

Abstract
Genetic analysis in Drosophila melanogaster has been widely used to identify a system of genes that control cell growth in response to insulin and nutrients. Many of these genes encode components of the insulin receptor/target of rapamycin (InR/TOR) pathway. However, the biochemical context of this regulatory system is still poorly characterized in Drosophila. Here, we present the first quantitative study that systematically characterizes the modularity and hormone sensitivity of the interaction proteome underlying growth control by the dInR/TOR pathway. Applying quantitative affinity purification and mass spectrometry, we identified 97 high confidence protein interactions among 58 network components. In all, 22% of the detected interactions were regulated by insulin affecting membrane proximal as well as intracellular signaling complexes. Systematic functional analysis linked a subset of network components to the control of dTORC1 and dTORC2 activity. Furthermore, our data suggest the presence of three distinct dTOR kinase complexes, including the evolutionary conserved dTTT complex (Drosophila TOR, TELO2, TTI1). Subsequent genetic studies in flies suggest a role for dTTT in controlling cell growth via a dTORC1- and dTORC2-dependent mechanism.
Keywords: cell growth, InR/TOR pathway, interaction proteome, quantitative mass spectrometry, signaling
Introduction
The control of cellular growth requires an intricate system of molecular mechanisms that orchestrate processes such as translation, cellular proliferation, apoptosis and autophagy in response to the nutritional and humoral environment of the cell. Genetic screens in Drosophila have frequently been used to identify key regulators of cell growth. Subsequent analyses of their functional relationships led to pathway models such as the insulin receptor and target of rapamycin (InR/TOR) pathway. However, a detailed molecular understanding of how a particular growth phenotype results from a specific genotype has remained largely elusive.
Biological processes, including cell growth, emerge from molecular networks involving proteins, which stably and/or transiently interact to form protein complexes that in turn organize in extended interaction networks. In most cases, it is therefore conceivable that growth phenotypes are the result of mutations affecting the function of proteins and thereby perturbing network states. The systematic analysis of the composition and dynamics of such protein interaction networks is therefore highly relevant for a systems level understanding on the molecular organization of cell growth control. Affinity purification coupled with tandem mass spectrometry (AP–MS/MS) is the most advanced method to characterize protein complexes and it has already revealed important insights into the modular organization of the proteome in yeast (Gavin et al, 2002; Krogan et al, 2006) and other species (Butland et al, 2005; Kuhner et al, 2009).
Signaling networks are highly dynamic and assemble or disassemble in response to changes in the cellular environment. However, most existing models on protein interaction networks are typically represented as static entities due to lack of quantitative MS information. Subsequent functional studies of AP–MS data represent key steps to develop new hypotheses on how biological processes emerge from biochemical networks. In this regard, Drosophila could provide a major advantage as it offers a number of unique genetic tools to characterize functional relevance of identified network components. However, surprisingly little protein interaction information has so far been reported using systematic AP–MS in Drosophila.
In this study, we present the first systematic quantitative AP–MS/MS analysis on the Drosophila InR/TOR pathway, an evolutionarily conserved growth regulating signaling system that has been linked to a number of severe human diseases such as cancer or diabetes (Saltiel and Kahn, 2001; Biddinger and Kahn, 2006; Zoncu et al, 2011). We studied the complexes around 16 bait proteins including membrane proximal signaling proteins, intracellular regulators as well as known effectors of this pathway. In all, 22% of the high confidence interactions identified turned out to be regulated by insulin, suggesting that the network as a whole is substantially affected by the activity state of the InR. High-density protein interaction data revealed a highly modular organization of the Drosophila TOR kinase interactome consistent with the existence of several distinct TOR complexes. Subsequent systematic functional experiments using RNA interference against all network components suggest that a significant fraction of these components can be functionally linked to the control of dTORC1 and dTORC2 activity. Among these we identified two components of the dTTT complex, which when mutated caused a similar growth phenotype as dTOR mutant flies. Taken together, these results illustrate how quantitative AP–MS when combined with systematic functional analysis in Drosophila can reveal novel insights into the dynamic organization of regulatory networks for cell growth control in metazoans.
Results
Systematic analysis of the InR/TOR interaction proteome
We selected 16 bait proteins that were previously linked by genetic or biochemical evidence to growth control by the InR/TOR pathway. Affinity-tagged versions of these proteins were inducibly expressed in stably transfected Drosophila Kc167 cell lines. To study hormone-induced remodeling of signaling complexes at the pathway level, we performed APs of protein complexes under insulin-stimulated and -non-stimulated conditions. The experimental workflow is depicted in Figure 1A and bait proteins are listed in Supplementary Table 1.
Figure 1.

Overview on the experimental workflow and data processing. (A) Experimental workflow. In all, 16 genes linked to dInR/TOR pathway were PCR amplified from cDNA pools. By recombinatorial cloning, the Drosophila ORFs were transferred into an in-house designed expression vector for inducible expression of epitope-tagged bait proteins. Bait expression was induced and cells were starved overnight in 2% FBS. Half of the cell population was exposed to 100 nM insulin for 20 min. The cells were lysed in the presence of DSP cross-linker and bait proteins were purified by anti-HA AP. All AP–MS/MS experiments were performed as two independent replicates. (B) Data processing of the AP–MS/MS data set for detection of specific PPIs using the SAINT algorithm. Following database search, spectrum counts for each identified protein were extracted from the LC–MS/MS data. Based on normalized spectrum counts, SAINT models the spectrum count distribution of each bait–prey interaction and calculates scores based on the relative abundance of each hit. In all, 24 independently control AP–MS experiments (using GFP as a bait protein) were performed in parallel to model false interaction distribution. (C) SAINT scores (data from two biological replicates) were plotted from dTOR purified in the absence (x axis) or the presence (y axis) of insulin to illustrate the specificity increase by the SAINT filtering approach. (D) Overlap with orthologous PPI data and known Drosophila PPIs. The PPI data obtained in this study were compared with available literature and database information covering known D. melanogaster and related Homo sapiens interactions (see also Materials and methods).
To discriminate between true and false protein interactors from the initially generated protein list, we used the recently introduced Significance Analysis of INTeractome (SAINT) 2.0 algorithm (Choi et al, 2010) (Figure 1B; for details see Materials and methods). Stringent filtering resulted in a final high confidence protein interaction (HCPI) data set containing 58 network components and 97 interactions (Supplementary Tables 2, 3 and 4). Figure 1C illustrates the result of filtering raw data from dTOR purifications using the SAINT score.
We compared our HCPI data with known interaction information available from the literature and protein interaction databases for Drosophila, yeast and humans (Figure 1D; Supplementary Table 5). Twelve interactions were already known from previous Drosophila studies. Of the 85 remaining interactions, 39 had been previously described among human orthologs. In all, 10 of these conserved interactions were also represented in the known yeast interaction proteome. These highly conserved interactions constitute the TORC1 and TORC2 complexes in the respective species (for details see also Supplementary Table 5). Protein interactions from our data set that were conserved in human, but not in yeast, centered around the InR signaling components, which are absent in yeast. Previous large-scale yeast two-hybrid studies on the Drosophila interaction proteome captured only three of the detected interactions, highlighting the need for AP–MS-based interaction proteomics for cell signaling research in Drosophila. In addition to these known interactions, we also found 46 novel interactions, which provide interesting entry points into further functional studies on the signaling mechanism underlying cell growth control by the InR/TOR signaling system and will be discussed later in the text.
Identification of insulin-modulated changes in the InR/TOR interaction proteome by quantitative MS
To systematically analyze insulin-modulated changes in the InR/TOR interaction network, we purified all protein complexes from insulin-treated or -untreated cells and quantified the abundance of the interacting proteins using label-free quantification (Rinner et al, 2007) (Figure 2A). The measured profiles from two replicate experiments quantified 86 out of 97 interactions identified (for detailed information see Materials and methods). Based on the distribution of average enrichment factors (AEF) (Figure 2B), interactions were specified as insulin sensitive when their measured fold-change was at least 1.5 in two independent experiments. The validity of a 1.5-fold cutoff was carefully evaluated by statistical analysis of controlled dilution experiments (Supplementary Figure 1; Supplementary Table 9). Using these criteria, we identified a total of 22 insulin-regulated interactions, corresponding to 25% of the quantified InR/TOR interaction proteome (Figure 2D; Supplementary Tables 6 and 7).
Figure 2.

