

Abstract
The Bcl-2 19 kDa interacting protein (BNIP3) is a pro-cell-death BH3-only member of the Bcl-2 family. We previously found that BNIP3 is localized to the nucleus in the majority of glioblastoma multiforme (GBM) tumors and fails to induce cell death. Herein, we have discovered that nuclear BNIP3 binds to the promoter of the apoptosis-inducing factor (AIF) gene and represses its expression. BNIP3 associates with PTB-associating splicing factor (PSF) and HDAC1 (histone deacetylase 1) contributing to transcriptional repression of the AIF gene. This BNIP3-mediated reduction in AIF expression leads to decreased temozolomide-induced apoptosis in glioma cells. Furthermore, nuclear BNIP3 expression in GBMs correlates with decreased AIF expression. Together, we have discovered a novel transcriptional repression function for BNIP3 causing reduced AIF expression and increased resistance to apoptosis. Thus, nuclear BNIP3 may confer a survival advantage to glioma cells and explain, in part, why BNIP3 is expressed at high levels in solid tumors, especially GBM.
Introduction
Glioblastoma multiforme (GBM) is the most malignant form of brain cancer (Gurney and Kadan-Lottick, 2001). The median duration of survival for patients with GBM is <15 months even with aggressive treatment that usually consists of a combination of surgery, radiation, and chemotherapy such as temozolomide (Chamberlain, 2006). Primary GBM arise de novo, and are distinguished from secondary GBM that develop from lower-grade gliomas over time. A pathological characteristic of GBM is extensive regions of necrosis, which indicate areas of hypoxia (defined by <1% oxygen) (Gurney and Kadan-Lottick, 2001; Louis et al., 2007).
The Bcl-2 19 kDa interacting protein 3 (BNIP3) is a pro-cell-death Bcl-2 family member that is upregulated during hypoxia (Vande Velde et al., 2000). When BNIP3 is upregulated it induces caspase-independent cell death by inducing mitochondrial dysfunction (Vande Velde et al., 2000; Kim et al., 2002). BNIP3 is directly upregulated under hypoxic conditions by the transcription factor HIF-1 contributing to hypoxia-induced cell death (Bruick, 2000; Kothari et al., 2003; Bacon and Harris, 2004). Paradoxically, BNIP3 is expressed at high levels in viable cells within hypoxic regions of tumors (Sowter et al., 2001). This is partially caused by nuclear localization of BNIP3 in tumors where BNIP3 fails to associate with the mitochondria and promote cell death (Burton et al., 2006). However, the function of BNIP3 in the nucleus is unclear.
Apoptosis-inducing factor (AIF) is a mitochondrial flavoprotein that plays an important role in mitochondrial function (Modjtahedi et al., 2006). When cells are exposed to stress, AIF is released from the mitochondria, translocates to the nucleus and mediates caspase independent cell death (Susin et al., 1999). Increasing total AIF expression in cells leads to increased sensitivity to cell death whereas knockdown of AIF levels leads to protection from apoptosis in many different cell types (Joza et al., 2001; Porter and Urbano, 2006). During apoptotic signaling, AIF is cleaved removing its transmembrane domain (Porter and Urbano, 2006). Cleaved AIF leaves the mitochondria when permeabilization of the mitochondrial membrane occurs, and it then translocates to the nucleus. Chemotherapeutic agents such as etoposide and cisplatin induce nuclear AIF translocation in many cancer cell lines where it induces chromatin condensation and large-scale DNA cleavage (50 Kb fragments) (Susin et al., 2000; Huerta et al., 2007).
Herein, we describe a novel transcriptional repression activity for the Bcl-2 family member BNIP3. We have discovered that nuclear localized BNIP3 binds to the AIF promoter and represses its expression. In GBM tumors, we observe that nuclear BNIP3 expression correlates with lower levels of AIF expression. BNIP3-mediated repression of AIF expression prevents temozolomide-induced apoptosis. These discoveries may explain why cells that express high levels of BNIP3 remain viable within tumors and how BNIP3 functions within the nucleus to confer a survival advantage to cancer cells.
Materials and Methods
Cell culture and transfections
Human glioma cell lines U251 and U87 were cultured as reported previously (Burton et al., 2006). In transfection experiments, the HEK293 cell line was transfected with Lipofectamine (Invitrogen), and the U87 and U251 cell lines were transfected using Geneporter (GTS) as per the manufacturer's instructions. Stable cell lines were derived in U251 and U87 cells by transfecting with pSUPER shRNA BNIP3 or nontargeting shRNA control and pCDNA3 containing NLS tagged BNIP3 or vector alone control, and selecting with 1.5 mg/ml G418 (Invitrogen BRL).
Plasmids
A partial AIF promoter sequence was inserted into the pGL3 promoter vector and pGL3 control vector as well as a scrambled control using NheI and XhoI restriction enzymes. The AIF promoter region is as follows: [foward (Fwd)] 5+-CTAGCCTCTCCCGCGGCGGGCCTTCCCCATT-GGCCAGCCAAACACAACCGACTGCGGCAGCCAC-3+; and scrambled control: (Fwd) 5+-CTAGCGCCTCGCGCTACGGCCTGCCTAGATGCCTCGGCCAGGTGGTGAACGCCAGCCCACGATC-3+.
Western blotting
Cell lines (U251, U87, HEK293) and frozen primary GBM tissue samples obtained from the Brain Tumor Tissue Bank (BTTB; London, ON, Canada) were lysed for total proteins, membrane proteins, or nuclear proteins as previously published (Burton et al., 2006). The lysates (60 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were incubated with monoclonal antibodies against BNIP3 (1:1000, preferentially recognizes the 30 kDa protein), HDAC1 (1:1000, Upstate Biotech), AIF (1:1000, Cell Signaling), Caspase 8 (1:1000 Cell Signaling), His tag (1:200, Santa Cruz), or βactin (1:50, Sigma). The Western blots were visualized with chemiluminescence (NEN-DuPont).
Real-time RT-PCR
RNA was isolated from U251, U251shRNABNIP3, and U251NLS-BNIP3 cells with RNA-Bee (TEL-TEST) as per the manufacturer's instructions. Real-time PCR was performed with an iCycler using the Biorad iScript SYBR green kit to measure AIF and BNIP3 mRNA levels in U251, U251shRNABNIP3, and U251NLS-BNIP3 stable cells. Intensities of the samples were normalized to a control gene (cyclophilin) mRNA expression run in the same PCR.
Cell death assays
Acridine orange staining.
Cells were trypsinized, removed from the culture plates, and centrifuged in 15 ml sterile tubes. The cells were stained and cell death determined as described by Kothari et al. (2003).
Measurement of sub-G1 peaks with flow cytometry.
HEK293 cells were transfected with siRNA for AIF (Santa Cruz) or a nontargeting control siRNA with RNAIFect (Qiagen) and treated with 1.5 mm TMZ (Schering) for 24 h. Cells were then fixed with 70% ethanol overnight, washed 2× with 1×PBS and resuspended in 400 μl of propidium iodide stain (50 μg/ml) containing 1 μl of RNase (20 μg/ml stock). After a15 min incubation, cells were analyzed for DNA content on a FACS machine (FACSCaliber, BD Biosciences) using the FL2 filter and CellQuest Pro software (BD Biosciences).
Caspase 3 assay
Caspase 3 assays were performed as per the manufacturer's protocol using U251, U251shRNABNIP3, U251NLS-BNIP3 cells grown in six-well dishes treated with 2 mm temozolomide (TMZ) for 48 h or DMSO for control (Sigma-Aldrich). After treatment, cells were lysed with 100 μl of NP40 lysis buffer/well and pelleted as described above. Cell lysate (5 μl) was added to a microfuge tube containing 200 μl of 1× assay buffer containing caspase 3 substrate (acetyl-DEVD-AMC; acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumerin). Tubes were incubated in the dark for 2 h. Fluorescence was read using a fluorimeter (Molecular Devices) set at an excitation of 360 nm and an emission of 460 nm. The cleavage of DEVD in treated cells was compared with a negative control of reaction buffer only and to untreated cells.
Tumor immunostaining
Formalin-fixed paraffin-embedded (FFPE) primary GBM tumor section slides (BTTB) were processed for immunostaining as described (Burton et al., 2006). Slides were incubated with polyclonal anti-BNIP3 1:700 dilution or anti-AIF 1:1000 dilution (Cell Signaling). Fluorescence was visualized and captured with an Olympus BX51 epifluorescence microscope using a Photometrics Cool Snap CF camera.
