (separatrices are described in Box 2). The shape of the separatrix implied that control over type I versus II phenotypes was multi-factorial: DISC activity and ligand levels were determinative for some protein concentrations whereas XIAP and caspase-3 levels were important across the entire sampled space. To test these predictions experimentally, we placed four tumor cell lines on the DLE landscape, focusing on two-dimensional slices corresponding to the [XIAP]:[caspase-3] ratio. We found that the separatrix correctly predicted whether a cell line was type I or type II. In the case of T47D cells, the [XIAP]:[caspase-3] ratio placed them close to the separatrix and experiments confirmed a mixed type I and type II phenotype. We also extended our analysis to changes in rate constants, focusing on mutations that reduced the rate of XIAP-mediated ubiquitylation of caspase-3. When this reaction was blocked modeling predicted, and experiments confirmed, a phenotype distinct from either type I or II behavior in which snap-action control over cleavage of effector caspase substrates was lost. Based on these observations, we propose that DLE-based phase diagrams will prove generally useful in understanding multi-factorial control of cellular biochemistry in different cell types.
Modeling receptor-mediated apoptosis.
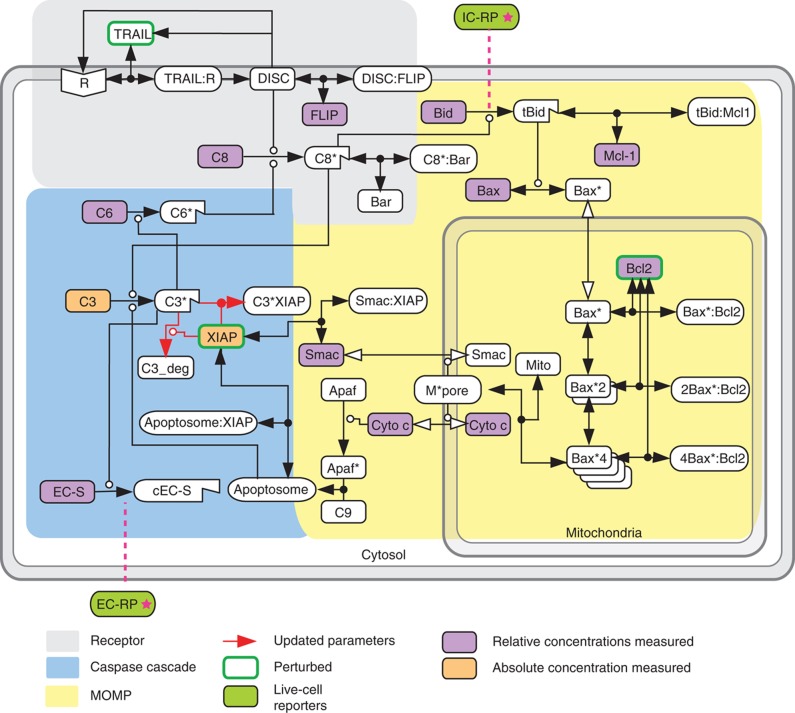 |
Box 1 Figure EARM1.4 network diagram. Schematic adapted from Albeck et al (2008b). The mass-action model used in the current paper, extrinsic apoptosis reaction model (EARM1.4), is closely related to previously published models that have been validated using live- and fixed-cell imaging, flow cytometry of caspase substrates, and biochemical analysis of normal and RNAi-treated HeLa cells (Albeck et al, 2008a, 2008b; Spencer et al, 2009). Model compartments are assumed to be well mixed and protein translocation is modeled as an elementary reaction in which proteins move between three compartments (cell surface, cytoplasm, and mitochondrial inter-membrane space). As in Spencer et al (2009), EARM1.4 includes protein synthesis (ks) and degradation (kdeg) reactions but some rate constant have been changed to improve goodness of fit (red arrows in Box 1 Figure summarize changes relative to Albeck et al (2008b); see Supplementary Tables SI–SIII for lists of model reactions and initial concentration and rate parameter values). Except for active caspase-3, the half-lives of proteins species are assumed to be 24 h, an approximation that is satisfactory because the model is relatively insensitive to changes in these rates. EARM1.4 successfully reproduces initiator and effector caspase dynamics in multiple cell types across a range of ligand doses and under various conditions of RNAi-mediated protein depletion (Albeck et al, 2008a, 2008b). EARM1.4 encompasses proteins activated by binding of TRAIL to DR4/5 receptors that ultimately cause cleavage of effector caspase substrates and cell death. EARM1.4 focuses on reactions regulating MOMP and caspase-3 activity; it does not model the formation of DISCs or the activation of caspase-8/10 in detail. However, two mechanisms negatively regulating caspase-8 are included in simplified form: competitive binding of FLIP and pro-caspase-8 to the DISC and reversible inhibition of processed caspase-8 by Bar. This level of detail is sufficient to explore the role of TRAIL, TRAIL receptor, and caspase-8/10 concentrations on downstream events, but it does not capture key events in receptor oligomerization. Two pathways link initiator and effector caspases. In the direct pathway, the sole mechanism required in type I cells, active caspase-8 cleaves pro-caspase-3; this activates the enzyme and allows it to cleave PARP. In the model, a positive feedback between effector and initiator caspases is mediated by caspase-6. Even though it is not yet completely clear how strong this feedback is in cells or whether proteins other than caspase-6 are involved, variation in caspase-6 levels in EARM1.4 can still be regarded as a means to alter the strength of a feedback mechanism that involves multiple proteins. Recent work suggests that this mechanism for caspase-8/10 activation is inefficient (Oberst et al, 2010; Wurstle et al, 2010) and feedback is therefore modeled as being weak in EARM1.4. In type II cells, MOMP, which also links initiator and effector caspases, is required for caspase-3 activation. MOMP-dependent translocation of cytochrome c from mitochondria to the cytosol leads to apoptosome formation and activation of caspase-9, another initiator caspase capable of cleaving pro-caspase-3. MOMP also causes cytosolic translocation of Smac, which binds to the effector caspase inhibitor XIAP, thereby inactivating it and relieving inhibition of caspase-3 activity. The analysis in this paper shows that the ratio of caspase-3 to XIAP levels is a major determinant of type I versus II behavior: when the ratio is low, XIAP is able to hold processed caspase-3 in check prior to MOMP. When the ratio is high, sufficient active caspase-3 accumulates to kill cells in the absence of MOMP. Many species in EARM1.4 are devoted to the regulation of MOMP but the process is nonetheless represented in a highly simplified manner. The pathway leading to MOMP begins with cleavage of Bid by active caspase-8 to form tBid. tBid can either be sequestered in the cytoplasm by an Mcl-1-like protein or bind to and activate Bax. Active Bax multimerizes and translocates to the mitochondrial outer membrane where it is inhibited by Bcl2 but once active, membrane-bound Bax levels exceed those of Bcl2, MOMP occurs, leading to translocation of Smac and cytochrome c to the cytosol, as described above. EARM1.4 does not attempt to model the full complexity of the Bcl2 protein family, instead representing all Bcl2 family activators by Bid, all anti-apoptotic family members by Bcl2 and Mcl-1 and all pore forming proteins by Bax. Nonetheless, a good match is observed between simulations and single-cell data on MOMP dynamics over a wide range of conditions (Albeck et al, 2008a, 2008b). Values for the initial concentrations of proteins in the model for which validated antibodies are available were determined by quantitative immunoblotting of extracts from HCT116 cells (see Box 1 Figure, purple, and Supplementary Table SII) and then adjusted for other cell lines using the ratios listed in Table I; for simplicity, all cells were modeled as having the same cell volume. When modeling the effect of deleting the RING domain of XIAP, the rate constant for the ubiquitylation of caspase-3 by XIAP (kc8) was set to zero. When cycloheximide was included in an experiment, synthesis terms in the corresponding simulations were set to 15% of their nominal values (Supplementary Table SII; Ceccarini and Eagle, 1976). Degradation rates were set assuming a half-life of 24 h as in Spencer et al (2009) except that the XIAP-independent rate of degradation of active caspase-3 (kr14) was set to fivefold higher to account for the observed instability of caspase-3 in HCT116ΔXIAP cells (BBA and PKS, unpublished data).
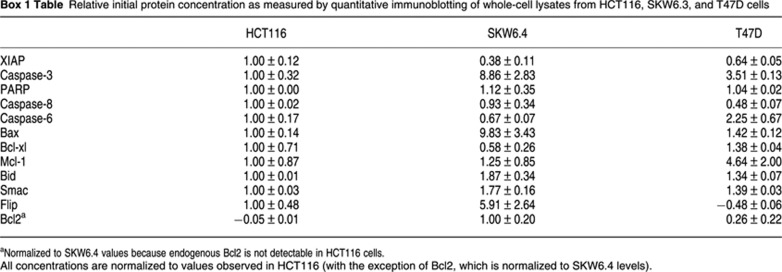 |
Model analysis using Lyapunov exponents.
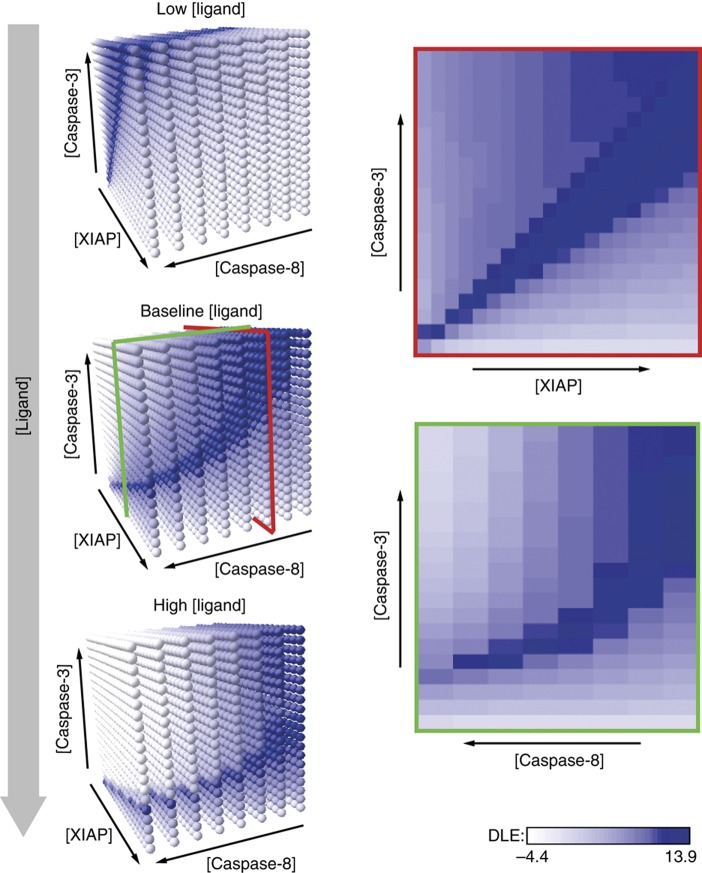 |
Box 2 Figure Multi-dimensional views of the phase space and a type I versus II DLE separatrix. DLE analysis is inherently multi-dimensional and phase diagrams can be visualized in different subspaces. On the left, three three-dimensional views are shown of a separatrix cutting through phase space for [XIAP]0, [caspase-3]0, and [caspase-8]0 at different concentrations of TRAIL. The two-dimensional phase diagrams on the right represent slices of the three-dimensional matrix under the baseline ligand condition (as indicated by the projected red and green outlines). The goal of this paper was to determine whether measured differences in protein concentrations could explain how cell lines sharing a common set of apoptotic regulators respond differently to death ligands. In some contexts, such a question could be approached using bifurcation analysis but this is challenging with a model as large as EARM1.4. More importantly, model steady states (as used for bifurcation analysis) are not reflective of the transient signals that determines whether cells live or die (see also Aldridge et al, 2006b). Lyapunov exponents are a simple means to compute the effects on model output arising from changes in initial protein concentrations. Because each protein present at t=0 gives rise to multiple species that differ in intracellular localization, extent of post-translational modification, incorporation into molecular complexes, etc., variation in initial protein concentrations generates unintuitive and complex changes in cellular biochemistry that vary from one cell type to the next. The critical dynamical variable in the current work is the level of cEC-S (species 23 ‘cPARP’ in EARM1.4); accumulation of cEC-S is assumed to reflect commitment to death (although this is not necessarily true in cell lines such as SKW6.4; see text for details). We represent the findings of our analyses using ‘phase space diagrams’ that depict outcomes across a range of protein concentrations observed in actual tumor cell lines. This approach assumes that rate constants and other critical model parameters remain the same across these cell lines. An exception to this rule involves the analysis of HCT116 cells carrying XIAPΔRING; in this case, we assumed that the rate of XIAP-mediated ubiquitylation of cleaved caspase-3 was zero (i.e. kc8=0). In principle, it would be possible to compute a phase diagram, or parameter-space diagram, for all rates in EARM1.4, but we have not yet implemented the algorithms to do this efficiently. A priori we expect some differences in initial protein concentrations to have a major impact on model outcome and others to have a smaller impact. To capture the magnitude of the deviation at a discrete point in time, we compute the multi-dimensional difference between model trajectories arising from neighboring sets of initial conditions. We quantify this difference by computing a DLE (Equation 1).