Quantitative analysis of insulin-sensitive PPIs. (A) Schematic overview on the experimental approach for monitoring insulin-sensitive bait–prey interactions using label-free MS and MS1 alignment (for details see Materials and methods). (B) Examples of dilution experiments in combination with label-free quantification and multiple LC–MS alignment. AP–MS analysis was performed on insulin-treated and -untreated Kc167 cell lines expressing Chico (dIRS) and p60 (Pi3K21B). Protein abundance profiles were normalized against the individual bait profiles of Chico and p60 (black lines). Bait–prey interactions showing changes in abundance upon insulin stimulation are presented as red (increase) and blue (decrease) profiles. Lines in green refer to unaffected protein interactions. Error bars indicate s.e.m. of the average MS1 signal intensity of an individual profile in a particular dilution. (C) Overview on enrichment factor distribution of all quantified protein interactions. The average enrichment factor (AEF) of every PPI were calculated from replicate experiments and plotted over the entire data set. All interactions with a SAINT score >0.8 are shown (see also Materials and methods). (D) List of all observed insulin-sensitive interactions. Pearson product-moment coefficient from linear regression analysis (r) as well as AEF calculated based on profile information between two replicates (Exp. A and B, respectively) are listed.
Membrane proximal signaling events around the activated InR complex illustrate how results from this quantitative approach can be used to specify insulin-sensitive and -insensitive interactions (Figure 2C). We found that the association of the InR substrate (IRS) homolog Chico with the regulatory phosphatidylinositol 3-(PI3) kinase subunit p60 (Pi3K21B) was highly induced upon insulin stimulation (Figure 2C, left panel). In contrast, the E3 ubiquitin ligase Mindbomb-2 (Mib-2) dissociated from Chico upon insulin treatment. Reciprocal experiments using p60 as a bait confirmed insulin inducible binding of p60 to Chico (Figure 2C, right panel) and revealed insulin-sensitive interactions with dInR and CG11063. The interaction of p60 with other binding partners including PDGF/VEGF receptor homolog Pvr, Lin19 and dp110 (Pi3K92E), was not affected by insulin. Insulin-sensitive remodeling of the InR/TOR interaction proteome was not restricted to the membrane proximal InR/PI3K complexes but expands also toward downstream pathway regulators, which are listed in Figure 2D and discussed in more detail below.
Overview on the Drosophila InR/TOR pathway interactome
We next assembled the interaction data (Supplementary Tables 2 and 6) into a quantitative network model to identify signaling modules and their changes upon insulin treatment (Figure 3). The term module is defined here as a group of proteins with high connectivity in the interaction network model due to complex formation or sharing of binding partners, indicating a related biochemical context. Overall, the resulting network was enriched for proteins with roles in cell signaling processes such as ubiquitin-dependent degradation, protein phosphorylation, GTPase signaling, transcription and translation. Using functional annotations and the obtained network topology, we grouped the network components into four functional modules: (1) the membrane proximal InR module, (2) the tuberous sclerosis complex (TSC), (3) the group of 14-3-3 complexes and (4) the TOR module. Selected interactions within these modules are described below in more detail.
Figure 3.

Modularity and insulin sensitivity of the dInR/TOR interaction proteome. High confidence PPI data are visualized using Cytoscape v2.8.1. Bait proteins are represented as hexagons, preys as circles. Names correspond to flybase gene symbols. Interactions that are increased or decreased upon insulin treatment are represented with arrow symbols and edge colors as indicated. Nodes have been color coded according to the functional classification given in the pie chart. Proteins are grouped into functional modules based on prior knowledge and network topology.
New molecular components within the Drosophila InR module
Besides the insulin-dependent interactions between the core components dInR, Chico and the PI3K subunits dp110 and p60, we identified a number of new interactions with potential interest for the regulation of this essential module in the InR/TOR pathway. Chico and the regulatory subunit of PI3K (p60) interacted with distinct E3 ligases. These previously undetected interactions suggest the involvement of several ubiquitin-dependent processes for membrane proximal signaling events. Specifically, Chico was found in association with the RING-finger domain containing E3 ligase Mib-2, an association that was decreased upon insulin treatment; the PI3K regulatory subunit p60 copurified with SkpA, Lin19, Pall, which have been shown to form an SCF (Skp1, Cullin, F-box) E3 ubiquitin ligase complex (Silva et al, 2007), consistent with the view that the entire SCF complex is associated with p60. In contrast to the Chico–Mib-2 interaction, the p60-SCFPal1 interaction was not affected by insulin.
Besides binding to the dInR, we found p60 also associated with Pvr, another receptor tyrosine kinase related to the human VEGF and PDGF receptor. In contrast to the p60–dInR interaction, which was strongly enhanced upon insulin, the p60–Pvr interaction was not affected by insulin. Thus, at least under the applied experimental conditions, insulin-induced recruitment of p60 to the dInR does not interfere with p60 binding to Pvr and maybe other RTKs. The catalytic subunit of PI3K dp110 was found in complexes with Rho-type GTPase exchange factor and the ARF GTPase-activating protein GIT (CG16728), suggesting a role of PI3K for specific GTPase controlled signaling events in Drosophila.
The Drosophila TSC
Next to the InR module, the TSC module is of particular importance for the regulation of TOR activity. dTSC1 and Gigas (dTSC2, gig), the Drosophila ortholog of human TSC2, as expected form heterodimers (van Slegtenhorst et al, 1998). It has been shown that TSC2 can repress the activity of the Ras-related GTPase Rheb via its associated GAP domain (GTPase-activating protein), resulting in the inactivation of TOR (Garami et al, 2003; Stocker et al, 2003). Consistently, we found Rheb stably associated with the dTSC complex. Remarkably, we identified another GAP domain protein, the gene product of CG6182, in complexes with dTSC1 and dTSC2. The mammalian ortholog of this protein, TBC1D7, associates with human TSC1, indicating that this interaction is conserved (Nakashima et al, 2007). It has been proposed that TBC1D7 may increase TORC1 activity toward S6K (S6 Kinase) via ubiquitin-mediated degradation of the TSC dimer (Nakashima et al, 2007). Recently, TBC1D7 expression has been associated with poor prognosis for pulmonary carcinogenesis (Sato et al, 2010).
Besides the association with GTPase signaling components, we found TSC1 in complexes with CG43143, the Drosophila homolog of NUAK1/ARK5, a so far poorly characterized kinase that is related to AMPK (adenosine monophosphate-activated protein kinase). The finding that the human TSC complex is regulated via AMPK (Inoki et al, 2003; Corradetti et al, 2004) further supports a potential new functional link between the NUAK1 homolog and the Drosophila TSC complex.
14-3-3 Complexes in the dInR/TOR pathway
14-3-3 proteins have been implicated in the regulation of a number of signaling pathways, including the InR/TOR pathway (Morrison, 2009). It is believed that phosphorylation-dependent binding of 14-3-3 proteins regulate stability, localization and activity of the bound proteins (Morrison, 2009). We found both 14-3-3ε and 14-3-3ζ in complexes with Chico, dTSC1, dTSC2, dRictor, dSIN1 and dFoxo. 14-3-3 complexes containing dFoxo or dSIN1 were most strongly affected by insulin stimulation. It was proposed that 14-3-3 binding sequesters Foxo transcription factors in the cytoplasm upon activation of the InR pathway (Brunet et al, 2002; Rinner et al, 2007). Binding of 14-3-3 proteins to the dTORC2 components dRictor or dSIN1, as shown here, has so far not been observed in Drosophila. dRictor has several predicted 14-3-3 binding sites and recent studies demonstrated 14-3-3 protein binding to Rictor in human cells (Dibble et al, 2009).
Drosophila TOR modules
The TOR kinase dTOR represents a central node in the growth control network. It integrates various signaling inputs including nutrient availability and insulin signaling, energy status and oxygen availability. Surprisingly little is known about the Drosophila TOR interaction proteome. To obtain a comprehensive representation of Drosophila TOR complexes, we selected the seven fly proteins as baits for AP–MS analysis that are homologous to human proteins implicated in the formation of TORC1 and TORC2 complexes (dTOR, dRaptor, dGβL/CG3004, dRictor, dSIN1) or act as downstream effectors (Thor/d4E-BP and dS6K). In addition, we included Unkempt (Unk) and the product of CG16908, two proteins that we identified in dTOR purifications and which have not been linked to mTORC1 or mTORC2 in previous studies. As a result, we obtained a highly interconnected subnetwork composed of established TOR signaling components known from studies in other species and proteins that have not been implicated in TOR signaling yet. All orthologous components of mTORC1 and mTORC2 complexes in Drosophila (dGβL, Lobe, dRaptor, dRictor and dSIN1) including the TORC1 substrates d4E-BP (Thor) and S6K were found to form complexes with dTOR.
