

Summary
Expression of the two critical virulence factors of Vibrio cholerae, toxin-coregulated pilus and cholera toxin, is initiated at the tcpPH promoter by the regulators AphA and AphB. AphA is a winged helix DNA-binding protein that enhances the ability of AphB, a LysR-type transcriptional regulator, to activate tcpPH expression. We present here the 2.2 Å X-ray crystal structure of full-length AphB. As reported for other LysR-type proteins, AphB is a tetramer with two distinct subunit conformations. Unlike other family members, AphB must undergo a significant conformational change in order to bind to DNA. We have found five independent mutations in the putative ligand-binding pocket region that allow AphB to constitutively activate tcpPH expression at the non-permissive pH of 8.5 and in the presence of oxygen. These findings indicate that AphB is responsive to intracellular pH as well as to anaerobiosis and that residues in the ligand-binding pocket of the protein influence its ability to respond to both of these signals. We have solved the structure of one of the constitutive mutants, and observe conformational changes that would allow DNA binding. Taken together, these results describe a pathway of conformational changes allowing communication between the ligand and DNA binding regions of AphB.
Introduction
Cholera is a diarrheal disease caused by oral ingestion of contaminated food or water harboring pathogenic strains of the Gram-negative bacterium, Vibrio cholerae. In order for V. cholerae O1 to cause disease, it must possess two genetic elements, the Vibrio pathogenicity island (VPI) (Karaolis et al., 1998) and the lysogenic CTX phage (Waldor and Mekalanos, 1996), both of which were acquired by horizontal gene transfer. The VPI contains the genes responsible for the synthesis and assembly of the essential colonization factor toxin-coregulated pilus (TCP) (Taylor et al., 1987) and the CTX phage encodes the cholera toxin (CT) genes (Waldor and Mekalanos, 1996). The expression of these virulence factors is controlled by a transcriptional cascade involving activator proteins encoded both within the VPI and the ancestral genome. ToxT is an AraC-type regulator encoded on the VPI that directly activates the expression of TCP and CT (Lowden et al., 2010; DiRita et al., 1991; Higgins et al., 1992; Champion et al., 1997). The expression of toxT is activated by cooperation between two homologous pairs of transmembrane regulatory proteins, TcpPH and ToxRS, encoded on the VPI and on the large chromosome, respectively (Carroll et al., 1997; Häse and Mekalanos, 1998; Miller et al., 1987; Higgins and DiRita, 1994; Krukonis et al., 2000). The activation of tcpPH expression by the regulatory proteins AphA and AphB, encoded in separate locations on the large chromosome, initiates the expression of the virulence cascade (Skorupski and Taylor, 1999; Kovacikova and Skorupski, 1999). AphA is a winged helix DNA binding protein (De Silva et al., 2005) that enhances the ability of AphB, a LysR-type regulator, to activate transcription at the tcpPH promoter (Kovacikova et al., 2004).
Pathogenic bacteria use multiple signal transduction pathways for sensing and responding to environmental stimuli that allow them to integrate horizontally acquired virulence genes on pathogenicity islands into the existing regulatory circuits of the cell in order to optimize survival both inside and outside of the host. The expression of the V. cholerae virulence cascade is influenced by a variety of environmental stimuli such as cell density, c-di-GMP, pH, temperature, oxygen tension, bile, fatty acids, bicarbonate and phosphate (Matson et al., 2007; Chatterjee et al., 2007; Marrero et al., 2009; Abuaita and Withey, 2009; Pratt et al., 2010). The mechanisms by which these stimuli regulate the expression of the virulence cascade are diverse and can either be direct through their influence on the activities of the virulence regulatory proteins themselves, or indirect through the influence of other processes that control the expression or activities of the virulence regulators such as the involvement of global regulatory proteins, regulatory RNAs, and proteolysis (Matson et al., 2007).
LysR-type transcriptional regulators (LTTRs) comprise one of the largest families of regulators in prokaryotes and members control the expression of an extremely diverse set of genes whose products are involved in a variety of biological processes such as metabolism, nitrogen fixation, oxidative stress responses, quorum sensing and virulence (Maddocks and Oyston, 2008). In addition to its ability to induce the expression of the virulence cascade, AphB activates the expression of a number of other genes in V. cholerae that have roles in pH homeostasis (Kovacikova et al., 2010). One of these is cadC, which encodes a member of the ToxR-like family of transmembrane transcriptional regulators that directly activates the expression of the cadBA operon encoding a lysine/cadaverine antiporter and lysine decarboxylase, respectively (Merrell and Camilli, 2000). Lysine decarboxylase plays an important role in an adaptive acid tolerance response in V. cholerae (Merrell and Camilli, 1999) and AphB plays a crucial role in this response by virtue of its ability to activate cadC expression (Kovacikova et al., 2010). AphB also activates the expression of a member of the ClC family of integral membrane proteins whose major action is to translocate chloride ions across cell membranes (Dutzler, 2006). This protein has previously been shown to confer mild resistance of V. cholerae to acid pH (Ding and Waldor, 2003).
The identification of additional AphB activated genes in V. cholerae has shed light on the specific signals that influence the expression of the virulence cascade through AphB. The expression of all the promoters directly activated by AphB was stimulated under aerobic conditions by lowering the pH and at neutral pH by reducing the levels of oxygen (Kovacikova et al., 2010). These results indicate that all of the promoters directly activated by AphB are responsive to two different environmental signals which are encountered by V. cholerae during the early stages of the infection process: low pH and anaerobiosis. Precisely how this occurs is not yet known, although it has been suggested that part of this response involves an oxygen-dependent modification of a cysteine at position 227 (Liu et al., 2011). LTTRs typically require the binding of a metabolically important small molecule coinducer to a pocket region in the C-terminal regulatory domain of the protein, which then triggers a conformational change that stimulates transcription (Maddocks and Oyston, 2008). However, it is not yet known whether AphB requires a coinducer to activate gene expression.
We have found that five independent substitutions to negatively charged residues in a region of the protein that corresponds to the ligand-binding pocket of other LTTR proteins allowed AphB to activate the expression of the tcpPH promoter at the non-permissive pH of 8.5. These mutations also increased the expression of tcpPH under aerobic conditions nearly equally to that under anaerobic conditions. These results indicate that the activity of AphB is responsive to intracellular pH as well as to anaerobiosis and that residues in the putative ligand-binding pocket of the protein influence its ability to respond to both of these signals. In order to gain further insights into the molecular mechanisms by which AphB responds to environmental stimuli to activate virulence gene expression, we have solved the full-length X-ray crystal structures of the wild-type AphB protein and the N100E “activated” variant to 2.2 Å and 2.5 Å resolution, respectively. These crystal structures reveal that the wild-type and N100E proteins are both tetramers, characteristic of many LTTR family proteins, but that their overall conformations differ from one another in their potential to bind to DNA and may represent two steps in the tcpPH transcriptional regulatory pathway.
Results and discussion
Overall structure of AphB
The 2.2 Å structure of AphB was solved by first crystallizing the Se-Met substituted C-terminal domain (residues 78-291), solving its structure using anomalous dispersion techniques, and then using the resulting structure as a molecular replacement model for data obtained from full-length AphB crystals (Table 1). In the wild-type AphB full-length crystal, the asymmetric unit contains a dimer (Fig. 1A), and a crystallographic two-fold rotation axis produces a tetramer. Characteristic of the LTTR family (Maddocks and Oyston, 2008; Schell, 1993), each AphB protomer comprises two domains: the N-terminal winged helix DNA-binding domain (residues 1-58) (Fig. 1B), and the C-terminal regulatory domain (residues 90-291) (Fig. 1C). The DNA-binding and regulatory domains are connected by a 30 amino acid linker helix (residues 59-89). Also similar to other LTTR proteins, the subunits adopt two distinct conformations, compact and extended, which assemble in an alternating manner via two distinct dimerization interfaces to form a tetramer (Fig. 2A and 2B).
