

Abstract
The scavenger receptor, class B, type I (SR-BI) binds high-density lipoprotein (HDL) and mediates selective delivery of cholesteryl esters (CEs) to the liver and steroidogenic cells of the adrenal and gonads. Although it is clear that the large extracellular domain (ECD) of SR-BI binds HDL, the role of ECD in the selective HDL-CE transport remains poorly understood. In this study, we used a combination of mutational and chemical approaches to systematically evaluate the contribution of cysteine residues, especially six cysteine residues of ECD, in SR-BI-mediated selective HDL-CE uptake, intracellular trafficking and SR-BI dimerization. Pretreatment of SR-BI overexpressing COS-7 cells with disulfide (S-S) bond reducing agent, β-mercaptoethanol (100 mM) or dithiothreitol (DTT) (10 mM) modestly, but significantly impaired the SR-BI mediated selective HDL-CE uptake. Treatment of SR-BI overexpressing COS-7 cells with the optimum doses of membrane permeant alkyl methanethiosulfonate (MTS) reagents, positively charged MTSEA or neutral MMTS that specifically react with the free sulfhydryl group of cysteine reduced the SR-BI-mediated selective HDL-CE uptake, indicating that certain intracellular free cysteine residues may also be critically involved in the selective cholesterol transport process. In contrast, use of membrane impermeant MTS reagent, positively charged MTSET and negatively charged MTSES showed no such effect. Next, the importance of eight cysteine residues in SR-BI expression, cell surface expression, dimer formation and selective HDL-derived CE transport was evaluated. These cysteine residues were replaced either singly or in pairs with serine and the mutant SR-BIs expressed in either COS-7 or CHO cells. Four mutations, C280S, C321S, C323S or C334S of the ECD, either singly or in various pair combinations, resulted in significant decreases in SR-BI (HDL) binding activity, selective-CE uptake, and trafficking to cell surface. Surprisingly, we found that mutation of the two remaining cysteine residues, C251 and C384 of the ECD, had no effect on either SR-BI expression or function. Other cysteine mutations and substitutions were also without any effect. Western blot data indicated that single and double mutants of C280, C321, C323 and C334 residues strongly favor dimer formation. However, they are rendered non-functional presumably due to mutation-induced formation of aberrant disulfide linkages resulting in inhibition of optimal HDL binding and, thus, selective HDL-CE uptake. These results provide novel insights about the functional role of four cysteine residues, C280, C321, C323 and C334 of SR-BI ECD domain in SR-BI expression and trafficking to cell surface, its dimerization, and associated selective CE transport function.
Introduction
Scavenger receptor class B, type I (SR-BI)2 is a cell surface glycoprotein of molecular mass ∼82 kDa that mediates selective uptake of HDL-derived cholesteryl esters (CE) (1-5), a process in which HDL-CE is taken into cells without parallel uptake and degradation of the HDL particle itself (6-8). It is expressed most abundantly in the liver and steroidogenic cells of the adrenal gland and ovary (9-12), where the selective uptake of HDL-CE is greatest. [Selective uptake is a major route for delivery of HDL-CE to steroidogenic tissues (for steroid hormone biosynthesis) and the liver (for bile acid synthesis) in rodents (1,2,7,8,13-17) and appears to be a major route for delivery in human steroidogenic cells as well (1,8,18,19)]. SR-BI expression in steroidogenic cells of the adrenal gland and gonads is regulated by trophic hormones (gonadotropins and adrenocorticotropic hormones) and their second messenger, cAMP, coordinately with the selective uptake and steroidogenesis (9-12,20,21). The functional expression of hepatic SR-BI, however, is primarily regulated post-transcriptionally via protein-protein interaction with PDZ (PSD-95, Discs large, ZO-1) domain containing protein, PDZK1 (22-24). It is of interest that in steroidogenic tissues, SR-BI is preferentially localized on microvilli (11,12,21) that form microvillar channels and constitute a microvillar compartment (11,12,21,25-27). It is in these microvillar channels that HDL particles are trapped in an effort to boost the efficiency of the selective HDL-CE transport process (25-27).
While significant progress has been made in understanding the regulation of SR-BI expression and function (1-5,23,24,28), relatively less is known about the structural requirement and contribution of various components of the SR-BI molecule in selective CE transport function. Previous studies have shown that the extracellular domain of SR-BI is essential for efficient HDL CE uptake, but the C-terminal domain is also critical for the optimum selective uptake process (29-31). Our recently published data provide evidence that the physical state of the SR-BI protein (i.e., monomeric, vs dimeric and higher order oligomeric forms of SR-BI), and architectural changes in the cell surface induced by the expression of SR-BI, also play major roles in the functional efficiency of the selective pathway (12,32,33). In this study we further examined the structure-function relationships and dynamics of SR-BI activity and have focused our efforts in determining the structural and functional contributions of cysteine residues within SR-BI. We elected to examine the contribution of cysteine residues for the following reasons: (a) cysteine residues are integral for inducing and maintaining the three-dimensional conformation in proteins by forming critical inter- and intra-molecular disulfide bond linkages (34,35); (b) sulfhydryl (SH) side chains of cysteines are polar similar to that of the hydroxyl group (OH) of serines and can participate in hydrogen bonding interactions and facilitate protein-protein interactions (36); (c) cysteine side-chains are preferred sites for various biological coupling and conjugation reactions such as palmitoylation, isoprenylation, disulfide crosslinking, and thiol-disulfide exchange which are known to play critical roles in intracellular protein trafficking, stability and/or activity (36-38); and (d) the SR-BI contains several cysteine residues that are highly conserved across the species and uniquely distributed within the different domains of the SR-BI molecule and as such are highly likely to contribute towards SR-BI structure and function (Table 1, Fig. 1).
Table 1. List of Primers.
| Primer Designation | Primer Sequence |
|---|---|
| C21S-Forward | 5′-CTAGGGCTGCTGAGTGCTGCGCTCG-3′ |
| C21S-Reverse | 5′-CGAGCGCAGCACTCAGCAGCCCTAG-3′ |
| C251S-Forward | 5′-CTATTGGCATTCGGAACAGAGCAACATGATCAATGGTAC-3′ |
| C251S-Reverse | 5′-GTACCATTGATCATGTTGCTCTGTTCCGAATGCCAATAG-3′ |
| C280S-Forward | 5′-CTTCAGCCCAGAAGCCAGCAGATCTATGAAGCT-3′ |
| C280S-Reverse | 5′-AGCTTCATAGATCTGCTGGCTTCTGGGCTGAAG-3′ |
| C321S-Forward | 5′-CTAATGAAGGCTTCAGCCCGTGCCGCGAG-3′ |
| C321S-Reverse | 5′-CTCGCGGCACGGGCTGAAGCCTTCATTAG-3′ |
| C323S-Forward | 5′-GCTTCTGCCCGAGCCGCGAGTCC-3′ |
| C323S-Reverse | 5′-GGACTCGCGGCTCGGGCAGAAGC-3′ |
| C334S-Forward | 5′-CAGAATGTCAGCACCAGCAGGTTTGGTGCGC-3′ |
| C334S-Reverse | 5′-GCGCACCAAACCTGCTGGTGCTGACATTCTG-3′ |
| C384S-Forward | 5′-GGGATCCCCATGAACAGTTCCGTGAAGATGC-3′ |
| C384S-Reverse | 5′-GCATCTTCACGGAACTGTTCATGGGGATCCC-3′ |
| C470S-Forward | 5′-TACCAACTGCGCAGCCAGGAGAAAAGCTTTTTATTTTG-3′ |
| C470S-Reverse | 5′-CAAAATAAAAAGCTTTTCTCCTGGCTGCGCAGTTGGTA-3′ |
| V3C-Forward | 5′-ACGCGAACATGGGCTGCAGCTCCAGGGCAC-3′ |
| V3C-Reverse | 5′-GTGCCCTGGAGCTGCAGCCCATGTTCGCGT-3′ |
| G453C-Forward | 5′-YGCTGCTGGGGCTTGGATGCCTCCTGC-3′ |
| G453C-Reverse | 5′-GCAGGAGGCATCCAAGCCCCAGCAGCA-3′ |
| Y462C-Forward | 5′-GCTTCTGGTGCCCATCATTTGCCAACTGCGCA-3′ |
| Y462C-Reverse | 5′-TGCGCAGTTGGCAAATGATGGGCACCAGAAGC-3′ |
Figure 1. Schematic diagram showing the position of rat SR-BI cysteine residues that were subjected to site-directed mutagenesis and location of artificially created cysteines.
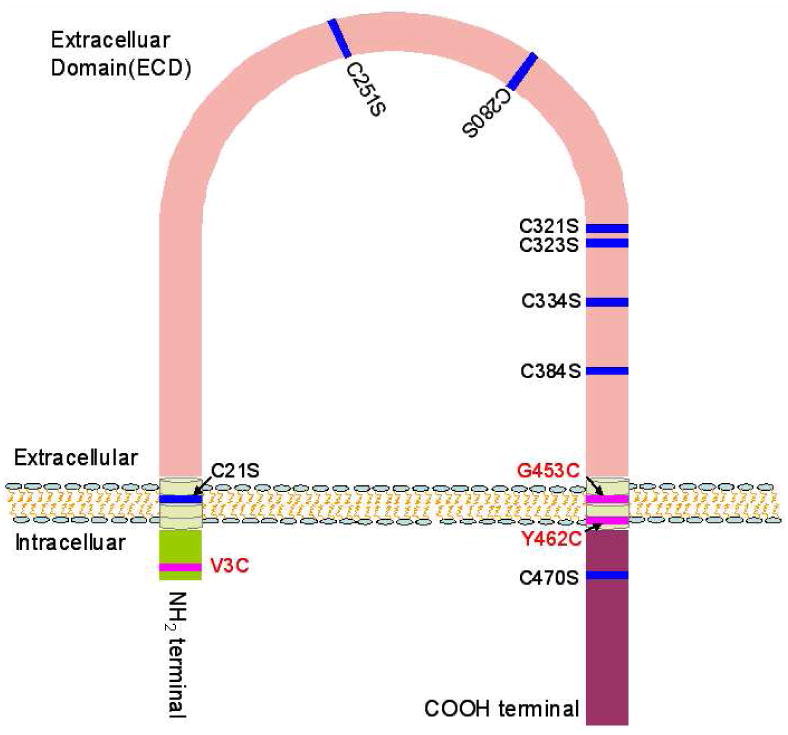
Its sequence contains a total of eight cysteine (C) residues (C21, C251, C280, C321, C323, C334, C384, and C470). Five residues (C280, C321, C323, C334, and C384) are clustered in the C-terminal half of the putative extracellular domain (ECD). The remaining three cysteine residues are equally distributed in the N-terminal transmembrane domain (C21), N-terminal half of the ECD (C251), and the C-terminal domain (C470). With the exception of C21, the remaining seven residues are highly conserved in other species including mouse, hamster, rabbit, pig, cow and human. Among the three artificially created cysteine residues, two (G453C and Y462C) were located in the C-terminal transmembrane domain, while the third one (V3C) was in the N-terminal domain.
We chose the rat SR-BI as a prototype for our studies because it contains more conserved cysteine sequences than SR-BI from any other species (Table 1). Its sequence contains a total of eight cysteine (C) residues (Fig. 1, Table 1). With the exception of C21, the remaining seven residues are highly conserved in other species including the mouse, hamster, rabbit, pig, cow and human. Five residues (C280, C321, C323, C334, and C384) are clustered in the C-terminal half of the putative extracellular domain (ECD). The remaining three cysteine residues are equally distributed in the N-terminal transmembrane domain (C21), N-terminal half of the ECD (C251), and the C-terminal domain (C470). Given that the extracellular domain contains six conserved cysteine residues, these could theoretically form up to three disulfide bonds and in turn, could help to stabilize the conformation of SR-BI, participate in its dimerization or contribute to SR-BI-mediated selective HDL-CE uptake. Using a combination of mutational and chemical approaches, we provide evidence that four cysteine residues, C280, C321, C323 and C334 of the extracellular domain (ECD) are crucial for preserving normal SR-BI (HDL) binding activity, selective-CE uptake, and trafficking to cell surface. The mutation of the two remaining cysteine residues, C251 and C384 of the ECD, has no effect on either SR-BI expression or function. Other cysteine mutations and substitutions were also without any effect.