Luciferase assays
U251, U251shRNABNIP3, U251NLS-BNIP3, U87, U87shRNABNIP3, and U87NLS-BNIP3 stable cells were transiently transfected with the pGL3 promoter luciferase reporter vector and the pGL3 promoter vector with the AIF promoter region that contained the candidate BNIP3 binding site identified by chromatin immunoprecipitation assay (ChIP). All cells were cotransfected with a β-galactosidase vector measured at 414 nm for control of transfection efficiency. Cells were lysed with cell culture lysis reagent (Promega) for 15 min at room temperature. Luciferase activity was measured by a Softmax Pro luminometer (Molecular Devices) for 10 s of relative light units. Results were normalized relative to β-gal activity. t tests were performed to determine significance (Shetty et al., 2005).
Electrophoretic mobility shift assay
Oligonucleotide probes (Invitrogen) were designed for the region of the AIF promoter to which BNIP3 was identified to bind by ChIP. Labeled double stranded probes were generated by annealing the complementary DNA for the wt and mutated sequence and 3+-end labeling with DIG-11-dUTP (Roche). The double-stranded DNA probe contained the potential BNIP3 consensus sequence, sense: 5+-CCCATTGGCCAGCCAAAC-3+ and antisense: 5+-GGAGGACAAAGAGCTGCAC-3+); mutated sense: 5+-CC-CATCGGCCATTCGAAC-3+ and antisense: 5+-GTTCGAATGGCC-GATGGG-3+. Binding reactions were performed as per the DIG gel shift kit protocol (Roche). The BNIP3 antibody and unlabeled “cold” probes were mixed with extracts before probe addition where indicated. The reaction mixtures were then loaded onto a 5% nondenaturing polyacrylamide gel, run at 180 V for 2 h and transferred onto a positively charged nylon membrane with the Biorad semidry transfer apparatus. Chemiluminescent detection reaction was performed as per the Roche kit protocol.
ChIP
ChIP assays were performed as outlined by Spencer et al. (2003) with the U251, U251shRNABNIP3, and U251NLS-BNIP3 cells using the BNIP3, HDAC1, or acetylated histone H3 primary antibodies to immunoprecipitate protein:DNA complexes. DNA was precipitated and resuspended in 20 μl of double-distilled H2O. PCR used primers specific to the AIF promoter region (5+-CCCATTGGCCAGCCAAAC-3+ and 5+-GGAGGACAAAGAGCTGCAC-3+). As a negative control, primers specific for DR4 intron 1 were used (Shetty et al., 2005). For each time point, 1% of the chromatin was assayed for equal loading (input). Input was conducted for each condition tested.
Pulse-field gel electrophoresis
U251, U251shRNABNIP3, and U251NLS-BNIP3 stable cells were treated with 2 mm TMZ for 48 h or DMSO for control, and 1 ng/ml TRAIL (Axxora) for 24 h. Genomic DNA was extracted using QIAamp DNA mini kit (Qiagen) and run on a 1% megabase agarose (Biorad) gel using the Bio-Rad CHEF-DR II pulse-field gel apparatus. The chamber was filled with 0.5% TBE buffer that was maintained at temperatures <14°C. The run parameters were set at 250 V with 10 s pulse for 30 h. When the run was complete the gels were stained in a 0.5 μg/ml ethidium bromide solution for 30 min. and destained in ddH2O for 3 h. DNA was visualized by placing the gel on a UV transilluminator (254–360 nm). Results were quantified by densitometry.
Results
Identification of a BNIP3 binding site in the promoter region of PDCD8
We have previously identified that BNIP3 is expressed under normoxic conditions in primary human astrocytes and glioma cell lines, but it is primarily localized to the nucleus (Burton et al., 2006). Crosslinking cellular proteins to DNA using formaldehyde or cisplatin in U251 glioma cell lines revealed that BNIP3 is bound to DNA with either agent (data not shown). Similar results were observed in U87 glioma cells (data not shown). To identify the specific DNA sequences bound to BNIP3 in glioma cells, a ChIP assay was performed using a polyclonal BNIP3 antibody to immunoprecipitate BNIP3:DNA complexes in U251 cells. After generation of a ChIP DNA library, the library was sequenced and run through BLAST (NCBI) to generate a list of potential BNIP3 binding sites or response elements in promoter regions, introns and exons of specific genes (supplemental Table 1, available at www.jneurosci.org as supplemental material). A potential BNIP3 binding site was identified in the promoter of multiple genes listed in Table 1. One of these genes that we focused on in this study is the AIF gene (PDCD8). This potential binding site is 5 kb upstream of the ATG start site (Fig. 1A). To confirm the putative binding site in vivo, the isolated DNA from the ChIP was PCR-amplified with oligonucleotide primers specific for the region of the AIF promoter that was identified from the ChIP DNA library (Fig. 1B). The specificity of the ChIP assay was confirmed by using U251 cells stably expressing high BNIP3 levels in the nucleus through insertion of a nuclear localization signal (NLS) in the cDNA of BNIP3 within a mammalian expression vector (U251NLS-BNIP3). Using these stably transfected cells, there was increased BNIP3 binding to the AIF promoter, whereas no binding was observed in the negative control. In addition, DNA from the ChIP assay was subjected to PCR with primers specific for a gene not identified by the ChIP library (DR4 intron 1); this failed to reveal any binding of BNIP3 (data not shown). To confirm that BNIP3 binds to the AIF promoter in vitro, an electrophoretic mobility shift assay (EMSA) was performed using an 18 bp double stranded DNA probe for the AIF promoter sequence that was identified to bind BNIP3 by the ChIP assay. When this oligonucleotide probe was incubated with recombinant BNIP3 proteins, a specific band shift was observed with the wild-type (wt) BNIP3 protein (Fig. 1C, lane 5), and the N-terminal BNIP3 fragment (Fig. 1C, lane 2) but not when a scrambled probe was used (Fig. 1C, lanes 4 and 7). The PEST domain of BNIP3 did not shift the oligonucleotide probe, suggesting that this fragment does not interact with DNA (Fig. 1D, lane 6). Binding specificity was confirmed using a 20-fold excess unlabeled (cold competitor) probe to compete out the shifted band (Fig. 1C, lane 3). The BNIP3-DNA complex was supershifted with a monoclonal BNIP3 antibody (Fig. 1D, lane 4) but not with a control IgG antibody (anti-HA) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material) further demonstrating that this protein-DNA complex contains BNIP3. In addition, a specific band could be detected from U251 nuclear lysates incubated with the wt probe (supplemental Fig. 3A, lane 2, available at www.jneurosci.org as supplemental material), that could be competed out using 20-fold excess cold competitor probe (supplemental Fig. 3A, lane 3, available at www.jneurosci.org as supplemental material).
Table 1.
Potential BNIP3 binding sites identified by ChIP library that contain regions of homology

Figure 1.

BNIP3 binds to a response element in the AIF promoter. A, Schematic representation of a BNIP3 binding site upstream of the AIF gene (PDCD8). Numbers above the sequence correspond to sequence numbers from NCBI gi14787430. Numbers below the sequence correspond to the position from the AIF transcription start site. This binding site was identified using a modified ChIP library protocol. B, U251 parental cells and U251 cells stably expressing NLS-BNIP3 were subjected to a ChIP assay with antibodies directed against BNIP3. Sample without added antibody was used as a negative control, whereas genomic DNA was used as the input control. Precipitated DNA was analyzed by PCR using primers specific for an upstream region of the AIF gene, including the region of the candidate BNIP3 binding site. All experiments were repeated three times. C, Recombinant wt BNIP3 and N-terminal (N-term) BNIP3 were incubated with a dsDNA probe specific for the identified BNIP3 binding site in the AIF promoter. Controls used: a scrambled probe (lane 4) and unlabeled probe (“cold competition”) (lanes 3 and 6). Arrows indicate the BNIP3-DNA complexes and the free probe. D, Recombinant wt BNIP3, Delta-CC mutant BNIP3, and PEST BNIP3 were each incubated with a dsDNA probe specific for the BNIP3 binding site in the AIF promoter. A monoclonal BNIP3 antibody was used to supershift the DNA-protein complexes, and HA antibody was used as a control antibody. Arrows indicate the BNIP3-DNA complexes, the antibody-BNIP3-DNA complexes, and the free probe.
