

Abstract
Male rat fetuses exposed to certain phthalate esters (PEs) during sexual differentiation display reproductive tract malformations due to reductions in testosterone (T) production and the expression of steroidogenesis- and INSL3-related genes. In the current study, we used a 96-well real-time PCR array containing key target genes representing sexual determination and differentiation, steroidogenesis, gubernaculum development, and androgen signaling pathways to rank the relative potency of several PEs. We executed dose-response studies with diisobutyl (DIBP), dipentyl (DPeP), dihexyl (DHP), diheptyl (DHeP), diisononyl (DINP), or diisodecyl phthalate (DIDP) and serial dilutions of a mixture of nine phthalates. All phthalates, with the exception of DIDP, reduced fetal testicular T production. Several genes involved in cholesterol transport, androgen synthesis, and Insl3 also were downregulated in a dose-responsive manner by DIBP, DPeP, DHP, DHeP, DINP, and the 9-PE mixture. Despite speculation of peroxisome proliferator activated receptor (PPAR) involvement in the effects of PEs on the fetal testis, no PPAR-related genes were affected in the fetal testes by exposure to any of the tested PEs. Furthermore, the potent PPARα agonist, Wy-14,643, did not reduce fetal testicular T production following gestational day 14–18 exposure, suggesting that the antiandrogenic activity of PEs is not PPARα mediated. The overall sensitivity of the fetal endpoints (gene expression or T production) for the six phthalates from most to least was Cyp11b1 > Star = Scarb1 > Cyp17a1 = T production > Cyp11a1 = Hsd3b = Insl3 > Cyp11b2. The overall potency of the individual phthalates was DPeP > DHP > DIBP ≥ DHeP > DINP. Finally, the observed mixture interaction was adequately modeled by the dose-addition model for most of the affected genes. Together, these data advance our understanding of the collective reproductive toxicity of the PE compounds.
Keywords: mixture toxicity, antiandrogen, PPAR, phthalate risk assessment
Phthalate esters (PEs) are widely used compounds found in a variety of consumer products. Many of the PEs have been extensively characterized as developmental reproductive toxicants. In utero exposure to these PEs during the sexual differentiation period of rat development leads to reproductive tract malformations in androgen- and INSL3-dependent tissues (Barlow and Foster, 2003; Foster, 2006; McKinnell et al., 2005; Mylchreest et al., 1999; Parks et al., 2000; Scott et al., 2008; Welsh et al., 2008; Wilson et al., 2004), among other effects. Many studies have focused on elucidation of the mode of action behind this reproductive developmental toxicity and provided detailed histological, morphological, hormone level, and gene and protein expression level data (Andrade et al., 2006; Barlow et al., 2003; Bowman et al., 2005; Howdeshell et al., 2008; Johnson et al., 2007; Lahousse et al., 2006; Mahood et al., 2005; Plummer et al., 2007). Nevertheless, the proximate molecular target of phthalate action in the developing male fetus remains unidentified.
The adult male reproductive tract malformations associated with gestational exposure to phthalates during sexual differentiation include reduced anogenital distance (AGD), retained nipples, undescended testes, and decreased organ weights or agenesis of the epididymis, gubernaculum, glans penis, prostate, seminal vesicles, vas deferens, and testes (Gray et al., 2000; Mylchreest et al., 1998, 1999). Normal development of these tissues is dependent upon Leydig cell (LC) hormones including androgens and INSL3. Therefore, a causal link has been drawn between the phthalate-induced reduction of testosterone (T) production during the critical developmental programming period and the postnatal malformations (Barlow and Foster, 2003; Hannas et al., 2011a; Mylchreest et al., 1998, 2000; Parks et al., 2000). Available fetal testis gene and protein expression data display effects on steroidogenic enzymes involved in testosterone synthesis including cytochrome P450 (CYP1) side-chain cleavage (P450scc), 3β-hydroxysteroid dehydrogenase (3β-HSD), and CYP17α (Barlow et al., 2003; Foster et al., 1983; Howdeshell et al., 2008; Johnson et al., 2007; Lehmann et al., 2004; Thompson et al., 2005). Other related genes affected by in utero phthalate exposure include peripheral benzodiazepine receptor (Bzrap1), scavenger receptor class B type 1 (Scarb1), and steroid acute regulatory protein (Star), which are collectively involved in cholesterol uptake and transport into the cell and mitochondria (Gazouli et al., 2002; Plummer et al., 2007; Thompson et al., 2004). Finally, the gene product for insulin-like hormone three (INSL3), a hormone involved in normal testicular descent, is reduced by in utero phthalate exposure (Borch et al., 2006; McKinnell et al., 2005; Wilson et al., 2004).