Table 1.
Data collection and refinement statistics
| Wild-type AphB | N100E AphB | |
|---|---|---|
| Data collection | ||
| Space group | I222 | C121 |
| Cell dimensions | ||
| a, b, c (Å) | 77.5, 94.6, 59.6 | 268.7, 54.5, 103.8 |
| β (°) | 90.0 | 101.0 |
| Resolution (Å) | 89.8 -2.2 (2.3-2.2) | 48.1-2.7(2.8-2.7) |
| Rmeas | 0.089(0.416) | 0.077(0.472) |
| I/σI | 13.1(4.1) | 21.08(4.67) |
| Completeness (%) | 99.6 (99.4) | 99.7 (99.4) |
| Redundancy | 4.9 (4.9) | 7.4 (7.6) |
| Refinement | ||
| Resolution (Å) | 48.1-2.2 (2.3-2.2) | 48.1-2.7 (2.8-2.7) |
| No. reflections | 49,907 | 39,763 |
| Rwork/ Rfree | 0.208/0.234 (0.264/0.305) | 0.201/0.237 (0.271/0.312) |
| No. atoms | ||
| Protein | 4631 | 9302 |
| Water | 349 | 200 |
| B-factors | ||
| Protein | 48.5 | 53.7 |
| Water | 43.8 | 37.8 |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.008 | 0.002 |
| Bond angles (°) | 1.11 | 0.634 |
Highest resolution shell is shown in parenthesis
Fig. 1. Ribbon diagram of AphB.
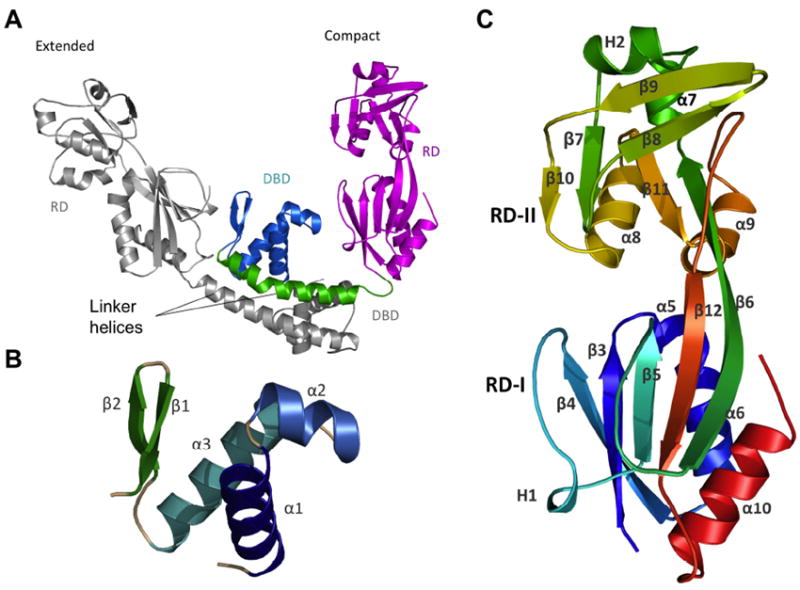
(A) The asymmetric unit of the full-length AphB structure contains one extended subunit (grey) and one compact subunit (color) that dimerize through their linker helices to form a coiled-coil. In the compact subunit, the DNA-binding domain (DBD, cyan) and linker helix (green) are folded in toward the regulatory domain (RD, magenta), whereas in the extended subunit, the DNA-binding domain and linker helix point away from the RD. (B) The DNA binding domain (residues 1-58) of AphB consists of a winged helix-turn-helix motif. (C) The regulatory domain of AphB contains two subdomains: RD-1 (residues 90-159 and residues 262-291) and RD-II (residues 160-261) colored from N-terminal residue (S90, blue) to the C-terminus (red). All figures were created using MacPyMOL (DeLano, 2002).
Fig. 2. Ribbon diagram of the AphB tetramer.
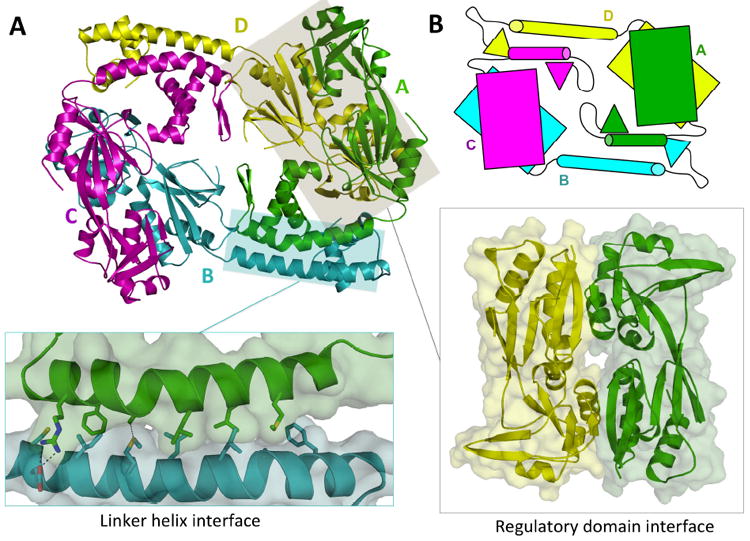
(A) The AphB tetramer is composed of four subunits, two in the extended (yellow, cyan) conformation and two in the compact (magenta, green) conformation. The N-terminal dimerization interface (left inset), along the linker helices of subunit A and B, is composed of polar, charged and hydrophobic residues. The head-to-tail interaction of the regulatory domain of subunit A with the regulatory domain of subunit D forms the second dimerization interface (right inset). (B) Schematic showing the overall fold of AphB, which is formed by four subunits, each composed of a regulatory domain (rectangles) connected to a DNA-binding domain (triangles) via a linker helix (cylinders).
N-terminal DNA-binding domain
The DNA-binding domain of AphB (Fig. 1B) shares a high degree of sequence similarity with other LTTR DNA-binding domains (Fig. S1) and folds into a typical winged helix motif (Maddocks and Oyston, 2008) that is composed of three α-helices (α1 to α3) followed by two β-strands that form the wing. The DNA-binding domains of protomer A and B of wild-type AphB align closely, having an RMSD of 1.04 Å over 58 α-carbons. In winged-helix transcription factors, helix α3 serves as the recognition helix that interacts with the major groove of the target DNA (Aravind et al., 2005). Three positively charged residues (R34, R35 and Q37) are located on the surface of the α3 recognition helix of AphB and may contact the phosphate backbone in the AphB-DNA complex. Three polar residues on α3 (T31, T33 and S42) could participate in hydrogen-bonding with the DNA backbone or contribute to specificity via hydrogen bonds to the bases lining the major groove. In the wing region, two arginine residues, R50 and R53, are conserved among LTTRs and appear to have an important role in DNA binding. Mutations of these residues in the LTTRs OxyR (R50W) and CrgA (R55Q) abolish DNA binding in electrophoretic mobility shift assays (Kullik et al., 1995; Sainsbury et al., 2009).