Bioinformatic analysis using the Coils program (ch.EMBnet.org) was used to identify a domain in the BNIP3 protein that may bind to DNA since there are no “classical” DNA binding domains within the BNIP3 protein. A potential coiled-coil region was predicted from amino acids 100–120 that contains multiple positive charged amino acids. We deleted five amino acids from this region (amino acids IERRK) in the wt BNIP3 protein (BNIP3 CC mutant protein) and found that that this mutant had significantly less affinity for the oligonucleotide probe of the AIF promoter (Fig. 1D, lane 5, available at www.jneurosci.org as supplemental material). These data support that BNIP3 specifically binds to the promoter region of AIF gene in situ, as well as in vitro.
BNIP3 is bound to the AIF promoter and represses its expression in vitro
To determine whether the BNIP3 binding site in the AIF promoter is important for the regulation of AIF gene transcription, a 60 bp fragment of the AIF promoter containing the putative BNIP3 binding site was cloned into a luciferase reporter gene construct driven by a minimal CMV promoter (vector+AIF promoter) to test whether this region can modulate expression of a reporter gene in vitro. When this vector+AIF promoter was transfected into U251 cells, luciferase expression was significantly decreased compared with vector alone and to scrambled controls (Fig. 2A). To determine whether nuclear BNIP3 is important in repression of AIF gene transcription, we transfected isogenic U251 cell lines stably expressing NLS-BNIP3 (higher levels of BNIP3 in the nucleus compared with parental), and short hairpin (sh) RNA against BNIP3 in a mammalian expression vector (lowers levels of BNIP3 in the nucleus compared with parental) with the luciferase constructs described above. We observed that the expression of luciferase with the vector +AIF promoter remained repressed in cells containing higher nuclear BNIP3 expression (U251NLS-BNIP3). In contrast, shRNA-mediated knockdown of BNIP3 expression (U251shRNABNIP3) resulted in a significant increase in luciferase expression compared with controls (Fig. 2A). Similar results were found using the glioma cell line U87 (data not shown). As a positive control, a luciferase reporter construct with a powerful SV40 enhancer (vector+SV40 enhancer) was transfected into U251 cells (Fig. 2B). This increased luciferase activity compared with vector alone. When the vector + SV40 enhancer and AIF promoter were cotransfected into U251 cells, there was an increase in luciferase activity, indicating that the enhancer abrogates the repressive effect of BNIP3 on expression of the AIF promoter region (Fig. 2B). These data strongly implicate that BNIP3 interacts with this region of the AIF promoter to mediate repression of AIF gene expression.
Figure 2.
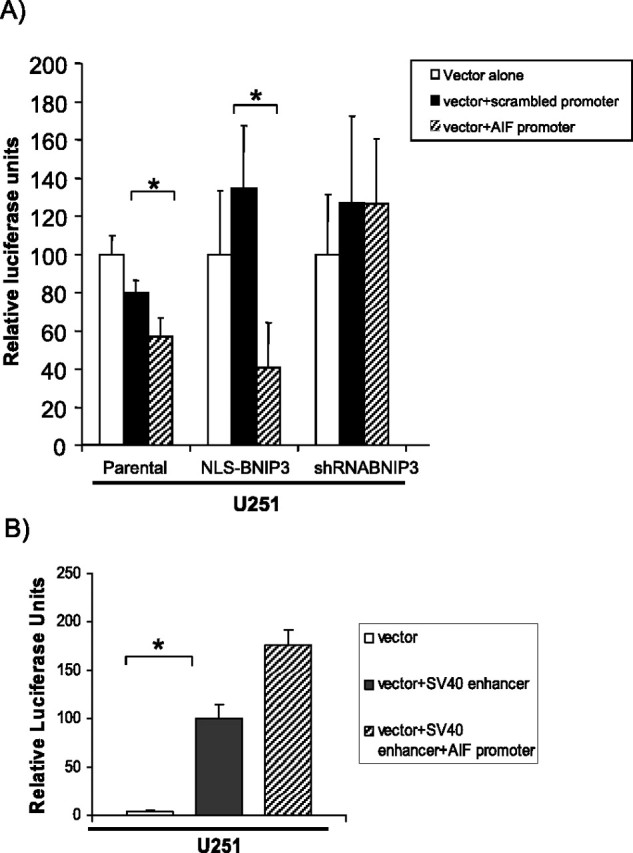
The AIF promoter region containing the BNIP3 binding site represses expression of a luciferase reporter construct when BNIP3 is present in the nucleus of glioma cells. A, A 60 bp DNA fragment containing the candidate BNIP3 binding site (spanning nucleotides 5212–5272 upstream of the AIF start, see Fig. 1A) was cloned into pGL3-p, a luciferase reporter vector containing a minimal SV40 promoter (pGL3-pAIF). U251 parental, U251NLS-BNIP3, and U251shRNABNIP3 stable cells were transiently transfected with pGL3-p (vector alone), pGL3-pAIF (vector+AIF promoter), and pGL3-pscrambled (vector+scrambled promoter). Luciferase activity was measured by a Softmax Pro luminometer. Results were normalized relative to β-gal activity. Error bars represent the SE determined from a minimum of three independent experiments. t tests were performed to determine significance; *p < 0.001, representing statistical significance between pGL3-p vector (control)-transfected cells and the pGL3-p vector containing the AIF promoter. B, The pGL3 vector containing the SV40 promoter and enhancer and the pGL3 vector containing the SV40 promoter and enhancer and the specific AIF promoter region were transfected into U251 cells as a positive control.
Nuclear BNIP3 represses the expression of AIF mRNA and protein
We found that BNIP3 specifically binds to the AIF promoter and is involved in repressing its expression in vitro. We then sought to determine whether AIF mRNA and protein expression is regulated by nuclear BNIP3 in U251 and U87 cell lines. We measured AIF protein and mRNA levels in the U251 and U87 stably transfected cell lines expressing different levels of BNIP3 (NLS-BNIP3 and shRNA BNIP3). We found that the NLS-BNIP3 stable cell lines had significantly less AIF mRNA (Tables 2, 3) and protein (Fig. 3A) expression than parental controls. Concurrently, shRNA BNIP3 stable cells had significantly increased AIF mRNA (Tables 2, 3) and protein levels (Fig. 3A) than parental controls in both U87 and U251 cell lines. U251 stable cells were fixed and immunostained with an AIF antibody to substantiate that AIF protein levels were increased in cells with low levels of nuclear BNIP3 expression (Fig. 3B). These results demonstrate that nuclear BNIP3 negatively regulates the expression of AIF in mammalian cells.
Table 2.
Relative AIF mRNA levels
| Average | SD | t test | |
|---|---|---|---|
| U251 NLS-BNIP3 | 88.13 | 4.34 | p < 0.023 |
| U251 parental | 100 | ||
| U251 shRNABNIP3 | 120.18 | 10.08 | p < 0.042 |
Table 3.
Relative BNIP3 mRNA levels
| Average | SD | t test | |
|---|---|---|---|
| U251 parental | 100 | ||
| U251 shRNABNIP3 | 58.19 | 14.6 | p < 0.019 |
Figure 3.