Experimental Procedures
Materials
Iodine-125I radionucleotide (carrier free, ∼629 GBq/mg; ∼17 Ci/mg) and [1α, 2α (N)-3H] cholesteryl oleolyl ether (1.78 TBq/mmol; 48.0 Ci/mmol) were obtained from GE Health Care/Amersham (Arlington Heights, IL). Molecular biology reagents were purchased from Roche (Basel, Switzerland), New England Biolabs (Ipswich, MA), Strategene (Agilant Technologies, La Jolla, CA), Bio-Rad (Hercules, CA) and Qiagen (Valencia, CA). Cell culture growth media and antibiotics were obtained from either Sigma-Aldrich Corp. (St Louis, MO) or Invitrogen (Life Technologies, Carlsbad, CA). Fetal calf serum was supplied by HyClone Laboratories (Logan, UT). Rabbit polyclononal antibody (NB400-134) against a peptide from the extracellular domain (amino acid residues 230-380) of SR-BI/SR-BII was purchased from Novus Biologicals (Littleton, CO). Rabbit polyclonal antibody raised against a peptide to the carboxyl terminus of mouse SR-BI (amino acid residues 489-509) was prepared as described previously (20); this antibody cross-reacts with mouse, rat, and human SR-BI. Rabbit anti-calnexin polyclonal antibody (ER marker), goat anti-mouse IgG Alexa Fluor488 and goat anti-rabbit IgG Alexa Fluor568 was obtained from Abcam (Cambridge, MA). The sulfhydryl-specific alkylating MTS reagents, MTSEA, MTSES, MTSET, and MMTS were purchased from Toronto Research Chemicals, Inc., (Toronto, Canada). Cholesteryl BODIPY FL C12 (BODIPY-CE) was supplied by Molecular probes/Life Technologies Corporation, (Carlsbad, CA). All other reagents used were of analytical grade.
Site-directed Mutagenesis of SR-BI
The role of disulfide bonds and cysteine residues of the rat SR-BI in SR-BI expression and function was examined by systematically mutating to serine each of the cysteine residues within the extracellular and transmembrane domains. These mutations generated a complete series of SR-BI mutants having either single C21S, C251S, C280S, C321S, C323S, C334S, C384S, and C470S or paired C21S/C251S, C251S/C384S, C280S/C323S, C280S/C334S, C251S-C321S, C280S-C321S, C334S-C384S, C321S/C323S, and C323S/C334S mutations. Three artificial cysteine residues, V3C, G453C or Y462C were also introduced in the native rat SR-BI via mutagenesis. In addition, double and triple mutants, G453C/Y462C, G453C/Y462C/C470S, and G453C/Y462C/C470A were created. All receptor mutations were integrated into the coding sequence of a rat SR-BI construct that was placed downstream of the constitutive cytomegalovirus enhancer/promoter in the eukaryotic expression vector pcDNA6/V5-His as described previously (32). Single–codon mutations were introduced into this rat SR-BI-V5-His/pcDNA template in whole-plasmid PCRs using the QuickChange® II site-directed mutagenesis kit supplied by Stratagene Inc., La Jolla, CA and appropriate sets of complementary forward and reverse mutagenic primers listed in Table 1. Multiple codon exchanges were introduced into SR-BI-V5coding sequences possessing an appropriate single mutation through successive rounds of QuickChange mutagenesis. Production of the correct mutations and absence of coding errors in the SR-BI mutant constructs was confirmed by automated DNA sequencing. The primers for various cysteine mutations were designed using QuickChange Primer Design Program (Stratagene online software) and synthesized by Elim Biopharmaceuticals Inc., (Hayward, CA).
Cell Culture and Transfection
Cell Culture and Transfection—two different cell lines, COS-7 and CHO were employed. They were obtained from the American Type Culture Collection (Manassas, VA), maintained as monolayers in DMEM containing 10% fetal bovine serum (Thermo Scientific Hyclone, Logan, UT) 2 mM glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (complete culture medium) in a humidified atmosphere consisting of 5% CO2 and 95% air, at 37°C. The COS-7 or CHO cells were seeded into 35-mm culture dishes at a density of 6 × 105 cells/well. When cells reached 70-80% confluence, they were transiently transfected with 2 μg of native (wild-type) or one of the various cysteine mutant SR-BI plasmid DNAs in 250 μl of Opti-MEM I and, 6 μl of Lipofectamine™ Transfection Reagent in 200 μl of Opti-MEM I according to the manufacturer's instructions. After 4h, the dishes were washed with complete culture medium and following the addition of 2 ml of the same medium to each dish, the cells were allowed to grow for an additional 44 h. As a control, some cells were transfected with empty vector (control). All experiments were carried out 48 h after transfection.
Fluorescence-activated Cell Sorting (FACS) Analysis
Fluorescence-activated Cell Sorting (FACS) Analysis—detection of native and various cysteine mutant forms of SR-BI protein on the cell surface of the transfected CHO cells was performed by flow cytometry using a rabbit polyclonal anti-SR-BI/SR-BII antibody. CHO cells transiently transfected with either native rat SR-BI-V5 plasmid or one of the various SR-BI-V5 cysteine mutant constructs were washed twice with phosphate buffered saline (PBS) and, subsequently, cell suspension was incubated with an anti SR-BI/SR-BII antibody (1:100 dilution of the stock antibody) on ice for 30 min. After washing twice with PBS, cells were incubated with Alexa Fluoro®488-conjugated anti-rabbit IgG (BD-Pharmingen™, San Jose, CA) (1:1000 dilution) for an additional 30 min on ice. Subsequently, cells were washed twice with PBS, resuspended in PBS supplemented with 1% of bovine serum albumin (BSA) and analyzed by multi-color flow cytometry (BD FACSCalibur™ flow cytometer, Sparks, MD) at the VA Palo Alto Flow Cytometry Core Facility. The data were analyzed using the accompanying software.
Uptake and Internalization of HDL-derived Cholesteryl Esters
Transiently transfected CHO or COS-7 cells were incubated with [125I]DLT/[3H]COE-hHDL3 (100 μg protein/ml) for 5h at 37°C. At the end of incubation, the dishes were washed and then solubilized in 2 ml of 0.1N NaOH. One-ml aliquots were precipitated with an equal volume of 20% trichloroacetic acid (TCA) to determine acid soluble and insoluble [125I] radioactivity or extracted with organic solvents to determine [3H]-radioactivity (13). Endocytic uptake is calculated from the TCA soluble [125I] label only. The difference between total and TCA soluble radioactivity is taken as the surface associated [125I]-hHDL3. Since both [125I] and [3H] labels are on the same particle, the surface bound [125I] is also equal to the surface bound [3H]. Thus, total [3H] minus surface bound [3H] equals the total amount of [3H] internalized. To calculate ‘selective’ uptake of CE, soluble [125I] radioactivity is subtracted from soluble [3H] radioactivity. Finally, to calculate the mass of CE internalized, these values are divided by a protein:cholesterol ratio of hHDL3 (i.e., 2.70).
For some studies, HDL-CE uptake was also measured using a fluorometric technique (39). Transiently transfected CHO or COS-7 were incubated with recHDL-BODIPY-CE (50 μg protein/ml) for 1-3h at 37°C and following incubation, dishes were rapidly washed 5 times in phosphate buffered saline-0.1% bovine serum albumin at 0-4°C, and extracted with hexaneisopropyl alcohol (3:2 v/v) as described previously (39). In each case a portion of hexaneisopropyl alcohol extract was transferred to a fluorescent plate and fluorescence was measured at the excitation wavelength of 488 nm and emission wavelength of 520 nm using a Fluorescent plate reader (Spectra MAX Gemini, Molecular Devices). The results are expressed as arbitrary units/mg protein or μg DNA and represent total BODIPY-CE uptake by the transiently transfected cells.
PFO-, SDS- or Triton X-100-PAGE/Western Blotting
PFO-, SDS- or Triton X-100-PAGE/Western Blotting—we used a detergent-PAGE electrophoresis technique to determine the relative efficacy of three detergents, PFO, SDS and Triton-X-100 on monomer, dimer and oligomer forms of SR-BI. Among these, PFO being a weak detergent, gently disrupts membranes, while maintaining the oligomeric structure of membrane proteins before and during electrophoresis (40). CHO cells transiently transfected with rat SR-BI-V5 for 36h were lysed by sonication in lysis buffer (common components: 125 mM Tris-HCl, pH 6.8, 5% glycerol, 10 μg/ml leupeptin, 1 mM PMSF, 20 μg/ml aprotenin and 5 μg/ml pepstatin) containing 2% of NaPFO, SDS or Triton X-100 and clarified by centrifugation. Likewise, a highly purified double-membraned plasma membrane preparation from luteinized rat ovary (25) was solubilized in lysis buffer containing 2% SDS and centrifuged briefly to sediment insoluble material. In some cases, SDS extracts were treated with 50 mM dithiothreitol (DTT) at 95°C for 2 min. Suitable aliquots (10-15 μg protein) of each sample were loaded on SDS-free gels, electrophoretically separated using Tris-Glycine buffer without detergent, transferred to Immobilon-P membrane (Millipore, Bedford, MA), and probed with rabbit anti-peptide (C-terminal peptide) rat SR-BI antibody (20) followed by a horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma). The bands were visualized using ECL Plus™ Western blotting detection reagents (GE Healthcare, Piscataway, NJ).
Western Blotting Analysis
Expression levels of native (wild-type) and cysteine mutant SR-BI receptors were estimated by Western blot analysis of total cellular lysates. Transiently transfected cells were washed twice with phosphate buffered saline (PBS, pH 7.4) and lysed in cell lysis buffer (125 mM Tris-HCl, pH 6.8, 2% SDS, 5% glycerol, 10 μg/ml leupeptin, 1 mM PMSF, 20 μg/ml aprotenin and 5 μg/ml pepstatin). The protein content of cell lysates was determined using a modification (41) of the procedure of Lowry et al (42). SDS-PAGE/Western blotting measurements were performed both under reducing and non-reducing conditions. Aliquots of samples (10 μg) were mixed with 0.01% bromophenol blue (non-reducing conditions) or 3 μg samples were mixed with 0.01% bromophenol blue and 50 mM DTT (reducing conditions). All samples were heated at 95°C for 5 min, subjected to SDS-PAGE (4-20% pre-made protein gels, Thermo Fisher Scientific, Rockford, IL) and transblotted to nitrocellulose membranes. Expression of V5-tag was detected using a 1:5000 dilution of anti-V5-tag monoclonal antibody (Invitrogen Life Technologies, Carlsbad, CA). In-house generated polyclonal rabbit anti-peptide (C-terminal peptide) rat SR-BI antibody (20) was used at a dilution of 1:1000 to detect the expression of SR-BI protein itself. Gel-loading of samples was detected using a 1:1000 dilution of anti-β-actin monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Following incubation of the membranes with the primary antibodies, detection of bands was performed using IRDye® 680 Goat anti-Rabbit IgG (H + L) and IRDye® Goat anti-Mouse IgG (H + L) secondary antibodies and Odyssey® Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Quantification of the infrared signals was performed using the Odyssey System Software. The quantitative numbers shown under the 2% SDS plus (+) 50 mM DTT bands (Figs. 7A and 7B) represent expression levels of SR-BI normalized to β-actin, i.e., SR-BI/β-actin ratio, where native (wild-type) SR-BI/β-actin ratio = 1.00. The numbers shown under the 2% SDS and minus (-) 50 mM DTT bands represent the % expression of SR-BI dimers. These numbers were derived by dividing intensity of SR-BI dimer band with the combined intensities of SR-BI dimer + monomer bands = % Dimer
Figure 7. Western analysis of wild type and mutant oligomeric forms of SR-BI proteins.