 |
where DLE(t, x0) is the exponent at time t and x and x0 are vectors that describe the concentrations of model species at time t and t=0, respectively. λmax is the maximum eigenvalue of the matrix. DLEs were computed over a grid of initial conditions whose range was chosen based on observed protein concentrations in tumor cell lines, thereby generating a multi-dimensional matrix of DLEs. The most informative regions of the matrix are those with high DLE values, which denote regions in which small changes in initial conditions result in dramatic changes in model output. Contiguous high DLE values define separatrices, membrane-like surfaces that separate regions of parameter space giving rise to different cEC-S dynamics. Separatrices are analogous to boundary layers in a physical phase diagram, for example between liquid, solid, and gas phases of water over a range of values for temperature and pressure. In the current work, we seek to distinguish type I and type II phenotypes in extrinsic apoptosis and therefore perform simulations with Bcl2 levels set at a level sufficiently high to block MOMP (the nominal value for [Bcl2]0 in HCT116 cells is 2 × 104 mol/cell; over-expression was modeled as [Bcl2]0=2 × 105 mol/cell). Initial conditions resulting in type I phenotypes were defined as those in which simulated final cEC-S levels were similar in the presence and absence of Bcl2 over-expression; conditions resulting in type II phenotypes where those in which Bcl2 over-expression blocked cEC-S accumulation. Based on preliminary analysis with a coarse grid, we concentrated on detailed DLE analysis of six model species. This gave rise to a six-dimensional matrix but, for clarity, the figures in this paper represent two-dimensional projections. Different cell lines were placed on these projections based on measuring protein levels by quantitative immunoblotting (Box 1 Table; Supplementary Figure S1) with cell-to-cell variability estimated by flow cytometry (Box 2 Table). Cell-to-cell variability was denoted on the phase diagrams by representing initial protein concentrations in each cell line as an ellipsoid (Figure 1B). Proteins other than those analyzed here also vary from one cell line to the next but the apparent success of DLE analysis suggests that these differences are not major determinants of extrinsic apoptosis in the cell lines we have studied, under our experimental conditions (alternatively, co-variation in other determinants may effectively cancel out their effects). Nonetheless, we expect that the levels of many additional proteins will contribute to TRAIL sensitivity; it will be possible to analyze these proteins using updated models and DLE phase diagrams with additional dimensions. The idea of analyzing dynamical systems with respect to their states at a future point in time has recently re-emerged as an important technique in diverse areas of natural science. For example, methods fundamentally similar to Lyapunov exponent analysis have been used to study the structure of fluid flow in the waters of Monterey Bay and other complex habitats. The ‘Lagrangrian coherent structures’ calculated using Lyapunov exponents define boundaries between currents that specify where pollutants will collect, exemplifying the wide applicability of DLE-style analysis (Coulliette et al, 2007).
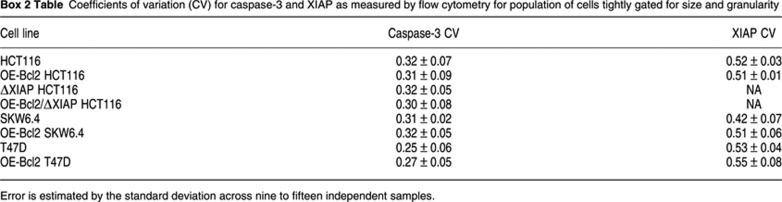 |
Results
A DLE-based phase map predicts the [XIAP]:[caspase-3] ratio to control type I versus II phenotypes
To determine which proteins control the choice between type I and type II phenotypes during apoptosis, we computed DLEs for a previously published ordinary differential equation model describing key reactions in TRAIL-mediated extrinsic apoptosis (EARM1.4; see Box 1). Bcl2 over-expression is the standard method for distinguishing type I and type II cells experimentally (Scaffidi et al, 1998): type I cells over-expressing Bcl2 die when exposed to death ligand but type II cells do not. Simulations were therefore performed in the presence of sufficient Bcl2 to block MOMP (this level was conservatively set to a value lower than the level of Bcl2 present in cell lines stably transfected with Bcl2 over-expression constructs; Supplementary Figure S1A). DLE analysis was performed by allowing the initial concentrations (i.e. concentrations prevailing prior to TRAIL exposure) of six proteins (receptor, ligand, caspase-3, caspase-6, caspase-8, and XIAP) to vary in discrete steps above and below nominal values (see Box 1 and Supplementary Table SII). The magnitude of the variation and the step size were determined by the sensitivity of cleaved effector caspase substrate (cEC-S) levels to changes in protein concentrations: greater sensitivity mandated smaller steps and biochemical plausibility set the upper limit at 106 molecules/cell (higher than the average concentration of any apoptotic regulator we have measured in any cell line). DLE analysis generated a multi-dimensional phase diagram relating initial protein concentrations to cell fate 8 h after TRAIL addition. Quantitative immunoblotting of cell extracts relative to recombinant protein standards was used to estimate the absolute concentrations of apoptotic regulators in tumor cell lines and flow cytometry was used to estimate cell-to-cell variability within a single line (Box 1 Table). Specific cell lines were then placed on the landscape of initial conditions (Figure 1A and B; Supplementary Figure S1). No model adjustment or refitting was performed when cell lines were added to the landscape; only the concentrations of proteins were changed to match measured values.
Figure 1.
A DLE phase diagram predicts that XIAP deletion in HCT116 cells switches them from type II to type I. (A) Dynamics. Simulation of EC substrate cleavage in TRAIL-treated OE-Bcl2 HCT116 (green) and OE-Bcl2/ΔXIAP HCT116 (red) cells using EARM1.4. (B) Phase diagram. DLE values for OE-Bcl2 conditions and 50 ng/ml TRAIL at 8 h. The DLE is visualized in a slice of phase space corresponding to caspase-3 levels of 1 × 103–106 molecules/cell and XIAP levels of 0–6 × 104 molecules/cell in linear scale. High DLEs demarcate a separatrix (magenta) that separates cells exhibiting type I behavior (that die despite Bcl2 over-expression) or type II behavior (that survive). Ovals denote the positions in phase space of HCT116 parental (green), ΔXIAP HCT116 (red), and SKW6.4 (cyan) cells, based on measurements of protein levels (Box 1 Table, Box 2 Table, and Supplementary Figure S1). (C) Protein levels. XIAP, caspase-3, and PARP concentrations in two cell lines as measured by quantitative immunoblots, relative to levels found in HCT116 cells; HCT116 (green, set to 1) and SKW6.4 (cyan) cells; immunoblot images are shown in Supplementary Figure S1A. Source data is available for this figure in the Supplementary Information.

As in any phase diagram, the most interesting regions of phase space are those in which small changes in parameter values cause large changes in outcome; in Lyapunov exponent analysis, these give rise to separatrices having high DLE values (Box 2). A separatrix was clearly apparent in two-dimensional slices of phase space representing [XIAP] versus [caspase-3] (in the following discussion ‘caspase-8’ or ‘caspase-3’ refers to the inactive pro-forms of the enzymes; in the few cases where we mean the active form, we specifically refer to active or cleaved caspase). Simulation predicted that cells having a high ratio of [XIAP]:[caspase-3] would survive TRAIL exposure in the presence of over-expressed Bcl2, thereby exhibiting type II behavior. However, cells with a low [XIAP]:[caspase-3] ratio should die, thereby exhibiting type I behavior (Figure 1A and B). We observed that SKW6.4 B-cell lymphoma and HCT116 colon carcinoma lines, which show prototypical type I and type II behaviors, respectively (Scaffidi et al, 1998; Ozoren and El-Deiry, 2002; Algeciras-Schimnich et al, 2003; Siegel et al, 2004), lay on opposite sides of the computed separatrix (Figure 1B and C; Supplementary Figure S1), consistent with the known behavior of these cells. SKW6.4 cells occupied a position in phase space at which even very high XIAP concentrations would not be expected to yield type II death (Figure 1B and C). We tested this prediction experimentally by showing that even 20-fold XIAP over-expression did not alter the fraction of cells that died in response to TRAIL exposure (Supplementary Figure S1D). In contrast, HCT116 cells occupied a position in phase space at which deletion of XIAP should switch them from type II to type I.
Loss of XIAP in type II HCT116 cells converts them to a type I phenotype
To explore the function of XIAP in HCT116 cells, we used retroviral infection to stably over-express Bcl2 in parental HCT116 cells and in a congenic line in which XIAP had been deleted by gene targeting (hereafter OE-Bcl2/ΔXIAP HCT116 cells; see Table I for a glossary of cellular and molecular reagents; Cummins et al, 2004). Quantitative immunoblotting confirmed that Bcl2 concentrations were similar in OE-Bcl2 and OE-Bcl2/ΔXIAP HCT116 lines (Figure 2A; Supplementary Figure S1A). Cells were then treated with TRAIL, FasL, or TNF and the extent of apoptosis monitored using three complementary methods: flow cytometry with antibodies specific for the cleaved form of the caspase-3/7 substrate PARP (cPARP; hereafter referred to as cEC-S), live-cell microscopy of cells expressing fluorescent reporters of initiator or effector caspases (IC-RP or EC-RP; Albeck et al, 2008a), and clonogenic survival assays. Although each assay has distinct strengths and weaknesses (Galluzzi et al, 2009), in our hands all three assays yielded quantitatively similar estimates of the probability and timing of extrinsic cell death (Albeck et al, 2008a) and corresponded well with other measures of apoptosis such as annexin V staining (Gaudet et al, 2005). Experiments were initially performed in the presence of the protein synthesis inhibitor cycloheximide (CHX), which simplified modeling because TRAIL-induced protein synthesis could be largely ignored (see Albeck et al, 2008b), but experiments were repeated in the absence of CHX. The results were similar (see below), and we conclude that our findings are not unduly influenced by low-dose CHX treatment.