The resulting network topology is consistent with the existence of evolutionary conserved dTORC1 and dTORC2 complexes in Drosophila, for example, when the dTORC1-specific protein dRaptor was used as a bait, only TORC1 proteins, but no TORC2 proteins, such as dRictor and dSIN1, were identified by shotgun proteomics. Likewise, AP–MS analysis of dRictor purifications did not reveal any dTORC1-specific components. The only exception was found when dSIN1 was used as a bait. Besides the expected dTORC2 proteins dTOR, dRictor and dGβL, we also found dRaptor. This may suggest that under the applied conditions, a fraction of dSIN1 may exist in complexes containing the TORC1 protein dRaptor.
From the identified core components, Lobe, the Drosophila ortholog of human PRAS40, was consistently reduced in dTOR, dRaptor, dGβL or Thor/d4E-BP purifications from insulin-treated cells.
Among the proteins that showed increased complex formation with dTOR upon insulin signaling, we identified the dTORC1 substrates d4E-BP, dS6K as well as Unk, a poorly characterized ring-finger protein with a proposed role in ubiquitin-mediated protein degradation (Lores et al, 2010). Insulin increased complex formation of Unk with a number of dTORC1 components, suggesting that Unk represents a novel insulin-sensitive component of dTORC1.
A specific set of proteins that were not linked to the canonical TOR complexes TORC1 and TORC2 were found in dTOR purifications. These include LqfR (liquid facets related), Pontin, Reptin, Spaghetti and the gene product of CG16908. We confirmed these interactions by reciprocal purification using CG16908 as bait. However, none of the dTORC1/2 components besides dTOR was identified in CG16908 purifications, indicating that these proteins form dTOR complexes distinct from dTORC1 and dTORC2. Sequence analysis revealed that LqfR and CG16908 encode proteins with homologies to human and Tel2 (see Supplementary Figure 2) and Tti1 (Kaizuka et al, 2010). Tel2 can form complexes with Tti1 and phosphatidylinositol 3-kinase-related protein kinases including TOR in yeast and mammalian cells (Hurov et al, 2010; Kaizuka et al, 2010). Consistent with this notion, we found besides dTOR also Nipped-A, the Drosophila homolog of the human PIK-related kinase TRAPP in CG16908 purifications. Based on the observed network topology and the homology to the proteins observed in human Tel2 complexes, we refer to this complex as dTTT complex (dTOR–dTel2–dTti1). In contrast to the dTORC1 interactions, we found that the level of dTTT components was not affected in dTOR purifications following insulin treatment.
Taken together, the high-density interaction data obtained in this study are consistent with the model that dTOR can form at least three major complexes (dTORC1, dTORC2 and dTTT), of which only dTORC1 appears to be affected by insulin.
Quantitative analysis dTOR complexes using directed MS
We next used a directed MS approach in combination with label-free quantification to estimate the relative distribution of dTOR complexes in Drosophila cells (Schmidt et al, 2008; Olsen et al, 2009) (Figure 4A). We used the average intensity of the three most intense peptide ions per protein (TOP3) to measure protein abundances in dTOR, CG3004/dGβL, dRaptor, dRictor purifications as proposed previously (Silva et al, 2006; Malmstrom et al, 2009).
Figure 4.

Analysis of dTOR complexes by quantitative AP–MS. (A) Workflow of AP–MS analysis for dTOR complex analysis. Peptide precursor m/z values obtained from tryptic in silico digest of dTOR core components were used for inclusion list-LC/MS/MS experiments. Based on all successfully identified peptide features, label-free quantification was performed and average signal intensities of the three most intense peptides (TOP3) were calculated to represent protein abundances. (B) Abundance distribution of proteins identified in dTOR and dGβL (upper panel), dRictor and dRaptor purifications (lower panel) relative to the bait. The average TOP3 signal intensity was used to infer protein abundances within individual AP–MS/MS experiments (data are listed in Supplementary Table 10). The average signal intensities of dTOR core components in each of the indicated purifications were calculated relative to corresponding bait intensity (set to 10E5) from four purification experiments and are shown in log scale in the bar chart. Error bars represent standard deviation in log scale.
dGβL represents the most abundant dTOR interactor (Figure 4B; see also Supplementary Table 10). This is not surprising as GβL is a common component of both mTORC1 and mTORC2 in human cells (Zoncu et al, 2011). dRaptor was close in abundance to dGβL but 10-fold more abundant than the dTORC2 components dRictor and dSIN1, suggesting that dTORC1 is the most abundant dTOR complex in Drosophila Kc167 cells. A similar abundance pattern for dTORC1 and dTORC2 components was observed when dGβL was used as a bait, confirming the results from the dTOR analysis.
In dRaptor purifications, the dTORC1 components dTOR, dGβL and Lobe were the most abundant proteins. We also detected the dTORC2 proteins dRictor and dSIN1 in dRaptor purifications. However, dRictor levels were less than an estimated 1% compared with the amount of the dTOR, Lobe and dGβL. Similarly, when dRictor was analyzed as a bait, we found besides the expected dTORC2 components dSIN1, dGβL and dTOR also evidence for the presence of dTORC1 components dRaptor and Lobe. The amounts were about two orders of magnitude below the amount of dRictor. These results suggest that under the experimental conditions applied including chemical cross-linking and targeted analysis, a small fraction of dTORC1 and dTORC2 components could be detected in the same complexes.
Based on our quantitative results, it appears that both dTTT components LqfR and CG16908 associate with dTOR to similar amounts, which are a bit higher than the amounts of dTORC2 component dRictor (Figure 4B, upper left). Both proteins, however, were not detected in the samples using either dGβL, dRaptor or dRictor as bait proteins, suggesting that dTTT represents a novel complex independent of the canonical dTORC1 and dTORC2. Overall, the quantitative data from the dTOR purifications indicate that dTORC1 is the most abundant dTOR complex we have identified in Kc167 cells (for details see Supplementary Table 10).
Systematic functional analysis of the dInR/TOR interaction proteome
We next wanted to study the potential roles of the identified network components for controlling the activity of the dInR/TOR pathway by systematic RNAi depletion experiments. As a functional readout, we applied quantitative western blotting to measure the changes in abundance of phosphorylated substrates of dTORC1 (Thor/d4E-BP, dS6K) and dTORC2 (dPKB) in RNAi-treated cells. Following hierarchical clustering of the obtained RNAi phenotypes (mean intensity of at least two independent experiments compared with EGFP control RNAi, for details see also Supplementary Figure 4), we were able to identify 16 proteins (out of 58) whose depletion caused an at least 50% increase or decrease in the levels of phosphorylated d4E-BP, S6K and/or PKB compared with control GFP RNAi. As expected, the known components of the dInR/Tor pathway were found among these proteins. For instance, depletion of dTOR itself or its binding partner dGβL/CG3004 strongly reduced the phosphorylation of dS6K, d4E-BP and dPKB. Likewise, we found that depletion of dRaptor or Rheb, two positive pathway regulators acting downstream of dPKB, reduced the phosphorylation of the two dTORC1 substrates d4E-BP and dS6K, while phosphorylation of the dTORC2 substrate dPKB was increased. Depletion of negative regulators acting downstream of dPKB such as dTSC1 or dTSC2 resulted in the opposite phenotypes. Phosphorylated dPKB was also strongly reduced after depletion of positive pathway regulators acting upstream of dPKB (Chico, p60, dp110) or dRictor as reported previously (Sarbassov et al, 2005). Importantly, besides these established pathway components, we also found several novel regulators within the dInR/TOR interaction network (Figure 5A). For example, RNAi against the novel insulin-regulated dTORC1 component Unkempt resulted in enhanced phosphorylation of the dTORC1 substrate d4E-BP (and to a lesser extent also of dS6K), which suggests a negative role for Unkempt on dTORC1 activity. In contrast, depletion of CG16908 and LqfR (see below) caused hypo-phosphorylation of all dTOR substrates similar to dTOR itself, suggesting a positive role for the dTTT complex on dTOR activity. A representative western blot underlying these results is shown in Figure 5B.
Figure 5.