C-terminal regulatory domain
The overall α/β fold of the regulatory domain (Fig. 1C) of AphB is similar to that of other LTTR regulatory domains (Stec et al., 2006). The regulatory domain contains two subdomains: RD-I (residues 90-159 and residues 262-291) and RD-II (residues 160-261), which are connected by two extended antiparallel β-strands (β6 and β12) (Fig. 1C). The putative ligand-binding pocket is located at the interface of RD-I and RD-II as in other LTTRs (Stec et al., 2006). An interesting feature of the regulatory domain is a β-hairpin structure in RD-II (β8 and β9), a structural motif that appears in only a subset of LTTRs: AphB, CrgA (Sainsbury et al., 2009), and a putative LTTR from Sinorhizobium meliloti (PDB ID: 3MZ1 (Tan et al., 2010)). Sequence similarity searches using BLAST (Altschul et al., 1990) between AphB and all structures in the PDB show CrgA and 3MZ1 to be the top two matches. In other LTTR structures, β8 and β9 are replaced by an α-helix followed by a loop (Muraoka et al., 2003; Monferrer et al., 2010). This “contact helix” is believed to be important in the activation of LTTR tetramers as the contact helices interact in the closed, inactive state, but do not interact in the open, active state (Monferrer et al., 2010; Devesse et al., 2011). There is no such interaction observed in AphB or CrgA, suggesting these proteins use alternate structural mechanisms for activation.
Formation of the tetramer
The wild-type AphB tetramer, formed by the subunits A, B, C and D, has approximate dimensions of 108 Å × 87 Å × 64 Å (Fig. 2A). The tetramer can be described as a dimer of dimers, whereby each identical dimer is composed of two protomers (AB and CD) in different conformations: compact (A and C) and extended (B and D). The compact and extended subunits differ from one another in the angle between the linker helix and the RD, which is 85° in the compact subunits and 150° in the extended subunits (Fig. 1A). The conformational variability in the AphB subunits can be attributed to the flexible loop connecting the linker helix to the regulatory domain.
The tetramer assembles via two distinct dimerization interfaces (Fig. 2A). The N-terminal dimerization interface (1142 Å2) is primarily made up of an antiparallel coiled-coil of the two linker helices, held together by hydrophobic, polar and ionic interactions (Fig. 2A, left inset). The C-terminal dimerization interface (1414 Å2) forms between regulatory domains, which pack against each other in a head-to-tail manner (Fig. 2A, right inset). To form the AphB tetramer, each compact subunit interacts with the regulatory domain of one extended subunit and with the linker helix of the other extended subunit (Fig. 2A and 2B).
Sedimentation equilibrium analytical ultracentrifugation studies confirm that full-length AphB forms a tetramer in solution under physiological conditions as well as in the buffer used for crystallization. Gel filtration chromatography confirms that AphB is a tetramer under physiological conditions (Fig. S2).
Comparison with other full length LTTR structures
The crystal structures of five other full-length LTTRs (CbnR, CrgA, ArgP, TsaR and BenM) have been determined (Muraoka et al., 2003; Sainsbury et al., 2009; Zhou et al., 2010; Monferrer et al., 2010; Ruangprasert et al., 2010) and of these five structures, CbnR, ArgP and TsaR are tetrameric in their crystal forms. The remaining two LTTRs, CrgA and BenM, form an octamer of compact subunits and an indiscrete network of subunits in their crystal forms, respectively. The tetrameric LTTRs have been further classified into open (TsaR and ArgP) and closed (CbnR) forms (Ruangprasert et al., 2010), with the closed form exhibiting an additional head-to-head interaction between the regulatory domain subunits at the contact helix. Using this classification, the AphB tetramer is most similar to the open tetramers, but with a unique arrangement of the DNA-binding domains (see below).
A striking difference is apparent between the position of the DNA-binding domains of AphB when compared to those of CbnR, ArgP and TsaR. Superposition of the compact subunits of CbnR, ArgP and TsaR shows that the DNA binding and regulatory domains of these three proteins are located on opposite sides of the linker helix whereas, in AphB, these domains are located on the same side of the linker helix (Fig. 3A). This conformation in AphB is stabilized by a number of polar interactions between the DNA-binding domains of the compact subunits A and C and the regulatory domains of the subunits D and B, respectively. The hydroxyl of S22 (subunit A) forms hydrogen bonds with Y192 (subunit D) and N129 (subunit D). Additionally, M26 (subunit A) accepts a hydrogen bond from N216 (subunit D). Interestingly, the S22 residue is unique to AphB when compared to the sequences of existing full length LTTR proteins that have been crystallized (Fig. S1). The positioning of the DNA-binding domains in AphB leads to formation of a DNA-binding surface that is remarkably different from other LTTR tetramers. In CbnR, ArgP and TsaR, the DNA-binding domains project outward and form a V-shaped surface along one face of the tetramer, in a conformation that could bind DNA, most likely in a bent conformation (Muraoka et al., 2003; Monferrer et al., 2010) (Fig. 3B). In contrast, the DNA-binding domains of AphB are close to the body of the tetramer and form a flatter interface, suggesting AphB binds to more linear DNA (Fig. 3B).
Fig. 3. Comparison of AphB with other LysR-type regulators.
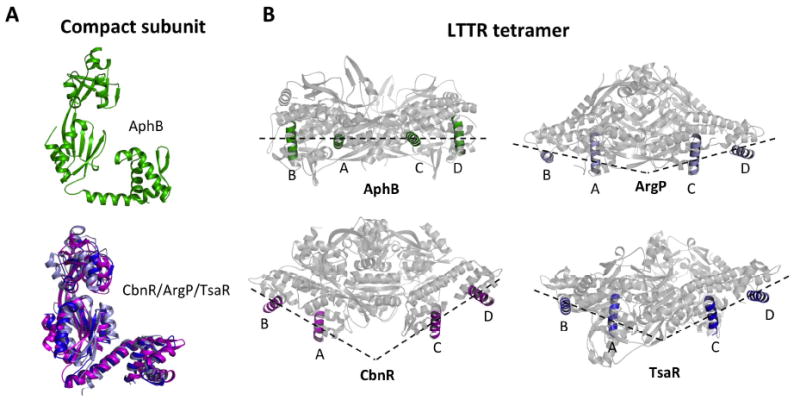
(A) The AphB wild-type (green) compact subunit showing that the DNA-binding and regulatory domains are located on the same side of the linker helix (top). Superposition of the linker helices of the compact subunits of ArgP (silver) and TsaR (blue) onto that of CbnR (magenta), showing that the DNA-binding and regulatory domains are located on opposite sides of the linker helix (bottom). The RD of CbnR was first superposed with that of AphB WT, in the same orientation as seen at top, to maintain the same perspective. (B) The recognition helices of the AphB tetramer (green) fall on a single plane and form a flat DNA-binding interface whereas the recognition helices of CbnR (magenta), ArgP (silver) and TsaR (blue) form a V-shaped DNA-binding interface. AphB was rotated 90° from the orientation shown in Fig. 2 and then the CbnR, ArgP and TsaR recognition helices were superposed with those of AphB.
The unique orientation of the DNA binding domains in AphB also results in a shorter distance between the two central DNA recognition helices in the AphB tetramer. Superposition of the recognition helix of subunit A of AphB with those of CbnR, ArgP and TsaR, shows that the distance between the two central recognition helices (A and C) in AphB is 38 Å whereas it is 58 Å, 49 Å and 49 Å, respectively, in the other three proteins (Fig. S3A). The recognition helices of the AB and CD dimers, however, are positioned at a similar distance (d’, 24 Å) from one another as observed in the other LTTR tetramers. As the d’ distance is defined largely by the linker helix length and is directly related to the spacing of the T-N11-A consensus motif (Schell, 1993), which are conserved among LTTR family members, it is not surprising that it is nearly the same in all four tetramers.