Table 1. Cellular and molecular reagents.
| Name | Description | Reference |
|---|---|---|
| HCT116 cells | HCT 116 human colon carcinoma cells, parental | |
| ΔXIAP HCT116 cells | HCT 116 human colon carcinoma cells, with XIAP gene knockout | Cummins et al (2004) |
| OE-Bcl2 HCT116 cells | HCT 116 human colon carcinoma cells, stably over-expressing Bcl2 | This study |
| OE-Bcl2/ΔXIAP HCT116 cells | HCT 116 human colon carcinoma cells, with XIAP gene knockout and stably over-expressing Bcl2 | This study |
| SKW6.4 cells | SKW6.4 human B lymphoma cells | |
| OE-Bcl2 SKW6.4 cells | SKW6.4 human B lymphoma cells, stably over-expressing Bcl2 | This study |
| T47D cells | T47D human breast carcinoma cells | |
| OE-Bcl2 T47D cells | T47D human breast carcinoma cells, stably over-expressing Bcl2 | This study |
| CHY | mCherry | |
| CHY-XIAP | Fusion of mCherry at the N-terminus of full-length XIAP | This study |
| CHY-XIAPΔRING | Fusion of mCherry at the N-terminus of XIAP with a deletion of the RING domain (a.a. 437–497) | This study |
| IC-RP | FRET-based initiator caspase reporter | Albeck et al (2008a) |
| EC-RP | FRET-based effector caspase reporter | Albeck et al (2008a) |
| cEC-S | Cleaved effector caspase substrates | This study |
| cIC-S | Cleaved initiator caspase substrates | This study |
Figure 2.
XIAP deletion in HCT116 cells switches them from type II to type I. (A) Time courses of cell death in cell lines having Bcl2 at normal (WT, blue lines) or over-expressed levels (OE-Bcl2, orange lines) following exposure to TRAIL (50 ng/ml), FasL (100 ng/ml), or TNF (100 ng/ml). The percentages of cells positive for cleaved EC substrate (cEC-S) was determined by flow cytometry with antibodies specific for cPARP (Supplementary Figure S2). Because of the nature of the targeted epitope, these antibodies can recognize other cleaved EC substrates in addition to cPARP, and we therefore refer to these as measures of ‘cleaved EC substrate.’ Bar plots to the right show Bcl2 expression in each cell line, relative to SKW6.4 (set to 1; Supplementary Figure S1). We assayed parental HCT116 colorectal carcinoma cells (top), HCT116 cells carrying a homozygous deletion of the XIAP gene (Cummins et al, 2004) (middle), and the SKW6.4 EBV-transformed B lymphocyte line (bottom). (B) Clonogenic survival. Bar plots showing the relative number of clones obtained in HCT116-derived cells (left) and SKW6.4 and OE-Bcl2 SKW6.4 cells (right) following TRAIL treatment, normalized to a CHX alone control. Average numbers of clones obtained are indicated (TRAIL-treated/control). (C) EC substrate cleavage. Fraction of OE-Bcl2 or OE-Bcl2/ΔXIAP HCT116 cells positive for EC substrate cleavage 6 h after TRAIL addition following transfection with plasmids expressing mCherry alone (CHY) or mCherry fused to XIAP (CHY-XIAP). TRAIL was added 18 h following transfection and EC substrate cleavage assayed by flow cytometry. Data are shown for cells expressing CHY-XIAP at endogenous (blue) and 20-fold over-expression levels (yellow; see Supplementary Figure S5A for quantification). All experiments were performed in the presence of 2.5 μg/ml of CHX (responses to CHX or TRAIL alone are shown in Supplementary Figures S3 and S4). Error bars report the standard deviation of biological triplicates from one representative experiment. Source data is available for this figure in the Supplementary Information.

As predicted by simulation, TRAIL, FasL, or TNF exposure killed 80–95% of WT, ΔXIAP, and OE-BCL2/ΔXIAP HCT116 cells within 6 h but killed only ∼5% of OE-Bcl2 HCT116 cells (as assayed in the presence of CHX; Figure 2A and B). Bcl2 over-expression in ΔXIAP HCT116 cells phenocopied Bcl2 over-expression in OE-Bcl2 SKW6.4 cells (which are naturally type I): nearly 100% of OE-Bcl2 SKW6.4 cells died in response to TRAIL or FasL within 6 h of ligand exposure (SKW6.4 cells do not respond by apoptosis to TNF, although they do express TNFR1 and TNFR2 (Grell et al, 1994) (Figure 2A and B). When we repeated experiments in the absence of CHX, we obtained similar results except in the case of clonogenic survival assays for TRAIL-treated SKW6.4 cells. While a similar fraction of TRAIL-treated SKW6.4 and OE-Bcl2 SKW6.4 cells were positive for cIC-S and cEC-S, over-expression of Bcl2 increased the fraction of SKW6.4 cells that survived TRAIL exposure as assessed by clonogenic assays, seemingly contrary to their designation as type I cells (Supplementary Figure S4). Maas et al (2010) have also observed that blocking MOMP in SKW6.4 cells makes them less sensitive to TRAIL in clonogenic assays. We conclude that while EARM1.4 correctly predicts whether effector caspases are activated in TRAIL-treated SKW6.4 and HCT116 in the presence and absence of Bcl2 over-expression, the model is unable to predict the effect of Bcl2 on SKW6.4 survival. Factors unaccounted for in EARM1.4 must control whether SKW6.4 cells actually live or die in response to TRAIL. However, with respect to comparing models and experiments, effector caspase activation is the key variable in our analysis.
The analysis of HCT116 cells described above makes three assumptions: (i) that comparison of parental and ΔXIAP HCT116 cells is not confounded by secondary mutations; (ii) that Bcl2 levels in ΔXIAP HCT116 cells are as high in ΔXIAP as in parental HCT116 cells and therefore sufficient to block MOMP in both cell lines; and (iii) that OE-Bcl2/ΔXIAP HCT116 cells, like the type II parental cells, undergo rapid and complete effector caspase activation. The first concern arises because, like most cancer cell lines, HCT116 cells are genetically unstable (LeBlanc et al, 2002). We therefore asked whether ectopic XIAP expression would rescue the XIAP deletion phenotypically. Conducting this complementation experiment was complicated by the well known difficulty of stably over-expressing full-length XIAP in cells (Silke et al, 2005). Transient transfection with XIAP-expressing plasmids is effective but has the secondary effect of sensitizing cells to extrinsic cell death. If this sensitization is taken into account, the complementation results were as expected: 95% of OE-Bcl2/ΔXIAP HCT116 cells transfected with a plasmid expressing mCherry alone (CHY) were dead 6 h after TRAIL exposure, whereas expression of mCherry-XIAP (CHY-XIAP) at levels that approximated those in the parental lines (as measured by flow cytometry; Supplementary Figure S5A), reduced the level of apoptosis to that of control CHY-expressing OE-Bcl2 HCT116 cells (∼30%; Figure 2C); moreover, at higher CHY-XIAP levels, even fewer cells died (Figure 2C, yellow bars). We conclude that ΔXIAP HCT116 cells exhibit a type I apoptosis phenotype because of XIAP deletion, not secondary mutations.
To determine whether Bcl2 was expressed in ΔXIAP HCT116 cells at levels sufficient to block MOMP, we assayed for mitochondrial permeabilization in TRAIL-treated cells. It is important to note that the distinguishing feature of type I cells is not that mitochondria remain intact following apoptosis, but rather that MOMP happens after effector caspases are activated making MOMP a consequence rather than a cause of caspase activation (Scaffidi et al, 1998; Barnhart et al, 2003; Maas et al, 2010). HCT116 cells were exposed to 50 ng/ml TRAIL and then fixed and stained with anti-cytochrome c antibodies to measure MOMP and anti-cEC-S antibodies to monitor effector caspase activity. After a 1 h TRAIL treatment, when the fraction of cEC-S-positive cells was ∼50%, 97% of these cells had cytoplasmic cytochrome c, as expected for type II cells (Figure 3). In contrast, 80% of cEC-S-positive OE-Bcl2/ΔXIAP HCT116 cells exhibited mitochondrial cytochrome c, as expected for type I cells. Some OE-Bcl2/ΔXIAP HCT116 cells were positive for both cytochrome c translocation and cEC-S but this does not necessarily indicate type II behavior because caspase-3/7 can trigger MOMP late in the process of killing type I cells (Ricci et al, 2004). We interpret these data as showing that Bcl2 is over-expressed at a sufficiently high level to block MOMP prior to caspase-3/7 activation in most if not all OE-Bcl2/ΔXIAP HCT116 cells (Figure 3B). However, in the ΔXIAP HCT116 cell line we observe that only 20% of cells with high cEC-S levels also exhibited a staining pattern in which cytochrome c is mitochondrial (Figure 3A). Thus, when Bcl2 is present at normal levels in ΔXIAP HCT116 cells, they preferentially die using an MOMP-dependent pathway whereas when Bcl2 is over-expressed they can still die, but use a MOMP-independent pathway.
Figure 3.
ΔXIAP HCT116 cells undergo MOMP prior to caspase-3/7 activation unless Bcl2 is over-expressed. (A) Fixed-cell imaging of WT (top), OE-Bcl2 (middle), and OE-Bcl2/ΔXIAP (bottom) HCT116 cells stained for cleaved EC substrates and cytochrome c 1 h after addition of 50 ng/ml TRAIL+2.5 μg/ml of CHX (scale bar=20 μm). Examples of cells positive for EC substrate cleavage in which cytochrome c has been translocated to the cytoplasm (an indicator of MOMP) are denoted by cyan arrowheads; cells positive for EC substrate cleavage in which cytochrome c is still restricted to mitochondria are denoted by yellow asterisks. Bar plots report the percentages of cEC-S-positive cells where cytochrome c is cytoplasmic (blue) or mitochondrial (yellow). Error bars report the standard deviation of biological triplicates from three wells counting ∼200–600 cells/well from one representative experiment (see Materials and methods). (B) Pathway usage. Schematic interpretations of the results in Figure 2 and (A) above. Degraded caspase-3 is indicated with a dotted outline. Bcl2 over-expression inhibited mitochondrial permeabilization in XIAP-deficient HCT116 cells (OE-Bcl2/ΔXIAP HCT116 cells; right). Source data is available for this figure in the Supplementary Information.

Apoptosis in both type I ΔXIAP HCT116 and type II parental HCT116 cells involves rapid and complete cleavage of effector caspase substrates
Under normal conditions, apoptosis in type II cells is an all-or-none process in which cleavage of effector caspase substrates, once initiated, proceeds rapidly to completion (Tyas et al, 2000; Vaughan et al, 2002). Is this also true in SKW6.4 and OE-Bcl2/ΔXIAP HCT116 cells? Live-cell imaging of TRAIL-treated OE-Bcl2 SKW6.4 cells transfected with a construct that measures effector caspase activity (EC-RP; Albeck et al, 2008a) revealed a preponderance of the switch-like, all-or-none behavior previously described for type II apoptosis in HeLa cells (Figure 4A). Live-cell imaging confirmed similarly rapid and complete EC-RP cleavage in nearly all TRAIL-treated WT, ΔXIAP and OE-Bcl2/ΔXIAP HCT116 cells (Figure 4B; Supplementary Figure S2) and in all cases complete EC-RP cleavage was concomitant with morphological manifestations of cell death (not shown). Thus, TRAIL-mediated apoptosis in OE-Bcl2/ΔXIAP HCT116 cells is distinct from the half-dead state created when XIAP levels are reduced by RNAi in Bcl2 over-expressing HeLa cells (Albeck et al, 2008a); in that case fractional activation of effector caspases is observed and only a subset of cells die.