Functional analysis of dInR/TOR pathway interaction proteome for the regulation of dTOR activity. (A) Effect of RNAi-mediated depletion of network components on the phosphorylation levels of S6K, PKB and d4E-BP. Identified network components were depleted by RNAi in Drosophila Kc167 cells as indicated next to the heat map. Total cell extracts were analyzed by immunoblotting and the band intensities obtained with anti-P-d4E-BP, anti-P-S6K and anti-P-PKB were quantified (see Materials and methods). The band intensities observed in total extracts of cells treated with RNAi against EGFP were set to 100%. Each frame represents the average signal intensity of at least two independent experiments. For further information, see Supplementary Figure 4. (B) Knockdown experiments of lqfR and CG16908 in Kc167 cells. Kc167 cells were treated with dsRNA as indicated and whole-cell lysates were analyzed after insulin stimulation. Individual or simultaneous depletion of LqfR or CG16908 in Kc167 cells severely reduced the phosphorylation of S6KT398 (P-S6K), PKBS505 (P-PKB) and d4E-BPT37/47 (P-d4E-BP/Thor). Depletion of Tor served as positive control and depletion of EGFP as negative control. The anti-α-tubulin (Tub) staining showed equal loading. Numbers on the right side indicate molecular weight markers (in kDa).
In summary, systematic RNAi-based phenotypic profiling revealed key regulators within the InR/TOR interaction proteome that represent promising new entry points for future genetic and biochemical experiments toward an integrated model on InR/TOR signaling.
dTTT components are required for cell growth in vivo
Given the role of the dTTT complex as a positive regulator for dTOR activity detected in Kc167 cells using RNAi, we next tested whether it also plays a role in TOR-mediated cell growth in vivo. Depletion of both dTTT components, CG16908 and LqfR, in the Drosophila eye by the expression of short hairpin UAS constructs using the ey-GAL4 system resulted in a substantial decrease in eye size (Figure 6A). Likewise, FLP-FRT-mediated mitotic recombination resulted in CG16908 and LqfR mutant clones with a similar reduced growth phenotype as observed in dTOR mutant clones (Figure 6B). In conclusion, the combined biochemical and genetic analysis revealed dTTT as a dTOR-containing complex that is required for the activity of both dTORC1 and dTORC2 and thus plays a critical role in controlling cell growth.
Figure 6.

Regulation of cell growth by LqfR/Tel2 and CG16908/Tti1. (A) Knockdown experiments of lqfR and CG16908 in the Drosophila eye. ey-GAL4 was used to drive expression of short hairpin UAS constructs in the Drosophila eye. Depletion of lqfR (UAS-lqfRRNAi) or CG16908 (UAS-CG16908RNAi) resulted in a severe reduction of eye size, suggesting that both proteins might act as positive growth regulators. A short hairpin construct against CG1315, which does not affect eye size, was used as control (control). (B) Flp-FRT-based mutagenesis of lqfR and CG16908. Flp-FRT-mediated recombination was used to create mutant clones of Tor, lqfR or CG16908 (white tissue) surrounded by wild-type tissue (red tissue) in the Drosophila eye. In comparison to control clones (white tissue, first image; the FRT chromosome carries no mutation), clones mutant for lqfR (white tissue, third image) or CG16908 (white tissue, fourth image) are severely smaller and show a growth disadvantage similar to clones mutant for Tor (white tissue, second image), suggesting a positive growth regulating function of lqfR and the product of CG16908. Exact genotypes are (1) y w ey-Flp; FRT82B, cl3R3w+/FRT82B, (2) y w ey-Flp; FRT40A, cl2L3 w+/FRT40A, Tor2L1, (3) y w ey-Flp; FRT82B, cl3R3w+/FRT82B, lqfRΔ117, (4) y w ey-Flp; FRT82B, cl3R3w+/FRT82B, CG16908MB01483.
Discussion
Information processing by cellular signaling pathways requires the induced assembly and disassembly of specific protein–protein interactions (PPIs) that constitute signaling complexes. These in turn further associate into dynamic molecular interaction networks. Here, we present the first quantitative AP–MS/MS analysis of the insulin-regulated interaction proteome constituting the Drosophila InR/TOR signaling pathway, an evolutionary conserved pathway for the control of cell growth.
The data revealed (1) a systematic data set on the dInR/TOR interaction proteome; (2) new network components, which suggest novel functional links of the canonical InR/TOR pathway to other signaling systems; (3) a set of 22 hormone-sensitive interactions that provide insights into the molecular mechanism of intracellular information processing in response to insulin and (4) a first detailed analysis of TOR complexes in Drosophila, which revealed dTTT, a TOR complex required for dTORC1 and dTORC2 activity and cell growth in vivo.
Systematic analysis of the Drosophila InR/TOR signaling interaction proteome
Our biochemical knowledge on signaling complexes within the metazoan InR/TOR pathway primarily has been inferred from separate studies on signaling complexes from human cells often analyzed in different cell types under varying conditions. However, changes in cellular signaling complexes upon insulin signaling are likely to occur in a concerted fashion, acting at multiple sites within the pathway. To date, no systematic study has been performed to allow for a coherent analysis and an integrated view on the InR/TOR interaction proteome and its response to insulin. Drosophila offers a wide range of genetic tools, which have been successfully used in the past to identify critical pathway components and their relationships underlying cell growth control. In contrast, only few Drosophila studies have been reported to infer the interactions of the genetically defined signaling proteins with other proteins. Such studies are, however, highly needed as proteins almost exclusively carry out their function as part of multiprotein complexes formed by specific and tightly regulated PPIs. Once identified, fly genetics could in turn allow for the effective in vivo functional validation of new protein interactions to generate novel hypotheses on the mechanisms underlying cell growth by the InR/TOR network.
We therefore embarked on a systematic AP–MS/MS analysis of the insulin-sensitive dInR/TOR interaction proteome to gain systems level insights on the molecular basis of insulin signaling in Drosophila cells. Applying stringent statistical filtering, we identified 97 high confidence PPIs, of which 22 are modulated by insulin. How does this information compare to existing data? When compared with interaction data available for Drosophila (http://www.droidb.org), we found only 12 overlapping interactions. Forty-nine interactions from our study overlapped with orthologous interactions found in humans (n=39) and yeast (n=10). In contrast, only three of these 49 conserved interactions have been found in previously published large-scale yeast two-hybrid screens (Giot et al, 2003; Stanyon et al, 2004), illustrating the advantage of the AP–MS/MS approach over previously used technologies to retrieve high quality protein interaction data on a larger scale for the Drosophila proteome.
Signaling systems linked to the dInR/TOR interaction proteome
Given its broad role in controlling cell growth in response to a variety of environmental conditions including insulin and the availability of nutrients and energy, it is likely that signaling by the InR/TOR network may involve hitherto undiscovered mechanisms of regulation and/or may extend toward other concurrently active signaling systems. The presented InR/TOR interaction proteome consists of a number of proteins that have specific roles in cell signaling. These include proteins involved in G-protein signaling, protein phosphorylation, transcription, translation and ubiquitin-dependent degradation. A number of these proteins have not yet been associated with the canonical pathway model and thus may link the InR/TOR pathway to other signaling systems including the JNK/MAPK (MSN, Pellino) and LKB1/AMPK (dNUAK1) pathways. In addition, the presented interactions may also point to novel signaling mechanisms such as ubiquitin-dependent degradation within the dInR module (Mib-2, SkpA, Pall, Lin19) or the dTOR module (Unkempt, Cul-3, COP9). These new linkages provide specific routes for further hypothesis driven genetic experiments to address their functional importance for growth control by the InR/TOR pathway.
Insulin regulation of the InR/TOR interaction proteome
Most cellular processes strongly depend on the coordinated formation and disassembly of protein complexes. Using a recently developed method for label-free quantitative MS analysis of protein complexes (Rinner et al, 2007), we found that 25% of the quantified interactions are regulated by insulin (22% of all identified PPIs). Insulin led to both the induction and dissociation of signaling complexes. Major hormone-induced rearrangements in the interaction network could be observed within membrane proximal InR/Chico/PI3K complexes, 14-3-3 containing complexes and in dTORC1, but not in dTORC2 complexes. For the dInR complexes, the observed changes reflect to a large extent the situation also observed for the human InR complex, where InR once activated recruits and phosphorylates IRS which in turn allows recruitment of PI3K via SH2 domains present in the regulatory PI3K subunit p85 (Virkamaki et al, 1999). Furthermore, we found that the ring-finger E3 ubiquitin ligase Mib-2 dissociates from Chico complexes upon insulin signaling. This raises the interesting question whether Mib-2 plays a role in negatively regulating Chico levels via ubiquitin-dependent degradation in the absence of insulin.