In addition to the linear DNA binding interface in AphB, we observe a significant difference from CbnR, ArgP, and TsaR in that the relative orientations of the DNA binding domains in AphB appear to preclude binding to DNA due to steric clashes. In CbnR, ArgP, and TsaR, the DNA recognition helices are oriented such that they are accessible to the major groove and could bind to bent DNA without significant structural rearrangements. In contrast, the position and orientation of α2 and α3 in AphB would block DNA binding. This finding suggests that the tetramer must undergo a significant conformational change in order for DNA binding to occur (discussed in detail below).
Putative ligand-binding cavity contains pH and oxygen-sensitive residues
A putative ligand-binding cavity is found at the interface of RD-I and RD-II of the AphB regulatory domain (Fig. 4A). The pockets are essentially identical in each protomer with a volume of approximately 292 Å3. The surface of the pocket is lined mainly by hydrophobic and polar amino acid residues, namely: P98, N100, L101, N128, V144, Y192, P193, L194, L220, P237, and M240 (Fig. 4A). The presence of a single positively charged residue, R262, indicates that the binding pocket likely accommodates an amphipathic, anionic moiety.
Fig. 4. Mutational analysis of the putative ligand-binding cavity of AphB.
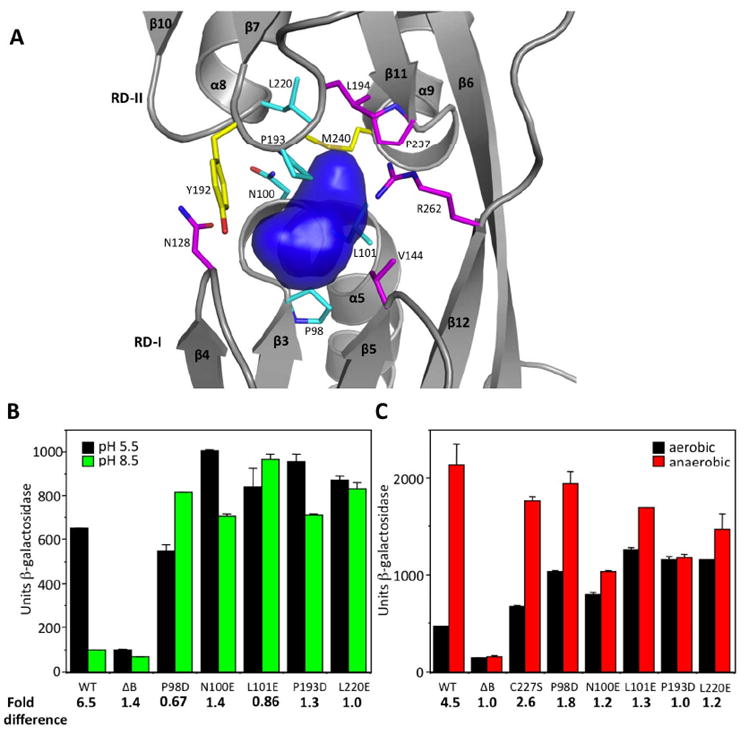
(A) Surface representation of the putative ligand-binding cavity of AphB (blue) generated with SURFNET (Laskowski, 1995). Residues lining the cavity were substituted with negatively-charged amino acids. Most of these substitutions resulted in loss (magenta) or gain (cyan) of function, whereas two (yellow) did not affect activity. (B) Influence of the ligand-binding pocket substitutions (P98D, N100E, L101E, P193D, L220E) on the expression of the tcpPH promoter in V. cholerae at pH 5.5 (black bars) and pH 8.5 (green bars). (C) The same ligand-binding pocket substitutions as in (B) and C227S on the expression of the tcpPH promoter in V. cholerae at neutral pH under aerobic (black bars) and anaerobic conditions (red bars).
Although it is not yet known whether AphB responds to a specific coinducer molecule, AphB has been shown to activate virulence gene expression in response to low pH and low oxygen tension (Kovacikova et al., 2010). As shown in Fig. 4B and 4C, in the presence of wild-type AphB expressed from its own promoter on a low copy plasmid, the expression of tcpPH decreased 6.5 fold as the pH was raised from 5.5 to 8.5 and increased 4.5 fold at neutral pH when the oxygen was removed. Since it has been shown for other LysR homologs that ligand binds in the pocket region that lies between RD-I and RD-II (Ezezika et al., 2007; Devesse et al., 2011) and mutations that influence ligand binding have been isolated in this region of the protein (Lochowska et al., 2001; Lang and Ogawa, 2009), we subjected the residues that comprise the surface of the AphB pocket to site-directed mutagenesis. We anticipated that many of these altered proteins would be inactive, as an introduced negative charge on the side chain would prevent binding of a negatively charged ligand. However, we found that independently changing 5 residues in this pocket to negative charges (P98D, N100E, L101E, P193D, L220E) completely restored the ability of AphB to activate the expression of the tcpPH promoter at the non-permissive pH of 8.5 (Fig. 4B) and also increased the expression of the promoter under aerobic conditions at neutral pH nearly equally to that under anaerobic conditions (Figure 4C). For each of these mutants, the levels of AphB protein under the various conditions were similar to that of wild-type. Changing some of these residues to polar, or non-polar residues (i.e. L101N, P193A) destroyed the ability of AphB to activate expression of the tcpPH promoter. Also, a number of other residues lining the binding pocket, such as N128E, V144E, L194E, P237D, and R262E rendered the protein stable but nonfunctional when changed to a negative charge whereas Y192E or M240E functioned similar to wild-type. These results indicate that certain residues within the ligand-binding pocket of AphB, when changed to a negative charge, are capable of rendering the activity of the protein nearly constitutive with respect to pH and oxygen.
It has recently been suggested that an oxygen-dependent modification of one of the three cysteine residues in AphB (C227) influences the ability of the protein to multimerize and activate gene expression and that this modification is reversible under reducing conditions (Liu et al., 2011). As the AphB structure shows the sulfhydryl group of C227 to be completely buried and solvent inaccessible, we altered it to serine in order to examine its effects on structure and function. Our results indicate that both wild-type AphB and the C227S variant are tetrameric under both reducing and non-reducing conditions by gel filtration (Fig. S2). Thus, C227 is not essential for determining the oligomeric state of the protein. As shown in Fig. 4C, in the presence of the C227S mutation, the expression of tcpPH was slightly induced under aerobic conditions whereas, in the presence of P98D, N100E, L101E, P193D or L220E, the aerobic induction of tcpPH expression was more pronounced. In contrast to the ligand-binding pocket mutations, AphB containing the C227S mutation did not differ from wild-type in its ability to respond to pH (data not shown).
The residues P98, N100, and L101 are located in the loop between β3 and α5 whereas P193 is located in the loop between β7 and β8 and L220 is at the beginning of α8 (Fig. 4A). The constitutive proteins containing the substitutions at these positions do not differ from wild type in their ability to tetramerize as determined by gel filtration or to bind to DNA in vitro as determined using electrophoretic mobility shift assays (Fig. S4). As P98 and N100 are in positions analogous to S99 and A101 of TsaR and V97 and S99 of BenM (Fig. S1), which have been shown to participate in direct contacts with ligand (Monferrer et al., 2010; Ezezika et al., 2007), it is possible that these residues play a similar role for AphB. In order to gain insights into how the changes in the ligand-binding pocket increase the ability of AphB to activate transcription at high pH and in the presence of oxygen, we have determined the crystal structure of one of the AphB variants, N100E (see below).