Figure 4.

EC substrate cleavage dynamics are similar in SKW6.4 and HCT116 cells with and without XIAP. Traces of FRET signal (EC-RP signal, as measured by background-corrected CFP/YFP ratio; Albeck et al, 2008a) for an effector caspase reporter based on live-cell imaging of (A) OE-Bcl2/SKW6.4 and (B) WT, ΔXIAP, and OE-Bcl2/ΔXIAP HCT116 cells expressing EC-RP following exposure to 50 ng/ml TRAIL + 2.5 μg/ml of CHX.
We have previously shown that apoptosis in type II HeLa cells involves an extended pre-MOMP delay in which initiator but not effector caspases are active (Albeck et al, 2008a). The delay is most easily visualized in two-dimensional flow cytometry plots in which the horizontal axis corresponds to caspase-3 cleavage (cIC-S; a measure of initiator caspase activity), and the vertical axis corresponds to PARP cleavage (cEC-S; a measure of effector caspase activity (see Albeck et al, 2008a for a detailed comparison of flow cytometry and live-cell imaging assays)). One hour after TRAIL addition, HCT116 cells had shifted from the lower left to the lower right quadrant of the plot, representing accumulation of cells in a pre-MOMP delay state with initiator caspases active and effector caspases inactive. By 6 h, cells shifted to the upper right quadrant, representing a state in which both initiator and effector caspases were active, at which point cells died (Figure 5A, blue and red arrows). As expected, 90–95% of OE-Bcl2 HCT116 cells accumulated in the lower right quadrant without evidence of effector caspase activity or death (Figure 5A). In contrast, TRAIL-treated ΔXIAP and OE-Bcl2/ΔXIAP HCT116 cells jumped directly from the lower left to the upper right quadrants (Figure 5A, orange arrow), confirming model-based predictions that they would die without delay in a pre-MOMP state. For SKW6.4 cells (which do express some XIAP), we observe some delay in progression to death—little caspase activity is observed 1 h after treatment—but flow cytometry data at subsequent times suggest that initiator and effector caspases are activated simultaneously, as they are in ΔXIAP and OE-Bcl2/ΔXIAP HCT116 cells (Figure 5B, 6 h). In addition, some SKW6.4 and OE-Bcl2 SKW6.4 cells were found in the upper left quadrant, suggesting that even relatively low activation of caspase-3 can trigger significant cleavage of effector caspase substrate in some cells (Figure 5B, orange bars).
Figure 5.
XIAP deletion in HCT116 cells leads to loss of the pre-MOMP delay. Two-dimensional flow cytometry of HCT116 (A) and SKW6.4 (B) cell lines with different genotypes following exposure to 50 ng/ml TRAIL + 2.5 μg/ml of CHX for 0–6 h, as indicated. The x axis represents levels of initiator caspase substrate cleavage (cIC-S; as determined using an antibody specific for cleaved caspase-3) and the y axis represents effector caspase substrate cleavage (as determined using a cPARP antibody). Bar plots above show percentages of cells with inactive initiator and effector caspases (green), active initiator and inactive effector caspases (blue), and active initiator and effector caspases (red) based on the gating denoted with colored dashed lines in (A, upper left). SKW6.4 have an additional population with low initiator caspase activity and high effector caspase activity (orange bars) denoted by orange dashed lines in (B, left). For OE-Bcl2 HCT116 cells (A, upper right), light bars represent data from a 24-h time point. Error bars report the standard deviation of biological triplicates from one representative experiment. Source data is available for this figure in the Supplementary Information.

Increasing XIAP levels results in graded transitions from type I to type II apoptosis and ultimately to TRAIL resistance
The DLE analysis in Figure 1 was performed over the range of XIAP concentrations that naturally occur in tumor cell lines we have studied. However, it is possible to over-express XIAP to significantly higher levels and EARM1.4 predicts that this should block apoptosis in cells such as HCT116 that have intermediate levels of caspase-3 (a prediction previously made by Rehm et al (2006) in the context of HeLa cells). Thus, as levels of ectopically expressed XIAP in ΔXIAP HCT116 cells increase from zero through the concentration found in parental cells (∼3 × 104 molecules/cell) to a very high level (∼6 × 105 molecules/cell), we should first observe a type I phenotype, then a type II phenotype, and then TRAIL resistance (cell survival in the absence of Bcl2 over-expression). To test this prediction, variable levels of CHY-XIAP or CHY alone were expressed in ΔXIAP and OE-Bcl2/ΔXIAP HCT116 cells and responses to TRAIL were monitored by flow cytometry using an antibody against cPARP (cEC-S; Figure 6A; Supplementary Figure S5). Accounting for the sensitizing effects of transfection itself (Figures 2C and 6A), we observed ΔXIAP and OE-Bcl2/ΔXIAP HCT116 cells to be equally sensitive to TRAIL at very low CHY-XIAP levels, as predicted for a type I phenotype (Figure 6A and B, green region) but as CHY-XIAP approached wild-type levels (Figure 6A and B, black arrows), a type II phenotype came to predominate (Figure 6B, blue region); at yet higher CHY-XIAP concentrations, both OE-Bcl2/ΔXIAP and ΔXIAP HCT116 cells were protected from TRAIL (Figure 6A and B; gray region). Notably, the transition from type I to II phenotypes was sufficiently gradual that the two phenotypes co-existed over a range of XIAP expression levels, as predicted from DLE analysis.
Figure 6.
XIAP levels control the fraction of HCT116 cells that exhibit type I versus type II phenotype. (A) EC substrate cleavage. Cell death (percentage positive) in TRAIL-treated ΔXIAP (blue) and OE-Bcl2/ΔXIAP (red) HCT116 cells expressing mCherry alone (CHY; left) or mCherry fused to full-length XIAP (CHY-XIAP; right), as measured by cEC-S by flow cytometry. Untreated transfected cells are shown in light blue and red (NT). Percentages of cells positive for cEC-S (measured with anti-cPARP) and CHY protein intensity (594 nm fluorescence) were measured by flow cytometry (representative raw data are shown in Supplementary Figure S5). The average endogenous XIAP level is denoted with an arrow (see Supplementary Figure S5 for quantification) and asterisks indicate the responses of untransfected cells. Error bars report the standard deviation of biological triplicates from one representative experiment. (B) Percentages of cells that were protected from apoptosis specifically by Bcl2 over-expression, as a function of CHY-XIAP levels. Percentages of protected cells were calculated by subtracting the percentage of cEC-S-positive cells in OE-Bcl2/ΔXIAP HCT116 cells from those of ΔXIAP cells and delineate type I, type II, and apoptosis-resistant regions. Source data is available for this figure in the Supplementary Information.

T47D, a cell line lying near the separatrix, exhibits a mix of type I and type II behaviors
To determine if type I and type II phenotypes might also co-exist in cell populations not subjected to genetic manipulation, we used immunoblotting to identify cell lines that straddled the separatrix in the [XIAP] versus [caspase-3] phase space (Figure 7A; Supplementary Figure S1; Box 1 Table). T47D human breast carcinoma cells have this property and have previously been reported to be type I based on ligand sensitivity but to cluster with type II cell lines by transcriptional profiling (Algeciras-Schimnich et al, 2003). We observed that over-expression of Bcl2 in T47D cells protected only ∼50% from either TRAIL- or FasL-mediated apoptosis (Figure 7B and C). One explanation for this result is that survivors have higher levels of Bcl2 than dying cells (hypothesis H1 in Figure 7D). Alternatively, the difference between dying cells and survivors could reflect another difference, such as the [XIAP]:[caspase-3] ratio (hypothesis H2). Because Bcl2 levels cannot be reliably measured in dying cells, we used flow cytometry to compare the concentration of Bcl2 in surviving cells with that of the starting cell population. No significant difference was observed over the 6h period in which the fraction of dying cells increase from 0 to ∼75% (Figure 7D). These data argue that Bcl2 levels were similar in dying and surviving cells and thus that some cells required MOMP for death and others did not (i.e. H1 is correct and H2 is wrong). In support of this idea, immunofluorescence of TRAIL-treated OE-Bcl2 T47D cells provided clear evidence of EC substrate cleavage in the absence of MOMP: at least 20% of cEC-S-positive cells were MOMP negative (Figure 7E). From these data we conclude that a fraction of T47D cells indeed exhibited a type I phenotype whereas others exhibited a type II phenotype and required MOMP for death. In preliminary experiments, we have observed similar behavior in MCF10A cells, an immortalized line of human breast epithelial cells that also straddles the separatrix (D Flusberg, SG, and PKS, unpublished data).
Figure 7.
Individual T47D cells can exhibit either a type I or type II phenotype. (A) Phase-space diagram. Ovals representing variation in [XIAP] and [caspase-3] in HCT116, T47D, and SKW6.4 cells (as measured by flow cytometry) are mapped onto the phase diagram. (B) Time courses of EC substrate cleavage in T47D cells having Bcl2 at normal (WT, blue lines) or over-expressed levels (OE-Bcl2, orange lines) following exposure to TRAIL (50 ng/ml), FasL (100 ng/ml), or TNF (100 ng/ml) in the presence of 2.5 μg/ml of CHX (responses to CHX or TRAIL alone are shown in Supplementary Figures S3 and S4). Percentages of cells positive for EC substrate cleavage were determined by flow cytometry. Bar plots to the right show Bcl2 expression in each cell line, relative to WT SKW6.4 (set to 1, see Figure 2A and Supplementary Figure S1). Error bars report the standard deviation of biological triplicates from one representative experiment. (C) Survival. Clonogenic assay of WT and OE-Bcl2 T47D cells following 6 h of TRAIL treatment, percentages were obtained by normalizing to CHX alone control. Average numbers of colonies are indicated above the graph (TRAIL-treated/control). Error bars report the standard deviation of biological triplicates from one representative experiment. (D) Bcl-2 levels in surviving cells. Schematic showing two possible hypotheses (H1 and H2) to explain why only a subset of T47D cells are protected from death ligand by Bcl2 over-expression. Assuming an untreated population in which Bcl2 levels vary (indicated with different shades of orange) it is possible that only cells with high Bcl2 levels survive TRAIL treatment (upper model, H1); alternatively, cell survival might be determined by other, unknown, factors such that Bcl2 levels in survivors end up similar to those in the untreated cell population (lower model, H2). Bar plots show the percentages of cells that are negative for EC substrate cleavage (lower panel, left) and mean Bcl2 expression levels in these cells (lower panel, right), as measured by flow cytometry in parental T47D cells (blue) and in OE-Bcl2 T47D cells prior to TRAIL treatment (red) and following 50 ng/ml TRAIL + 2.5 μg/ml of CHX exposure for 1–6 h (orange; right). Error bars report the standard deviation of biological triplicates from one representative experiment, the same trends were observed in multiple experiments. (E) Fixed-cell images of WT (top) and OE-Bcl2 (bottom) T47D cells stained for EC substrate cleavage and cytochrome c 3.5 h after 50 ng/ml TRAIL + 2.5 μg/ml of CHX addition (scale bar=20 μm). Arrowheads (cyan) denote cells positive for both EC substrate cleavage and cytoplasmic translocation of cytochrome c and asterisks (yellow) denote cells positive for EC substrate cleavage but having cytochrome c restricted to mitochondria. Bar plots report the percentages of EC substrate cleavage positive cells where cytochrome c is cytoplasmic (blue) or mitochondrial (yellow). Error bars report the standard deviation of biological triplicates from three to five wells, counting ∼150–700 cells/well (see Materials and methods). Source data is available for this figure in the Supplementary Information.