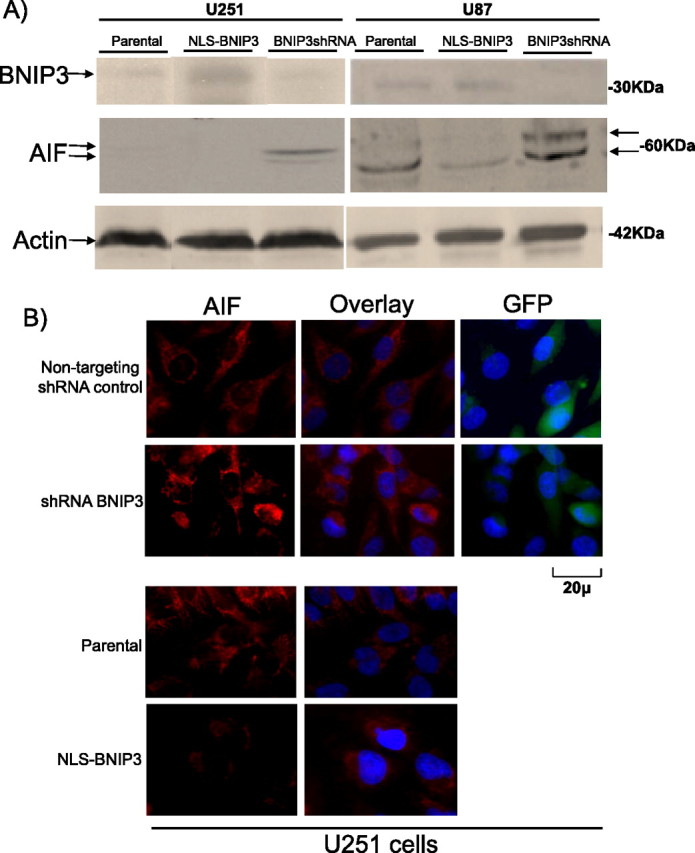
AIF protein is downregulated by nuclear BNIP3 in glioma cells. A, U251 parental, U251shRNABNIP3, U251NLS-BNIP3, U87 parental, U87NLS-BNIP3, and U87shRNABNIP3 stable cells were Western blotted for AIF expression and the blots were stripped and reprobed with antibodies against BNIP3 and actin for loading control. Arrows denote the BNIP3, AIF, and actin proteins where indicated. B, U251 parental, U251 shRNA nontargeting control, U251shRNABNIP3, and U251NLS-BNIP3 stable cells grown on glass coverslips were fixed and immunostained with an antibody to AIF (red), and costained with DAPI for DNA (blue). Images were captured on an epifluorescence microscope. Green staining for GFP was used as a control to indicate stable expression of the pSUPER shRNA plasmid constructs. All experiments were repeated three times.
Decreased AIF protein expression leads to resistance to temozolomide-induced cell death
AIF plays an important role in caspase-independent cell death in many cancer cell types when treated with different chemotherapeutic agents (Porter and Urbano, 2006). To establish the functional consequences in glioma cells with altered AIF levels as a result of nuclear BNIP3, we treated the U251 and U87 stable cell lines that have different nuclear BNIP3 levels with TMZ, an oral chemotherapeutic agent approved for the treatment of newly diagnosed and recurrent GBM (Fig. 4). Cells with high levels of nuclear BNIP3 were more resistant to TMZ-induced cell death (Fig. 4A). Correspondingly, cells with low levels of nuclear BNIP3 expression were more sensitive to TMZ-induced cell death (Fig. 4A). Hence, increasing BNIP3 levels in the nucleus reduced chemotherapy-induced cell death in glioma cells. The mechanism underlying this alteration in drug sensitivity could be caused by the ability of BNIP3 to alter AIF expression. It has been previously established that increased levels of AIF protein are associated with higher AIF-induced DNA cleavage and increased apoptosis (Loeffler et al., 2001; Stambolsky et al., 2006). Higher AIF levels in BNIP3 knockdown cells may lead to an increase in sensitivity to TMZ-induced apoptosis because of an increase in AIF-induced DNA fragmentation. Subsequently, a functional assay for AIF-dependent DNA cleavage was conducted using the U251 stable cell lines that were treated with TMZ. Genomic DNA was isolated and run using pulse-field gel electrophoresis to detect large-scale DNA fragmentation. This approach was used because AIF is known to preferentially induce 50 kb DNA fragmentation that is distinct from caspase-induced DNA fragmentation. We discovered that cells with high nuclear BNIP3 and low AIF expression contain less AIF-induced DNA fragmentation than parental controls when treated with TMZ. In cells that have low nuclear BNIP3 and high AIF expression, increased AIF-induced DNA fragmentation was detected compared with parental controls (Fig. 4B). This result confirms that nuclear BNIP3 regulated AIF-induced DNA fragmentation is caused by a caspase-independent mechanism and that AIF is contributing to TMZ-induced apoptosis.
Figure 4.
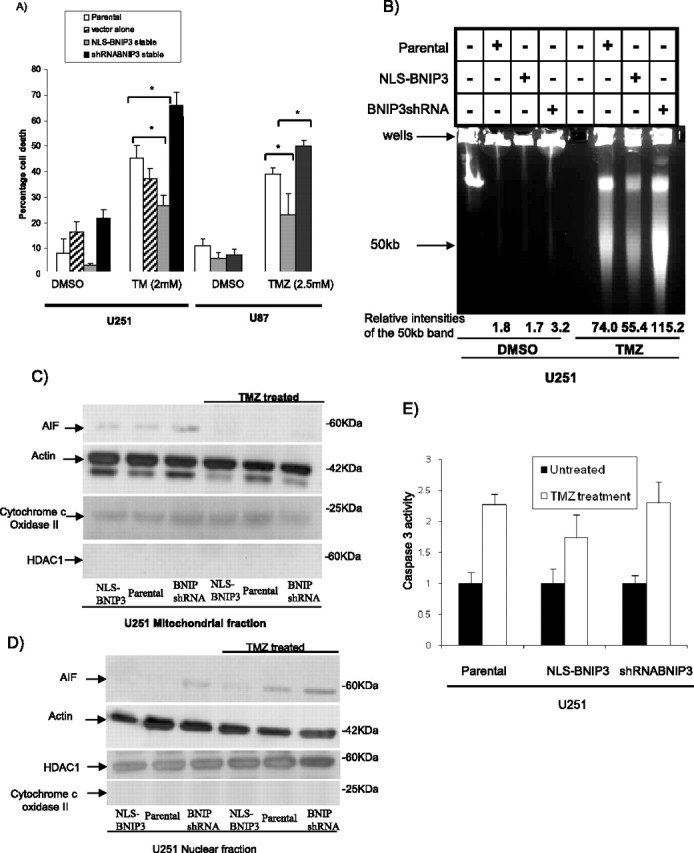
Induction of cell death and AIF-induced large-scale DNA fragmentation is blocked by nuclear BNIP3. A, U251 parental, U251 shRNA nontargeting control, U251shRNABNIP3, and U251NLS-BNIP3 stable cells were treated with 2 mm TMZ for 48 h or with DMSO for control. U87 parental, U87NLS-BNIP3, and U87shRNABNIP3 stable cells were treated with 2.5 mm TMZ for 48 h. Percentage cell death was determined by acridine orange staining. B, U251 parental, U251shRNABNIP3, and U251NLS-BNIP3 stable cells were treated with TMZ as above, and genomic DNA was extracted and then run on a Bio-Rad pulse-field gel apparatus. The lower arrow indicates approximately where the 50 kb DNA fragments migrate. This experiment was repeated three times and results were quantified by densitometry. C, Mitochondrial fractions were isolated from U251 parental, U251shRNABNIP3, and U251NLS-BNIP3 that were untreated, or treated with TMZ as above. The lysates were Western blotted for AIF, HDAC1 (a nuclear protein), and cytochrome c oxidase II (a mitochondrial protein). D, Nuclear fractions were isolated from U251 parental, U251shRNABNIP3, and U251NLS-BNIP3 that were untreated, or treated with TMZ as above. The lysates were Western blotted for AIF, HDAC1, and cytochrome c oxidase. E, U251 parental, U251shRNABNIP3, and U251NLS-BNIP3 stable cells were treated with 100 ng/ml TMZ for 24 h. Relative caspase 3 activity was measured as outlined in Materials and Methods.
To determine whether AIF release from mitochondria is affected by nuclear BNIP3 after TMZ treatment, TMZ-treated U251 cells were fractionated into nuclear and mitochondrial lysates and Western blotted for AIF expression. We found that AIF protein expression in the nucleus increased after TMZ treatment (Fig. 4D). Western analysis was also used to investigate whether AIF protein localizes similarly in U251 stable cell lines containing altered levels of BNIP3 in the nucleus, compared with parental controls when subjected to drug treatment. AIF was visualized in the mitochondria in all three cell lines (U251, U251NLS-BNIP3, and U251shRNABNIP3) when untreated, and subsequently was released from the mitochondria when cells were treated with TMZ (Fig. 4C). Nuclear fractions of U251 and U251NLS-BNIP3 cell lines have low levels of AIF in untreated conditions, whereas U251shRNABNIP3 cells have higher levels corresponding to high expression of AIF. When the cells were treated with TMZ, AIF translocation to the nucleus was increased. Cells with increased nuclear BNIP3 levels showed reduced TMZ-induced AIF nuclear translocation and cells having low nuclear BNIP3 showed the highest TMZ-induced AIF nuclear translocation (Fig. 4D). Cytochrome c oxidase and HDAC1 antibodies were used as controls for the mitochondrial and nuclear fractions, respectively. To confirm that nuclear BNIP3 regulated AIF-induced DNA fragmentation is caused by a caspase-independent mechanism, caspase 3 activity was measured in the U251 stable cell lines and parental controls after TMZ treatment. There was activation of caspase 3 activity after TMZ treatment but there was no statistical difference in caspase 3 activity between U251, U251NLS-BNIP3 and U251shRNABNIP3 cell lines (Fig. 4E).