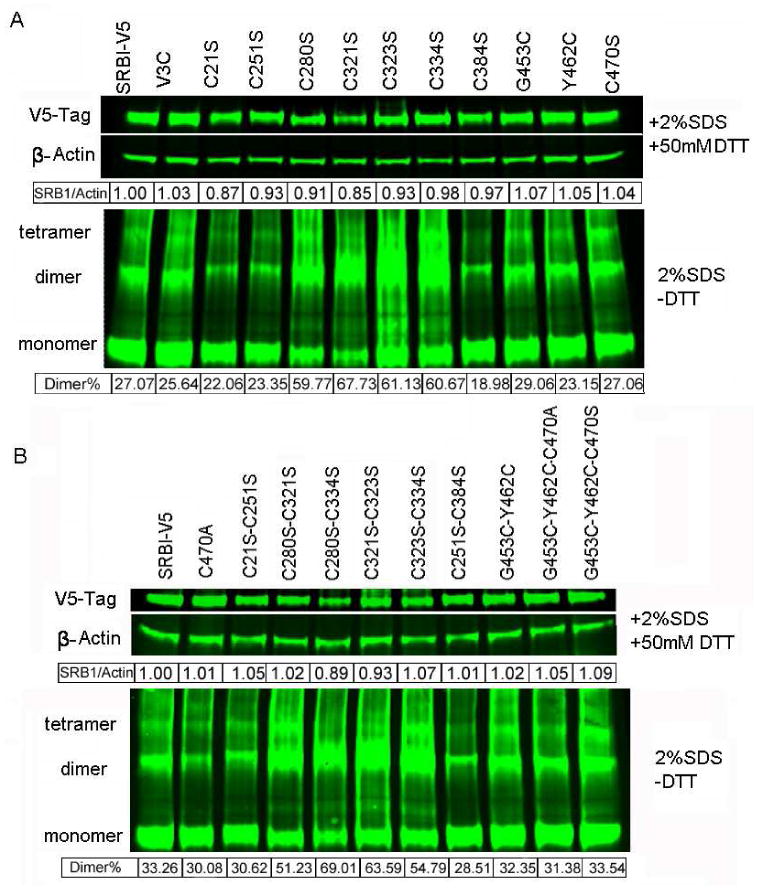
CHO cells were transiently transfected with empty vector (Mock), V5-tagged rat SR-BI, or the indicated mutants. After 36h of transfection, cell lysates were prepared and aliquots of 10 μg samples were mixed with 0.01% bromophenol blue (non-reducing conditions) or 3 μg samples were mixed with 0.01% bromophenol blue and 50 mM DTT (reducing conditions) and subjected to Western blot analysis as described under “Materials and Methods.” Expression of V5-tagged rat SR-BI was detected using a 1:5000 dilution of anti-V5-tag monoclonal antibody (Invitrogen Life Technologies, Carlsbad, CA). Gel-loading of samples was detected using a 1:1000 dilution of anti-β-actin monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Following incubation of the membranes with the primary antibodies, detection of bands was performed using IRDye® 680 Goat anti-Rabbit IgG (H + L) and IRDye® Goat anti-Mouse IgG (H + L) secondary antibodies and Odyssey® Infrared Imaging System. A, Blots showing the total protein levels of wild type, single cysteine mutant and introduced cysteine mutant SR-BIs both under reducing (SR-BI monomer) and non-reducing conditions (SR-BI dimer and oligomers). B, Blots showing the total protein levels of wild type, and various double and triple cysteine mutant SR-BIs both under reducing (SR-BI monomer) and non-reducing (SR-BI dimer and oligomers) conditions. The blots shown are representative of three independent experiments. Note, that the amount of total cell lysate protein applied in each case under reducing conditions was only one-third to that of the respective amounts applied to gels under non-reducing conditions. This was in an effort to prevent overloading of monomeric SR-BI protein from the DTT treated samples. Detection and quantification of the infrared signals were performed using the Odyssey System Software. Quantification of the infrared signals was performed using the Odyssey System Software. The quantitative numbers shown under the 2% SDS plus (+) 50 mM DTT bands (Fig. 7A and 7B) represent expression levels of SR-BI normalized to β-actin i.e., SR-BI/β-actin ratio, where native (wild-type) SR-BI/β-actin ratio = 1.00. The numbers shown under the 2% SDS and minus (-) 50 mM DTT bands represent % expression of SR-BI dimers. These numbers were derived by dividing intensity of SR-BI dimer band with the combined intensities of SR-BI dimer + monomer bands = % Dimer
Confocal Immunofluorescence Microscopy
CHO cells were seeded into six-well tissue culture plates containing sterilized poly-D-lysine (Sigma) treated 25-mm round glass coverslips and transiently transfected with 1.0 μg native (wild-type) or mutant (C280S, C321S, C323S, C334S, and C280S/C334S) SR-BI-V5 DNA according to the aforementioned transfection protocol. After 48h, transfected cells were washed with phosphate buffered saline, fixed for 15 min in a freshly prepared solution of 4% paraformaldehyde in PBS, washed twice with PBS and then permeabilized with 0.1% Triton X-100 in PBS for 10 min. After incubating with blocking solution (5% normal goat serum and 5% non-fat dry milk in PBS) for 1h, the cells were incubated with anti-V5 mouse monoclonal antibody (SR-BI-V5 fusion protein) or antipeptide-SR-BI rabbit polyclonal antibody (20) together with anti-calnexin rabbit polyclonal antibody (ER membrane marker) overnight at 4°C. Cells were then washed and treated with goat anti-mouse IgG Alexa Fluor488 and goat anti-rabbit IgG Alexa Fluor568 in blocking solution for 45 min at room temperature, and after washing, coverslips were mounted on microscope slides using Fluoromount G (Fisher Scientific, Pittsburgh, PA). All steps were carried out at room temperature. The labeled cells were viewed and photographed with a Laser-scanning confocal microscope (Zeiss LSM 510 Meta Confocal Microscope, Zeiss, Jena, Germany). Images were collected on the confocal microscope using LSM510 software and assembled into figures using Photoshop software (Adobe, San Jose, CA).
Treatment of SR-BI Expressing COS-7 cells with Sulfhydryl Reagents, Methanethiosulfonate compounds, β-Mercaptoethanol and Dithiothreitol
To study the effect of MTS compounds on selective HDL-CE uptake, COS-7 cells transiently overexpressing SR-BI (48 h post-transfection) were treated with either vehicle (control), MTSEA (positively charged), MTSET (positively charged), MTSES (negatively charged) or MTSM (neutral) (final concentration 1 mM) at 37°C for 5 min. The reaction was stopped by washing the cells with ice-cold PBS. Cells were then incubated in the culture medium containing 10 μg/ml of HDL-BODIPY-CE at 37°C for 2h. After washing cells with cold PBS, total intracellular fluorescence was quantified.
Lipoprotein preparation
Human apoE-free high-density lipoprotein3 (hHDL3) was isolated as described previously (31). hHDL3 was used exclusively because it is not recognized by the LDL (B/E) receptor-mediated endocytic pathway. For uptake and internalization studies, hHDL3 preparations were conjugated with residualizing labels, i.e., [125I]-labeled dilactitol tyramine (DLT) and [3H]cholesteryl oleolyl ether (COE) (13). Reconstituted (rec) HDL-BODIPY-cholesteryl ester particles (rec-HDL-BODIPY-CE) were used for the fluorometric determination of HDL-CE uptake and for confocal microscopic localization of HDL-derived cellular CEs. recHDL-BODIPY-CE was prepared as described previously (39). Protein content of the HDL preparations was determined by a modification (43) of the procedure of Lowry et al (42).
Statistical Analysis
Results are expressed as means ± SE of at least three independent experiments. Differences between two groups were evaluated using the Student's t test and the GraphPad Prism version 4.0 for Windows (GraphPad Software Inc., San Diego, CA); differences among multiple groups were evaluated with a one-way analysis of variance followed by a post hoc Dunnett's test. P values of <0.05 were considered to be statistically significant.
Results
Homo-oligomeric SR-BI
Initially we determined the effects of three different detergents (SDS, Triton X-100 and NaPFO) and DTT, a sulfhydryl reducing agent, on the stability and effective separation of multimeric forms of SR-BI by polyacrylamide gel electrophoresis (PAGE)-Western blotting. Treatment of cells or isolated membranes with SDS results in efficient solubilization of membrane bound proteins and often solubilized protein maintain their oligomeric forms following resolution by SDS-PAGE (44-46). The poly(oxyethylene) series of nonionic detergents such as Triton X-100 is widely used for solubilization of membrane proteins because of its inoffensiveness toward membrane proteins with little or no undesirable effect on the stability of protein oligomers (47). The weak detergent, NaPFO is also known to gently disrupt the membranes while at the same time helps to maintain weak interactions between proteins as well as the oligomeric state of proteins before and during electrophoresis (48). CHO cells transiently overexpressing rat SR-BI were solubilized in 2% SDS, 2% Triton X-100 or 2% NaPFO, and suitable aliquots subjected to polyacrylamide gel electrophoresis as described above. Similarly, SR-BI dimer enriched double membraned luteinized plasma membranes were solubilized in 2% Triton X-100-SDS, exposed to ± 50 mM DTT and subjected PAGE. As shown in Fig. 2, Western blot analysis of the detergent extracts from rat SR-BI overexpressing CHO cells resolved in non-reducing SDS-PAGE indicates the presence of monomeric, dimeric and tetrameric immunoreactive bands. Interestingly, all three detergents yielded almost identical results suggesting that oligomeric forms of SR-BI are relatively resistant to the types and the presence of detergent (Fig. 2A). In contrast, pretreatment of SDS extracts with the DTT (reducing conditions) led to a complete dissociation of the large oligomeric forms of SR-BI to a single monomeric band suggesting that the higher order complexes potentially require disulfide bonding. Similar results were obtained using luteinized ovary plasma membranes, i.e., extract prepared in 2% SDS alone showed the presence of monomeric, dimeric and oligomeric forms of SR-BI bands, whereas a single monomer band was visible when a DTT treated SDS sample was used.
Figure 2. Analysis of multimerization of rat SR-BI.
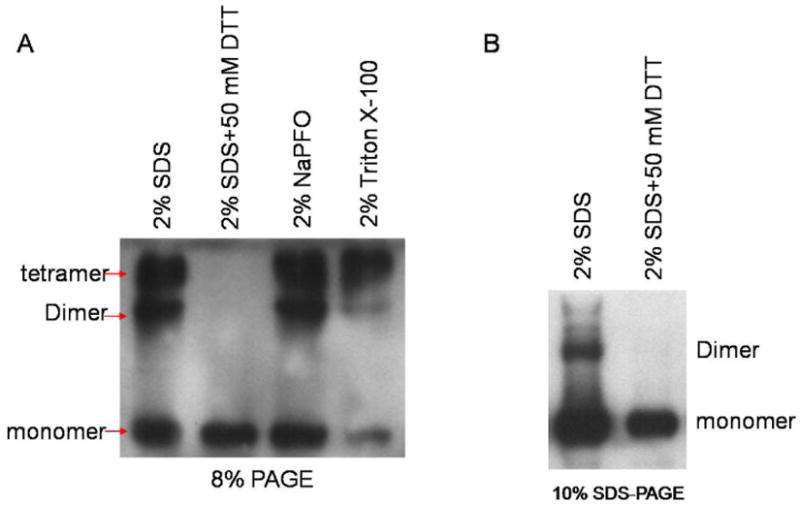
A, CHO cells transiently transfected with rat SR-BI-V5 plasmid for 36h were lyzed in lysis buffer containing 2% SDS, NaPFO or Triton X-100. After heating samples with or without 50 mM DTT at 95°C for 2 min, suitable aliquots (15 μg protein) were separated on 8% PAGE and immunoblotted with rabbit anti-peptide (C-terminal peptide) rat SR-BI antibody as described under “Materials and Methods.” B, Highly purified double-membraned plasma membrane preparations from luteinized rat ovaries were solubilized in lysis buffer containing 2% SDS, heated at 95°C for 2 min in the presence and absence of 50 mM DTT and suitable aliquots subjected to 10% PAGE. Following transfer, membranes were immunoblotted with rabbit anti-peptide (C-terminal peptide) rat SR-BI antibody probed with rabbit anti-peptide (C-terminal peptide) rat SR-BI antibody (20) followed by a horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma). The bands were visualized using ECL Plus™ Western blotting detection reagents (GE Healthcare, Piscataway, NJ) as described under “Materials and Methods.” Arrows indicate the bands corresponding to monomers, dimer and teramer forms of SR-BI. The blot shows that rat SR-BI dimers or oligomers are resistant to types and the presence of detergent, but sensitive to sulfhydryl reducing agent, dithiothreitol.
Effects of Disulfide Bond Reducing Agents and Sulfhydryl Recative Alkyl Methanethiosulfonate (MTS) Reagents on SR-BI-mediated Selective HDL-CE Uptake
The rat SR-BI contains a total of 8 cysteine residues of which 6 conserved residues in the extracellular domain have the potential to form three inter-disulfide bonds in SR-BI monomer or intra-disulfide bonds in SR-BI dimers and high order oligomers (Table 2). To assess whether any disulfide (S-S) bond present in the extracellular domain is involved in SR-BI function, we examined the effects of chemical (β-mercaptoethanol or dithiothreitol) reduction of SR-BI on selective HDL-CE uptake (36). We reasoned that if critical disulfide (S-S) bonds present within the extracellular domain of the SR-BI are required for selective CE transport, then chemical reduction of cell surface SR-BI should be associated with a defect in SR-BI function. Thus, selective HDL-CE uptake measurement was performed on SR-BI overexpressing COS-7 cells pre-treated with or without the optimum concentrations of β-mercaptoethanol or dithiothreitol, which breaks sulfhydryl (S-S) bonds and maintains cysteine residues in a reduced state. As shown in Fig. 3, exposure of SR-BI overexpressing COS-7 cells to 100 mM β-mercaptoethanol or 10 mM dithiothreitol (DTT) for 30 min followed by quick washes modestly, but significantly impaired, the SR-BI mediated selective uptake of HDL-BODIPY-CE. Under the experimental conditions employed, DTT was modestly more effective than β-mercaptoethanol in causing reduction of selective HDL-BODIPY-CE uptake. Western blot analysis of cell lysates from β-mercaptoethanol- or DTT-pretreated cells demonstrated near complete absence of dimer and oligomer forms of SR-BI and an elevated pool of SR-BI monomer indicative of reduction of S-S bonds to SH. These results thus suggest that the optimum functioning of SR-BI-mediated selective HDL-CE uptake requires the presence of certain disulfide bonds.