Multi-factorial control over type I versus II behavior
The experiments described above focus on the [XIAP]:[caspase-3] plane in phase space but our DLE analysis covers a six-dimensional space. When we examined successive slices of the [XIAP]:[caspase-3] plane, varying ligand, receptor, or caspase-8 concentrations, we observed that as these increased the position of the separatrix swung downward (toward the [XIAP] axis) effectively reducing the size of the type II region (Figure 8A). When we exposed T47D and OE-Bcl2 T47D cells to TRAIL over a 12-fold range of concentrations, we observed a fivefold increase in the fraction of cells exhibiting a type I phenotype between low and high doses of TRAIL (Figure 8B). Thus, the DLE-based phase space appears to correctly predict at least one other variable regulating type I versus II death. Moreover, the observation that type I death is favored at higher ligand doses is consistent with previous data suggesting the importance of FasL dose and oligomeric state in controlling type I fate (Scaffidi et al, 1998, 1999b; Schmitz et al, 1999; Algeciras-Schimnich et al, 2003; Barnhart et al, 2003; Rudner et al, 2005), although EARM1.4 is not yet sufficiently detailed to compute directly the impact of ligand oligomerization.
Figure 8.
The proportion of type I and type II T47D cells is dependent on TRAIL concentration. (A) Positions of the separatrix in the [XIAP] versus [caspase-3] dimensions under conditions in which ligand, caspase-8, or receptor levels were changed as indicated (fold decrease or increase from the baseline; see Materials and methods). The type I region widens as the separatrix shifts toward the [XIAP] axis. (B) TRAIL dose response. Graphs show the fraction of dying T47D cells that are WT or OE-Bcl2 (lower panel) as calculated from a dose-response curve to TRAIL concentration + 2.5 μg/ml CHX (upper panel). Error bars report the standard deviation of biological triplicates from one representative experiment. Source data is available for this figure in the Supplementary Information.

Extending phase space analysis to rate constants
We sought to extend our analysis of changes in protein concentrations to changes in activity that arise from mutation. XIAP inhibits caspase-3/7 through two distinct mechanisms: one involving competitive inhibition of caspase-3/7 via binding of XIAP to the caspase catalytic cleft with an affinity of ∼1 nM (Eckelman et al, 2006) and the second in which XIAP promotes active caspase-3/7 ubiquitylation and proteosome-mediated protein degradation (Yang et al, 2000; Suzuki et al, 2001). This second mechanism is regulated by the E3 ubiquitin ligase activity of XIAP, and it can be blocked by deleting or mutating the XIAP RING domain without affecting binding of XIAP to the caspase-3/7 catalytic site (Yang et al, 2000). Modeling suggests that XIAP-mediated caspase-3 degradation is important for maintaining a pre-MOMP delay in type II cells (Albeck et al, 2008a). This prediction is consistent with data showing that, in the absence of XIAP E3 ligase activity, murine cells are more sensitive to FasL-mediated killing, even though XIAP protein levels increase (Schile et al, 2008).
To explore the role of XIAP E3 activity in type II versus type I behavior, we transfected OE-Bcl2/ΔXIAP HCT116 cells with mCherry-tagged XIAP mutants deleted for the RING domain (residues 438–497, XIAPΔRING) or carrying a mutation in a highly conserved RING-domain residue (XIAP-H467A; Yang et al, 2000). We observed that acquisition of a type II phenotype, in which effector substrate cleavage was blocked by Bcl2 over-expression, required significantly higher concentrations of XIAPΔRING than full-length XIAP (the effect is particularly evident at the 24-h time point; Figure 9A; Supplementary Figure S5B). Because XIAP and XIAPΔRING were tagged with mCherry, it was possible to measure the amount of recombinant protein at a single-cell level and account for any differences in protein levels; this is important because RING-domain deletion significantly alters XIAP half-life and expression levels (Schile et al, 2008). Strikingly, the rapid cleavage of effector caspase substrates observed in OE-Bcl2/ΔXIAP HCT116 cells (by live-cell imaging of cells co-transfected with EC-RP) was disrupted in cells expressing intermediate amounts of XIAPΔRING. In these cells, cleavage of an EC-RP reporter occurred gradually over a period of hours (Figure 9B). This phenotype corresponds to neither type I nor type II behavior but instead to a loss of ‘snap-action’ effector caspase activation. The loss of this ‘all-or-none’ control was also evident in flow cytometry data: many cells expressing intermediate level of mCherry-XIAPΔRING exhibited partial EC substrate cleavage (Supplementary Figure S5C, bottom row).
Figure 9.
Deletion of the XIAP RING domain disrupts the dynamics of effector caspase activation. (A) EC substrate cleavage in OE-Bcl2/XIAP HCT116. Cell death (percentage positive) in TRAIL-treated OE-Bcl2/ΔXIAP HCT116 cells expressing mCherry alone (CHY; dark gray), mCherry fused to full-length XIAP (CHY-XIAP; blue), or mCherry fused to an XIAP truncation lacking the RING domain (excluding residues 437–497, CHY-XIAPΔRING; red). Untreated mCherry-transfected cells are shown in light gray (CHY (NT)). EC substrate cleavage and CHY protein intensity (594 nm fluorescence) were measured by flow cytometry (representative raw data are shown in Supplementary Figure S6C). Average endogenous XIAP level is denoted with an arrow and the responses of untransfected cells indicated with asterisks. As discussed in the text, transfection itself sensitizes cells HCT116 cells to TRAIL, and it is therefore necessary to compare the red and blue data to the control data in light gray. Error bars report the standard deviation of biological triplicates from one representative experiment. (B, C) Dynamics of EC-RP cleavage as determined by live-cell microscopy (B) and predicted by simulation (C) for TRAIL-treated OE-Bcl2/ΔXIAP HCT116 cells transfected with mCherry alone (CHY; left), mCherry fused to full-length XIAP (CHY-XIAP; center), or mCherry fused to a RING-domain deletion mutant of XIAP (CHY-XIAPΔRING; right). The level of mCherry fluorescence is reported using a scale from yellow to green, as indicated. (D) Simulated duration of cleaved EC substrate rise in OE-Bcl2/XIAP HCT116. 2D phase space maps showing how the duration of the rise in cleaved EC substrate for wild-type XIAP (left) and XIAPΔRING (right) is dependent on the concentration of the XIAP and caspase-3 species in model simulations. For XIAPΔRING, the rate parameter for XIAP E3 ligase activity toward its caspase-3 substrate is set to zero. Black lines represent the demarcation between type I versus II phenotypes as determined by fraction of cleaved EC substrate in these simulations, with type I cells mapping above and type II cells below (Supplementary Figure S8). Source data is available for this figure in the Supplementary Information.

To extend the mapping of phase space to proteins with mutations that affect reaction rates, we generated a reduced version of EARM1.4 that focuses on reactions involving XIAP and caspase-3 (Supplementary Figure S7; Supplementary Tables SIV–SVI). When the dynamics of effector caspase substrate cleavage were simulated using the reduced model assuming either (i) no XIAP, (ii) wild-type XIAP at normal levels, or (iii) E3 ligase-deficient XIAP (present at wild-type levels), we observed a good match to data. The model correctly predicted that when MOMP was blocked (by Bcl2 over-expression) cleavage of effector caspase substrates should be rapid and complete in the absence of XIAP and fully blocked in the presence of wild-type XIAP. However, when the rate constant for XIAP E3 ligase activity was set to zero (kc8=0, the assumed consequence of RING-domain deletion) cleavage of caspase-3 substrates lost its normal ‘snap-action’ property at intermediate amounts of XIAP (Figure 9C). To map EC substrate cleavage dynamics in the two-dimensional phase space of [caspase-3] versus [XIAP], we computed the duration of the rise in cEC-S in the presence of full-length or truncated XIAP. When cells expressed XIAPΔRING, but not when they expressed the full-length protein, we observed a dramatic increase in the time required for completion of PARP cleavage near the type I versus type II separatrix (Figure 9D). We conclude that the RING domain of XIAP is indeed necessary for holding caspase-3/7 in an inactive state during the pre-MOMP delay in type II HCT116 cells. When XIAPΔRING is present at concentrations similar to normal levels of full-length XIAP, a breakdown in snap-action control of effector caspase activity is predicted and observed. Over-expression of XIAPΔRING can compensate for this defect at least partly, perhaps explaining the relative normalcy of mice expressing E3 ligase-deficient XIAP, which is substantially more abundant than wild-type XIAP (because of the increased stability of XIAP lacking an E3 ligase domain; Schile et al, 2008).
Discussion
In this paper, we study cells undergoing receptor-mediated apoptosis and ask how MOMP-independent type I and MOMP-dependent type II phenotypes are controlled. Using a previously developed ODE-based model of extrinsic apoptosis, we compute DLEs and generate a multi-dimensional landscape that relates the concentrations of six key regulatory proteins to the likelihood of type I versus type II phenotypes. We then measure protein concentrations in tumor cell lines and place them on the DLE landscape. Each cell line is represented as an ellipsoid whose midpoint corresponds to the population-average protein concentration (as determined from calibrated immunoblots) and whose axial dimensions are determined by cell-to-cell variation in protein concentrations (as determined by flow cytometry). We find that control over type I and type II phenotypes is multi-dimensional with a particularly strong dependence on the ratio of XIAP to pro-caspase-3 concentrations. A separatrix sharply demarcates the two phenotypes in the [XIAP]:[caspase-3] landscape meaning that a relatively small change in the ratio of the two proteins can cause a sudden switch from type I to type II behavior. We have validated these predictions in three ways. First, we demonstrate that HCT116 cells, which are exclusively type II but are predicted to be sensitive to XIAP levels, can be switched to type I if XIAP is deleted by homologous recombination. Second, we show that SKW6.4 cells, which are normally type I and are predicted to be insensitive to XIAP levels, remain type I when XIAP is over-expressed. Finally, we show that both T47D breast cancer cells and ΔXIAP HCT116 expressing intermediate levels of XIAP lie near the separatrix such that some cells in a population exhibit a type I behavior and others a type II behavior, a striking example of cell-to-cell variability in phenotype.