The N100E variant of AphB appears to be competent for DNA binding
The crystal structure of the N100E variant of AphB was solved using molecular replacement. The N100E protein crystallized under different conditions, and in a different crystallographic space group, than the wild-type protein. Like wild-type AphB, the N100E protein forms a tetramer of subunits A, B, C and D. However, the N100E AphB asymmetric unit is a tetramer, rather than a dimer related by a crystallographic two-fold symmetry axis as is the case for wild-type. This change in symmetry derives from the structural rearrangements that are caused by the N100E substitution (see below).
Introduction of the glutamate side chain at position 100 in the binding pocket of AphB allows formation of a salt bridge with R262 in the compact subunits, and a superposition of the wild-type and N100E proteins on the RD-II reveals that this interaction draws the β3-α5 loop 1.3 Å closer to the β12 strand that connects RD-I and RD-II (Fig. 5A). This causes a movement between the two lobes of the regulatory domain, displacing RD-I with respect to RD-II by 1.8 Å (subunit A) to 3 Å (subunit C) at the attachment point of the N-terminal domain (Fig. 5B). At this point there are two glycine residues (G88 and G91) that allow a rigid body movement of the N-terminal domain dimer with respect to the regulatory domain. As a result, the helix-turn-helix motifs of the extended subunits rotate by about 26° (Fig. 5C) resulting in a significant reorientation of the DNA-binding domains such that α2 would no longer block DNA binding and α3 is oriented to better interact with the major groove of DNA (Figs. 5D, 5E). This movement also increases the distance between the two central recognition helices (A and C) by ~4.5 Å, however, they are still closer to each other than in other LTTR proteins (Fig. S3B) and the DNA-binding interface retains its linear arrangement.
Fig. 5. Structural changes in the AphB N100E variant.
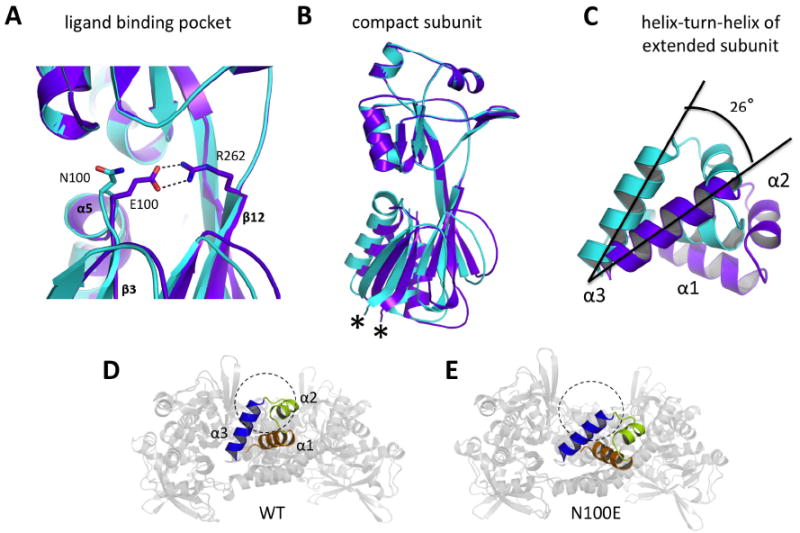
(A) Formation of a salt bridge between E100 and R262 in the regulatory domain of N100E moves the β3-α5 loop 1.3 Å closer to β12. Wild-type AphB, cyan; N100E, purple. (B) The RD-I of N100E moves by up to 3Å at the attachment point of the linker helix (denoted by *) and this movement is relayed to the entire N-terminal domain dimer, which moves as a rigid body. Colors are the same as in (A). (C) Rigid body movement of the N-terminal domain dimers causes a displacement of the DNA-binding domains. The α3 helices of the extended subunits of wild-type and N100E align on their C-terminal ends and the entire helix-turn-helix motif of N100E aligns to that of wild-type upon a 26° counterclockwise rotation. This angle was determined manually, by orienting the WT and N100E recognition helices in MacPymol such that they were on XY plane and measuring the angle between them with a protractor. These results were verified by rotating the N100E helix-turn-helix about the Z-axis (centered on the C-terminal ends of the recognition helices) by -26° in MacPymol. Colors are the same as in (A). (D) View down the DNA-binding groove of the wild-type AphB tetramer showing outer recognition helix (α3) (blue) and the outer α2 helix (lime green) would preclude DNA-binding (DNA cross-section shown as dashed circle) due to steric clashes. (E) View down the DNA-binding groove of the N100E AphB tetramer showing the outer α3 helix (blue) is now poised to interact with the major groove of DNA (DNA cross-section shown as dashed circle) and the α2 helix (lime green) has moved to a position where steric clashes with DNA will not occur.
Interestingly, the conformational change resulting from the N100E substitution in AphB does not disrupt the dimerization interfaces, rather the hinges formed by G88 and G91 in the compact and extended subunits allow the small conformational changes initiated in the regulatory domain of the compact subunits to be amplified into a significant rearrangement of the DNA-binding domains. These rearrangements occur in both dimers, but for reasons that are unclear, one dimer changes to a greater degree than the other resulting in an asymmetric tetramer. This is in contrast to wild-type AphB, where the two dimers comprising the tetramer are symmetrical. Variability in the symmetry of other LTTR tetramers has been observed: TsaR and ArgP are symmetric, whereas the native CbnR is asymmetric (Monferrer et al., 2010; Zhou et al., 2010; Muraoka et al., 2003). While it is unclear whether these other tetramers are in active or inactive conformations, the differences observed between wild-type and N100E AphB suggest that a change in symmetry of the tetramer may be associated with activation of LTTR proteins. Such a change in symmetry makes sense from a structural perspective, as proteins in solution often form symmetric oligomers. However, the DNA binding sites for AphB at the tcpPH promoter are inherently asymmetric (AphB occupies a primary binding site centered at -60 and appears to interact weakly with a non-consensus binding site further downstream in the promoter centered at -17 (Kovacikova & Skorupski, 2001)). Because of this, a shift from a symmetric to an asymmetric tetramer could represent a transition from an inactive conformation (wild-type) to a conformation that favors transcriptional activation (N100E).
A variety of evidence indicates that many LTTR proteins undergo an effector-mediated conformational change, which alters the protein-DNA complex and facilitates recruitment of RNA polymerase to the promoter to activate gene expression (Maddocks and Oyston, 2008; Schell, 1993). In the model for BenM, ligand binding moves the two DNA binding domain dimers toward one another altering the pattern of occupancy at the promoter, which favors transcriptional activation (Ezezika et al., 2007). Mutations that result in an effector independent phenotype are thought to bypass the need for the effector by inducing conformational changes in the structure of the protein DNA complex that mimic the presence of ligand (Maddocks and Oyston, 2008; Schell, 1993) or affect the dipole moments, and thereby the position of the helices, in the regulatory domain (Ezezika et al., 2007). The results shown here indicate that wild-type AphB, crystallized in the absence of an effector, is sterically unable to bind to DNA whereas the N100E variant undergoes a series of conformational changes that alter the position of its DNA binding domain, possibly mimicking a ligand-bound conformation, resulting in constitutive activation of transcription. These results provide a structural mechanism to explain how changes in the ligand binding domain of AphB can influence DNA binding and transcriptional activation.