Table 2. List and position of cysteine residues in mouse, rat, hamster, rabbit, pig, cow, dog, Northern tree shrew and human SR-BI.
| Species | ND | NTD | ECD | ECD | ECD | ECD | ECD | ECD | CTD | CTD | CD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse (9) | - | C21 | C251 | C280 | C321 | C323 | C334 | C384 | - | C462 | C470 |
| Rat (8) | - | C21 | C251 | C280 | C321 | C323 | C334 | C384 | - | - | C470 |
| Hamster (8) | - | C21 | C251 | C280 | C321 | C323 | C334 | C384 | C453 | - | C470 |
| Rabbit (8) | - | C21 | C251 | C280 | C321 | C323 | C334 | C384 | - | - | C470 |
| Pig (8) | - | - | C251 | C280 | C321 | C323 | C334 | C384 | C453 | - | C470 |
| Cow (8) | - | - | C251 | C280 | C321 | C323 | C334 | C384 | C453 | - | C470 |
| Dog (12)* | - | - | C251 | C280 | C321 | C323 | C334 | C384 | C453 | - | C470 |
| Tree Shrew (9) | - | C21 | C251 | C280 | C321 | C323 | C334 | C384 | C453 | - | C470 |
| Human (11) | C3 | C21 | C251 | C280 | C321 | C323 | C334 | C384 | C453 | C462 | C470 |
The predicted dog SR-BI sequence indicates the presence of 4 additional cysteine residues at positions 45, 53, 92, and 100 in the ECD. Positions of cysteine residues obtained from protein sequences using GenBank™ Accession numbers NM_016741, NP_113729.1, U11453, NM_001082788, NM_213967, DAA20570, XM_543366, EU379936, and NM_001082959 for mouse, rat, hamster, rabbit, pig, cow, dog, Northern tree shrew and human, respectively. ND, N-terminal domain; NTD, N-terminal transmembrane domain; ECD, extracellular domain; CTD, C-terminal transmembrane domain; CD, C-terminal domain
Figure 3. Effects of reducing agents and MTS reagents on the ability of SR-BI to mediate selective HDL-CE transport.
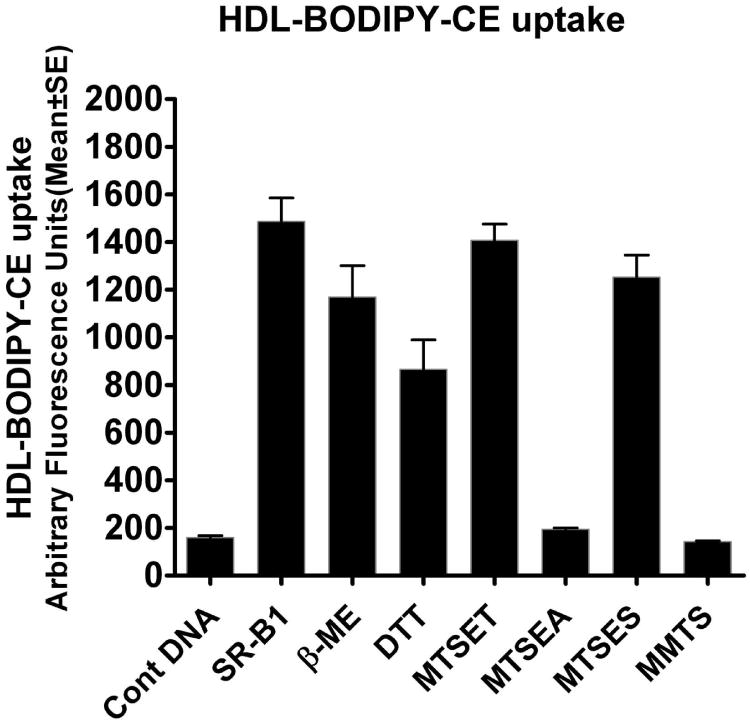
Intact COS-7 cells transiently expressing rat SR-BI were preincubated with freshly prepared DTT (10 mM) or β-mercaptoethanol (100 mM) at 37°C for 30 min. Likewise, some SR-BI overexpressing COS-7 cell dishes were pre-treated with freshly prepared MTSEA, MTSET, MTSES or MMTS (1 mM) for 5 min. After a few washes, the intact cells were then incubated for 5 min at 37°C with HDL-BODIPY-CE, and selective HDL-BODIPY-CE uptake by the COS-7 cells was quantified by the fluorescence measurement as described under “Experimental Procedures.” Each bar represents the Mean ± SE of data obtained from at least three independent experiments. As can be seen no significant change in HDL-BODIPY-CE uptake was observed when SR-BI expressing COS-7 cells were treated with disulfide (S-S) bond reducing agent β-mercaptoethanol or DTT. In contrast, treatment of SR-BI expressing cells with cysteine (SH) reactive alkyl methanethiosulfonate (MTS) reagents, MTSEA, MTSET and MMTS completely blocked the SR-BI-mediated selective HDL-BODIPY-CE uptake, whereas cells were relatively insensitive to treatment with the negatively charged MTSES. SR-BI-V5 alone vs + β-mercaptoethanol (β-ME), p = 0.00715; SR-BI-V5 alone vs + dithiothreito (DTT), p = 0.00458; SR-BI-V5 alone vs + MTSEA, p = 0.00017; SR-BI-V5 alone vs + MTSES, p = 0.08908 (NS); SR-BI-V5 alone vs + MMTS, p = 0.0.00014; SR-BI-V5 alone vs + MTSET p = 0.316155 (NS).
The contribution of cysteine (SH) residues in SR-BI mediated selective HDL-CE uptake was also determined with the use of alkyl methanethiosulfonate (MTS) reagents that specifically react with the free sulfhydryl group of cysteine (49), and which have been used widely to probe the role of cysteine residues in the structure/function of a number of proteins including ion channels, transporters and receptors (50-56). We reasoned that if an endogenous cysteine residue in SR-BI is involved in ligand (HDL) binding, covalent modification with MTS reagents will potentially block the HDL binding to SR-BI and, consequently, inhibition of selective delivery of HDL-CE. Here we utilized four cysteine reactive MTS reagents, MTSEA, MTSET, MTSES and MMTS to determine their effects on SR-BI-mediated selective HDL-CE uptake in SR-BI overexpressing COS-7 cells. Among these, the water-soluble and largely positively charged MTSEA is the most commonly used MTS reagent, which converts the neutral cysteine side-chain to a positively charged group resembling a lysine side chain. At the extracellular pH of 7.4 in the incubation medium, a significant fraction of MTSEA is unprotonated and membrane permeable (57). MTSET like MTSEA although it carries a fixed positive charge, is not expected to cross the cell membrane. MTSES being negatively charged does not enter into the cell interior. Thus, both MTSET and MTSES are membrane-impermeant. The MMTS is a small and uncharged (neutral) MTS compound, which crosses the cell membrane readily, and thus, could potentially interact with accessible residues on either the cytoplasmic or the extracellular domain of the SR-BI. Fig. 3 shows that treatment of SR-BI overexpressing COS-7 cells with optimum doses of MTSEA and MMTS completely blocked SR-BI-mediated selective HDL-BODIPY-CE uptake, indicating that free cysteine residues are critically involved in the selective cholesterol transport process. Treatment with a negatively charged MTSES, however, had no significant effect on the selective delivery of HDL-BODIPY-CE to the cell interior of SR-BI expressing COS-7 cells. Likewise, the positively charged, but membrane impermeable MTSET also showed no effect on selective uptake of HDL-BODIPY-CE in SR-BI expressing COS-7 cells.
Construction of SR-BI Constructs Having Single, Paired and Triple Cysteine Mutations or Introduced Cysteine Residues
Construction of SR-BI Constructs Having Single, Paired and Triple Cysteine Mutations or Introduced Cysteine Residues—given that sulfhydryl reducing agents, β-mercaptoethanol and dithiothreitol and three MTS reagents had a significant effect on selective HDL-CE transport function, we directly and individually tested the role of 8 cysteine residues including 6 highly conserved residues in the extracellular domain on SR-BI function. To accomplish this, we constructed SR-BI mutant receptors, each of which contained a serine substituted for one of the eight cysteines (C21, C251, C280, C321, C323, C334, C384 and C470) in rat SR-BI and assessed the functional implications of losing these individual cysteines. In addition, we examined the effects of replacing certain pairs of cysteine residues on the expression and function of SR-BI (Table 3). We used serine instead of alanine substitution because of the better structural match between the alcohol and sulfhydryl side chains of serine and cysteine, respectively (58). To establish specificity, some key observations were also confirmed by employing alanine substitution of certain cysteine residues. We also tested the function of artificially creating one of the three cysteine residues in rat SR-BI that additionally exist in human SR-BI/CLA-I (Table 2). Finally, two triple mutants were created in which Gly453 was mutated to cysteine, Tyr462 to Ser and Cys470 to Ala (first mutant) or Ser (second mutant).
Table 3. Effects of cysteine mutations and substitutions of SR-BI on selective HDL-CE uptake by COS-7 or CHO cells.
| SR-BI mutants | COS-7 cells Selective HDL-[3H]COE uptake (ng HDL/mgprotein) | CHO cells Selective HDL-BODIPY-CE uptake (fluorescence units) |
|---|---|---|
| Vector alone# | 105.25 ± 14.77 | 106.23 ± 3.53 |
| WT* | 500.24 ± 36.92 | 363.22 ± 37.38 |
| V3C | 617.49 ± 40.24 | 293.57 ± 31.84 |
| C21S | 591.71 ± 58.48 | 267.60 ± 15.37 |
| C251S | 525.25 ± 74.50 | 310.02 ± 43.30 |
| C280S | 370.41 ± 55.11 | 123.60 ± 3.49 |
| C321S | 344.61 ± 16.58 | 111.14 ± 15.60 |
| C323S | 240.53 ± 53.00 | 92.82 ± 5.00 |
| C334S | 340.20 ± 60.46 | 176.78 ± 16.34 |
| C384S | 675.55 ±106.20 | 242.00 ± 18.23 |
| G453C | 550.21 ± 28.62 | 324.33 ± 32.86 |
| Y462C | 605.00 ± 67.23 | 316.67 ± 36.39 |
| C470S | 565.20 ± 16.16 | 243.08 ± 24.01 |
| C470A | 663.85 ± 65.27 | 250.63 ± 14.11 |
| C21S/C251S | 643.79 ± 43.25 | 246.80 ± 8.00 |
| C251S/C384S | 320.10 ± 39.23 | 229.15 ± 17.33 |
| C280S/C323S | 275.32 ± 60.20 | 83.75 ± 1.72 |
| C280S/C334S | 365.00 ± 50.20 | 121.73 ± 8.06 |
| C321S/C323S | 216.88 ± 58.25 | 132.76 ± 21.00 |
| C323S/C334S | 547.29 ± 36.64 | 82.84 ± 1.04 |
| G453C/Y462C | 519.13 ± 54.58 | 184.24 ± 4.42 |
| G453C/Y462C/C470S | 530.20 ± 75.21 | 185.18 ± 6.57 |
| G453C/Y462C/C470A | 375.45 ± 46.10 | 204.85 ± 5.42 |
PcDNA6-V5HisB;
SR-BI-V5
COS-7 cells or CHO cells were transiently transfected with vector alone, V5-rat SR-BI or the indicated mutants. Thirty-six hours post-transfection, COS-7 cells were incubated with a culture medium containing 50 μg protein/ml of [125I-DLT]-[3H]COE-labeled hHDL3 for 5h. Following incubation, the cells were processed for the quantification of selectively internalized HDL-[3H]COE as described under “Materials and Methods.” Likewise, 36h after transfection, CHO cells were incubated with 10 μg protein/ml of recHDL-BODIPY-CE for 2hours and, subsequently, processed for the fluorometric determination of selectively internalized HDL-BODIPY-CE as detailed under “Materials and Methods.” The results are a Mean ± SE in at least three to four independent experiments.
Characterization of Cysteine Mutants
Characterization of Cysteine Mutants—initially fluorescence-activated cell scanning (FACS) analysis using a polyclonal anti-SR-BI/SR-BII extracellular domain specific antibody was used to assess the cell surface expression of native and mutant SR-BI proteins. As shown in Fig. 4, several single and paired mutations behaved differently with respect to cell surface expression. Single cysteine mutants C280S, C321S, C323S and C334S and paired cysteine mutants C280S/C321S, C280S/C334S, C321S/C323S and C323S/C334S of the extracellular domain exhibited decreases in cell surface expression of SR-BI ranging from ∼20% to ∼80%, when expressed in CHO cells. Likewise, CHO cells transfected with double cysteine mutants, C251S-C321S, C280S-C323S, and C334S-C384S showed only a 30-50% surface expression of the mutant SR-BI as compared to the expression of native (wild-type) SR-BI (data not shown). Two of the single cysteine mutants (C321S and C323S) showed about 40% cell surface expression of wild-type SR-BI, while C280S and C334S exhibited ∼50% and ∼60% expression of wild-type, respectively (Fig. 4A). A comparison of various double cysteine mutants indicated that two double mutants, C280S/C312S and C323S/C334S, gave the lowest cell surface expression of SR-BI (∼20% of wild-type) suggesting that these four cysteine residues are critically involved in the translocation of SR-BI from the cell interior to the cell surface. Interestingly, use of C280S/C334S and C321S/C323S double mutants showed somewhat improved cell surface expression of the SR-BI compared to the expression noted with the use of C321S and C323S mutants alone. Additional mutations targeting Cys-21(C21S), Cys-251(C251S), Cys-384 (C384S) and Cys-470 (C470S or C470A) or introduction of new cysteine residues (V3C, G453C, or Y462C) did not significantly alter the cell surface expression of SR-BI. Likewise, various combinations of other double and triple cysteine mutants also showed no affect on the cell surface localization of SR-BI (Fig. 4). To further confirm the reduced cell surface expression of representative single (C280S) and double (C280S/C321S) of mutant SR-BI, we also examined the surface expression of these mutants by the confocal immunofluorescence microscopy. Fig. 5(A) shows that, as expected, cell surface localization of the C280S and C280S/C321S SR-BI proteins monitored by the confocal microscopy were significantly reduced compared to wild type SR-BI. These results complement the FACS data (Figs. 4A) as well as provide additional confirmation for the reduced cell surface expression of these two cysteine mutant SR-BIs.