DLE-based phase diagrams predict that the [XIAP]:[caspase-3] ratio is not the only factor regulating type I versus type II behavior: the position of the separatrix in the [XIAP]:[caspase-3] plane shifts significantly with changes in the concentrations of ligand, receptor and pro-caspase-8. We test the dependence on ligand concentration experimentally and confirm that the type I region expands as ligand concentration increases. Thus, control over extrinsic cell death is multi-factorial and sensitivity to any single protein concentration (e.g. [XIAP]) is dependent on the concentrations of other proteins. Moreover, by analyzing cells in which the RING domain of XIAP has been deleted or mutated, we also show that phase space analysis can be extended to predicting the consequences of altering rate parameters. The E3 ligase activity of XIAP normally promotes caspase-3 ubiquitylation and consequent proteasome-dependent degradation. Modeling predicts that eliminating this reaction (by setting rate parameter kc8 to zero in EARM1.4) will prevent cells from sustaining the pre-MOMP delay characteristic of type II apoptosis. Experiments confirm this prediction and reveal that cells expressing XIAPΔRING lack normal ‘snap-action’ cleavage of effector caspase substrates. We propose that DLE-based phase diagrams represent a methodology for studying multi-factorial control over cell fate in a rational and systematic manner. In the current work, this approach allows us to reconcile a literature that finds XIAP and its E3 ligase activity to be either critical or irrelevant in the control of extrinsic apoptosis, depending on the experimental setting and cell type under study (Deveraux et al, 1997; Harlin et al, 2001; Bratton et al, 2002; MacFarlane et al, 2002; Sun et al, 2002; Cummins et al, 2004; Silke et al, 2005; Eckelman et al, 2006; Rehm et al, 2006; Schile et al, 2008). The strong context dependence of these XIAP phenotypes is explained (at least in principle) by the impact of other cellular factors known to vary significantly from one cell type to the next, such as caspase-3 levels, and by experimental variables, such as ligand dose and potency.
A key feature of DLE-based phase diagrams is that they reveal the dynamical consequences of variation in protein concentration that exists prior to treatment of cells with death ligand. In contrast to bifurcation analysis of steady states, computing DLEs maps phenotypes onto transient signals, which is important for a system in which steady states are not reached on any meaningful timescale (except when apoptosis pathways are inactive in cells not exposed to ligand). The ranges of protein concentrations over which DLEs are computed are derived from data from real cells in their natural states or following gene deletion and over-expression. Predictions then cover meaningful biological variation, and DLE phase diagrams allow us to determine when cell-to-cell variation in protein levels will be consequential. Because the process of gene expression and protein translation are inherently noisy (Lestas et al, 2010) even genomically identical cells exhibit variability in protein expression levels. For cells that map close to a separatrix, this inherent variability will lead to heterogeneous phenotypes. Cell lines that lie further from a separatrix are likely to behave more homogenously, as observed for SKW6.4 and parental HCT116 cells. In the future, it should be possible to extend the approach of mapping cell-to-cell variation onto phase diagrams to other forms of programmed cell death, such as autophagy and necroptosis.
Insight into the regulation of type I and type II cell death phenotypes in human cells
With regard to the regulation of type I and type II apoptosis phenotypes in human cells, our data support four conclusions. First, they demonstrate that deletion of XIAP in type II cells causes them to behave as type I cells while retaining the ability to rapidly and completely activate effector caspases when exposed to death ligands. Jost et al (2009) have observed a similar phenomenon in the livers of Bid−/− XIAP−/− mice by showing that hepatocytes switch from a type II to a type I phenotype. We predict that a key factor in the switch is the [XIAP]:[caspase-3] ratio: when it is low, type I behavior predominates and when it is high, type II behavior is prevalent. At very high [XIAP]:[caspase-3] ratios, apoptosis is blocked altogether.
We find no evidence that the levels of caspase-8/10 required to trigger apoptosis in parental and ΔXIAP HCT116 differ; both flow cytometry and live-cell imaging data suggest that all four HCT116-derived cell lines have similar initiator caspase activation levels and dynamics (Figure 4A; Supplementary Figure S2B). This leads us to speculate that, at least under our conditions, MOMP functions as a gatekeeper for activation of effector caspases rather than as an amplifier of initiator caspase; enhanced cleavage of pro-caspase-3 (e.g. by caspase-9) does not appear to be critical. Low [XIAP] and/or high [caspase-3] make MOMP unnecessary in type I cells, and cleavage of pro-caspase-3/7 by caspase-8/10 is sufficient to induce death. In type II HCT116 cells, by contrast, the key role of MOMP in extrinsic apoptosis is to cause Smac-dependent inhibition of XIAP. Consistent with this, Wilson et al (2009) recently showed that RNAi-mediated depletion of Smac but not caspase-9 (which is activated by cytochrome c released during MOMP) blocks apoptosis in HCT116 cells treated with anti-Fas antibody or TRAIL. Similarly, Shawgo et al (2009) showed that, following treatment with anti-Fas antibody, type II Jurkat cells do not require activation of caspase-9 by the apoptosome to die. We conclude that HCT116 cells can accumulate sufficient ‘latent’ caspase-3/7 an hour or so after TRAIL exposure to die via a direct, type I mechanism, but that XIAP imposes a block on this latent activity that must be relieved by MOMP, thereby enforcing type II behavior.
Second, we show that type I and type II phenotypes can co-exist within a cell population. The two phenotypes have hitherto been regarded as fundamentally distinct with type I behavior predominating in mesenchymal-like cells and type II behavior in epithelial-like cells (Algeciras-Schimnich et al, 2003). Indeed, all SKW6.4 cells are type I whereas all HCT116 cells are type II. However, in lines such as T47D or HCT116 expressing intermediate levels of XIAP, some cells are type I and others are type II. The relative frequency of type I and type II phenotypes is determined by the ligand dose, as predicted by simulation and consistent with previously reported but unexplained observations (Rudner et al, 2005). This phenotypic variability presumably arises from natural variation in the levels of proteins such as caspase-3 and XIAP. We and others have recently shown such variation to have a significant impact on how rapidly cells die (Rehm et al, 2009; Spencer et al, 2009) and modeling suggests that measured variation in protein levels is sufficient for cell lines to straddle the separatrix in the [XIAP] versus [caspase-3] phase space. Our findings may also explain why XIAP-null mice have few obvious defects (Harlin et al, 2001; Cummins et al, 2004): in the absence of XIAP, cells may simply alter the balance between type I and type II phenotypes.
Third, we predict and demonstrate experimentally that the E3 ubiquitin ligase domain of XIAP is necessary for effector caspase activity to be restrained prior to MOMP during extrinsic cell death. In the absence of this domain, effector caspase processing is gradual and normal snap-action cleavage of effector caspase substrates is not observed. We have previously observed fractional PARP cleavage and a ‘half-dead’ state in OE-Bcl2 HeLa cells in which XIAP levels were reduced with anti-XIAP siRNA or proteasome activity blocked with drugs (Albeck et al, 2008a). These data suggest that inhibition of MOMP in XIAPΔRING mice (Schile et al, 2008), by Bid deletion or Bcl2 over-expression for example, might generate a much stronger phenotype than either Bid deletion or Bcl2 over-expression, or XIAPΔRING mutation alone. XIAPΔRING cells require MOMP to promote snap-action effector caspase dynamics and therefore a state of partial activation could arise when MOMP is blocked in these cells. Partial effector caspase activation may be tumorigenic in vivo if caspase-activated DNases damage the genome (Vaughan et al, 2002, 2005; Lovric and Hawkins, 2010). More generally, these data highlight that the ability of XIAP to inhibit caspase-3 activity in two distinct ways—competitive inhibition and degradation—is important for its function. Protein degradation may play a key role in regulating the activation kinetics in other cell processes as competitive inhibition alone is likely to be insufficient to completely inactivate many enzymes (Albeck et al, 2008b).
Fourth, we demonstrate the utility of phase diagrams as a means to understand and predict how different cell types respond to death ligand. From these diagrams, it is clear that control over type I versus II behavior is multi-factorial with the ‘downstream’ proteins caspase-3 and XIAP playing the primary role in our studies. In contrast, elegant work by Peter and colleagues has shown ‘upstream’ events such as the efficiency of DISC assembly to be determinative of type I versus type II phenotypes in other contexts (Scaffidi et al, 1998; Barnhart et al, 2003). Such apparent discrepancies are common in the study of complex regulatory pathways but DLE-based phase diagrams promise to clarify the situation by providing predictive insight into multi-factorial control mechanisms. Providing a platform to reconcile discrepancies is particularly relevant because published studies use a variety of cell lines and ligands. Our findings do not contradict previous conclusions (Scaffidi et al, 1998; Barnhart et al, 2003); we simply explore a different region of phase space. The most interesting regions in this space lie along separatrices that divide cells into fundamentally different types. We have investigated some dimensions of this separatrix but we presume that other dimensions (such as FLIP levels (Scaffidi et al, 1999b; Lavrik et al, 2007; Wilson et al, 2009) or ligand and receptor multimerization (Scaffidi et al, 1999a; Siegel et al, 2004)) will also be important for understanding the control of type I versus II phenotypes. Moreover, it should also be noted that the computational and experimental analysis in the current paper follows the common practice of using Bcl2 over-expression to assay for type I death (a practice similar to using Bid−/− mice; Jost et al, 2009). However, our immunofluorescence data suggest that Bcl2 levels can also alter the fraction of cells that use MOMP in response to death ligand. Understanding in detail the flux through the caspase cascade and MOMP pathways in type I cells where MOMP is not inhibited will require further investigation. Development of more efficient software should make it feasible to compute and validate separatrices found in the full multi-dimensional phase space of extrinsic apoptosis. We also anticipate that the DLE methodology can be made more flexible by developing interactive software tools that enable normalization across several parameters, computation of the multi-dimensional difference between model trajectories using a subset of protein species, and modulation of specific rate parameters. However, more precise and detailed underlying models are required before we can study processes such as ligand or receptor oligomerization computationally (Scaffidi et al, 1998; Schmitz et al, 1999; Algeciras-Schimnich et al, 2003; Rudner et al, 2005; Lee et al, 2006).
Summary
Lyapunov exponent analysis has allowed us to generate a contextualized view of the regulation of type I versus type II behavior in apoptosis: multi-dimensional cell fate maps predict the behavior of multiple cell lines over multiple doses of death ligand and several changes in protein expression levels. The XIAP RING deletion examined here is an unusual mutation in that it completely eliminates one of the two reactions in which XIAP participates; most naturally occurring mutations have more subtle effects on reaction rates. Improved software and models should make it possible to analyze more subtle phenotypic changes associated with point mutations. Ultimately, we expect experimental validation of other dimensions in phase space to reconcile further discordant observations from the literature thereby generating a sound and predictive understanding of type I versus II control. Signaling phase diagrams based on computation and experiment hold promise as biochemical analogs of the classic polygenic models of Yule (1906) and Fisher (1918), which were developed to explain quantitative control of traits by multiple genes.
Materials and methods
Cell line derivation and maintenance
Parental and ΔXIAP HCT116 cells were obtained from B Vogelstein and F Bunz (Cummins et al, 2004). Parental and ΔXIAP HCT116 cells were maintained in McCoy's 5A media supplemented with 50 units/ml penicillin/streptomycin, 2 mM L-glutamine (Invitrogen), and 10% bovine calf serum (Hyclone) and SKW6.4 and T47D cells (ATCC) were maintained in RPMI supplemented with 50 units/ml penicillin/streptomycin, 2 mM L-glutamine (Invitrogen), and 10% bovine calf serum (Hyclone). OE-Bcl2 derivative cell lines were generated using a retrovirus produced by co-transfection of 293T cells (ATCC) with pCL-ampho and pBabe-Bcl2 (a gift from J Brugge). Stable OE-Bcl2 and OE-Bcl2/ΔXIAP HCT116 cells were selected with 5 μg/ml of puromycin, and stable OE-Bcl2 SKW6.4 and T47D cells were selected with 1 μg/ml and 2 μg/ml puromycin, respectively. Following selection, experiments were performed in clones for stable OE-Bcl2 and OE-Bcl2/ΔXIAP HCT116 cells and pools for stable OE-Bcl2 SKW6.4 and T47D cells.