In summary, like other members of the LTTR family, AphB forms a tetramer comprised of two distinct subunit conformations. However, the protein is different from most known LTTRs in that it appears to require a major conformational change in order for it to bind to DNA and that the shape of the interface formed by the DNA-binding domains is linear rather than curved/bent. The finding that five independent mutations in the putative ligand-binding pocket allow the protein to activate gene expression more efficiently at high pH and in the presence of oxygen indicates that AphB is capable of responding to two different environmental signals by a similar mechanism. Structural comparison of the wild-type and an “activated” variant of AphB illuminates a pathway of conformational changes in the protein, explaining how ligand binding could lead to activation of the AphB-tcpPH promoter complex.
Experimental procedures
Cloning, expression, and purification of AphB
The purification of full-length AphB using the IMPACT protein fusion and purification system (New England Biolabs) has been described (Kovacikova et al., 2004). The C-terminal end of AphB (AphB-CTD) corresponding to M78 – Q291 was PCR amplified from V. cholerae classical strain O395 using the forward primer RDB78Met containing a Nde1 site and the reverse primer CO2-34 (Kovacikova et al., 2004) containing a Sap1 site. After digestion, the DNA fragment was ligated into pTXB1 (New England Biolabs). The resulting plasmid, pRDS1, was transformed into E. coli strain ER2566. A single colony was incubated at 30 °C overnight in LB media containing 100 μg mL-1 ampicillin. A 1:100 dilution of this culture was made using fresh LB containing ampicillin and allowed to incubate at 30 °C with constant shaking (220 r.p.m.) until the optical density at 600 nm reached 0.8. The midlog culture at 30 °C was induced with 1 mM IPTG (Sigma). Upon induction, the temperature was reduced to 12 °C and the cells were allowed to incubate for an additional 20 hrs with continuous shaking. The sonicated, clarified AphB supernatant was loaded onto a chitin column (New England Biolabs) at a rate of 0.5 mL min-1 and equilibrated with Buffer A (20 mM Tris-HCl pH 8.0, 500 mM NaCl, and 1 mM EDTA) and AphB was allowed to bind. The column was then washed with 10 column volumes of buffer A and 5 column volumes of Buffer B (Tris-HCl pH 8.0, 1 M NaCl, and 1 mM EDTA) at a flow rate of 1 mL min-1 respectively. The column was then quickly washed with 3 column volumes of cleavage buffer (Buffer A containing 100 mM DTT) at a flow rate of 1 mL min-1 and incubated overnight at 12 °C. AphB was then eluted off the column using column buffer without DTT and the purest fractions determined by SDS-PAGE were pooled together and dialyzed overnight in 20 mM Tris-HCl pH 7.5, 10 mM NaCl, 1 mM EDTA and 0.1 mM DTT. The resulting protein was concentrated to 6 mg mL-1 using 5 kDa MWCO Vivaspin centrifugal concentrators (Sartorius) and dialyzed overnight in 20 mM Tris-HCl pH 7.6, 500 mM NaCl, 1 mM EDTA, and 0.1 mM DTT.
To express the selenomethionine (Se-Met) substituted AphB-CTD, E. coli methionine auxotrophic strain B854(DE3) was grown in minimal media with selenomethionine. The purification process was generally the same as for the native protein. Selenium for sulfur substitution was verified using MALDI-TOF mass spectroscopy with sinapinic acid matrix (Voyager-DE, Applied Biosystems).
For N100E AphB, the plasmid pWEL219 was transformed into E. coli BL21-Star cells. Overnight cultures from single colonies were diluted 1:500 into ZYM-5052 autoinducing medium containing ampicillin as described by (Studier, 2005). After 26 hours, cell pellets were harvested, resuspended in Buffer B (8.0, 1 M NaCl, and 1 mM EDTA) and stored at -80 °C. Thawed cells were lysed by sonication and the clarified supernatant was applied, by gravity, to a chitin column equilibrated with Buffer B. The column was washed with 5 column volumes of Buffer B, then 1.5 column volumes of cleavage buffer (20 mM Tris-HCl pH 8.0, 0.5 M NaCl, 1 mM EDTA and 100 mM DTT) and incubated overnight at 16°C. Protein was eluted in Buffer A (20 mM Tris-HCl pH 8.0, 0.5 M NaCl, 1 mM EDTA), concentrated to 5 mg mL-1 and dialyzed overnight in Buffer A to remove DTT.
Wild type and C227S AphB were also purified as C-terminal His-tagged constructs in order to prepare the proteins in the absence of DTT. Top10 cells were transformed with the plasmid pKAS256 or pGKK465, respectively, and overnight cultures from single colonies were grown and diluted 1:100 into LB. Cells were grown for 4 hours at 37°C before induction with 0.02% arabinose. After an additional 6 hours, cells were harvested and stored at -80 °C. Thawed cells were lysed by sonication and His-tagged protein from the clarified lysate was purified over a Ni-NTA superflow column, using an imidazole gradient to elute the bound protein. Purified protein was exchanged into PBS buffer over a HiTrap desalting column and concentrated to 2 mg/mL for size exclusion analysis.
Crystallization
Full-length wild-type AphB
Protein (5 mg mL-1) and 4 M sodium formate as the precipitant were mixed in a 1:1 ratio at room temperature with 0.01 M betaine monohydrate as an additive and crystallization occurred by vapor diffusion. Long rod-like tetragonal shaped crystals (form I) with a largest size of 0.8 mm × 0.2 mm × 0.2 mm appeared after 4 days. A second form (form II) of full-length wild-type AphB crystals of cuboid shape with a largest size of 0.2 mm × 0.2 mm × 0.1 mm appeared after 2-3 weeks incubation at room temperature in the same conditions as above. For both forms of full-length AphB crystals, 20-30% glycerol was used as the cryoprotectant before crystals were flash frozen in liquid nitrogen.
N100E AphB
Clusters of small crystals appeared in sitting drops of a 1:1 mixture of 5 mg mL-1 full length N100E AphB with 1M tri-Sodium citrate and 0.1 M Sodium cacodylate pH 6.5 after two days. Larger, single crystals were obtained by micro seeding, mounted on loops in cryoprotectant containing 30% sucrose and flash-frozen in liquid nitrogen. Although initial attempts to crystallize the N100E protein were performed under the wild-type crystallization conditions, N100E AphB did not form crystals under these conditions, nor did the wild-type protein crystallize under the N100E conditions.
Se-Met derivative AphB-CTD
Crystals were obtained as described above, by mixing equal volumes of proteins with 2M ammonium sulfate. Hexagonal long rod-like crystals appeared after 3-4 days of incubation at room temperature. Crystals were mounted on loops, cryoprotected in 20-30% glycerol and flash frozen in liquid nitrogen for X-ray data collection.
Data collection and structure solution
SeMet AphB-CTD
X-ray diffraction data was collected at NSLS beamline X6A. Crystals belonged to the trigonal space group P3221 with unit cell dimensions of a = 112.6 Å, b= 112.6 Å, c= 90.5 Å, and α = β = 90°, γ = 120° (Table 1). X-ray fluorescence scanning of the crystals at the Se edge was carried out to identify the optimal wavelengths for single wavelength anomalous dispersion (SAD) data collection. Two wavelengths (peak, 0.9789 Å; inflection, 0.9792 Å) were collected and processed using XDS (Kabsch, 2010). Only the first data set was used to calculate the initial electron density map to 2.2 Å resolution using the program SOLVE/RESOLVE (Terwilliger, 2003). The resulting map was of high quality and RESOLVE automatically modeled 60% of the main chain residues and 35% of the side-chains. The remaining residues were manually built in COOT (Emsley and Cowtan, 2004). The final refinement steps were performed in REFMAC5 (Murshudov et al., 1997) using TLS parameters. The solvent atoms were initially built using the program ARP/wARP (Perrakis et al., 1999) and later added or removed by manual inspection. The final Rwork and Rfree (calculated with 5% of the reflections omitted from the refinement) were 22.5% and 27.3%, respectively.