Figure 4. FACS analysis of CHO cells transiently overexpressing wild-type rat SR-BI or its cysteine mutants.
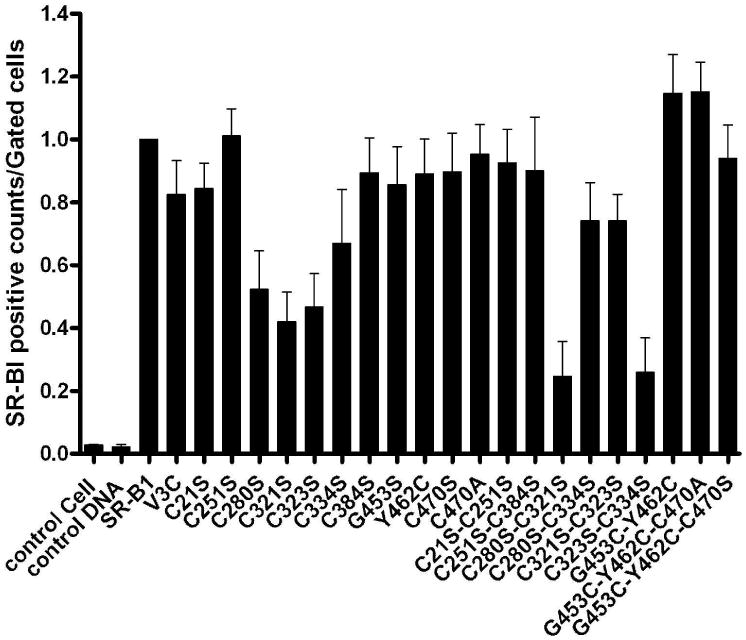

A, cell surface expression of rat SR-BI and its cysteine mutants or rat SR-BI with introduced cysteines. CHO cells were transiently transfected with empty vector (Mock), V5-tagged rat SR-BI, or the indicated mutants. The cells were then incubated with a primary polyclonal SR-BI (ECD) antibody on ice for 30 min. After washing with PBS, cells were incubated with Alexa Fluoro®488-conjugated anti-rabbit IgG for 30 min on ice. Cell surface expression of each construct was estimated by flow cytometry. The results are the Mean ± SE of three independent experiments. B, representative results showing expression of wild-type rat SR-BI or its various cysteine mutants as indicated. The number of cells was estimated by flow cytometry.
Figure 5. Comparison of the surface expression of the wild-type and SR-BI with a single cysteine mutation at residue 280 (C280S) or double mutations at residues 280 and 321 (C280S/C321S) by confocal immunofluorescence microscopy and FACS analysis.
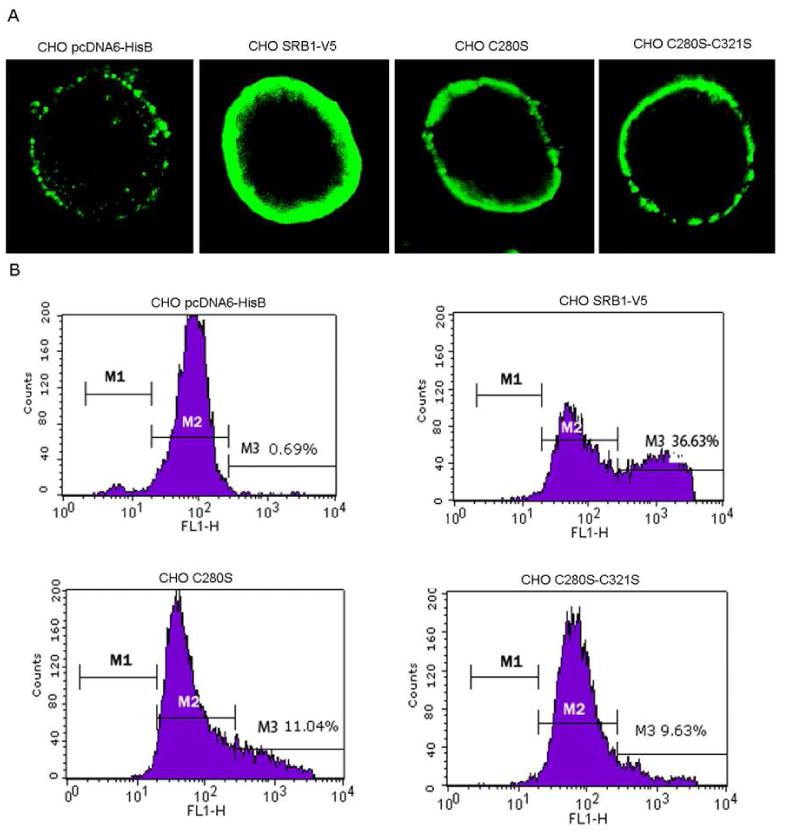
A, cell surface localization of wild-type, and single (C280S) and double (C280S/C321S) cysteine mutant SR-BI by confocal immunofluorescence microscopy. CHO cells were transiently transfected with V5-tagged SR-BI, C280S mutant SR-BI or C280S/C321S double mutant SR-BI construct and 36h after transfection, the cells were fixed, treated with primary and secondary antibodies, mounted and analyzed by confocal microscopy as described under “Materials and Methods.” B, cell surface expression of wild type and single (C280S) and double (C280S/C321S) cysteine mutants of SR-BI analyzed by flow cytometry. CHO cells were transiently transfected with either wild type SR-BI, C280S-SR-BI or C280S/C321S-SR-BI for 36h and subsequently analyzed for cell-surface expression by flow cytometry as described under Materials and Methods.
Because four single, C280S, C321S, C323S and C334S and double, C280S/C321S, C280S/C334S, C321S/C323S and C323S/C334S mutants showed significant decreases in their cell surface expression (Figs. 4 and 5), we wanted to determine whether such decreases were due to defective transport of proteins to the cell surface or simply due to differences in their protein expression. To address these two possibilities, we first examined the total intracellular expression and localization of some of these SR-BI mutants (C280S, C321S or C280S-C321S) as V5-tagged fusion protein by confocal immunofluorescence microscopy. Transiently transfected CHO cells were permeabilized and assayed for SR-BI expression using an anti-V5 antibody (Sr-BI-V5 fusion protein). As shown in Fig. 6, a very low level of diffuse fluorescence was observed when either mock- or control DNA transfected CHO cells were incubated with monoclonal mouse anti-V5 IgG antibodies. In contrast, staining of wild-type, C280S, C321S or C280S-C321S double mutant SR-BI transfected cells with anti-V5 antibodies resulted in fluorescence next to and surrounding part of the nucleus, a pattern typical of ER localization. As further evidence that overexpressed SR-BI proteins preferentially accumulate in the ER, SR-BI transfected cells express either native or mutant protein with an antibody specific for the ER membrane protein, calnexin, the calcium binding protein. As shown in Fig. 6, there was a co-localization of the SR-BI protein with calnexin in cells transfected with either native or one of the three mutant SR-BI constructs. Likewise, total intracellular SR-BI expression (as measured by the intensity of immunofluorescence) was not affected by cysteine mutation of C280S, C321S or both (Fig. 6). These results were further complemented by Western blotting analysis which showed equivalent expression of total SR-BI protein in cells transfected for wild-type or various mutant SR-BIs both under reducing and non-reducing conditions (Fig. 7A and 7B). Thus, the loss of cell surface expression seen with mutations in C280S, C321S, C280S-C321S, or other cysteine mutants (C323S, C334S, C280S/C334S, C321S/C323S and C323S/C334S) is not attributable to the decreased SR-BI protein expression or increased intracellular degradation. Since we were dealing with an overexpressing system, most of the expressed SR-BI protein was localized intracellularly with only a small fraction of even native (wild-type) SR-BI transported to the cell surface. When considered together, even a greater than 80% inhibition of cell surface localization of SR-BI should not be expected to alter intracellular levels in any significant way.
Figure 6. Comparison of cellular distribution of wild-type and SR-BIs with cysteine mutations at residue C280 or residues C280/C321.
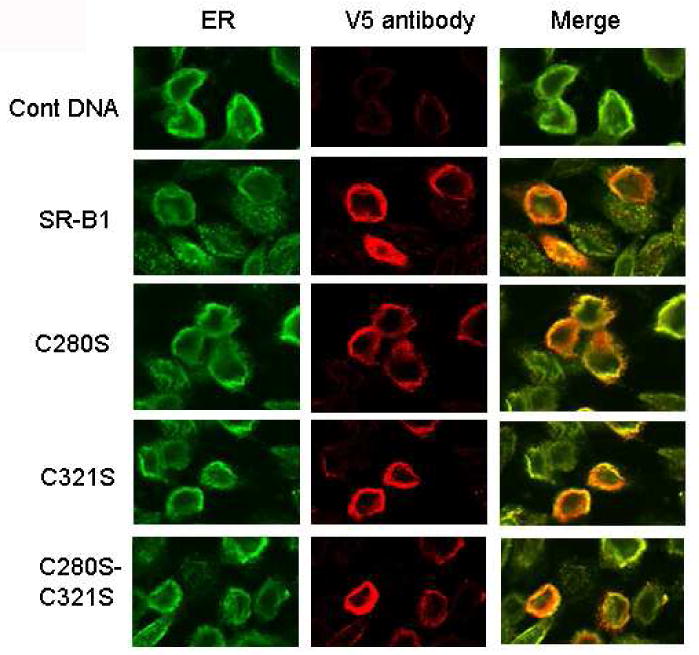
CHO cells transiently expressing wild-type V5-SR-BI, C280S V-5-SR-BI, C321 V-5-SR-BI or C280S/C321S V-5-SR-BI were fixed, permeabilized and simultaneously stained with anti-V5 (to detect the V5 epitope tagged SR-BI) and anti-calnexin (ER marker) antibodies followed by appropriate fluorescent linked secondary antibodies as summarized under Materials and Methods and, subsequently, analyzed by confocal microscopy. Green, V5 fluorescence from SR-BI (left); red, calnexin (middle) fluorescence; yellow, co-localization of the SR-BI and calnexin. Images shown are the representative images from 3-4 independent transient transfections.
Effects of cysteine mutations and substitutions on HDL-binding and selective HDL-CE uptake
Effects of cysteine mutations and substitutions on HDL-binding and selective HDL-CE uptake—to examine the role of cysteine residues on SR-BI-mediated HDL binding and selective HDL-CE uptake, we again employed cysteine mutation and substitution and overexpression strategies. As before, CHO cells were transiently transfected with the wild type or various SR-BI mutants and subsequently analyzed for HDL binding and/or selective HDL-CE using either radioactive ([125I-DLT]-[3H]COE-labeled hHDL3) or fluorescent (recHDL-BODIPY-CE) HDL preparations as a tracer ligand for SR-BI. Expression of single C280S, C321S, C323S and C334S and double, C280S/C321S, C280S/C334S, C321S/C323S and C323S/C334S cysteine mutants significantly reduced the HDL binding compared to that seen with the wild type SR-BI (Fig. 8B), consistent with their level of presentation at the cell surface (Fig. 4A, B). Similarly, expression of double cysteine mutants, C251S-C321S, C280S-C323S, and C334S-C384S cysteine caused a 50-60% reduction in cell surface binding of HDL (i.e., association of HDL with SR-BI) (data not shown). Expression of remaining mutants or constructs with cysteine substitutions including C251S and C384S exhibited HDL binding that was comparable to the binding seen with the CHO cells overexpressing wild type of SR-BI.
Figure 8. Effects of cysteine mutations and substitutions of SR-BI on HDL-binding activity and selective HDL-CE uptake by CHO cells.