To ascertain whether the alteration of sensitivity to TMZ was due, at least to some extent, to differences in AIF expression, we used siRNA against AIF to knock down its expression. AIF siRNA effectively knocked down endogenous AIF protein levels in HEK293 cells in a dose dependent manner (Fig. 5A). Cells were transiently transfected with siRNA targeting AIF or nontargeting siRNA and treated with TMZ for 16 and 24 h, and cell death was measured using two types of assays: quantification of cell death by DNA condensation was detected by acridine orange staining (Fig. 5B), and the proportion of cells that were in sub-G1 stage was detected by flow cytometry (Fig. 5C). AIF knockdown cells showed a significant decrease in TMZ-induced cell death after 16 and 24 h of treatment (Fig. 5B) compared with control and nontargeting siRNA-treated cells. In addition, AIF knockdown cells that were treated with TMZ had a significantly lower proportion of cells that were in the sub-G1 peak than the nontargeting siRNA-treated cells (Fig. 5C). These findings indicate that AIF plays an important role in the caspase-independent cell death induced by TMZ.
Figure 5.
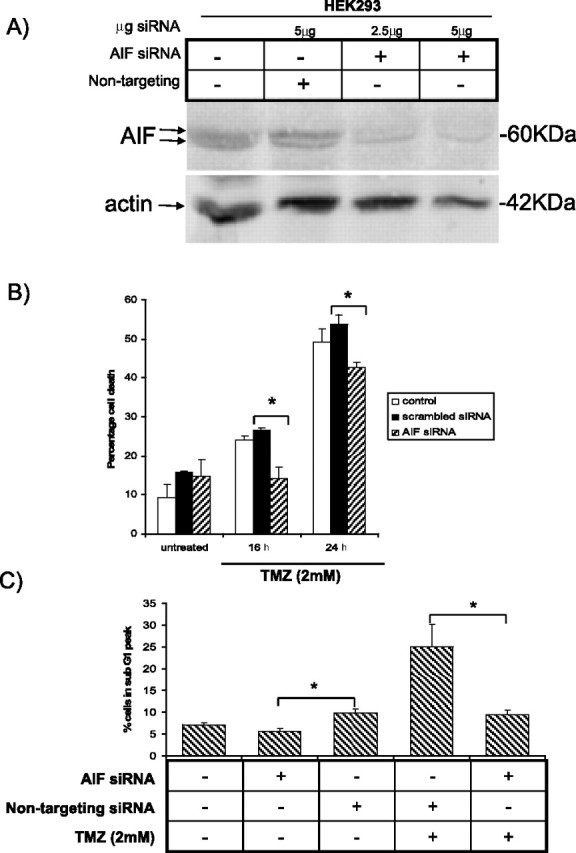
AIF knockdown protects HEK293 from TMZ-induced cell death. A, HEK293 cells were transfected with AIF siRNA (2.5 or 5 μg) or 2.5 μg of a nontargeting siRNA for control. Forty-eight hours after transfection, cells were harvested for Western blot analysis with the AIF antibody. The blot was stripped and reprobed with antibodies against actin for a loading control. B, HEK293 cells were transfected with 2.5 μg of AIF siRNA or nontargeting siRNA control. Forty-eight hours after transfection, cells were treated with 2 mm TMZ for 16 and 24 h, or with DMSO for control. Percentage cell death was determined by acridine orange staining. C, HEK293 cells were transfected with siRNA and treated as in B. Cells were stained with propidium iodide and the percentage of cells in sub-G1 was measured with flow cytometry. Error bars represent the SE determined from three independent experiments. *p < 0.02, representing statistical significance between AIF siRNA-transfected cells treated with TMZ and nontargeting siRNA control-transfected cells treated with TMZ.
Identification of a BNIP3 transcriptional repression complex
To identify BNIP3 binding proteins that could be part of a transcriptional repressor complex, isolated nuclear proteins were exposed to recombinant His-tagged BNIP3 bound to Ni2+-agarose beads. The proteins bound to BNIP3 were analyzed using liquid chromatography tandem mass spectroscopy (LC-MS/MS) to identify proteins that form complexes with nuclear BNIP3. This procedure revealed that His-tagged BNIP3 associated with the PTB-associated splicing factor, PSF (supplemental Table 2, available at www.jneurosci.org as supplemental material). PSF is known to form complexes with transcriptional corepressors such as Sin3A and HDAC1, and repress transcription of specific genes (Shav-Tal and Zipori, 2002). His-tagged BNIP3 pulldown assay and immunoprecipitations were performed with Western analysis to confirm that BNIP3 binds to PSF (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). Since PSF is known to bind with the histone deacetylase HDAC1 (Dong et al., 2007), HDAC1 was immunoprecipitated from U251 cells with an HDAC1 antibody and PSF was detected by Western blot (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Similar to other cell types, PSF associates with HDAC1 in glioma cells suggesting that BNIP3 could form a complex with HDAC1. His tagged pull-down and coimmunoprecipitation experiments were conducted to confirm that BNIP3 binds to HDAC1 (Fig. 6A,B). These experiments indicate that BNIP3 associates in a protein complex consisting of PSF and HDAC1 in the nucleus of glioma cells.
Figure 6.
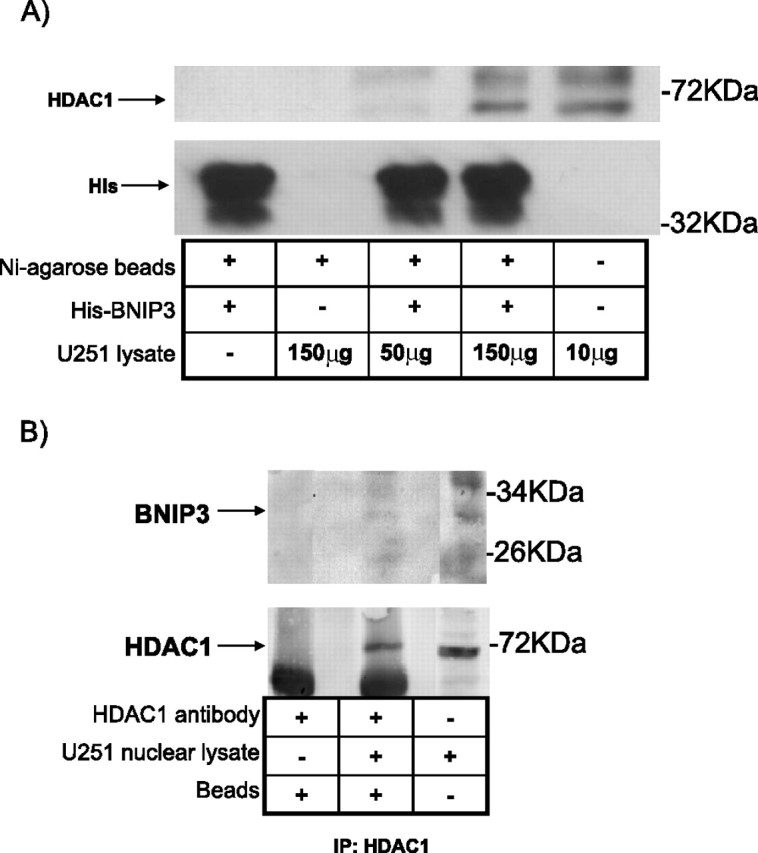
HDAC1 associates with PSF and BNIP3 by His pulldown and immunoprecipitation. A, His-tagged wt BNIP3 was bound to nickel (Ni) agarose beads and 50 or 150 μg of U251 cell lysate was added to columns containing either nickel agarose beads alone or nickel agarose beads with bound His-BNIP3. The beads were washed and proteins bound to His-BNIP3 were eluted with 500 mm imidazole. The resulting elution was Western blotted for HDAC1 expression. As a negative control, Ni agarose beads containing bound His-BNIP3 were also run on the gel. Lysate alone was used as a positive control. The blot was reprobed with an antibody against His tag for control. B, U251 cell lysate was immunoprecipitated (IP) with an anti-HDAC1 antibody and immunoblotted with anti-BNIP3 antibody. The positive control used was lysate alone and the negative control used antibody with IP beads alone. The blot was reprobed with an HDAC1 antibody.