Expression constructs
cDNAs coding for mCherry and for mCherry fused to full-length XIAP, XIAP with a RING-domain deletion, and XIAP with a RING-domain point mutation (CHY, CHY-XIAP, CHY-XIAPΔRING, and CHY-XIAP-H467A, respectively) were amplified by PCR (TaqPlus Precision PCR System, Stratagene), cloned in pExchange-1 (Stratagene) and sequence verified. CHY was amplified using forward primer (5′-CGCGCGGATCCGCCGCCATGGTGAGCAAGGGCGAGGAGGATAACATGGCC-3′) and reverse primer (5′-CCGCCGGAATTCTTACTTGTACAGCTCGTCCATGCCGCCGGTGGAGTGGCGGCCC-3′) and cloned in the BamHI and EcoRI sites to generate a vector to express CHY alone, or forward primer (5′-GCGGCGGCCGCGCCGCCATGGTGAGCAAGGGCGAGGAGGATAACATGGCC-3′) and reverse primer (5′-GCGGGATCCGCCGCCATGGTGAGCAAGGGCGAGGAGGATAACATGGCC-3′) and cloned in NotI and BamHI sites to generate a vector to express fusion proteins. XIAP, XIAPΔRING, and XIAP-H467A were amplified from pEBB-XIAP or pEBB-XIAPH467 obtained from Jon Ashwell (via Addgene.org), using forward primer (5′-GGCCGGATCCGCCGCCATGACTTTTAACAGTTTTGAAGGATCTAAAACTTGTGTACCTGC-3′), and reverse primer (5′-CCGCCGGAATTCTTACTTGTACAGCTCGTCCATGCCGCCGGTGGAGTGGCGGCCC-3′) for XIAP and XIAPH467 or reverse primer (5′-GGCCGAATTCTTAAGTACTAATCTCTTTCTGTAATGAAGTCTGACTTGACTC-3′) for XIAPΔRING (excluding residues 437–497). These PCR products were cloned in the BamHI and EcoRI sites of pExchange-1-CHY. Construction and use of the IC- and EC-FRET reporters (IC-RP and EC-RP) was previously described (Albeck et al, 2008a).
Cell treatments
TRAIL (SuperKiller TRAIL, Alexis Biochemicals) TNF (PeproTech), or FasL (SuperFasL Alexis Biochemicals) and CHX (Sigma-Aldrich) were added to cells in 1/20 of the culture volume at the indicated final concentrations. For treatment with death ligands, HCT116-derived cell lines were plated at 4 × 104 cells/cm2 1 day before treatment, T47D cells were also plated at 4 × 104 cells/cm2 2 days before treatment and SKW6.4 cells were plated at 4 × 105 cells/ml on the day of treatment.
For transient transfection with CHY-fusions with or without IC-RP or EC-RP, HCT116-derived cell lines were plated at 4 × 104 cells/cm2 2 days before treatment and transfected using Lipofectamine 2000 (Invitrogen) as per the manufacturer's recommendation 1 day before treatment. For transiently transfected cells, stimulus was added by changing the medium at the beginning of treatment.
SKW6.4-derived cell lines were transfected by electroporation (1 × 106 cells and 10 μg plasmid DNA in 400 μl, 950 μF, 250 mV with the GenePulserII (Bio-Rad)) 2 days prior to treatment and plated at 4 × 105 cells/ml before adding stimulus in 1/20 of the culture volume treatment.
Flow cytometric assay for initiator and effector caspase activity and distributions of protein levels
Preceding treatment (for distributions of protein levels) or following treatment (for assays of initiator and effector caspase activation), floating cells were collected and mixed with the trypsinized adherent cells before fixation in 4% paraformaldehyde in PBS, followed by permeabilization in 100% methanol. The cells were stored in methanol at −20°C for up to a week. Cells were washed with PBS + 0.1% Tween-20 and stained for 1 h with primary antibodies. Following washes in PBS + 0.1% Tween-20, cells were stained for 1 h with secondary antibodies if warranted. For assays of initiator and effector caspase activation cells were stained with custom Alexa Fluor 488-conjugated anti-cPARP (Asp214) monoclonal antibody (1:100 BD624020; BD Biosciences) for effector caspase activity measurements and PE-conjugated anti-cleaved caspase-3 antibody (1:100 BD550914; BD Biosciences) for initiator caspase activity measurements, and where indicated, with Alexa Fluor 405-conjugated mouse anti-Bcl2 (1:25 SC7382; Santa Cruz Biotechnologies). Note that because of the nature of the targeted epitopes, anti-cPARP and anti-cleaved caspase-3 antibodies recognize cleaved caspase substrates other than PARP or caspase-3, and we therefore refer to these as measures of ‘cleaved EC substrate’ and ‘cleaved IC susbstrate,’ respectively (cEC-S and cIC-S). For all flow cytometry experiments, single antibody stained controls were used for compensation. For distributions of protein levels, cells were stained with Alexa-647-conjugated α-XIAP clone 28 (610717; BD Biosciences) or Alexa-488-conjugated α-C3 (SC7272; Santa Cruz Biotechnology) before final washes and resuspension in PBS+0.1% Tween-20. All antibody dilutions were made in PBS+0.1% Tween-20+1% bovine serum albumin. Flow cytometry analysis was performed using the High Throughput Sampler on a BD LSRII instrument (BD Biosciences). Data were analyzed using FlowJo 8.8 (TreeStar) and custom software in MATLAB (The MathWorks).
Clonogenic assays
For clonogenic assays, HCT116-, T47D-, and SKW6.4-derived cells were treated in biological triplicates for 6 h with or without 2.5 μg/ml CHX alone and with or without 50 ng/ml TRAIL+2.5 μg/ml CHX as described above. After treatment, the cells were diluted in their respective culture medium (a minimum of 2400-fold). HCT116-derived lines were plated at ∼300 cells/6 cm dish, and colonies were counted after 12 days, T47D-derived lines were plated at ∼600 cells/6 cm dish, and colonies counted at 20 days. SKW6.4-derived lines were plated at ∼2 cells/well in one 96-well plate for each sample and growth-positive wells were counted after 21 days. For TRAIL+CHX treatment, we report the relative average number of colonies from biological triplicate samples, normalized by the average number of colonies obtained from the triplicate CHX alone samples. For TRAIL-only treatments, we normalized by the average number of colonies obtained from the triplicate mock-treated cells.
Live-cell imaging and image processing
Cells were imaged on a DeltaVision microscope (Applied Precision Inc.) at 3-min intervals, under ∼5% CO2 at 37°C. Time-lapse imaging data of IC-RP- or EC-RP-transfected cells were preprocessed in ImageJ to generate a mask and cells were then tracked by custom software in MATLAB (Jaqaman et al, 2008) that was reparameterized for HCT116 cells. Post-processing of the data was performed in MATLAB (The MathWorks). OE-Bcl2 SKW6.4 cells were seeded centrifuging onto Cell-Tak (BD Biosciences) coated chamber slides 18 h prior to imaging.
Fixed-cell imaging
Following plating and treatment in 96-well optical plates, cells were fixed in 4% paraformaldehyde in PBS, followed by permeabilization in 100% methanol. Cells were incubated in Odyssey Blocking Buffer (Licor) for 1 h at room temperature, then stained overnight at 4°C with FITC-conjugated anti-cleaved caspase-3 (1:25 BD559341; BD Biosciences), Alexa Fluor 647-conjugated anti-cPARP (1:250 BD558710; BD Biosciences) and Alexa Fluor 555-conjugated anti-cytochrome c (1:250 BD558700; BD Biosciences) in PBS+0.1% Tween-20+1% bovine serum albumin. After washing with PBS+0.1% Tween-20, cells were stained with Hoechst 33352 (4 μM; Invitrogen) and Whole Cell Blue dye (1:1000; Cellomics). To insure that all cells—including apoptotic cells—were visualized, the cells were centrifuged at every step (4 min at 1000 g). Images were obtained on a CellWorX station and a DeltaVision microscope (Applied Precision Inc.). The number of cells counted per well for each cell line was as follows: HCT116 (384, 175, 416), OE-Bcl2/ΔXIAP HCT116 (350, 236, 244), ΔXIAP HCT116 (174, 341, 225), T47D (608, 576, 691), and OE-Bcl2 T47D (157, 172, 221,187, 214).
Immunoblots
For immunoblot analysis, cells were lysed in sample buffer (112.5 mM Tris–Cl, pH 7.5, 4% SDS, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 1 μg/ml chymostatin) to generate whole-cell homogenates. Total protein concentration was determined using the bicinchoninic acid assay (Pierce). Equal amounts of lysates were run on 8% Tricine SDS–PAGE gels, and transferred to nitrocellulose. Membranes were incubated 1 h at room temperature in Odyssey Blocking Buffer (Licor) then probed with mouse monoclonal anti-caspase-8 (1:1000 clone 1C12 CST9746), rabbit polyclonal anti-caspase-6 (1:500 CST9762), rabbit polyclonal anti-PARP (1:500 CST9542), rabbit polyclonal anti-Bcl-xl (1:250 CST2762) (Cell Signaling Technologies), mouse monoclonal anti-caspase-3 (1:250 SC7272), mouse monoclonal anti-Bcl2 (1:100 SC7382), rabbit polyclonal anti-Bax (1:200 SC483), rabbit polyclonal anti-Mcl1 (1:200 SC819) (Santa Cruz Biotechnology), mouse monoclonal anti-XIAP (1:500 clone 28; BD610716; BD Biosciences), rabbit polyclonal anti-Smac (1:500 567365; Calbiochem), rat polyclonal anti-FLIP (1:1000; Alexis Biosciences), or rabbit polyclonal anti-Bid (1:250 HPA000722; Atlas Antibodies) overnight at 4°C. After washing, the antigen–antibody complexes were detected with IRDye800-conjugated donkey anti-mouse or anti-rabbit secondary antibodies as appropriate (1:5000, 610-732-124, and 612-732-120; Rockland Immunochemicals). Fluorescence was assessed on an Odyssey Scanner (Licor) and images were analyzed using the Odyssey software (Licor).
Physicochemical modeling of the apoptosis pathway
The apoptosis signaling model was built and run in MATLAB 2007b using the ode15s solver. This model is a variant of a published model (Albeck et al, 2008b) and includes the major features of both the direct and mitochondrial pathways downstream of death receptor activation (see Supplementary Methods and Supplementary Table SI for details). The model was reparameterized from the version published for HeLa cells (Albeck et al, 2008b) to parameters better suited for HCT116 cells, as described in Supplementary Tables SI–SIII and Box 1. The reduced model used for the analysis of E3 ligase-deficient XIAP focuses on the interactions between caspase-3 and XIAP and is parametrized with the same reaction rates as EARM1.4, as described in Supplementary Figure S7 and Supplementary Tables SIV–SVI.