Over the past several decades, there has been an ongoing debate with regards to the role of the peroxisome proliferator activated receptors (PPARs) in phthalate-induced reproductive toxicity. PPARs are nuclear receptors that play roles in regulating cellular differentiation, development, and metabolism, including cholesterol uptake and transport. Some groups have proposed that PEs interfere with testicular development and function or T production by interacting with PPARs (Bhattacharya et al., 2005; Boberg et al., 2008; Corton and Lapinskas, 2005; Gazouli et al., 2002; Plummer et al., 2007; Ward et al., 1998), potentially by interfering with the regulation of cholesterol transport (Borch et al., 2006). This hypothesis is based on data demonstrating that several antiandrogenic phthalate monoester metabolites are also activators of the PPAR receptors (Hurst and Waxman, 2003a,b; Lampen et al., 2003; Maloney and Waxman, 1999) and that many PPAR agonists reduce T production in vitro using LC cultures from adult rats (Biegel et al., 1995; Boberg et al., 2008; Borch et al., 2006; Gazouli et al., 2002; Liu et al., 1996). Ward et al. (1998) concluded that diethylhexyl phthalate (DEHP)–induced testicular toxicity in mice is PPARα-dependent because PPARα-null mice lacked testicular effects after 16 weeks of feeding on a DEHP-containing diet. These same mice, however, later developed delayed testicular toxicity through a PPARα-independent mechanism. Other groups have hypothesized that PPARβ/δ or PPARγ are potentially involved in phthalate-induced testicular toxicity (Corton and Lapinskas, 2005; Shipley and Waxman, 2004). Little data are available with regards to the expression of PPAR receptors and the potential role of phthalate-induced PPAR activation in the fetal testis during the sexual differentiation period. Therefore, in the current study, we also tested the hypothesis that PPARα activation with the potent agonist, Wy-14,643, during a critical period of sexual differentiation (gestational day, GD 14–18) would reduce fetal testicular T production, as is seen with phthalate exposure. Furthermore, we assessed expression of PPARα, β/δ, and γ, and a few PPAR-target genes in the fetal testes following phthalate exposure during GD 14–18.
To screen additional molecular pathways, which are potentially targeted by phthalates, we designed a custom PCR array containing genes that represent several candidate pathways involved in male sexual determination, differentiation, and development. Specifically, we targeted male testis cell differentiation and signaling, gubernaculum development, steroidogenesis, androgen receptor signaling and other nuclear receptors, Frizzled signaling; PPAR/RXR signaling, neurotropins, and inhibins and activins. Because many antiandrogenic phthalates downregulate expression of Cyp11a, Hsd3b, Scarb1, Cyp17a, and Insl3, we included these genes on the array as positive controls for the screening of additional less studied phthalates. Therefore, the results reported in the current study include dose-response data demonstrating that several phthalates, which have been shown to reduce fetal testicular testosterone production and also modulate expression levels of candidate target genes following 5-day exposure (GD 14–18).
Exposure data indicate that humans are exposed to multiple phthalates (Silva et al., 2004; Wittassek et al., 2007). Additionally, phthalate mixture-induced fetal and postnatal reproductive toxicity conforms to dose-additive mathematical models (Hotchkiss et al., 2004; Howdeshell et al., 2008; Rider et al., 2008), indicating that each PE in a mixture will contribute to the overall effect, if the total mixture dose exceeds the no observable adverse effect level. Together, these factors support the need to perform a cumulative risk assessment on these compounds. It has been proposed that dose-addition (DA) modeling is appropriate for use with compounds that all share a similar mechanism of action (Altenburger et al., 2000; Silva et al., 2002) or target a similar pathway/endpoint (Rider et al., 2010), and response addition (RA) is generally used for mixtures with chemicals that act independently or through different mechanisms of action (Greco et al., 1992). We previously demonstrated that the effects of a 9-PE mixture were better predicted using a DA model than an RA model for fetal ex vivo T production (Hannas et al., 2011b). Our current assessment will determine if this same 9-PE mixture similarly affects the fetal testicular genomic endpoints in a dose-additive manner, thereby further supporting cumulative risk assessment of this compound class.