BNIP3 recruits HDAC1 to the promoter region of AIF and represses AIF expression
To determine the presence of the HDAC1 protein at the AIF promoter in vivo in the previously identified region where BNIP3 binds, ChIP assays were performed by immunoprecipitating HDAC1 and amplifying this specific AIF promoter region. HDAC1 was associated with the AIF promoter in U251 cells (Fig. 7A, lane 2), but in cells with low nuclear BNIP3 levels (U251shRNABNIP3), decreased HDAC1 was associated with the AIF promoter, whereas cells with higher nuclear BNIP3 (NLS-BNIP3) had significantly increased HDAC1 associated with the AIF promoter (Fig. 7A). Using a ChIP assay to determine histone H3 acetylation status (a measure of the relative activities of histone acetyltransferases (HATs) and HDACs) of the AIF promoter in vivo, U251shRNABNIP3 cells were found to have higher acetylated histone H3 levels than parental U251 cells, and U251NLS-BNIP3 had lower levels of acetylated histone H3 (Fig. 7B). This finding suggests a mechanism whereby BNIP3 recruits HDAC1 to the promoter of AIF leading to deacetylation of histones and repression of transcription. U251 and U87 cells were then treated with the HDAC inhibitor valproic acid (VPA) (Fig. 7C). There was a significant increase in AIF expression after 24 and 48 h of treatment with VPA in both U87 and U251 cells, showing that HDACs are important in maintaining expression of AIF.
Figure 7.
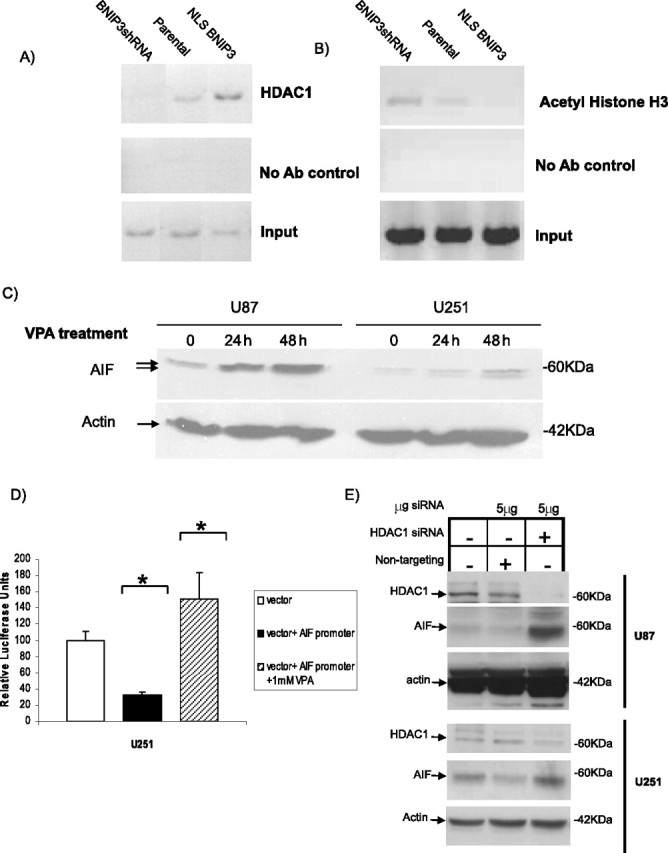
Nuclear BNIP3 recruits HDAC1 to the AIF promoter, leading to silencing of AIF protein expression. A, U251 parental, U251shRNABNIP3, and U251NLS-BNIP3 stable cells were subjected to ChIP using the HDAC1 antibody to immunoprecipitate protein-DNA complexes. Primers specific for the AIF promoter containing the candidate BNIP3 binding site were used for PCR. Sample without added antibody was used as a negative control, whereas genomic DNA was used as the input control. B, The same procedure as in A was repeated with an antibody specific for acetylated histone H3 to determine acetylation status of the specific AIF promoter region. C, U251 and U87 glioma cells were treated with 1 mm VPA. Cell lysates were prepared at 24 and 48 h, and samples were Western blotted for AIF expression. The blots were stripped and reprobed with antibodies against actin for a control. D, U251 cells were transiently transfected with the pGL3-p vector and the pGL3-pAIF vector. Cells transfected with pGL3-pAIF were also treated with 1 mm VPA for 24 h. Luciferase assays and data analysis were performed as described in Figure 2; *p < 0.01. E, U251 and U87 cells were transfected with 5 μg of HDAC1 siRNA or 5 μg of a nontargeting siRNA for control. Forty-eight hours after transfection, cells were harvested for Western blot analysis and probed with HDAC1 and AIF-specific polyclonal antibodies. The blots were stripped and reprobed with antibodies against actin for a loading control.
Luciferase assays were repeated, as in Figure 2, with the vector alone and vector+AIF promoter. U251 cells transfected with vector+AIF promoter were then treated with 1 mm VPA for 24 h to determine whether the promoter region of AIF continues to act as a repressor of transcription when HDACs are inhibited. We found that the AIF promoter region no longer represses transcription of the reporter gene when HDACs are inhibited suggesting that HDACs, as well as BNIP3, play an important role in regulating this region of the AIF promoter (Fig. 7D). U251 cells were then transfected with HDAC1 siRNA to confirm that the repression of transcription of AIF is due specifically to HDAC1 (Fig. 7E). When HDAC1 levels were knocked down, AIF protein expression increased. Overall, these results indicate that BNIP3 represses AIF expression by binding to the AIF promoter and recruiting HDAC1, a corepressor of transcription.
Nuclear localized BNIP3 in primary human GBM tumors correlates with low AIF expression
Nuclear BNIP3 has been detected in GBMs (Burton et al., 2006), lung (Giatromanolaki et al., 2004), and breast tumors (Sowter et al., 2003; Tan et al., 2007). Also, AIF expression has been observed to be decreased in several different tumors (Delettre et al., 2006; Lee et al., 2006). Both nuclear BNIP3 and AIF expression have been shown to play an important role in tumor progression but a relationship between the two has not yet been established. To determine whether nuclear BNIP3 correlated with levels of AIF expression in GBM tumors (WHO grade IV astrocytomas), we immunostained FFPE sections of primary GBM tumors with antibodies against BNIP3 and AIF. Tumors were counterstained for DNA using DAPI to identify the nucleus of the cells. To examine the localization of BNIP3 in GBM tumors, 38 tumors were graded according to whether BNIP3 was highly nuclear in localization, moderately nuclear, or with low/no nuclear staining. The same tumors were similarly graded for AIF expression. When grading of the tumors was analyzed by a χ2 test, tumors with high nuclear BNIP3 levels had corresponding low levels of AIF expression, whereas tumors with low nuclear BNIP3 levels had high AIF expression levels (p < 0.005) (Table 4). Two representative GBM tumors containing high and low nuclear BNIP3 expression, respectively, immunostained for AIF show that high nuclear BNIP3 correlated with lower AIF expression (Fig. 8A). Consistent with these findings, by immunoblotting of lysates obtained from unfixed frozen GBM samples matched to the FFPE tumor sections, nuclear BNIP3 expression was also associated with lower AIF expression (Fig. 8B). This provides strong evidence consistent with nuclear BNIP3 repression of the expression of the AIF gene in GBM tumors.
Table 4.
Primary GBM tumors that have high nuclear BNIP3 levels have low AIF expression
| BNIP3localization | AIF expression | |||
|---|---|---|---|---|
| Expressed(0–+) | Overexpressed(++) | Stronglyoverexpressed(+++) | Total | |
| Nuclear (+++) | 9 | 2 | 2 | 13 |
| Moderate nuclear (++) | 3 | 2 | 2 | 7 |
| Low nuclear (0–+) | 7 | 2 | 11 | 20 |
Chi-squared test: p < 0.005.
Figure 8.