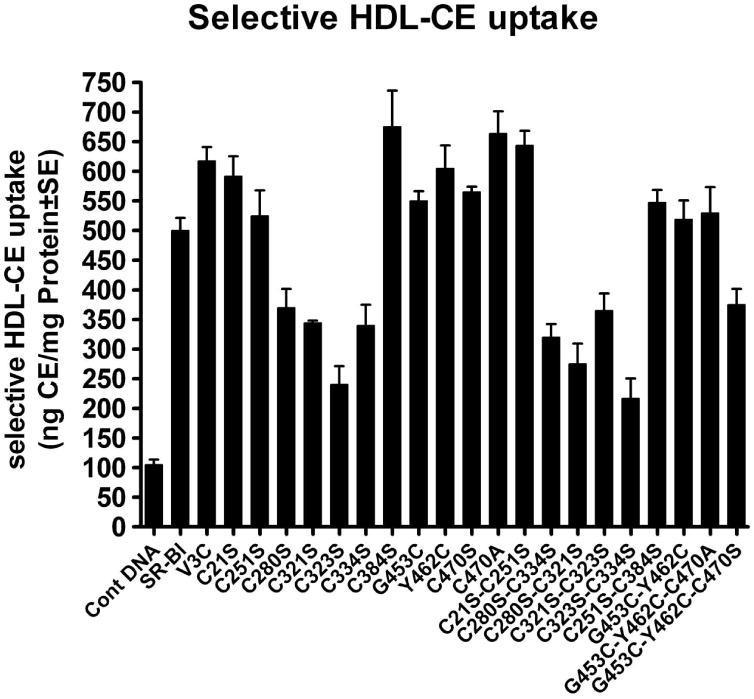
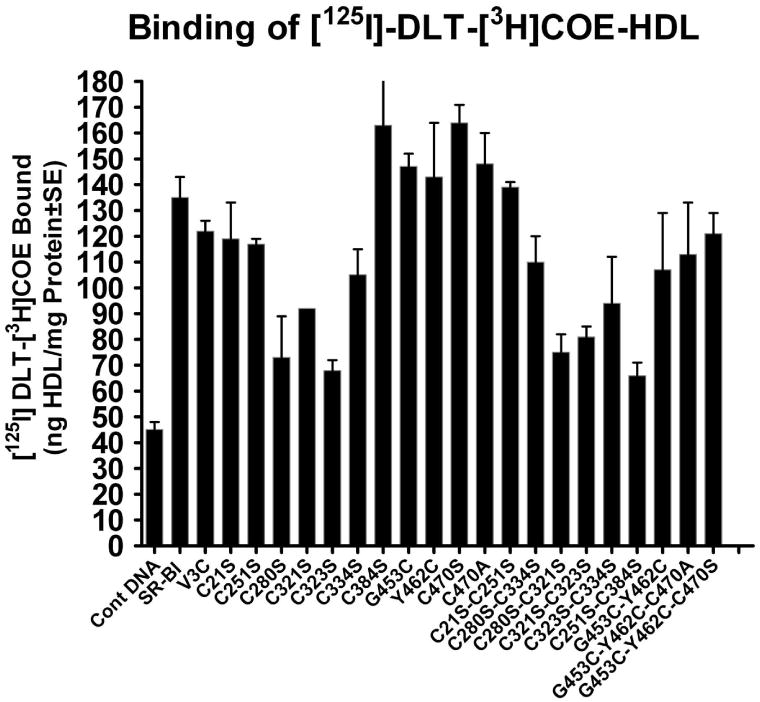
CHO cells were transiently transfected with vector alone, V5-rat SR-BI or the indicated mutants. Thirty-six hours post-transfection, cells were incubated with a culture medium containing 50 μg protein/ml of [125I-DLT]-[3H]COE-labeled hHDL3 for 5h. At the end of incubation, the cells were processed for the determination of [125I-DLT]-hHDL3 binding activity and the quantification of selectively internalized HDL-[3H]COE as described under “Materials and Methods.” The results are Mean ± SE at least three independent experiments and normalized to per unit of total expressed SR-BI. A, selective HDL-CE uptake and B,HDL binding activity.
We then assessed SR-BI-mediated selective HDL-CE uptake in CHO cells expressing wild type or one of the cysteine mutant or cysteine substitution constructs. Consistent with HDL binding, expression of single C280S, C321S, C323S and C334S or double C280S/C321S, C280S/C334S, C321S/C323S, C323S/C334S, C251S-C321S, C280S-C323S, and C334S-C384S resulted in a significant reduction of HDL-CE uptake as measured by the internalization of the HDL-derived tracer [3H]COE (Fig. 8A). Also, as with the HDL binding, the remaining mutants did not significantly impact the SR-BI mediated selective HDL-CE uptake. Complementary results were obtained when the relative efficacy of these various mutants was assessed in COS-7 and CHO cells using [125I-DLT]-[3H]COE-labeled hHDL3 and HDL-BODIPY-CE uptake as a ligand, respectively (Table 3).
Effects of cysteine mutations and substitutions on the relative expression of SR-BI monomers, dimers and oligomers
Our recently published data provide evidence that the physical state of the SR-BI protein (i.e., monomeric, vs. dimeric and higher order oligomeric forms of SR-BI), and architechtural changes in the cell surface induced by the expression of SR-BI, also play major roles in the functional efficiency of the selective pathway (12,32,33). To determine whether cysteine residues are important for SR-BI oligomerization, various cysteine mutants described above were tested by Western blotting for their ability to form SR-BI dimers and oligomers. Fig. 7A indicates that expression of single cysteine mutants, C21S, C251S and C384S in CHO cells inhibited, while C280S, C321S, C323S and C334S promoted SR-BI dimer formation. Mutation of cysteine C470S or introduction of new cysteine residues, V3C or Y462C, however, had no effect on SR-BI dimerization (Fig. 7A). Likewise, expression of double mutants, C251S/C384S, C280S/C321S, C280S/C334S, and C321S/C323S and triple mutants, G453C/Y462S/C470S caused increased formation of SR-BI dimers (Fig. 7B). Use of remaining double and triple mutants exhibited either no or very little modulatory effect on SR-BI dimerization. It is noteworthy that expression of four C280S, C321S, C323S or C334S mutants or their various paired combinations, which lead to a loss of selective HDL-CE uptake, strongly favor the SR-BI dimer formation. Thus, a complex but unresolved relationship exists between SR-BI dimer/oligomer formation and SR-BI-mediated HDL binding and associated selective HDL-CE uptake. Moreover, although C280, C321, C323 or C334 residues strongly favor dimer formation, they are rendered non-functional. However, they are rendered non-functional presumably due to formation of aberrant disulfide linkages resulting in inhibition of optimal HDL binding and hence, selective HDL-CE uptake.
Discussion
The SR-BI, an HDL receptor, mediates the bulk delivery of plasma lipoprotein-cholesteryl esters (CEs) to steroidogenic and liver tissues for product formation (steroid hormones and bile acids) or their elimination by a process known as ‘selective’ transport process. (2,3,5-7). This process differs from the classic endocytic, LDL receptor pathway in that exogenous circulating lipoproteins such as HDL and LDL contribute their CEs to cells without internalization of the intact lipoprotein particle (7,13,15,59). Thus, in the selective cholesterol uptake process, lipoprotein lipids enter cells unaccompanied by apolipoproteins. However, the information about the cellular events connected with and underlying mechanisms involved in selective CE transport remain poorly defined. In this study, we evaluated the importance of the eight cysteine (C) residues in the extracellular, transmembrane- and C-terminal domains of the rat SR-BI. Because seven out of eight cysteine (C) residues (C251, C280, C321, C323, C334, C384, and C470) of SR-BI are well-conserved across the species (Table 2), we suspected that they might play important roles in SR-BI expression, dimerization/oligomerization, and/or function (binding of HDL and SR-BI-mediated selective HDL-CE uptake). In addition, we determined the effects of artificially introduced cysteine residues on the structure and function of SR-BI. All of these studies were accomplished using a combination of site-directed mutagenesis and chemical modification of cysteine residues of the SR-BI. The latter employed disulfide bond reducing agents β-ME and DTT and sulfhydryl alkylating agents, like MTS reagents.
In the first set of studies, we found that treatment of SR-BI overexpressing cells with reducing agents (β-ME and DTT) caused small but significant reduction in selective HDL-CE, although DTT was somewhat more effective than β-ME (Fig. 3). Such a modest effect of β-ME and DTT on SR-BI function suggests that disulfide bonds either exist at low levels or contribute minimally towards the maintenance of the secondary structure of SR-BI. Another possibility could be that the structural changes that result from disrupting the accessible disulfide bonds in SR-BI may not be severe enough to cause a much greater impact on its function. Furthermore, one cannot rule out the possibility that potential SR-BI disulfide bonds are inaccessible to DTT and β-ME. A similar inaccessibility of DTT has been suggested for the disulfide bonds in channels and receptors such as the inwardly rectifying K+ channel, Kir 2.1, and P2X1, and P2X2 receptors (60-62). Although it has been clearly established that disulfide bonds are essential for the proper functioning of Kir2.1 channel, and P2X1, and P2X2 receptors, these channel and receptor proteins, however, are unaffected by reducing agents. This is also consistent with the findings that disulfide bonds of the mature proteins are often inaccessible to reducing agents unless the protein is rendered denatured (63,64).
We next examined the potential contribution of endogenous free cysteines of the native SR-BI in selective HDL-CE transport. This was accomplished using free cysteine (sulfhydryl) reactive neutral (MMTS), positively (MTSEA and MTSET) and negatively (MTSES) charged MTS reagents. Our studies show that neutral MMTS and positively charged MTSEA reagents greatly reduced the SR-BI-mediated selective HDL-CE uptake in SR-BI overexpressing COS-7 cells (Fig. 3). Based on these findings it would appear that certain endogenous cysteine residues participate in SR-BI-mediated selective HDL-CE uptake. Given that MTS reagents including MMTS and MTSEA primarily react with ionized thioles present in water-accessible regions (64-67), these results indicate that critical endogenous cysteine residues involved in the selective transport process must exist in a water accessible environment. Since MMTS and MTSEA (in the pH range of the functional assay, a significant fraction of MTSEA is neutral and able to permeate the lipid bilayer) can enter the cell quite readily, it is possible that they merely interact with the N-terminal transmembrane membrane domain cysteine 21 and/or C-terminal domain cysteine 470 and produce an inhibitory effect. However, using mutagenesis, we have shown that the functional behavior of C21S and C470S were, in fact, very similar to that exhibited by the wild-type SR-BI, and thus, are unlikely to be involved in the selective CE transport. In contrast, the insensitivity to membrane-impermeant positively and negatively charged MTSET and MTSES reagents, respectively, suggest either endogenous cysteine residues are inaccessible to these MTS reagents in the aqueous environment or that their alkylation does not hamper the SR-BI-mediated selective HDL-CE uptake. A second possibility is that MTSET or MTSES might “bypass” the crucial cysteine residues involved in the selective CE transport process. Similar differential sensitivity to various MTS reagents has also been described for several other systems. For example, Zhu et al (68) reported that two introduced cysteine mutants (A435C and T442C) in the TM1 region of human NBCel-A were highly inhibited by three MTS reagents, MTSES, MTSET and MTSEA. Furthermore, I441C was significantly inhibited by MTSET and MTSEA and A428 by MTSEA alone (68). Likewise, it was reported that treatment of COS-1 cells expressing human cannabinoid receptor (CB1) with MMTS or MTSEA leads to a dramatic reduction in agonist SR141716A binding to the receptor; such treatment, however, had no significant effect on agonist CP55940 binding to the CB1 receptor (69). Human AT1 receptors (70) and P2X2 (61) are shown to be insensitive to MTSEA and MTSET, respectively. On the other hand, it was reported that MTSET treatment of HEK cells transiently overexpressing glucagon receptor enhanced (instead of decreasing) glucagon potency for cAMP stimulation by two- to three-fold (71).
Our data with the use of sulfhydryl-reactive reagent, such as cell-impermeant DTT, raised the possibility that disulfide bond(s) exist in SR-BI and contribute to the selective HDL-CE (Fig. 3). In addition, the reduction of higher complexes by DTT suggested that ectodomain (or ECD) dimer/oligomer formation may be mediated, at least, in part, by disulfide bond formation via the six conserved cysteine residues found on the ECD of the SR-BI. To study directly the role of disulfide bonds and other cysteine residues, we employed single and paired cysteine mutagenesis coupled with Western blotting, confocal microscopy and functional studies to show that certain cysteine residues in the ECD of rat SR-BI form disulfide bonds, participate in SR-BI dimerization/oligomerization, facilitate normal trafficking of SR-BI to the cell surface, and contribute to the binding of HDL to SR-BI and SR-BI-mediated selective HDL-CE uptake.
As noted above and in the Results section, rat SR-BI, the prototype used here, contains eight cysteine (Cys) residues; while these residues are dispersed throughout the protein, they are mainly concentrated in the ECD. Because seven out of eight Cys residues including six in ECD (C251, C280, C321, C323, C334, and C384) of SR-BI are well-conserved across the species (Table 1), we reasoned that they might play an essential role in SR-BI expression and function as well as its dimerization/oligomerization. Substitution of cysteines with serine at positions C280S, C321S, C323S and C334S in extracellular domain caused significant reduction in ligand HDL binding to SR-BI and the ability of SR-BI to mediate the selective delivery of HDL-CE into the cell (Table 3, Fig. 8 A and B). The data suggest that these residues may directly participate in SR-BI-HDL interaction and SR-BI-mediated selective HDL-CE uptake and may also be involved in disulfide bond formation. Interestingly, despite the absolute conservation and occurrence of the C384 in the ECD, the C384 mutant (C384S) did not affect the function of SR-BI (Table 3, Fig. 8 A and B). Likewise, mutation of N-terminal domain C21 (C21S) and C-terminal domain C470 (C470S) to serine or mutation of N-terminal domain V3 (V3C) and C-terminal transmembrane domain G453 (G453C) and Y462 (Y462C) to cysteine residues also were without effect suggesting that they are not required for optimum HDL-binding to the SR-BI or SR-BI-mediated selective HDL-CE uptake (Table 3, Figs. 8 A and B).