Our previous studies (Hannas et al., 2011a,b) rank the potencies of certain antiandrogenic phthalates for reducing ex vivo fetal testicular T production when measured on GD 18 following gestational exposure during the sexual differentiation critical programming period of GD 14–18 (Carruthers and Foster, 2005; Welsh et al., 2008). We determined that dipentyl phthalate (DPeP) was the most potent, followed by diisobutyl phthalate (DIBP), and that diisononyl phthalate (DINP) was least potent. In the current study, we built upon these data by determining the dose-response profiles of diheptyl phthalate (DHeP), dihexyl phthalate (DHP), and diisodecyl phthalate (DIDP) for T production. Based on compound structure and postnatal data, we hypothesized the following relative potencies: DPeP > DHP > DIBP ≥ DHeP > DINP and that DIDP would have no effect on T production. Additional objectives of the current study were to, first, determine if the individual phthalates classified as positive for antiandrogenic activity are acting through a similar mechanism by identifying affected genes on the screening PCR array. Second, determine if the individual PE potency for reducing T production correlates with the potency for the reduction of gene expression. Third, use dose-response modeling to predict the ability of a 9-PE mixture to reduce expression of affected genes and test the model predictions.
MATERIALS AND METHODS
Animals.
Time-pregnant Sprague Dawley (SD) rats were purchased from Harlan Laboratories (Indianapolis, IN) and shipped on GD 1–2 (GD 0 = sperm positive). All rats were housed individually in 20 × 25 × 47 cm clear polycarbonate cages with laboratory-grade pine shavings for bedding. Environmental conditions were 22°C–23°C, 50–60% humidity, and 14L:10D light cycle (lights off at 9:00 p.m.). Animals were fed NIH 07 breeding diet for rats and were provided municipal drinking water (Durham, NC) ad libitum. Water provided to animals in the Environmental Protection Agency (EPA) Reproductive Toxicology Facility is filtered (5 μm), tested monthly for Pseudomonas, and tested every 4 months for a subset of pesticides, heavy metals, and other chemical contaminants. The animal use protocol for this study was approved by the National Health and Environmental Effects Research Laboratory’s Institutional Animal Care and Use Committee, and all studies were conducted at a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Doses and administration of chemicals.
Separate experiments were conducted to assess the fetal endocrine responses of rats to DPeP, DIBP, or DINP exposure (as detailed in Hannas et al., 2011a,b) and DHeP, DHP, and DIDP. DPeP, DHP, and DHeP have 5-, 6-, and 7-carbon side-chain lengths, respectively. DIBP, DINP, and DIDP are mixtures of various isobutyl, isononyl, and isodecyl esters, respectively. DPeP (CAS 131-18-0, lot # 1431420) and DIBP (CAS 84-69-5; Lot # 07425BJ) were purchased from Sigma. Two separate formulations of DINP (referred to as DINP 1 and DINP 2) were used because they differ in the percent content of different ester side-chain structures, with DINP 1 having more isodecanol than DINP 2 (15–25 vs. 0%, respectively) and less methyl octanols (50–20 vs. 35–40%, respectively), and we therefore wanted to determine if they differed in toxicity to the fetal LC. DINP 1 (CAS 68515-48-0) is manufactured by the “Polygas” process. DINP 2 (CAS 28553-12-0) is n-butene based. DINP 2 was a gift from BASF (Badische Anilin und Soda Fabrik; CAS 28553-12-0), and DINP 1 was purchased from Sigma (CAS 68515-48-0). DHeP (CAS 3648-21-3; Lot # 125AG) and DHP (CAS 84-75-3, Lot # 139AG) were obtained from the National Toxicology Program (NTP) at the National Institute of Environmental Health Sciences (NIEHS, Research Triangle Park, North Carolina) where the purity of all tested phthalates was verified at 100% by gas chromatography with flame ionization detection. DIDP (CAS 26761-40-0; Lot # 1379769 23008238) was purchased from Fluka (Buchs, Switzerland).