Wild-type full-length AphB
Form I crystals diffracted to 4.0 Å resolution and belong to the tetragonal space group P422, with unit-cell parameters a = 101.3 Å, b = 101.3 Å, c = 368.8 Å, α = β = γ = 90° (crystal packing, Fig. S5). Form II crystals belong to the orthorhombic space group I222 (a = 77.5 Å, b = 94.6 Å, c = 59.6 Å, and α = β = γ = 90°), and diffracted to 2.2 Å resolution. X-ray diffraction data for the full-length wild-type AphB form II was collected at Argonne National Laboratory (ANL) beamline GM/CA-CAT. Two copies of the AphB-CTD were then used as independent search models for the molecular replacement calculations using data range 2.2 to 48.8 Å using the program PHENIX (Adams et al., 2010). After a round of Autobuild (Terwilliger et al., 2008) the PHENIX suite was able to build 25% of the DNA-binding domain. There are two subunits of full-length AphB in the asymmetric unit, and biological unit is a tetramer formed by a two-fold rotation axis in the crystal structure. The remaining residues in the DNA-binding domain were built manually using COOT (Emsley and Cowtan, 2004) and the solvent molecules were assigned as above. No ions or buffer components were identified in the electron density. The final rounds of refinement were done in PHENIX, using parameters from the TLSMD server (Merritt, 2006a; 2006b) to yield a final Rwork of 20.8% and Rfree of 23.5%, (calculated with 5% of the reflections omitted from the refinement) using all data to 2.2 Å. Results were validated with MOLPROBITY and the structure ranks in the 88th percentile among proteins of similar resolution, with a MOLPROBITY score of 1.95.
N100E AphB
Diffraction data for N100E AphB was collected to 2.4 Å at NSLS beamline X6A at Brookhaven National Laboratory. Crystals belonged to the monoclinic space group C2, with unit cell dimensions of a = 268.7 Å, b= 54.5 Å, c= 103.8 Å, and α = γ = 90°, β = 101°. Data was processed in XDS (Kabsch, 2010) and structure determination was carried out using AutoMR in Phenix (McCoy et al., 2007). Four copies of the wild-type AphB regulatory domain (amino acids 88-290) and four copies of a portion of the N-terminal domain (amino acids 1-81) were used as search models for molecular replacement. The remaining residues were built manually in Coot (Emsley and Cowtan, 2004). Multiple rounds of refinement and rebuilding were carried out in Phenix.refine (Afonine et al., 2005) and Coot, respectively, using data to 2.7 Å. The final rounds of refinement were carried out using parameters from the TLSMD server (Merritt, 2006a; 2006b), as above, and with Ramachandran restraints, resulting in a final Rwork of 20.1% and Rfree of 23.7%. In MOLPROBITY validation results, this structure was assigned a MOLPROBITY score of 2.78, which is in the 71st percentile among structures of comparable resolution.
Construction of point mutations
Plasmid pGKK408 was constructed by PCR amplifying the aphB gene and its promoter from C6706str2 using primers CO2-6 and CO2-35 (Kovacikova et al., 2004) and ligating the resulting fragment into the low copy plasmid pKAS178 (Kovacikova et al., 2005) in the opposite orientation from the pBAD promotor. To avoid obtaining frameshifts in the run of 8 A’s in the beginning of the aphB coding sequence, codons corresponding to K23 and K24 were changed from AAA to AAG using overlapping PCR with primers CO2-52 and CO2-53 (Table S2). The nucleotide changes in the aphB coding sequence in pGKK408 were made using QuikChange XL-site directed mutagenesis (Stratagene) and the resulting plasmids were confirmed by DNA sequencing. The sequences of the oligonucleotides used to generate the nucleotide changes are listed in Table S2.
β-Galactosidase assays
β-galactosidase assays (Miller, 1972) were carried out with strains KSK725 (C6706str2 ΔlacZ3 tcpP-lacZ) and its ΔaphB derivative GK138 (Kovacikova and Skorupski, 1999) harboring the various aphB plasmids. Cultures grown to mid log phase (A600 = 0.2 – 0.3) under aerobic or anaerobic conditions as previously described (Kovacikova et al., 2010). Assays were in duplicate for each culture and the data are representative results from at least two separate experiments.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants AI072661 (to F.J.K.), Grant AI41448 (to K.S.), and Grant AI039654 (to R.K.T). We acknowledge Dr. Vivian Stojanoff and the X6A team at the NSLS, Brookhaven National Laboratory, as well as Dr. Robert Fischetti and the GM-CA/CAT team at the APS, Argonne National Laboratory, for their assistance in X-ray data collection. Research carried out at the X6A beam line was funded by the NIH (NIGMS) under agreement GM-0080. The NSLS at Brookhaven National Laboratory is supported by the U.S. D.O.E. under contract DE-AC02-98CH10886. The GM/CA CAT beamline has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). The use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract DE-AC02-06CH11357. We thank Dr. Dean Madden for help with SedAnal processing and Dr. Henry Higgs for assisting with use of the analytical ultracentrifuge. We enthusiastically thank Julia Kiernan for assistance in purification and crystallization of the N100E protein, and thank Dr. Oliver Baettig for feedback on the manuscript.