The parallel decrease in cell surface expression (Figs. 4 and 5) and the lack of effect of the mutations on the intrinsic SR-BI properties indicate that the primary cause of impaired SR-BI expression and function for the C280S, C321S, C323S, and C334S mutants is the routing of the SR-BI to the cell surface. Although at present we are unable to provide an exact explanation, two possibilities may be considered to explain the decreased localization of mutant SR-BI proteins at the cell surface: a) introduction of cysteine mutations resulted in misfolding of SR-BI proteins, which interfered with protein trafficking; and 2) reduced levels of cysteine mutant SR-BI proteins due their enhanced susceptibility to degradation. The latter possibility, however, seems unlikely given that both Western blotting and immunofluorescence quantification of total SR-BI protein showed equal expression of SR-BI in cells transfected with native (wild-type) or various cysteine mutant SR-BIs (Figs. 6 and 7). Therefore, the loss of HDL binding and selective cholesterol transport function seen with C280S, C321S, C323S, and C334S (Table 3 and Fig. 8) is not attributable to decreased SR-BI protein expression (synthesis) or increased intracellular degradation. When, considering the fact that these 4 cysteine residues (i.e., C280, C321, C323, and C334) are clustered within the ECD and that mutants of these cysteines were quite similar in their level of cell surface expression and failure to bind HDL and mediate selective HDL-CE uptake, it is most likely that they participate in inter-disulfide bond formation. Moreover, it is possible that mutation of these cysteine residues affects disulfide formation, in turn affecting folding, ultimately intracellular SR-BI trafficking, and therefore ligand (HDL) binding and function (selective HDL-CE uptake). Previous studies have shown that the extracellular domain of the CD36 scavenger receptor, a defining member of the SR-BI gene family, contains three disulfide bridges involving C313-C322, C243-C311, and C280-C333 cysteine residues (72). Given that ECDs of CD36 and SR-BI show very close sequence homology to each other, it would appear that SR-BI may also contain 3 disulfide bonds involving C323-C334, C251-C321 and C280-C384 cysteine residues. Additional experimental studies are underway to explore this likely possibility.
We further examined the possibility that paired cysteine mutations may rescue the loss of expression and function of SR-BI. We reasoned that simultaneous elimination of both cysteine partners (which form disulfide bonds) in any specific pair combination would minimize the structural alterations in SR-BI and restore the functional expression of SR-BI. However, this turned out not to be the case and double mutations of SR-BI involving C280S/C321S, C280S/C334S, C321S/C323S, C323S/C334S, C251S-C321S, C280S-C323S, and C334S-C384S also showed impaired cell surface expression, HDL binding and selective CE transport function (Figs. 4, 5 and 8, Table 3). A comparison of these various double cysteine mutants indicated that two double mutants, C280S/C321S and C323S/C334S, gave the lowest cell surface expression of SR-BI (∼20% of wild-type) suggesting that these four cysteine residues are not only involved in the maintenance of SR-BI structure but also important for optimal cell surface expression of SR-BI, and low cell surface expression is the primary cause of the impaired SR-BI function (Figs. 4 and 5).
We also studied the potential involvement of disulfide bonds in SR-BI dimer formation. To do this we expressed the mutated single and paired cysteine residue constructs as described above and analyzed for the presence of SR-BI monomer and dimer by SDS-polyacrylamide gel electrophoresis under non-reducing conditions following Western blotting. Expression of single cysteine mutants, C21S, C251S and C384S in CHO cells inhibited, while C280S, C321S, C334S and C323S and other double (C251S/C384S, C280S/C321S, C280S/C334S, and C321S/C323S) and triple (G453C/Y462C/C470S) mutants promoted SR-BI dimer formation (Fig. 7 A and B). Use of other double and triple cysteine mutants exhibited either no or very little influence on SR-BI dimer formation. Introduction of a new cysteine residue had either no effect (V3C) or inhibited (Y462C) SR-BI dimerization. It is noteworthy that expression of four single C280S, C321S, C323S or C334S mutants or their various paired combinations, which lead to a loss of selective HDL-CE uptake, strongly favor the SR-BI dimer formation. The dysfunctional SR-BI dimers observed with the use of these single and double mutants could potentially result due to the formation of aberrant disulfide linkages. In this regard, mutations of one or more of these cysteine residues that are essential for intra-disulfide bond formation and SR-BI dimerization could disrupt these bonds but at the same time other free cysteine residues may rearrange to form new disulfide bonds and cause enhanced SR-BI dimerization. However, they are rendered nonfunctional because these residues may also be important for optimal HDL binding and hence, selective HDL-CE uptake.
In conclusion, the results of this study provide evidence that four extracellular domain cysteine residues of SR-BI, C280, C321, C223 and C334 are essential for SR-BI structure including dimerization, its optimal expression at the cell surface and HDL binding and selective CE transport function. In addition, based on MTS studies, certain free cysteine residues especially those localized in the transmembrane and C-terminal domains, also play an important role in SR-BI function. These results provide important insights into the functional role of cysteines on SR-BI dimerization, cell surface expression, and function. While this manuscript was being finalized for submission to this journal for publication, a report by Papale et al (73) appeared providing experimental evidence about the potential role of the intramolecular disulfide bonds in the extracellular domain of SR-BI in SR-BI-mediated selective cholesterol transport. Although, this publication also emphasizes the importance of four ECD cysteines in SR-BI function, some notable differences exist between this report and the results of our study. For example, the major finding of Papale et al (73) is that mutation of any four of the cysteine residues, C280S, C321S, C323S or C334S of the extracellular domain of SR-BI leads to impaired HDL binding and subsequent inhibition of selective HDL-CE uptake. Furthermore, such inhibition in SR-BI binding activity and cholesterol transport occurred without impacting the cell surface expression of mutant SR-BI proteins. While our studies reached the same conclusion, we provide extensive evidence to suggest that mutation of any of the four cysteine residues, C280S, C321S, C323S or C334S, results in a mutant SR-BI protein whose surface expression is greatly reduced and as a consequence results in inhibition of both HDL binding as well as selective HDL-CE uptake. This inhibition in surface transport occurs because mutations likely result in misfolding of proteins, which interferes with SR-BI trafficking. Interestingly Western blotting and immunofluorescent measurement for total SR-BI protein (Figs. 6 and 7) showed equivalent expression of SR-BI in cells transfected with native or various cysteine mutant SR-BIs. Thus, as discussed above, loss of SR-BI HDL binding activity and selective cholesterol transport function is not due to decreased SR-BI protein expression or increased intracellular degradation, but results from reduced cell surface expression of the mutant SR-BI proteins. At present, we are unable to provide a n exact explanation about the observed discrepancy between our results and those of Papale et al (73). However, a close visual inspection of Western blot data shown in Fig. 6 of the Papale et al report (73) revealed that total expression of C280S, C321S, C323S or C334S mutant protein may be less than 50% of the wild-type SR-BI although no quantitative data accompanied such blots. To what extent such potential reduction in total mutant SR-BI protein levels may have contributed to the observed loss of SR-BI ligand binding activity and selective cholesterol transport was not apparent. Our studies further demonstrate that introduction of cysteine mutations can result in aberrant SR-BI dimer formation, which are also likely to interfere with selective HDL-CE uptake function (SR-BI dimers formed from native [wild-type] SR-BI are the ones that mediate selective cholesterol transport). Finally, with the use of various charge specific MTS reagents, we provide evidence that free cysteine residues also play a role in SR-BI function.
Footnotes
The abbreviations used are: CE, cholesteryl ester; CHO, Chinese-hamster ovary; PBS, phosphate buffered saline; COE, cholesteryl oleolyl ether; COS-7, African green monkey kidney cells; CTD, C-terminal domain; DMEM, Dulbecco's modified Eagle's medium; DTT, dithiothreitol; DLT, dilactitol tyramine; ECD, extracellular domain; FACS, fluorescence- activated cell sorting; FITC, fluorescein isothiocyanate; HDL, high-density lipoprotein; β-ME, β-mercaptoethanol; MMTS, methyl methanethiosulfonate; MTSEA, 2-aminoethyl methanethiosulfonate hydrobromide; MTS, methanethiosulfonate; MTSES, Sodium (2-sulfonatoethyl) methanethiosulfonate; MTSET, [2-(trimethylammonium)ethyl] methanethiosulfonate bromide; NHERF3, Na+/H+-exchanger regulatory factor 3; PDZ, Postsynaptic density protein PSD95-Drosophila homolog Discs-large and tight junction protein Zona occludens-1; PDZK1, PDZ domain containing protein kidney 1; recHDL-BODIPY-CE, recombinant HDL-BODIPY-CE; SDS, sodium dodecyl sulfate; NaPFO, sodium perfluorooctanoate; SR-BI, scavenger receptor, class B, type I phosphate-buffered saline; TMD, transmembrane domain
References
- 1.Azhar S, Reaven E. Scavenger receptor class BI and selective cholesteryl ester uptake: partners in the regulation of steroidogenesis. Mol Cell Endocrinol. 2002;30:1–26. doi: 10.1016/s0303-7207(02)00222-8. [DOI] [PubMed] [Google Scholar]
- 2.Azhar S, Leers-Sucheta S, Reaven E. Cholesterol uptake in adrenal and gonadal tissues: the SR-BI and ‘selective’ pathway connection. Front Biosci. 2003;8:s998–1029. doi: 10.2741/1165. [DOI] [PubMed] [Google Scholar]
- 3.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 4.Connelly MA, Williams DL. SR-BI and cholesterol uptake into steroidogenic cells. Trends Endocrinol Metab. 2003;14:467–472. doi: 10.1016/j.tem.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 6.Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apolipoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A. 1983;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reaven E, Chen YD, Azhar S. Morphological evidence that high density lipoproteins are not internalized by steroid-producing cells during in situ organ perfusion. J Clin Invest. 1984;74:1384–1397. doi: 10.1172/JCI111549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gwynne JT, Mahaffee DD. Rat adrenal uptake and metabolism of high density lipoprotein cholesteryl ester. J Biol Chem. 1989;264:8141–8150. [PubMed] [Google Scholar]
- 9.Landschulz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest. 1996;98:984–995. doi: 10.1172/JCI118883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rigotti A, Edelman ER, Seifert P, Iqbal SN, DeMattos RB, Temel RE, Williams DL. Regulation by adrenocorticotropic hormone of the in vivo expression of scavenger receptor class B type I (SR-BI), a high density lipoprotein receptor, in steroidogenic cells of the murine adrenal gland. J Biol Chem. 1996;271:33545–33549. doi: 10.1074/jbc.271.52.33545. [DOI] [PubMed] [Google Scholar]
- 11.Reaven E, Nomoto A, Leers-Scheta S, Temel R, Williams DL, Azhar S. Expression and microvillar localization of scavenger receptor, class B, type I (a high density lipoprotein receptor) in luteinized and hormone-desensitized rat ovarian models. Endocrinology. 1998;139:2847–2856. doi: 10.1210/endo.139.6.6056. [DOI] [PubMed] [Google Scholar]
- 12.Azhar S, Nomoto A, Reaven E. Hormonal regulation of adrenal microvillar channel formation. J Lipid Res. 2002;43:861–871. [PubMed] [Google Scholar]
- 13.Azhar S, Stewart D, Reaven E. Utilization of cholesterol-rich lipoproteins by perfused rat adrenals. J Lipid Res. 1989;30:1799–1810. [PubMed] [Google Scholar]
- 14.Pieters MN, Schouten D, Bakkeren HF, Esbach B, Brouwer A, Knook DL, van Berkel TJ. Selective uptake of cholesteryl esters from apolipoprotein-E-free high-density lipoproteins by rat parenchymal cells in vivo is efficiently coupled to bile acid synthesis. Biochem J. 1991;280:359–365. doi: 10.1042/bj2800359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reaven E, Tsai L, Azhar S. Cholesterol uptake by the ‘selective’ pathway of ovarian granulosa cells: early intracellular events. J Lipid Res. 1995;36:1602–1617. [PubMed] [Google Scholar]