Within the dTOR module, we noted several interactions that are insulin sensitive. These include the insulin-induced dissociation of the Drosophila PRAS40 homolog Lobe from dTOR. In human cells, PRAS40 binds to mTOR in an insulin-dependent manner and may act as a negative regulator of TORC1 in the absence of insulin (Sancak et al, 2007; Vander Haar et al, 2007). Our data suggest that this mechanism is conserved in Drosophila cells. Three different proteins became recruited to dTOR complexes upon insulin treatment: the two dTORC1 substrates d4E-BP, dS6K and Unkempt, a novel dTORC1-associated protein identified in our study. Enhanced recruitment of dS6K and d4E-BP to dTOR complexes could represent a plausible mechanism to enhance phosphorylation of these substrates upon insulin signaling. 4E-BP plays an evolutionary conserved role in suppressing translation via complex formation with the eukaryotic translation initiation factor 4E (eIF-4E) when nutrients or insulin are low. In human cells, it has been shown that mTOR can phosphorylate 4E-BP1, which causes dissociation from eIF-4E and subsequent release of translational suppression (Mamane et al, 2006). Consistent with this model, we found complex formation between d4E-BP and deIF-4E to be decreased when cells were stimulated with insulin. Insulin increased the binding of the ring-finger protein Unkempt to dTORC1. Unkempt has not been linked to TOR signaling in other species. The observed insulin-regulated recruitment of Unkempt to dTORC1 represents a specific entry point for further functional studies on the control of dTOR signaling by this novel dTORC1 component.
Taken together, systematic label-free quantification as performed in this study reliably revealed insulin-regulated changes in several signaling modules. The observed changes demonstrate the evolutionary conservation of signaling dynamics observed in human cells but also point to new insulin controlled interactions, suggesting new molecular mechanism in insulin signal transduction.
TORC1 and TORC2 complexes in Drosophila
TOR is an evolutionary conserved regulator of cell growth and has been studied in a wide range of species. Over the years, building on the data mainly obtained from biochemical studies in human and yeast cells, it has been proposed that TOR forms two major conserved complexes TORC1 and TORC2 (Wullschleger et al, 2006; Bhaskar and Hay, 2007). The detailed composition of Drosophila TOR complexes has not been analyzed. In this study, we used high-density protein interaction data from multiple reciprocal AP–MS/MS experiments involving new quantification methods to determine dTOR complex composition and estimated the relative abundance of dTOR complexes in Drosophila Kc167 cells using directed MS. Overall, the quantitative data from the dTOR purifications indicate that dTORC1 is the most abundant dTOR complex in KC167 cells (for details see Supplementary Table 10). The identified complexes show similarities but also differences to the TOR complexes described in humans (Hurov et al, 2010; Zoncu et al, 2011). dTORC1, the most abundant complex, contains all Drosophila orthologs of mTORC1 components (dTOR, dRaptor, Lobe, dGβL). DEPTOR, another protein associated with mTORC1, is not encoded by the Drosophila genome. Instead, we found Unkempt, a poorly characterized ring-finger protein, which binds dTORC1 in response to insulin. In addition, our systematic RNAi experiments suggest that Unkempt, similar to TSC1/2, may act as a negative regulator of dTORC1 activity in Drosophila cells. No biochemical or functional links to dTOR have been established yet. However, Unkempt has been reported to be among the strongest transcriptionally upregulated genes following inhibition of dTOR with rapamycin in Drosophila S2 cells (Guertin et al, 2006). These results, together with our data, hint at a regulatory feedback relationship between Unkempt and dTOR.
Likewise, dTORC2 contains all orthologs mTORC2 components except DEPTOR and PROTOR, for which no orthologs are found in the Drosophila genome. Both DEPTOR and PROTOR appear during higher vertebrate evolution (they are absent in the two sequenced Ciona species, string-db.org). These findings suggest that TORC1 and TORC2 are composed of evolutionary ancient core complexes, which acquired further functionality during metazoan evolution by the association of additional proteins such as DEPTOR, PROTOR or Unkempt in the case of dTORC1.
Our quantitative analysis using directed MS showed that the majority of dTORC1 and dTORC2 complexes exist as two separate complexes. We also obtained evidence from multiple independent purifications that a small fraction of dTORC1-specific proteins such as dRaptor are associated with TORC2 components dSIN1 and dRictor, which was not observed in previous studies on the corresponding human or yeast TOR complexes. In contrast to previous studies, we used chemical cross-linking and directed proteomics to enhance the sensitivity in our analysis, which may account for the observed differences. Genetic experiments in Drosophila and biochemical experiments in human cells suggest that TOR complexes can exist as dimers (Takahara et al, 2006; Zhang et al, 2006; Yip et al, 2010). Our AP–MS results could be explained by the existence of a small fraction TORC1/TORC2 heterodimers.
dTTT, a conserved dTOR complex required for dTORC1/dTORC2 activity and cell growth
This study revealed the existence of a third dTOR complex here referred to as dTTT. Based on orthology information (see Supplementary Table 8) and reciprocal AP–MS and coimmunoprecipitation data (see Supplementary Figure 3), we propose that dTTT contains, besides dTOR, the gene product of CG16908, LqfR, Pontin, Reptin and Spaghetti. These proteins were specifically identified in dTOR and CG16908 purifications but not in purifications using any of the other dTORC1 and dTORC2 complex components, strongly suggesting that they form complexes independent of dTORC1 and dTORC2. Sequence inspection revealed that CG16908 and LqfR represent orthologs of Tti1 and Tel2 from fission yeast and human cells. In agreement with our results on the Drosophila complex, previous studies in fission yeast and human cells showed complex formation between Tti1, Tel2 and PIK-related kinases including TOR (Hayashi et al, 2007; Hurov et al, 2010; Kaizuka et al, 2010). Recent studies on human Tel2 complexes demonstrated the presence of human Pontin and Reptin (Horejsi et al, 2010; Takai et al, 2010), which together with our results suggests that Tel2-TOR containing complexes are probably larger than originally described. What is the role of Tel2 complexes for TOR signaling? We found that flies mutated or silenced for CG16908 or LqfR showed reduced growth similar to dTOR mutant flies. We also showed that the presence of CG16908 or LqfR is required for substrate phosphorylation by both dTORC1 and dTORC2 complexes, suggesting that the observed growth phenotypes result from lowered dTORC1 and dTORC2 activities in cells lacking functional dTTT complexes. The detailed mechanisms, how dTTT may affect dTORC1 and dTORC2 activity is not clear yet, but may involve the regulation of dTOR protein levels. It has been shown that mice lacking the TTT component Tel2 have decreased the levels of mTOR (Takai et al, 2007). However, it remains to be seen whether silencing of the dTTT components CG16908 and LqfR cause a similar decrease in dTOR protein levels in Drosophila cells. Recent biochemical studies in mammalian cells proposed a model in which Tel2 complexes are necessary for the proper folding of mTOR (Takai et al, 2010). The biochemical details are largely unknown, but it has been proposed that Tel2 may act as a scaffold to coordinate HSP90 or the R2TP/prefoldin chaperones in the assembly of PIK-related kinase complexes (Horejsi et al, 2010; Takai et al, 2010). The R2TP/prefoldin complex contains RPAP3, the human ortholog of Spaghetti, which we also found associated with dTOR complexes, and which shares components with the prefoldin containing URI/RMP complex previously linked to nutrient signaling controlled by mTOR (Gstaiger et al, 2003).
In conclusion, this work represents the first systematic quantitative AP–MS/MS analysis of the insulin-regulated interaction proteome of the Drosophila InR/TOR signaling pathway. Given the high degree of evolutionary conservation of this central growth control pathway, this study represents a valuable framework for future focused studies using genetic and biochemical approaches in a variety of species.