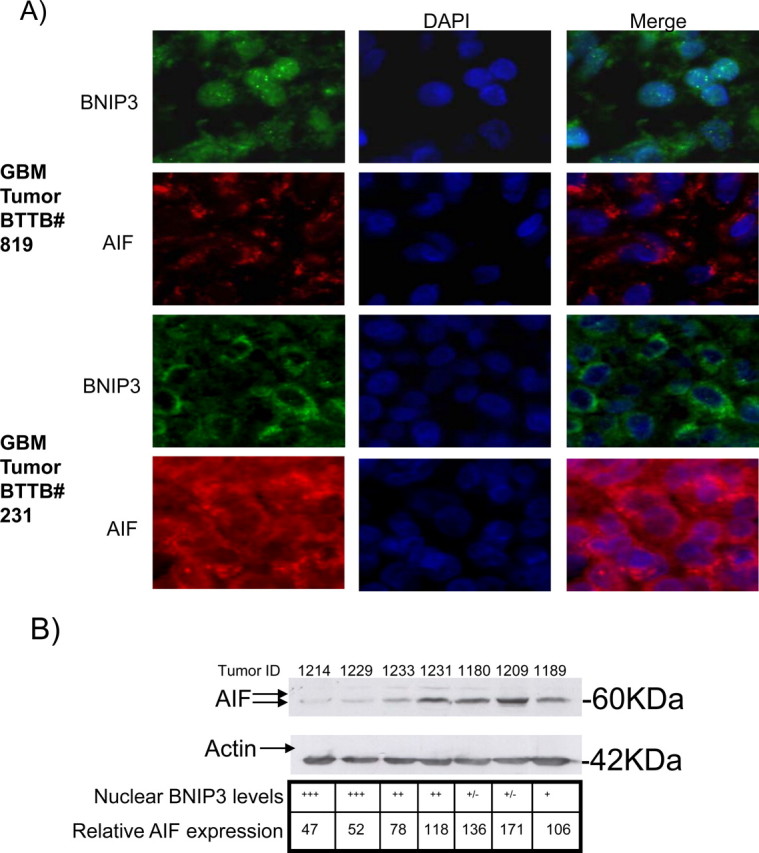
Primary GBM tumors that have high nuclear BNIP3 levels have low AIF expression. A, Formalin-fixed paraffin- embedded sections of representative primary GBM tumors sections were immunostained with antibodies against BNIP3 (green) and AIF (red). DNA was stained with DAPI (blue), and the slides were analyzed on an Olympus epifluorescence microscope. B, Representative matched frozen GBM tumor tissues were lysed to extract total protein and analyzed by Western blot for AIF expression. The blots were stripped and reprobed with actin antibodies for a loading control. The grading for nuclear BNIP3 levels (determined by immunofluorescence) is indicated for each tumor. Nuclear staining was graded as follows: +++ for high nuclear staining, ++ for moderate nuclear staining, + for low nuclear staining, and +− for undetectable nuclear staining. Three independent experiments were quantified by densitometry with Quantity One (Bio-Rad), and averaged to obtain AIF expression relative to actin loading controls.
Discussion
We have identified that nuclear BNIP3 acts as a transcriptional repressor binding to the promoter region of the AIF gene, thereby preventing apoptosis. Indeed, the region where BNIP3 binds contains a sequence that is homologous to a consensus repressor signal for neural-specific genes (Karadsheh and Delpire, 2001). Novel mechanisms have also been proposed for the function of other Bcl-2 family members in the nucleus (Zinkel et al., 2007). A recent study identified that Bcl-2 inhibits transcription factor activity in the nucleus and alters DNA repair enzyme expression (Massaad et al., 2004; Wang et al., 2008), contributing to altered DNA repair and genomic instability in cancer cells with high levels of Bcl-2 expression.
Solid tumors, especially highly proliferative, invasive tumors such as GBMs, are constantly subjected to metabolic stress. The tumor cells experience nutrient and oxygen deprivation because of loss of vasculature and undergo stringent selection, in which genetic and epigenetic changes occur to establish a phenotype that can survive under these conditions. We have discovered that normal astrocytes localize BNIP3 to the nucleus preventing its prodeath function (Burton et al., 2006). BNIP3 is also detected at very low levels in the nucleus of astrocytes in normal brain sections. This mechanism is exploited in GBM tumors, where upregulation of BNIP3 in the nucleus occurs in a large subset of these tumors (60% of tumors examined displayed some nuclear staining, with 32.5% having exclusive nuclear staining). Nuclear localization of BNIP3 is also reported to occur in lung (Giatromanolaki et al., 2004), breast (Tan et al., 2007), and cervical tumors (Leo et al., 2006), as well as in focal brain ischemia (Schmidt-Kastner et al., 2004). In ductal carcinoma in situ (DCIS) of the breast, 25% of tumors exhibited nuclear BNIP3 staining and 6% had primarily nuclear BNIP3 (Tan et al., 2007). When nuclear BNIP3 was used as a variable to determine a difference in patient outcome, nuclear BNIP3 was significantly correlated with a shorter disease free survival (Tan et al., 2007). In lung tumors, 15% of tumors exhibited nuclear BNIP3 staining and 5% had primarily nuclear BNIP3. It was observed that nuclear localization of BNIP3 occurred in a subset of cases that had a particularly poor prognosis (Giatromanolaki et al., 2004). These studies, in combination with the unique repressor function for BNIP3, suggest an important mechanism for how cancer cells survive stress and become resistant to cancer therapy.
Proteins that interact with BNIP3 have been implicated to play a role in its function. BNIP3 dimerization is necessary for its cell death function, whereas BNIP3 binding to BCL-2 and BCL-XL, suppress its ability to induce cell death (Ray et al., 2000). When BNIP3 is localized to the membrane it binds to Rheb and blocks the mTOR pathway, inhibiting cell growth when cells are under hypoxic conditions (Li et al., 2007). We have identified PSF as a BNIP3 binding partner in the nucleus as well as other proteins that are contained in complexes that bind to RNA/DNA (supplemental Table 2, available at www.jneurosci.org as supplemental material). PSF is a transcriptional corepressor, associating with Sin3A and HDAC1, which leads to the repression of target genes such as the androgen receptor and P450scc (Shav-Tal and Zipori, 2002). We also found that HDAC1 associates with BNIP3 and participates in repression of AIF expression. This suggests a model where HDAC1 and PSF form a complex with BNIP3 directly or indirectly resulting in transcriptional repression. The association of BNIP3 with PSF and HDAC1 could also serve as a nuclear localization mechanism for BNIP3. BNIP3 does not contain a canonical nuclear localization signal but is found in the nucleus of glioma cells. PSF and HDAC1 contain nuclear localization signals and could maintain BNIP3 in the nucleus.
The transcription factors regulating AIF expression are largely unknown but a recent study indicated that p53 induces AIF expression through binding to p53 response elements in the AIF gene (Stambolsky et al., 2006). In H1229 cells, AIF knockdown partially protected against p53-induced cell death and when p53 was expressed, cisplatin-treated cells had increased DNA fragmentation induced by AIF (Stambolsky et al., 2006). Our findings indicate that BNIP3 regulates AIF expression resulting in reduced TMZ-induced cell death due, in part, to decreased AIF-induced DNA cleavage. In GBM tumors, decreased AIF levels may provide a survival advantage because GBM tumor cells would then be more resistant to stress such as chemotherapy that releases AIF from the mitochondria. However, these cells would still maintain the essential role of AIF in the mitochondria. This reduction of nuclear AIF could also drive the selection of tumor cells that have adapted mechanisms to localize BNIP3 to the nucleus. When caspase-dependent cell death is inhibited and AIF is knocked down, DNA condensation is blocked when these cells are treated with cell-death-inducing agents such as staurosporine, etoposide and glutamate (Porter and Urbano, 2006). Recent reports have observed that AIF is downregulated in many tumor types (Delettre et al., 2006), and depletion of the AIF protein in malignant skin, colon, lung, and breast cells contributes to chemoresistance, supporting AIF pro-cell-death functions in the context of cancer (Huerta et al., 2007). A HER2 antibody-AIF protein chimeric molecule significantly prolonged survival of mice with tumors that over-express HER2 (Yu et al., 2006). Indeed, nuclear BNIP3 repressed AIF expression to a greater extent than other apoptotic proteins as determined by a cDNA microarray analysis (data not shown). Together, BNIP3 repression of AIF expression could render tumors resistant to caspase independent apoptosis.