DLE computation
The DLE analysis was performed as described in Aldridge et al (2006a), with the phase space region searched limited to caspase-3, caspase-8, caspase-6, ligand, receptor, XIAP, and EC substrate. Bcl2 over-expression conditions were used (10 × , sufficient to block MOMP) and initial conditions were sampled evenly from the following ranges: caspase-3 (1 × 103–1 × 106 molecules/cell sampled linearly with 50 and 20 concentrations in Figures 1B and 7A and in Figure 8A, respectively), XIAP (0–6 × 104 molecules/cell sampled linearly with 50 and 20 concentrations in Figures 1B and 7A and in Figure 8A, respectively), caspase-6 (0–2 × 104 molecules/cell sampled linearly with 50 and 20 concentrations in Figures 1B and 8A, respectively), caspase-8 (1500–8500 sampled linearly with eight concentrations), ligand (1.6, 40, 1 × 103, 2.5 × 104, and 1 × 105 a.u. where 1 × 103 is equivalent to treatment with 50 ng/ml of TRAIL), and receptor (100, 5 × 103, 5 × 104, and 5 × 105 molecules/cell). Initial concentrations used for the EC substrate PARP were 1 000 000 and 1 000 001 molecules/cell; this nominal range allowed us to include EC substrate cleavage as a readout for apoptosis in the DLE calculation without having the EC substrate concentration affect the value of the DLE across those two conditions. The baseline phase space slice through XIAP and caspase-3, unless otherwise noted, is 1 × 104 molecules/cell for caspase-6, 3500 molecules/cell for caspase-8, 1 × 103 a.u. for ligand (for 50 ng/ml of TRAIL), 5 × 103 molecules/cell for receptor. The simulations and DLEs were computed in MATLAB 2007b (The MathWorks) and were visualized in Spotfire software. The simulation and DLE computation code is available in the Supplementary Materials.
Supplementary Material
Acknowledgments
We thank J Albeck, J Bachman, J Burke, D Flusberg, T Maiwald, and S Spencer for helpful discussions and K Jaqaman and G Danuser for advice on image processing. This work was funded by NIH Grant GM68762 to PKS and CA139980 to PKS and SG. BBA was funded by a DOE Computational Science Graduate Fellowship.
Author contributions: BBA, SG, DAL, and PKS designed, conducted, and analyzed the experiments and computation and all authors contributed to the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Albeck JG, Burke JM, Aldridge BB, Zhang M, Lauffenburger DA, Sorger PK (2008a) Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell 30: 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, Sorger PK (2008b) Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol 6: 2831–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK (2006a) Physicochemical modelling of cell signalling pathways. Nat Cell Biol 8: 1195–1203 [DOI] [PubMed] [Google Scholar]
- Aldridge BB, Haller G, Sorger PK, Lauffenburger DA (2006b) Direct Lyapunov exponent analysis enables parametric study of transient signalling governing cell behaviour. Syst Biol (Stevenage) 153: 425–432 [DOI] [PubMed] [Google Scholar]
- Algeciras-Schimnich A, Pietras EM, Barnhart BC, Legembre P, Vijayan S, Holbeck SL, Peter ME (2003) Two CD95 tumor classes with different sensitivities to antitumor drugs. Proc Natl Acad Sci USA 100: 11445–11450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart BC, Alappat EC, Peter ME (2003) The CD95 type I/type II model. Sem Immunol 15: 185–193 [DOI] [PubMed] [Google Scholar]
- Bratton SB, Lewis J, Butterworth M, Duckett CS, Cohen GM (2002) XIAP inhibition of caspase-3 preserves its association with the Apaf-1 apoptosome and prevents CD95- and Bax-induced apoptosis. Cell Death Differ 9: 881–892 [DOI] [PubMed] [Google Scholar]
- Ceccarini C, Eagle H (1976) Some paradoxical effects of inhibitors of protein synthesis on protein turnover in cultured human cells. In Vitro 12: 346–351 [DOI] [PubMed] [Google Scholar]
- Coulliette C, Lekien F, Paduan JD, Haller G, Marsden JE (2007) Optimal pollution mitigation in monterey bay based on coastal radar data and nonlinear dynamics. Environ Sci Technol 41: 6562–6572 [DOI] [PubMed] [Google Scholar]
- Cummins JM, Kohli M, Rago C, Kinzler KW, Vogelstein B, Bunz F (2004) X-linked inhibitor of apoptosis protein (XIAP) is a nonredundant modulator of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis in human cancer cells. Cancer Res 64: 3006–3008 [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304 [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL (2006) Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 7: 988–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissing T, Conzelmann H, Gilles ED, Allgower F, Bullinger E, Scheurich P (2004) Bistability analyses of a caspase activation model for receptor-induced apoptosis. J Biol Chem 279: 36892–36897 [DOI] [PubMed] [Google Scholar]
- Fisher RA (1918) The Correlation between relatives under the supposition of Mendelian inheritance. Trans R Soc Endinburgh 52: 399–433 [Google Scholar]
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD et al. (2009) Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 16: 1093–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet S, Janes KA, Albeck JG, Pace EA, Lauffenburger DA, Sorger PK (2005) A compendium of signals and responses triggered by prodeath and prosurvival cytokines. Mol Cell Proteomics 4: 1569–1590 [DOI] [PubMed] [Google Scholar]
- Grell M, Krammer PH, Scheurich P (1994) Segregation of APO-1/Fas antigen- and tumor necrosis factor receptor-mediated apoptosis. Eur J Immunol 24: 2563–2566 [DOI] [PubMed] [Google Scholar]
- Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB (2001) Characterization of XIAP-deficient mice. Mol Cell Biol 21: 3604–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G (2008) Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods 5: 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, Strasser A, Kaufmann T (2009) XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460: 1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrik IN, Golks A, Riess D, Bentele M, Eils R, Krammer PH (2007) Analysis of CD95 threshold signaling: triggering of CD95 (FAS/APO-1) at low concentrations primarily results in survival signaling. J Biol Chem 282: 13664–13671 [DOI] [PubMed] [Google Scholar]
- LeBlanc H, Lawrence D, Varfolomeev E, Totpal K, Morlan J, Schow P, Fong S, Schwall R, Sinicropi D, Ashkenazi A (2002) Tumor-cell resistance to death receptor—induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med 8: 274–281 [DOI] [PubMed] [Google Scholar]
- Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schutze S, Peter ME, Chan AC (2006) The role of receptor internalization in CD95 signaling. EMBO J 25: 1009–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestas I, Vinnicombe G, Paulsson J (2010) Fundamental limits on the suppression of molecular fluctuations. Nature 467: 174–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai AG (2008) Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nat Rev Cancer 8: 121–132 [DOI] [PubMed] [Google Scholar]
- Lovric MM, Hawkins CJ (2010) TRAIL treatment provokes mutations in surviving cells. Oncogene 29: 5048–5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas C, Verbrugge I, de Vries E, Savich G, van de Kooij LW, Tait SW, Borst J (2010) Smac/DIABLO release from mitochondria and XIAP inhibition are essential to limit clonogenicity of Type I tumor cells after TRAIL receptor stimulation. Cell Death Differ 17: 1613–1623 [DOI] [PubMed] [Google Scholar]
- MacFarlane M, Merrison W, Bratton SB, Cohen GM (2002) Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem 277: 36611–36616 [DOI] [PubMed] [Google Scholar]
- Newsom-Davis T, Prieske S, Walczak H (2009) Is TRAIL the holy grail of cancer therapy? Apoptosis 14: 607–623 [DOI] [PubMed] [Google Scholar]
- Oberst A, Pop C, Tremblay AG, Blais V, Denault JB, Salvesen GS, Green DR (2010) Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem 285: 16632–16642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozoren N, El-Deiry WS (2002) Defining characteristics of types I and II apoptotic cells in response to TRAIL. Neoplasia 4: 551–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rateitschak K, Wolkenhauer O (2010) Thresholds in transient dynamics of signal transduction pathways. J Theor Biol 264: 334–346 [DOI] [PubMed] [Google Scholar]
- Rehm M, Huber HJ, Dussmann H, Prehn JH (2006) Systems analysis of effector caspase activation and its control by X-linked inhibitor of apoptosis protein. EMBO J 25: 4338–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm M, Huber HJ, Hellwig CT, Anguissola S, Dussmann H, Prehn JH (2009) Dynamics of outer mitochondrial membrane permeabilization during apoptosis. Cell Death Differ 16: 613–623 [DOI] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773–786 [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y (2004) Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 5: 897–907 [DOI] [PubMed] [Google Scholar]
- Rudner J, Jendrossek V, Lauber K, Daniel PT, Wesselborg S, Belka C (2005) Type I and type II reactions in TRAIL-induced apoptosis—results from dose-response studies. Oncogene 24: 130–140 [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J 17: 1675–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C, Schmitz I, Krammer PH, Peter ME (1999a) The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 274: 1541–1548 [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME (1999b) Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J Biol Chem 274: 22532–22538 [DOI] [PubMed] [Google Scholar]
- Schile AJ, Garcia-Fernandez M, Steller H (2008) Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev 22: 2256–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz I, Walczak H, Krammer PH, Peter ME (1999) Differences between CD95 type I and II cells detected with the CD95 ligand. Cell Death Differ 6: 821–822 [DOI] [PubMed] [Google Scholar]
- Shawgo ME, Shelton SN, Robertson JD (2009) Caspase-9 activation by the apoptosome is not required for fas-mediated apoptosis in type II Jurkat cells. J Biol Chem 284: 33447–33455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RM, Muppidi JR, Sarker M, Lobito A, Jen M, Martin D, Straus SE, Lenardo MJ (2004) SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol 167: 735–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Kratina T, Chu D, Ekert PG, Day CL, Pakusch M, Huang DC, Vaux DL (2005) Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc Natl Acad Sci USA 102: 16182–16187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK (2009) Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459: 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM (2002) Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem 277: 11345–11351 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Nakata K, Reed JC, Takahashi R (2001) X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J Biol Chem 276: 27058–27063 [DOI] [PubMed] [Google Scholar]
- Tyas L, Brophy VA, Pope A, Rivett AJ, Tavare JM (2000) Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Rep 1: 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan AT, Betti CJ, Villalobos MJ (2002) Surviving apoptosis. Apoptosis 7: 173–177 [DOI] [PubMed] [Google Scholar]
- Vaughan AT, Betti CJ, Villalobos MJ, Premkumar K, Cline E, Jiang Q, Diaz MO (2005) Surviving apoptosis: a possible mechanism of benzene-induced leukemia. Chem Biol Interact 153–154: 179–185 [DOI] [PubMed] [Google Scholar]
- Wilson TR, McEwan M, McLaughlin K, Le Clorennec C, Allen WL, Fennell DA, Johnston PG, Longley DB (2009) Combined inhibition of FLIP and XIAP induces Bax-independent apoptosis in type II colorectal cancer cells. Oncogene 28: 63–72 [DOI] [PubMed] [Google Scholar]
- Wurstle ML, Laussmann MA, Rehm M (2010) The caspase-8 dimerization/dissociation balance is a highly potent regulator of caspase-8, -3, -6 signaling. J Biol Chem 285: 33209–33218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877 [DOI] [PubMed] [Google Scholar]
- Yule GU (1906) On the theory of inheritance of quantitative compound characters on the basis of Mendel's laws—a preliminary note. In Third International Conference on Genetics, pp 140–142 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.