Materials and methods
Expression constructs and cell line generation
Drosophila ORFs of interest were PCR amplified from Drosophila cDNA pools using the Phusion Polymerase (Finnzymes), sequenced and inserted into a Gateway compatible entry vector (pENTR-D-TOPO, Invitrogen). Using LR recombination, the ORFs were transferred from the entry vector into in-house designed expression vectors allowing inducible expression of the bait protein fused to a triple hemagglutinin (HA) affinity-tag from the Metallothionein promoter. For generation of inducibly expressing cell lines, the expression construct was transfected using the Transfecten reagent (Qiagen) to Drosophila Kc167 cells cultivated in Schneiders S2 medium (Invitrogen) containing 10% FBS and 50 μg/ml penicillin, 50 μg/ml streptomycin. In all, 10 μg/ml Blasticidin was used for selection for 5 weeks. Cell pools were tested for positive expression by western blotting using anti-HA antibodies (Covance). For bait expression, the cells were exposed to 600 μM CuSO4 overnight.
Affinity purification
Prior to AP, Kc167 cells were grown in shaking flasks in Schneider S2 medium containing 10% FBS. The cells were serum starved in 2% FBS overnight and bait expression was induced using 600 μM CuSO4 for at least 16 h. Cells were either treated with 100 nM insulin for 20 min or left untreated before harvest. For AP, the cell pellets were lysed on ice for 30 min in 10 ml HNN (50 mM HEPES pH 7.5, 5 mM EDTA, 250 mM NaCl, 0.5% NP-40, 1 mM PMSF, 50 mM NaF, 1.5 mM Na3VO4, protease inhibitor cocktail (Roche), 3 mM DSP) using a tight-fitting Dounce homogenizer. Following cell lysis, reactive DSP was quenched by adding 1 ml 1M Tris, pH 7.5. Insoluble material was removed from the lysate by centrifugation and the supernatant was precleared using 100 μl Protein A-Sepharose (Sigma) for 1 h at 4 °C on a rotating shaker. After removal of the Protein A-Sepharose, 100 μl anti-HA agarose (Sigma) was added to the extracts and incubated for 4 h at 4 °C on a rotating shaker. Immunoprecipitates were washed 4 × with 20 bedvolumes of lysis buffer and 3 × with 20 bedvolumes of buffer without detergent and protease inhibitor. The proteins were released from the beads by adding 3 × 150 μl 0.2 M Glycine, pH 2.5. Following neutralization using 100 μl 1 M NH4CO3, the eluates were treated with 5 mM TCEP for 30 min at 37 °C and alkylated with 10 mM Iodacetamide for 30 min at RT in the dark. For tryptic digest, 1 μg trypsin was added to the eluate and incubated at 37 °C overnight. The tryptic digest was acidified to pH<3 using TFA and purified using C18 Microspincolumns (Harvard Apparatus) according to the protocol of the manufacturer. Dried peptides were resolved in 0.1% formic acid containing 1% acetonitrile and injected into the mass spectrometer.
LC–MS/MS analysis
LC–MS/MS analysis of affinity-purified samples was performed on an LTQ-FT-ICR mass spectrometer (Thermo Electron), which was connected to an online electrospray ion source. Peptide separation was carried out using an Eksigent Tempo nano LC System (Eksigent Technologies) equipped with a RP-HPLC column (75 μm × 15 cm) packed in-house with C18 resin (Magic C18 AQ 3 μm; Michrom BioResources) using a linear gradient from 96% solvent A (0.15% formic acid, 2% acetonitrile) and 4% solvent B (98% acetonitrile, 0.15% formic acid) to 35% solvent B over 60 min at a flow rate of 0.3 μl/min. The data acquisition mode was set to obtain one high-resolution MS scan in the ICR cell at a resolution of 100 000 full width at half maximum (at m/z 400) followed by MS/MS scans in the linear ion trap of the three most intense ions (overall cycle time of 1 s). To increase the efficiency of MS/MS attempts, the charged state screening modus was enabled to exclude unassigned and singly charged ions. Only MS precursors that exceeded a threshold of 150 ion counts were allowed to trigger MS/MS scans. The ion accumulation time was set to 500 ms (MS) and 250 ms (MS/MS) using a target setting of 106 (for MS) and 104 (for MS/MS) ions. After every sample, a peptide mixture containing 200 fmol of [Glu1]-Fibrinopeptide B human (Sigma, Buchs) was analyzed by LC–MS/MS to constantly monitor the performance of the LC–MS/MS system.
For directed mass spectrometry, LC–MS/MS experiments were carried out on an Orbitrap Velos mass spectrometer coupled online to a nano-LC systems (Proxeon Biosystems) and an electrospray ion source (Proxeon Biosystems). LC settings (flow rates and buffer composition) were identical to those described before. Survey full MS spectra were acquired in the Orbitrap with a resolution of 60 000 full width at half maximum (at m/z 400), followed by MS/MS spectra acquired in the linear ion trap of the five most intense ions. Another five MS/MS spectra were triggered on target m/z derived from in silico digests of dTOR core components (dTOR, dRaptor, dRictor, CG3004/dGβL, dSIN1, Lobe, dS6k, Thor/d4E-BP, CG16908, Unkempt, LqfR) using trypsin as protease. The charge state screening modus was enabled to exclude singly charged and uncharged ions. General settings were similar to FT-MS measurements, except CID-based fragmentation was triggered when the precursor exceeded 500 ion counts. The dynamic exclusion duration was set to 15 s. The ion accumulation time was set to 300 ms (MS) and 50 ms (MS/MS). All MS raw data can be accessed via the following ftp site:
ftp://ftp:a@ftp.peptideatlas.org/pub/PeptideAtlas/Repository/PAe001966/ and ftp://ftp:a@ftp.peptideatlas.org/pub/PeptideAtlas/Repository/PAe001967/
MS2 peptide assignment
Acquired MS2 scans were searched against the Drosophila Flybase database version 5.7 using the SORCERER-SEQUEST (TM) search algorithm, which was run on the SageN Sorcerer (Thermo Electron). Data represented in Supplementary Table 9 were searched using MASCOT against a decoy database. In silico trypsin digestion was performed after lysine and arginine (unless followed by proline) tolerating two missed cleavages in fully tryptic peptides. Database search parameters were set to allow phosphorylation (+79.9663 Da) of serine, threonine and tyrosine as a variable modification and carboxyamidomethylation (+57.021464 Da) of cysteine residues as fixed modification. Furthermore, a variable modification of lysine residues (+145.01975) from the carboxyamidomethylated cleaved DSP cross-linker was considered. The fragment mass tolerance was set 0.5 Da and the precursor mass tolerance to 10 p.p.m. Search results were evaluated on the Trans Proteomic Pipeline using Peptide Prophet (v3.0) and Protein Prophet (Keller et al, 2002; Nesvizhskii et al, 2003). For SEQUEST searches, a minimum peptide probability corresponding to <5% false discovery rate (FDR) was required for protein identification. For MASCOT searches, only peptides with a scores of 31 corresponding to a 5% FDR were accepted.
Filtering for specific interaction partners
SAINT (Breitkreutz et al, 2010; Choi et al, 2010) was used to assign confidence scores to observed PPIs. SAINT performs statistical modeling of the quantitative (in this work, using normalized spectral counts) bait–prey association matrix. It generates distributions for true and false interaction and reports the probability score for classification into the two categories. To take advantage of the control purifications (using GFP as a bait protein) generated in parallel with experimental purifications using bait proteins, the data were analyzed using SAINT 2.0 version of the algorithm (Choi et al, 2010). In SAINT 2.0, the false interaction distribution for each prey protein is learned with the help of the quantitative prey abundance data observed in control purifications. After simultaneously learning both true and false interaction distributions from the data, the method determines whether the observation of a prey protein in a particular experimental purification indicates true interaction based on that prey's abundance measurement relative to the prey-specific false and true interaction distributions (Figure 1B) using Bayes’ rule:
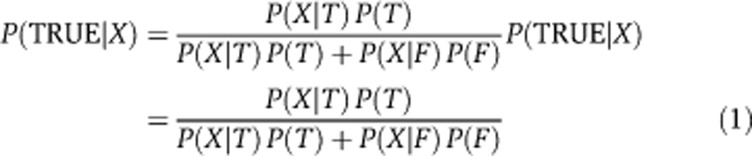 |
Because bait proteins were profiled in two biological replicates for each condition (insulin-treated and -untreated AP–MS experiments), the final SAINT score is computed as an average of the individual probabilities across the replicates. Bait–prey interactions are sorted in a decreasing order of SAINT scores. The FDR associated with a threshold can be approximated from the probabilities in the selected set of interactions (see also Figure 1B):
 |
In running this data set, the quantitative data for both conditions were pooled into a single data set, where identical baits in different conditions were treated as independent baits, and SAINT was applied to this data. High confidence interactions were selected to meet the requirement that the local FDR is controlled at (1–x) × 100% (posterior probability ⩾x). In the first step, network components were defined based on a SAINT posterior probability of 0.99. In addition to the SAINT filtering, which removed the majority of contaminants, we excluded 17 known contaminant proteins (Supplementary Table 3), resulting in the identification of 58 high confidence network components. Protein interactions between high confidence network components were included in the network model, if SAINT probability was at least 0.8. The filtered protein interaction data from this publication have been submitted to the IMEx (http://www.imexconsortium.org) consortium through IntAct (Aranda et al, 2010) and assigned the identifier IM-15821.