Pregnant rats were weight ranked and assigned to dosage groups (generally n= 3–4) to minimize differences in means and variance among treatment groups. Sample sizes were based on power calculations performed on previous similarly conducted studies which demonstrated 3–4 litters per dosage group with three individual males/litter for T production, and the remaining males’ testes pooled for messenger RNA (mRNA) analyses was sufficient for detecting significant dose-related reductions in T production and gene expression.
In the current study, rats were dosed orally on a daily basis during GD 14–18 (covering a critical programming period for male sexual differentiation) with increasing doses of DINP, DHP, DHeP, DIDP, DPeP, or DIBP in separate blocks (one block per PE with approximately 15 pregnant rats per block) except for the two DINP formulations, which were examined in three blocks. The dosage range for each PE was selected based on a combination of preliminary screening of each compound in our laboratory at a single high dosage level, dosage levels used in Howdeshell et al. (2008), and postnatal effect studies (Andrade et al., 2006; Borch et al., 2006; Foster et al., 1983; Gray et al., 2000; Saillenfait et al., 2008, 2009). Dosage levels were as follows: corn oil (vehicle control), 11, 33, 100, or 300 mg DPeP/kg/day; vehicle control, 100, 300, 600, or 900 mg DIBP, DHeP, or DHP/kg/day; and vehicle control, 500, 750, 1000, or 1500 mg DINP or DIDP/kg/day. All phthalates were administered in corn oil (2.5 ml vehicle/kg bw).
An additional study was conducted to determine if gene expression changes in the PPARα pathway were correctly identified with our custom arrays. We dosed pregnant rats with the well-characterized PPARα agonist Wy-14,643 (CAS 50892-23-4, purchased from Cayman Chemical, Ann Arbor, MI). In this block of the study, we also used DIBP (CAS 84-69-5; Lot # 07425BJ; Sigma) as a positive control for changes in testicular gene expression and to directly compare with the effects of Wy-14,643 on the maternal liver. Rats were dosed with 0, 50, 100, or 200 mg Wy-14,643/kg bw/day or 500 mg DiBP/kg bw/day in corn oil from GD 14–18, as detailed above.
Fixed ratio-dilution study with a mixture of 9-PEs.
Harlan SD dams were dosed orally on each of GD 14–18 with one of six dilutions (0, 8, 17, 33, 67, and 100% of the top dose of 650 mg/kg/day total phthalate) of a mixture of DEHP, diisoheptyl phthalate (DIHP), DIBP, di-n-butyl phthalate (DBP), DHeP, DPeP, DHP, benzyl butyl phthalate (BBP; CAS 85-68-7; Lot No. 03405JH), and dicyclohexyl phthalate (DCHP; CAS 84-61-7; Lot No. 17518JB). The top dose of the mixture consisted of 10 mg DPeP/kg/day and 80 mg/kg/day of each of the other phthalates. The mixture ratio of the phthalates was designed such that each phthalate would contribute equally to the effects of the mixture on fetal testicular ex vivo T production, if the phthalates behaved in a dose-additive manner. The treatment groups contained 8, 3, 4, 7, 4, and 3 litters, respectively. ED50 and Hill slope values were determined from dose-response studies with DEHP, DHP, DIBP, DBP, DPeP (Hannas et al., 2011a,b, Howdeshell, Lambright, Furr, Evans, Foster, Wilson, and Gray, unpublished data), DHP, and DHeP, whereas BBP and DCHP were assumed equipotent to the mean values for all of the other PEs in the mixture.
Fetal necropsies.