Glossary
The abbreviations used are:
- LTTRs
LysR-type transcriptional regulators
- CT
cholera toxin
- TCP
toxin co-regulated pilus
- SAD
single anomalous dispersion
- PDB
protein data bank
- RMSD
root mean square deviation
Footnotes
The atomic coordinates and structure factors (PDB IDs 3SZP and 3T1B) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, http://www.rscb.org
References
- Abuaita BH, Withey JH. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun. 2009;77:4111–4120. doi: 10.1128/IAI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P, Afonine P, Bunkoczi G. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P, Grosse-Kunstleve R, Adams PD. The Phenix refinement framework. CCP4 newsletter. 2005 July; Contribution 8. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Carroll P, Tashima K, Rogers M. Phase variation in tcpH modulates expression of the ToxR regulon in Vibrio cholerae. Mol Microbiol. 1997;25:1099–1111. doi: 10.1046/j.1365-2958.1997.5371901.x. [DOI] [PubMed] [Google Scholar]
- Champion GA, Neely MN, Brennan MA, DiRita VJ. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol Microbiol. 1997;23:323–331. doi: 10.1046/j.1365-2958.1997.2191585.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Dutta PK, Chowdhury R. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun. 2007;75:1946–1953. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. Crystal structure of the virulence gene activator AphA from Vibrio cholerae reveals it is a novel member of the winged helix transcription factor superfamily. J Biol Chem. 2005;280:13779–13783. doi: 10.1074/jbc.M413781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. The PyMOL molecular graphics system 2002 [Google Scholar]
- Devesse L, Smirnova I, Lönneborg R, Kapp U, Brzezinski P, Leonard GA, Dian C. Crystal structures of the DntR inducer binding domains in complex with salicylate offer insights into the activation of LysR-type transcriptional regulators. Mol Microbiol. 2011;81:354–367. doi: 10.1111/j.1365-2958.2011.07673.x. [DOI] [PubMed] [Google Scholar]
- Ding Y, Waldor MK. Deletion of a Vibrio cholerae ClC channel results in acid sensitivity and enhanced intestinal colonization. Infect Immun. 2003;71:4197–4200. doi: 10.1128/IAI.71.7.4197-4200.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRita VJ, Parsot C, Jander G, Mekalanos JJ. Regulatory cascade controls virulence in Vibrio cholerae. Proc Natl Acad Sci USA. 1991;88:5403–5407. doi: 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R. The ClC family of chloride channels and transporters. Curr Opin Struct Biol. 2006;16:439–446. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ezezika OC, Haddad S, Clark TJ, Neidle EL, Momany C. Distinct effector-binding sites enable synergistic transcriptional activation by BenM, a LysR-type regulator. J Mol Biol. 2007;367:616–629. doi: 10.1016/j.jmb.2006.09.090. [DOI] [PubMed] [Google Scholar]
- Häse CC, Mekalanos JJ. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci USA. 1998;95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DE, Nazareno E, DiRita VJ. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J Bacteriol. 1992;174:6974–6980. doi: 10.1128/jb.174.21.6974-6980.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DE, DiRita VJ. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol Microbiol. 1994;14:17–29. doi: 10.1111/j.1365-2958.1994.tb01263.x. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta cryst. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci USA. 1998;95:3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova G, Lin W, Skorupski K. Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol Microbiol. 2005;57:420–433. doi: 10.1111/j.1365-2958.2005.04700.x. [DOI] [PubMed] [Google Scholar]
- Kovacikova G, Lin W, Skorupski K. The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J Bacteriol. 2010;192:4181–4191. doi: 10.1128/JB.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova G, Lin W, Skorupski K. Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol Microbiol. 2004;53:129–142. doi: 10.1111/j.1365-2958.2004.04121.x. [DOI] [PubMed] [Google Scholar]
- Kovacikova G, Skorupski K. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol. 1999;181:4250–4256. doi: 10.1128/jb.181.14.4250-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova G, Skorupski K. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol. 2001;41:393–407. doi: 10.1046/j.1365-2958.2001.02518.x. [DOI] [PubMed] [Google Scholar]
- Krukonis ES, Yu RR, DiRita VJ. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane localized transcriptional activators on a single promoter. Mol Microbiol. 2000;38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- Kullik I, Stevens J, Toledano MB, Storz G. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for DNA binding and multimerization. J Bacteriol. 1995;177:1285–1291. doi: 10.1128/jb.177.5.1285-1291.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GH, Ogawa N. Mutational analysis of the inducer recognition sites of the LysR-type transcriptional regulator TfdT of Burkholderia sp. NK8. Appl Microbiol Biotechnol. 2009;83:1085–1094. doi: 10.1007/s00253-009-1960-5. [DOI] [PubMed] [Google Scholar]
- Laskowski RA. SURFNET: A program for visualizing molecular surfaces, cavities and intermolecular interactions. J Mol Graph. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci USA. 2011;108:810–815. doi: 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochowska A, Iwanicka-Nowicka R, Plochocka D, Hryniewicz MM. Functional dissection of the LysR-type CysB transcriptional regulator. Regions important for DNA binding, inducer response, oligomerization, and positive control. J Biol Chem. 2001;276:2098–2107. doi: 10.1074/jbc.M007192200. [DOI] [PubMed] [Google Scholar]
- Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, Kull FJ. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc Natl Acad Sci USA. 2010;107:2860–2865. doi: 10.1073/pnas.0915021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks SE, Oyston PCF. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- Marrero K, Sánchez A, Rodríguez-Ulloa A, González LJ, Castellanos-Serra L, Paz-Lago D, Campos J, Rodríguez BL, Suzarte E, Ledón T, et al. Anaerobic growth promotes synthesis of colonization factors encoded at the Vibrio pathogenicity island in Vibrio cholerae El Tor. Res Microbiol. 2009;160:48–56. doi: 10.1016/j.resmic.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Matson JS, Withey JH, DiRita VJ. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun. 2007;75:5542–5549. doi: 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell DS, Camilli A. The cadA gene of Vibrio cholerae is induced during infection and plays a role in acid tolerance. Mol Microbiol. 1999;34:836–849. doi: 10.1046/j.1365-2958.1999.01650.x. [DOI] [PubMed] [Google Scholar]
- Merrell DS, Camilli A. Regulation of Vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcriptional regulators. J Bacteriol. 2000;182:5342–5350. doi: 10.1128/jb.182.19.5342-5350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Cryst. 2006a;D62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Merritt E. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006b;39:109–111. [Google Scholar]
- Miller J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; NY: 1972. pp. 352–355. [Google Scholar]
- Miller VL, Taylor RK, Mekalanos JJ. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell. 1987;48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- Monferrer D, Tralau T, Kertesz MA, Dix I, Solà M, Usón I. Structural studies on the full-length LysR-type regulator TsaR from Comamonas testosteroni T-2 reveal a novel open conformation of the tetrameric LTTR fold. Mol Microbiol. 2010;75:1199–1214. doi: 10.1111/j.1365-2958.2010.07043.x. [DOI] [PubMed] [Google Scholar]
- Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J Mol Biol. 2003;328:555–566. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Pratt JT, Ismail AM, Camilli A. PhoB regulates both environmental and virulence gene expression in Vibrio cholerae. Mol Microbiol. 2010;77:1595–1605. doi: 10.1111/j.1365-2958.2010.07310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruangprasert A, Craven SH, Neidle EL, Momany C. Full-length structures of BenM and two variants reveal different oligomerization schemes for LysR-type transcriptional regulators. J Mol Biol. 2010;404:568–586. doi: 10.1016/j.jmb.2010.09.053. [DOI] [PubMed] [Google Scholar]
- Sainsbury S, Lane LA, Ren J, Gilbert RJ, Saunders NJ, Robinson CV, Stuart DI, Owens RJ. The structure of CrgA from Neisseria meningitidis reveals a new octameric assembly state for LysR transcriptional regulators. Nucleic Acids Res. 2009;37:4545–4558. doi: 10.1093/nar/gkp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- Skorupski K, Taylor RK. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol. 1999;31:763–771. doi: 10.1046/j.1365-2958.1999.01215.x. [DOI] [PubMed] [Google Scholar]
- Stec E, Witkowska-Zimny M, Hryniewicz MM, Neumann P, Wilkinson AJ, Brzozowski AM, Verma CS, Zaim J, Wysocki S, Bujacz GD. Structural basis of the sulphate starvation response in E. coli: crystal structure and mutational analysis of the cofactor-binding domain of the Cbl transcriptional regulator. J Mol Biol. 2006;364:309–322. doi: 10.1016/j.jmb.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Tan K, Xu X, Cui H, Chin S, Savchenko A, Edwards A, Joachimiak A. The crystal structure of a possible transcription regulator protein from Sinorhizobium meliloti 1021 2010 [Google Scholar]
- Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci USA. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. 2008;D64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- Zhou X, Lou Z, Fu S, Yang A, Shen H, Li Z, Feng Y, Bartlam M, Wang H, Rao Z. Crystal structure of ArgP from Mycobacterium tuberculosis confirms two distinct conformations of full-length LysR transcriptional regulators and reveals its function in DNA binding and transcriptional regulation. J Mol Biol. 2010;396:1012–1024. doi: 10.1016/j.jmb.2009.12.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.