Detection of insulin-sensitive protein interaction and analysis of dTOR complexes using label-free quantification
Differences in complex composition between stimulation conditions were quantified based on the ratio of MS1 signal intensity under insulin-stimulated and -unstimulated conditions. In order to increase significance of apparent differences, a three-step dilution series was measured by mixing tryptic peptides from insulin-starved samples with peptides from the corresponding insulin-stimulated samples as described in Rinner et al (2007). MS1 signal intensities obtained for each peptide mapping to a specific protein were grouped according to the dilution factor (0, 30, 100% insulin-treated sample). The median signal intensity of the 10 most intense peptide precursors of the individual bait proteins were used to calculate factors that were used to normalize prey MS1 abundance profiles relative to the bait abundance. Individual peptide dilution profiles were accepted for further analysis when aligned MS1 features were detected at least twice within the profile. Otherwise, the individual peptide profile was discarded from analysis. In cases where only two data points were observed, the profile was extrapolated to cover a full profile. On each valid peptide profile, a linear regression from dilution factor to MS1 signal intensity was performed to determine the difference between the theoretical and observed protein abundance profile. The Pearson product-moment correlation coefficient (r) was used to represent the quality of profiles, indicating insulin-regulated interactions. Data points differing by >2 s.d. from the average intensity for a specific dilution point were discarded as outliers before linear regression. Profiles showing a linear regression with r>0.5 were accepted. The slope inclination of the profile indicates the enrichment of an interaction between insulin-stimulated and -unstimulated condition. Protein profiles were considered as changed when the enrichment factor of the same bait–prey protein profile in both replicate experiment were >1.5 (enriched) or <0.67 (depleted). The validity of a 1.5-fold cutoff was evaluated using triplicate experiments followed by t-test analysis (see Supplementary Figure 1 and Supplementary Table 9).
For analysis of dTOR complexes, defined dTOR core components (dTOR, dRaptor, dRictor, CG3004/dGβL, SIN1, Lobe, CG16908, Unkempt, LqfR) were in silico digested. Predicted double and triply charged tryptic fragment m/z were used as target masses for inclusion list LC–MS/MS analysis. Profile mzXML data for each AP experiment (four purifications per bait protein from insulin-treated and -untreated cells) were used for label-free quantification using Progenesis software Version 3.0 (Nonlinear Dynamics Limited). The raw data were first normalized by the Median TIC. In a second step, the three most intense peptide signal intensities of aligned peptides matching to dTOR core components were extracted from LC–MS maps (TOP3) and the average signal intensity of the TOP3 peptides for each protein in each AP–MS experiment was calculated. The average TOP3 intensities of each protein were then normalized by the average TOP3 intensity of the respective bait protein in each AP–MS experiment. The data were further processed with the Spotfire Decision Site program (TIBCO).
Analysis of protein interaction data
Protein interaction data from AP–MS/MS experiments were assembled into PPI network models using Cytoscape 2.6.1 (http://www.cytoscape.org). GO annotations have been retrieved from PANTHER (http://www.pantherdb.org). To identify known interactions, all PPI data were compared against the information available from the Biogrid database (http://www.thebiogrid.org) version 2.0.53 and DroID (http://www.droidb.org) version 4.0. Human and yeast orthologs have been retrieved from the Ensemble database using biomart version 0.7 (http://www.biomart.org) and Blast searches (http://www.expasy.ch/tools/blast). Orthologous interactions have been searched using DroID (http://www.droidb.org), the yeast biogrid database (http://www.biogrid.org) and an in-house database containing 61 263 human protein interactions from various databases (BIND, DIP, IntAct, HPRD) and published literature (Ramani et al, 2005; Rual et al, 2005; Stelzl et al, 2005).
Fly genetics
The UAS hairpin lines 25 707 (UAS-lqfRRNAi), 47 096 (UAS-CG1315RNAi) and 16908R-1 (UAS-CG16908RNAi) were obtained from the Vienna Drosophila RNAi Center and the National Institute of Genetics (Japan), respectively. CG16908MB01483 (Metaxakis et al, 2005) and the GAL4 driver line ey-GAL4 (Hazelett et al, 1998) were from the Bloomington Drosophila Stock Center. The alleles lqfRΔ117 (Lee et al, 2009) and TOR2L1 (Oldham et al, 2000) as well as the FRT insertions FRT40A and FRT82B (Xu and Rubin, 1993) and the lines y w ey-FLP; FRT40A, w+, cl2L3/CyO and y w ey-FLP; FRT82B, w+, cl3R3/TM6B, Tb, Hu, y+ (Newsome et al, 2000) have been described. Lines carrying mutations on FRT chromosomes were established by meiotic recombination.
For Figure 6A, ey-GAL4 females have been crossed to males carrying the different UAS transgene insertions. For Figure 6B, y w ey-FLP; FRT40A, w+, cl2L3/CyO or y w ey-FLP; FRT82B, w+, cl3R3/TM6B, Tb, Hu, y+ females have been crossed to males of the following lines: (1) y w; FRT82B/TM6B, Tb, Hu, y+, (2) y w; FRT40A, Tor2L1/CyO, (3) y w; FRT82B, lqfRΔ117/TM6B, Tb, Hu, y+, (4) y w; FRT82B, CG16908MB01483/TM6B, Tb, Hu, y+.
Generation of dsRNA
Gene fragments fused to T7 promoters were amplified by PCR (for primer sequences see Supplementary Table 11) and subjected to in vitro transcription using the Ambion Megascript Kit.
Transfection of dsRNA into Drosophila KC cells and cell lysis
In all, 106 Kc cells were plated in 1 ml serum-free medium (six-well plate) and incubated with 10 μg dsRNA. After 30 min, serum-containing medium was added. Five days after transfection, the cells were stimulated with 40 μg insulin (Sigma) for 10 min, washed with PBS and lysed on ice for 30 min in 30 μl lysis buffer (120 mM NaCl, 50 mM Tris–HCl (pH 8), 20 mM NaF, 1 mM EDTA, 6 mM EGTA, 15 mM Na4P2O7, 1 mM benzamidine, 1% NP-40) supplemented with 30 mM para-nitrophenylphosphate and 30 mM β-glycerolphosphate. After centrifugation, the whole-cell lysates were analyzed by SDS–PAGE.
Immunoblotting and quantification of band signal intensities
Hybond ECL membranes (GE Healthcare) and Immobilon Western detection reagent (Millipore) were used for immunoblotting. The following antibodies were used: rabbit anti-phospho-Drosophila p70 S6K (Thr398; Cell Signaling) at 1:9.000, rabbit anti-phospho-AKT (Ser473; Cell Signaling) at 1:9.000, rabbit anti-phospho-4E-BP1 (Thr37/46; Cell Signaling) at 1:1.000 and mouse anti-α-tubulin (DM1A; Sigma) at 1:100.000.
Scanned images of the immunoblots were processed with ImageJ for quantification of band signal intensities. Therefore, individual bands were selected by a rectangle and the total signal intensity was determined after correcting each pixel within the rectangle by a background intensity value. This value was obtained by calculating the average pixel intensity of a region directly above or below the selected rectangle. Subsequently, the signal intensities of the bands corresponding to P-S6K, P-PKB and P-d4E-BP were further corrected for loading differences by normalization with the corresponding tubulin bands. For the heat map in Figure 5, the signal intensities of the bands corresponding to P-S6K, P-PKB and P-d4E-BP were averaged for each RNAi experiment after normalization to EGFP RNAi experiments, which were set to 100%.
Note: The two bands, which are recognized by the anti-phospho-PKB antibody, represent the two phosphorylated PKB isoforms and have been quantified simultaneously.