- 16.Mardones P, Quiñones V, Amigo L, Moreno M, Miquel JF, Schwarz M, Miettinen HE, Trigatti B, Krieger M, VanPatten S, Cohen DE, Rigotti A. Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class B type I-deficient mice. J Lipid Res. 2001;42:170–180. [PubMed] [Google Scholar]
- 17.Wiersma H, Gatti A, Nijstad N, Kuipers F, Tietge UJ. Hepatic SR-BI, not endothelial lipase, expression determines biliary cholesterol secretion in mice. J Lipid Res. 2009;50:157–180. doi: 10.1194/jlr.M800434-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azhar S, Tsai L, Medicherla S, Chandrasekher Y, Giudice L, Reaven E. Human granulosa cells use high density lipoprotein cholesterol for steroidogenesis. J Clin Endocrinol Metab. 1998;83:983–991. doi: 10.1210/jcem.83.3.4662. [DOI] [PubMed] [Google Scholar]
- 19.Briand O, Lestavel S, Pilon A, Torpier G, Fruchart JC, Calvey V. SR-BI does not require raft/caveola localization in the human adrenal cell line NCI-H295R. Biochim Biophys Acta. 2003;163:42–50. doi: 10.1016/s1388-1981(02)00354-2. [DOI] [PubMed] [Google Scholar]
- 20.Azhar S, Nomoto A, Leers-Sucheta S, Reaven E. Simultaneous induction of an HDL receptor protein (SR-BI) and the selective uptake of HDL-cholesteryl esters in physiologically relevant steroidogenic cell model. J Lipid Res. 1998;39:1616–1628. [PubMed] [Google Scholar]
- 21.Reaven E, Zhan L, Nomoto A, Leers-Sucheta S, Azhar S. Expression and microvillar localization of scavenger receptor class B, type (SR-BI) and selective cholesteryl ester uptake in Leydig cells from testis. J Lipid Res. 2000;41:343–356. [PubMed] [Google Scholar]
- 22.Ikemoto M, Arai H, Feng D, Tanaka K, Aoki J, Dohmae N, Takio K, Adachi H, Tsujimoto M, Inoue K. Identification of a PDZ-domain-containing protein that interacts with the scavenger receptor class B type I. Proc Natl Acad Sci U S A. 2000;97:6538–6543. doi: 10.1073/pnas.100114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silver D. SR-BI and protein-protein interactions in hepatic high density lipoprotein metabolism. Rev Endocr Met Disord. 2004;5:327–333. doi: 10.1023/B:REMD.0000045104.38104.8e. [DOI] [PubMed] [Google Scholar]
- 24.Kocher O, Krieger M. Role of the adaptor protein PDZK1 in controlling the HDL receptor SR-BI. Curr Opin Lipidol. 2009;20:236–241. doi: 10.1097/MOL.0b013e32832aee82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reaven E, Shi XY, Azhar S. Interaction of lipoproteins with isolated ovary plasma membranes. J Biol Chem. 1990;265:19100–19111. [PubMed] [Google Scholar]
- 26.Reaven E, Boyles J, Spicher M, Azhar S. Evidence for surface entrapment of cholesterol-rich lipoproteins in luteinized ovary. Arteriosclerosis. 1988;8:298–309. doi: 10.1161/01.atv.8.3.298. [DOI] [PubMed] [Google Scholar]
- 27.Reaven E, Spicher M, Azhar S. Microvillar channels: a unique plasma membrane compartment for concentrating lipoproteins on the surface of rat adrenal cortical cells. J Lipid Res. 1989;30:1551–1560. [PubMed] [Google Scholar]
- 28.Hu J, Zhang Z, Shen WJ, Azhar S. Cellular cholesterol delivery, intracellular processing, and utilization for biosynthesis of steroid hormones. Nutr Metab. 2010;7:47. doi: 10.1186/1743-7075-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu X, Trigatti B, Xu S, Acton S, Babitt J, Krieger M. The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J Biol Chem. 1998;273:26338–26348. doi: 10.1074/jbc.273.41.26338. [DOI] [PubMed] [Google Scholar]
- 30.Connelly MA, Klein SM, Azhar S, Abumrad NA, Williams DL. Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J Biol Chem. 1999;274:41–47. doi: 10.1074/jbc.274.1.41. [DOI] [PubMed] [Google Scholar]
- 31.Parathath S, Sahoo D, Dralington YF, Peng Y, Collins HL, Rothblat GH, Williams DL, Connelly MA. Glycine 420 near the C-terminal transmembrane domain of SR-BI is critical for proper delivery and metabolism of high density lipoprotein cholesteryl ester. J Biol Chem. 2004;279:24976–24985. doi: 10.1074/jbc.M402435200. [DOI] [PubMed] [Google Scholar]
- 32.Reaven E, Cortez J, Leers-Sucheta S, Nomoto A, Azhar S. Dimerization of the scavenger receptor class B type I: Formation, function, and localization in diverse cells and tissues. J Lipid Res. 2004;45:513–528. doi: 10.1194/jlr.M300370-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Reaven E, Nomoto A, Cortez Y, Azhar S. Consequences of over-expression of rat scavenger receptor, SR-BI, in an adrenal cell model. Nutr Metab (Lond) 2006;3:43. doi: 10.1186/1743-7075-3-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mamathambika BS, Bardwell JC. Disulfide-linked protein folding pathways. Annu Rev Cell Dev Biol. 2008;24:211–235. doi: 10.1146/annurev.cellbio.24.110707.175333. [DOI] [PubMed] [Google Scholar]
- 35.Trivedi MV, Laurence JS, Siahann TJ. The role of thiols and disulfides on protein stability. Curr Protein Pept Sci. 2009;10:614–625. doi: 10.2174/138920309789630534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stitham J, Gleim SR, Douville K, Arehart E, Hwa J. Versatility and differential roles of cysteine residues in human prostacyclin receptor structure and function. J Biol Chem. 2006;281:37227–37236. doi: 10.1074/jbc.M604042200. [DOI] [PubMed] [Google Scholar]
- 37.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–72. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 38.Nagahara N, Matsumura T, Okamoto R, Kajihara Y. Protein cysteine modifications: (1) medical chemistry for proteomics. Curr Med Chem. 2009;16:4419–4444. doi: 10.2174/092986709789712880. [DOI] [PubMed] [Google Scholar]
- 39.Reaven E, Tsai L, Azhar S. Intracellular events in the “selective” transport of lipoprotein-derived cholesteryl esters. J Biol Chem. 1996;271:16208–16217. doi: 10.1074/jbc.271.27.16208. [DOI] [PubMed] [Google Scholar]
- 40.Ramjeesingh M, Huan LJ, Grami E, Bear CE. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J. 1999;342:119–123. [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson GL. A simplification of the protein assay method of Lowry et al which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 42.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 43.Markwell MAK, Hass SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simply protein determination in membrane and lipoprotein samples. Anal Chem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 44.Wu JJ, Guidotti G. Construction and characterization of a monomeric insulin receptor. J Biol Chem. 2002;277:27809–27817. doi: 10.1074/jbc.M200367200. [DOI] [PubMed] [Google Scholar]
- 45.Salahpour A, Bonin H, Bhalla S, Petäjä-Repo U, Bouvier M. Biochemical characterization of β2-adrenergic receptor dimers and oligomers. Biol Chem. 2003;384:117–123. doi: 10.1515/BC.2003.012. [DOI] [PubMed] [Google Scholar]
- 46.Ivanov AV, Gable ME, Askari A. Interaction of SDS with Na+/K+-ATPase: SDS-solubilized enzyme retains partial structure and function. J Biol Chem. 2004;279:29832–29840. doi: 10.1074/jbc.M401986200. [DOI] [PubMed] [Google Scholar]
- 47.Silvius JR. Solubilization and functional reconstitution of biomembrane components. Annu Rev Biophys Biomol Struct. 1992;21:323–348. doi: 10.1146/annurev.bb.21.060192.001543. [DOI] [PubMed] [Google Scholar]
- 48.Ramjeesingh M, Huan LJ, Garami E, Bear CE. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J. 1999;342:119–123. [PMC free article] [PubMed] [Google Scholar]
- 49.Stauffer DA, Karlin A. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 1994;33:6840–6849. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- 50.Gandhi CS, Clark E, Loots E, Pralle A, Isacoff EY. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron. 2003;40:515–525. doi: 10.1016/s0896-6273(03)00646-9. [DOI] [PubMed] [Google Scholar]
- 51.Dodd JR, Christie DL. Substituted cysteine accessibility of the third transmembrane domain of the creatine transporter: defining a transport pathway. J Biol Chem. 2005;280:32649–32654. doi: 10.1074/jbc.M506723200. [DOI] [PubMed] [Google Scholar]
- 52.Fay JF, Dunham TD, Farrens DL. Cysteine residues in the human canabinoid receptor: only C257 and C264 are required for a functional receptor, and steric bulk at C386 impairs antagonist SR141716A binding. Biochemistry. 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 53.Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem. 2007;282:20180–20190. doi: 10.1074/jbc.M611761200. [DOI] [PubMed] [Google Scholar]
- 54.Beck EJ, Yang Y, Yaemsiri S, Raghuram V. Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem. 2008;283:4957–4966. doi: 10.1074/jbc.M702235200. [DOI] [PubMed] [Google Scholar]
- 55.Xu W, Sanz A, Pardo L, Liu-Chen LY. Activation of the μ opioid receptor involves conformational rearrangements of multiple transmembrane domains. Biochemistry. 2008;47:10576–10586. doi: 10.1021/bi800381v. [DOI] [PubMed] [Google Scholar]
- 56.Hou Z, Cherian C, Drews J, Wu J, Matherly LH. Identification of the minimal functional unit of the homo-oligomeric human reduced folate carrier. J Biol Chem. 2010;285:4732–4740. doi: 10.1074/jbc.M109.086033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holmgren M, Liu Y, Xu Y, Yellen G. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 1996;35:797–804. doi: 10.1016/0028-3908(96)00129-3. [DOI] [PubMed] [Google Scholar]
- 58.Lisenbee CS, Dong M, Miller LJ. Paired cysteine mutagenesis to establish the pattern of disulfide bonds in the functional intact secretin receptor. J Biol Chem. 2005;280:12330–12338. doi: 10.1074/jbc.M414016200. [DOI] [PubMed] [Google Scholar]
- 59.Pittman RC, Knecht TP, Rosenbaum MS, Taylor CA. A nonendocytic mechanism for the selective uptake of high density lipoprotein-associated esters. J Biol Chem. 1987;262:2443–2450. [PubMed] [Google Scholar]
- 60.Cho HC, Tsushima RG, Nguyen TT, Guy HR, Backs PH. Two critical cysteine residues implicated in disulfide bond formation and proper folding of Kir 2.1. Biochemistry. 2000;39:4649–4657. doi: 10.1021/bi992469g. [DOI] [PubMed] [Google Scholar]
- 61.Clyne JD, Wang LF, Hume RI. Mutational analysis of the conserved cysteines of the rat P2X2 purinoceptor. J Neurosci. 2002;22:3873–3880. doi: 10.1523/JNEUROSCI.22-10-03873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ennion SJ, Evans RJ. Conserved cysteine residues in the extracellular loop of the human P2X1 receptor from disulfide bonds and are involved in receptor trafficking to the cell surface. Mol Pharmacol. 2002;61:303–311. doi: 10.1124/mol.61.2.303. [DOI] [PubMed] [Google Scholar]
- 63.Creighton TE. Disulfide bonds and protein stability. BioEssays. 1988;8:57–63. doi: 10.1002/bies.950080204. [DOI] [PubMed] [Google Scholar]
- 64.Tatu U, Braakman I, Helenius A. Membrane glycoprotein folding, oligomerization and intracellular transport: effects of dithiothreitol in living cells. EMBO J. 1993;12:2151–2157. doi: 10.1002/j.1460-2075.1993.tb05863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA. Reactivity of small thiolate anions and cysteine-25 in papain toward methyl methanethiosufonate. Biochemistry. 1986;25:5595–5601. doi: 10.1021/bi00367a038. [DOI] [PubMed] [Google Scholar]
- 66.Javitch JA, Shi L, Liapakis G. Use of the substituted cysteine accessibility method to study the structure and function of G protein-coupled receptors. Methods Enzymol. 2003;343:137–156. doi: 10.1016/s0076-6879(02)43131-x. [DOI] [PubMed] [Google Scholar]
- 67.Fay JA, Dunham FD, Farrens DL. Cysteine residues in the human cannabinoid receptor: only C257 and C264 are required for a functional receptor, and steric bulk at C386 impairs antagonist SR141716A binding. Biochemistry. 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 68.Zhu Q, Azimov R, Kao L, Newman D, Liu W, Abuladze N, Pushkin A, Kurtz I. NBCe1-A transmembrane segment 1 lines the ion translocation pathway. J Biol Chem. 2009;284:8918–8929. doi: 10.1074/jbc.M806674200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fay JF, Dunham TD, Farrens DL. Cysteine residues in the human cannabinoid receptor: only C257 and C264 are required for a functional receptor, and steric bulk at C386 impairs antagonist SR141716A binding. Biochemistry. 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 70.Domazet I, Martin SS, Holleran BJ, Morin ME, Lacasse P, Lavigne P, Escher E, Leduc R, Guillemette G. The fifth transmembrane domain of angiotensin II type 1 receptor participates in the formation of ligand-binding pocket and undergoes a counterclockwise rotation upon receptor activation. J Biol Chem. 2009;284:31953–31961. doi: 10.1074/jbc.M109.051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prévost M, Vertongen P, Raussens V, Roberts DJ, Cnudde J, Perret J, Waelbroeck M. Mutational and cysteine analysis of the glucagon receptor N-terminal domain. J Biol Chem. 2010;285:30951–30958. doi: 10.1074/jbc.M110.102814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2(72):re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Papale GA, Hanson PJ, Sahoo D. Extracellular disulfide bonds support scavenger receptor class B type-1-mediated cholesterol transport. Biochemistry. 2011;50:6245–6254. doi: 10.1021/bi2005625. [DOI] [PMC free article] [PubMed] [Google Scholar]