BNIP3 transcriptional repression of AIF was also reversed by HDAC inhibitors. We found that the HDAC inhibitor VPA and HDAC1 knockdown experiments resulted in an increase of AIF expression, suggesting that HDAC inhibitors could be used to treat tumors with nuclear BNIP3 expression. Indeed, HDAC inhibitors have been shown to induce apoptosis in glioma cells (Komata et al., 2005). Hence, HDAC inhibitors might mediate de-repression of BNIP3 transcriptional activity and provide a novel target for development of treatments for chemotherapy resistant GBM tumors.
Overall, we have discovered that nuclear BNIP3 participates in a multiprotein complex with HDAC1 and PSF to downregulate AIF expression in glioma cells, leading to resistance to TMZ-induced cell death, due in part to reduced AIF-dependent DNA fragmentation in glioma cells. These results provide a novel and potentially important mechanism underlying how nuclear BNIP3 may repress gene expression thereby resulting in a survival advantage to tumor cells. This could explain why expression of nuclear BNIP3 is increased in solid tumors, especially those with a poor prognosis such as GBM.
Footnotes
This work was supported by a CancerCare Manitoba Foundation and Canadian Institutes of Health Research Grant MOP 64330 to D.D.E. and S.B.G. S.B.G. holds a Manitoba Research Chair and T.R.B. was supported by a graduate scholarship from the Terry Fox Foundation awarded by the National Cancer Institute of Canada.
References
- Bacon AL, Harris AL. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann Med. 2004;36:530–539. doi: 10.1080/07853890410018231. [DOI] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton TR, Henson ES, Baijal P, Eisenstat DD, Gibson SB. The pro-cell death Bcl-2 family member, BNIP3, is localized to the nucleus of human glial cells: implications for glioblastoma multiforme tumor cell survival under hypoxia. Int J Cancer. 2006;118:1660–1669. doi: 10.1002/ijc.21547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain MC. Treatment options for glioblastoma. Neurosurg Focus. 2006;20:E2. doi: 10.3171/foc.2006.20.4.12. [DOI] [PubMed] [Google Scholar]
- Delettre C, Yuste VJ, Moubarak RS, Bras M, Robert N, Susin SA. Identification and characterization of AIFsh2, a mitochondrial apoptosis-inducing factor (AIF) isoform with NADH oxidase activity. J Biol Chem. 2006;281:18507–18518. doi: 10.1074/jbc.M601751200. [DOI] [PubMed] [Google Scholar]
- Dong X, Sweet J, Challis JR, Brown T, Lye SJ. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol Cell Biol. 2007;27:4863–4875. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giatromanolaki A, Koukourakis MI, Sowter HM, Sivridis E, Gibson S, Gatter KC, Harris AL. BNIP3 expression is linked with hypoxia-regulated protein expression and with poor prognosis in non-small cell lung cancer. Clin Cancer Res. 2004;10:5566–5571. doi: 10.1158/1078-0432.CCR-04-0076. [DOI] [PubMed] [Google Scholar]
- Gurney JG, Kadan-Lottick N. Brain and other central nervous system tumors: rates, trends, and epidemiology. Curr Opin Oncol. 2001;13:160–166. doi: 10.1097/00001622-200105000-00005. [DOI] [PubMed] [Google Scholar]
- Huerta S, Heinzerling JH, Anguiano-Hernandez YM, Huerta-Yepez S, Lin J, Chen D, Bonavida B, Livingston EH. Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells: roles of Fas, Apaf-1, NFkappaB, IAPs, Smac/DIABLO, and AIF. J Surg Res. 2007;142:184–194. doi: 10.1016/j.jss.2006.12.551. [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zúñiga-Pflücker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- Karadsheh MF, Delpire E. Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J Neurophysiol. 2001;85:995–997. doi: 10.1152/jn.2001.85.2.995. [DOI] [PubMed] [Google Scholar]
- Kim JY, Cho JJ, Ha J, Park JH. The carboxy terminal C-tail of BNip3 is crucial in induction of mitochondrial permeability transition in isolated mitochondria. Arch Biochem Biophys. 2002;398:147–152. doi: 10.1006/abbi.2001.2673. [DOI] [PubMed] [Google Scholar]
- Komata T, Kanzawa T, Nashimoto T, Aoki H, Endo S, Kon T, Takahashi H, Kondo S, Tanaka R. Histone deacetylase inhibitors, N-butyric acid and trichostatin A, induce caspase-8- but not caspase-9-dependent apoptosis in human malignant glioma cells. Int J Oncol. 2005;26:1345–1352. doi: 10.3892/ijo.26.5.1345. [DOI] [PubMed] [Google Scholar]
- Kothari S, Cizeau J, McMillan-Ward E, Israels SJ, Bailes M, Ens K, Kirshenbaum LA, Gibson SB. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene. 2003;22:4734–4744. doi: 10.1038/sj.onc.1206666. [DOI] [PubMed] [Google Scholar]
- Lee JW, Jeong EG, Soung YH, Kim SY, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. Immunohistochemical analysis of apoptosis-inducing factor (AIF) expression in gastric carcinomas. Pathol Res Pract. 2006;202:497–501. doi: 10.1016/j.prp.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Leo C, Horn LC, Höckel M. Hypoxia and expression of the proapoptotic regulator BNIP3 in cervical cancer. Int J Gynecol Cancer. 2006;16:1314–1320. doi: 10.1111/j.1525-1438.2006.00394.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, You M, Guan KL. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282:35803–35813. doi: 10.1074/jbc.M705231200. [DOI] [PubMed] [Google Scholar]
- Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, Brothers G, Penninger JM, Kroemer G. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad CA, Portier BP, Taglialatela G. Inhibition of transcription factor activity by nuclear compartment-associated Bcl-2. J Biol Chem. 2004;279:54470–54478. doi: 10.1074/jbc.M407659200. [DOI] [PubMed] [Google Scholar]
- Modjtahedi N, Giordanetto F, Madeo F, Kroemer G. Apoptosis-inducing factor: vital and lethal. Trends Cell Biol. 2006;16:264–272. doi: 10.1016/j.tcb.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Porter AG, Urbano AG. Does apoptosis-inducing factor (AIF) have both life and death functions in cells? Bioessays. 2006;28:834–843. doi: 10.1002/bies.20444. [DOI] [PubMed] [Google Scholar]
- Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Aguirre-Chen C, Kietzmann T, Saul I, Busto R, Ginsberg MD. Nuclear localization of the hypoxia-regulated pro-apoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res. 2004;1001:133–142. doi: 10.1016/j.brainres.2003.11.065. [DOI] [PubMed] [Google Scholar]
- Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett. 2002;531:109–114. doi: 10.1016/s0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- Shetty S, Graham BA, Brown JG, Hu X, Vegh-Yarema N, Harding G, Paul JT, Gibson SB. Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol Cell Biol. 2005;25:5404–5416. doi: 10.1128/MCB.25.13.5404-5416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C, Harris AL. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol. 2003;201:573–580. doi: 10.1002/path.1486. [DOI] [PubMed] [Google Scholar]
- Spencer VA, Sun JM, Li L, Davie JR. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods. 2003;31:67–75. doi: 10.1016/s1046-2023(03)00089-6. [DOI] [PubMed] [Google Scholar]
- Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prévost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EY, Campo L, Han C, Turley H, Pezzella F, Gatter KC, Harris AL, Fox SB. BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin Cancer Res. 2007;13:467–474. doi: 10.1158/1078-0432.CCR-06-1466. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Gao F, May WS, Zhang Y, Flagg T, Deng X. Bcl2 negatively regulates DNA double-strand-break repair through a nonhomologous end-joining pathway. Mol Cell. 2008;29:488–498. doi: 10.1016/j.molcel.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CJ, Jia LT, Meng YL, Zhao J, Zhang Y, Qiu XC, Xu YM, Wen WH, Yao LB, Fan DM, Jin BQ, Chen SY, Yang AG. Selective proapoptotic activity of a secreted recombinant antibody/AIF fusion protein in carcinomas overexpressing HER2. Gene Ther. 2006;13:313–320. doi: 10.1038/sj.gt.3302672. [DOI] [PubMed] [Google Scholar]
- Zinkel SS, Hurov KE, Gross A. Bid plays a role in the DNA damage response. Cell. 2007;130:9–10. doi: 10.1016/j.cell.2007.06.035. author reply 10–11. [DOI] [PubMed] [Google Scholar]