Supplementary Material
Supplementary figures S1-4
Acknowledgments
TG was supported by a TH grant of the ETH Zürich and a fellowship from the Roche Research Foundation. OR was supported by fellowships of the Deutsche Forschungsgemeinschaft and the Roche Research Foundation. This work was supported by grants from SystemsX.ch (WingX), the Swiss National Science Foundation and by grants from F Hoffmann-La Roche Ltd (Basel, Switzerland) provided to the Competence Center for Systems Physiology and Metabolic Disease (AW and AS).
Author contributions: TG, OR and RBS designed the experiments, analyzed the data, prepared the figures and wrote the manuscript; AW, MAJ, KK, IJ and HS contributed to reagents; HC and AIN performed statistical analysis using SAINT; TG and AS carried out LC–MS analysis; HS, EH and RA contributed to study design and commented on the manuscript; MG designed the study, analyzed the data and wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K et al. (2010) The IntAct molecular interaction database in 2010. Nucleic Acids Res 38: D525–D531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar PT, Hay N (2007) The two TORCs and Akt. Dev Cell 12: 487–502 [DOI] [PubMed] [Google Scholar]
- Biddinger SB, Kahn CR (2006) From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol 68: 123–158 [DOI] [PubMed] [Google Scholar]
- Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras AC, Nesvizhskii AI, Tyers M (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328: 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB (2002) 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol 156: 817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433: 531–537 [DOI] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, Nesvizhskii AI (2010) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods 8: 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL (2004) Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 18: 1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Asara JM, Manning BD (2009) Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol 29: 5657–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11: 1457–1466 [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T et al. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147 [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, Vijayadamodar G, Pochart P, Machineni H, Welsh M, Kong Y, Zerhusen B, Malcolm R, Varrone Z, Collis A, Minto M et al. (2003) A protein interaction map of Drosophila melanogaster. Science 302: 1727–1736 [DOI] [PubMed] [Google Scholar]
- Gstaiger M, Luke B, Hess D, Oakeley EJ, Wirbelauer C, Blondel M, Vigneron M, Peter M, Krek W (2003) Control of nutrient-sensitive transcription programs by the unconventional prefoldin URI. Science 302: 1208–1212 [DOI] [PubMed] [Google Scholar]
- Guertin DA, Guntur KV, Bell GW, Thoreen CC, Sabatini DM (2006) Functional genomics identifies TOR-regulated genes that control growth and division. Curr Biol 16: 958–970 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hatanaka M, Nagao K, Nakaseko Y, Kanoh J, Kokubu A, Ebe M, Yanagida M (2007) Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 12: 1357–1370 [DOI] [PubMed] [Google Scholar]
- Hazelett DJ, Bourouis M, Walldorf U, Treisman JE (1998) Decapentaplegic and wingless are regulated by eyes absent and eyegone and interact to direct the pattern of retinal differentiation in the eye disc. Development 125: 3741–3751 [DOI] [PubMed] [Google Scholar]
- Horejsi Z, Takai H, Adelman CA, Collis SJ, Flynn H, Maslen S, Skehel JM, de Lange T, Boulton SJ (2010) CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol Cell 39: 839–850 [DOI] [PubMed] [Google Scholar]
- Hurov KE, Cotta-Ramusino C, Elledge SJ (2010) A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev 24: 1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590 [DOI] [PubMed] [Google Scholar]
- Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, Iemura S, Natsume T, Mizushima N (2010) Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem 285: 20109–20116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643 [DOI] [PubMed] [Google Scholar]
- Kuhner S, van Noort V, Betts MJ, Leo-Macias A, Batisse C, Rode M, Yamada T, Maier T, Bader S, Beltran-Alvarez P, Castano-Diez D, Chen WH, Devos D, Guell M, Norambuena T, Racke I, Rybin V, Schmidt A, Yus E, Aebersold R et al. (2009) Proteome organization in a genome-reduced bacterium. Science 326: 1235–1240 [DOI] [PubMed] [Google Scholar]
- Lee JH, Overstreet E, Fitch E, Fleenor S, Fischer JA (2009) Drosophila liquid facets-related encodes Golgi epsin and is an essential gene required for cell proliferation, growth, and patterning. Dev Biol 331: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lores P, Visvikis O, Luna R, Lemichez E, Gacon G (2010) The SWI/SNF protein BAF60b is ubiquitinated through a signalling process involving Rac GTPase and the RING finger protein Unkempt. FEBS J 277: 1453–1464 [DOI] [PubMed] [Google Scholar]
- Malmstrom J, Beck M, Schmidt A, Lange V, Deutsch EW, Aebersold R (2009) Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 460: 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N (2006) mTOR, translation initiation and cancer. Oncogene 25: 6416–6422 [DOI] [PubMed] [Google Scholar]
- Metaxakis A, Oehler S, Klinakis A, Savakis C (2005) Minos as a genetic and genomic tool in Drosophila melanogaster. Genetics 171: 571–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK (2009) The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol 19: 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Yoshino K, Miyamoto T, Eguchi S, Oshiro N, Kikkawa U, Yonezawa K (2007) Identification of TBC7 having TBC domain as a novel binding protein to TSC1-TSC2 complex. Biochem Biophys Res Commun 361: 218–223 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ (2000) Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127: 851–860 [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E (2000) Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 14: 2689–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E, Denisov E, Lange O, Remes P, Taylor D, Splendore M, Wouters ER, Senko M, Makarov A, Mann M, Horning S (2009) A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics 8: 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM (2005) Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol 6: R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinner O, Mueller LN, Hubalek M, Muller M, Gstaiger M, Aebersold R (2007) An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat Biotechnol 25: 345–352 [DOI] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S et al. (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437: 1173–1178 [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799–806 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903–915 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- Sato N, Koinuma J, Ito T, Tsuchiya E, Kondo S, Nakamura Y, Daigo Y (2010) Activation of an oncogenic TBC1D7 (TBC1 domain family, member 7) protein in pulmonary carcinogenesis. Genes Chromosomes Cancer 49: 353–367 [DOI] [PubMed] [Google Scholar]
- Schmidt A, Gehlenborg N, Bodenmiller B, Mueller LN, Campbell D, Mueller M, Aebersold R, Domon B (2008) An integrated, directed mass spectrometric approach for in-depth characterization of complex peptide mixtures. Mol Cell Proteomics 7: 2138–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva E, Au-Yeung HW, Van Goethem E, Burden J, Franc NC (2007) Requirement for a Drosophila E3-ubiquitin ligase in phagocytosis of apoptotic cells. Immunity 27: 585–596 [DOI] [PubMed] [Google Scholar]
- Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics 5: 144–156 [DOI] [PubMed] [Google Scholar]
- Stanyon CA, Liu G, Mangiola BA, Patel N, Giot L, Kuang B, Zhang H, Zhong J, Finley RL Jr (2004) A Drosophila protein-interaction map centered on cell-cycle regulators. Genome Biol 5: R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksoz E, Droege A, Krobitsch S, Korn B et al. (2005) A human protein-protein interaction network: a resource for annotating the proteome. Cell 122: 957–968 [DOI] [PubMed] [Google Scholar]
- Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E (2003) Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 5: 559–565 [DOI] [PubMed] [Google Scholar]
- Takahara T, Hara K, Yonezawa K, Sorimachi H, Maeda T (2006) Nutrient-dependent multimerization of the mammalian target of rapamycin through the N-terminal HEAT repeat region. J Biol Chem 281: 28605–28614 [DOI] [PubMed] [Google Scholar]
- Takai H, Wang RC, Takai KK, Yang H, de Lange T (2007) Tel2 regulates the stability of PI3K-related protein kinases. Cell 131: 1248–1259 [DOI] [PubMed] [Google Scholar]
- Takai H, Xie Y, de Lange T, Pavletich NP (2010) Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev 24: 2019–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A, Reuser A, Sampson J, Halley D, van der Sluijs P (1998) Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 7: 1053–1057 [DOI] [PubMed] [Google Scholar]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9: 316–323 [DOI] [PubMed] [Google Scholar]
- Virkamaki A, Ueki K, Kahn CR (1999) Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 103: 931–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: 471–484 [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM (1993) Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117: 1223–1237 [DOI] [PubMed] [Google Scholar]
- Yip CK, Murata K, Walz T, Sabatini DM, Kang SA (2010) Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell 38: 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Billington CJ Jr, Pan D, Neufeld TP (2006) Drosophila target of rapamycin kinase functions as a multimer. Genetics 172: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures S1-4