Dams were rapidly euthanized by decapitation on GD18, and fetuses were removed, anesthetized via hypothermia on ice, decapitated, and dissected under a Leica MZ6 dissecting microscope (Wetzlar, Germany). For all experiments, a single testis from the first three males identified in a litter were removed and used for ex vivo T production (data for DPeP, DIBP, and DINP, previously published in Hannas et al., 2011a,b). All remaining testes in each litter were pooled, immediately homogenized in TRI reagent (Sigma, St Louis, MO) on ice using a Kontes pestle homogenizer, and stored at −80°C until used to extract RNA. All necropsies began 1 h following administration of the final maternal dose and were conducted within a 2-h time frame between 08:00 and 10:00 a.m. Eastern Standard Time to avoid any potential confounding effects of fetal growth or time of day on the fetal endpoints.
Ex vivo fetal testicular T production.
Following removal, fetal testes were immediately transferred individually into a single well on a 24-well plate containing 500 μl M-199 media without phenol red (Hazelton Biologics, Inc., St Lenexa, KS), supplemented with 10% dextran-coated charcoal-stripped fetal bovine serum (Hyclone Laboratories, Logan, UT). Testes were incubated for 3 h at 37°C on a rotating platform. Following incubation, media was removed and stored at −80°C until used for T measurement. The level of T in the media samples was measured by radioimmunoassay according to the manufacturer’s instructions (Diagnostic Products Corporation Coat-A-Count kits; Siemens Corp., Los Angeles, CA). The intraassay coefficient of variation was 3.1% (based on variability of the standard curve), and the interassay coefficient of variation was 13.7%. Cross-reactivity of the T antibody with dihydrotestosterone (DHT) was 3.2%. The limit of detection was 0.2 ng/ml for T.
Fetal testis gene expression analysis.
RNA was extracted from the fetal testes homogenate as previously described (Wilson et al., 2004) and cleaned to eliminate any potential genomic DNA contamination using Qiagen RNeasy Mini Kit (Valencia, CA) with the on-column DNase treatment step according to the manufacturer’s instructions. RNA concentration and purity were determined with a NanoDrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE). An additional genomic DNA elimination reaction and complementary DNA (cDNA) synthesis were performed on the RNA samples using the SABiosciences RT2 First Strand Kit according to the kit instructions. For each individual sample, 300 ng of RNA was added to a single reaction, to be used across a 96-well array plate. The template cDNA was then added to RT2 SYBR Green qPCR Master Mix (SABiosciences, Fredrick, MD), and 25 μl was added to each well of the plate. The 96-well gene array plate (purchased from SABiosciences) was custom designed to contain 89 individual target genes, 3 housekeeping genes (beta-actin, beta-glucuronidase, and lactate dehydrogenase), an interassay control, a genomic DNA control, a reverse transcription control, and a positive PCR control (genes listed in Fig. 3). To verify interassay consistency in CT cycling, aliquots of fetal testicular cDNA from males of a corn oil (control)–treated dam were added to a well containing primers for the housekeeping gene beta-2 microglobulin on all plates.
FIG. 3.


Gene expression changes in fetal testes on GD 18 following 5-day in utero exposure to increasing doses of one of six phthalates (GD 14–18). Group number corresponds to listed groups below heat map. Control threshold cycle (CT) refers to the mean (± SEM) of control litter values across all phthalate blocks (n = 19) for comparison of relative expression. Control CT values ≥ 33 were considered nondetectable and represented by black boxes. Red and yellow boxes indicate downregulation with p < 0.01 and p < 0.05, respectively, and green boxes represent upregulation of the gene with p < 0.01. The number inside the boxes indicates the lowest dose at which a significant change from control was detected. Gene names shown in bold were those that were downregulated by all phthalates that were positive for reducing fetal testicular T production.
The PCR reaction was run on an iCycler iQ Real-Time Detection System (Bio-Rad, Hercules, CA) using the following cycling parameters: one cycle of 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min. Product purity was verified by melting curve analysis. The ΔCT value for each gene was determined by dividing the gene CT value by the mean CT value of three housekeeping genes. The 2−ΔΔCT method was used to analyze data and change in gene expression levels were reported as fold change (Tusher et al., 2001) or the ratio of the phthalate treated sample group to the respective control group. The sensitivity, specificity, and reproducibility of the gene array system are discussed in detail by Arikawa et al. (2011).
Mixture model predictions.
ED50 and Hill slope values derived from the individual phthalate dose-responsive effects on gene expression levels were used to calculate the mixture model response predictions for the 9-PE mixture on genes that were affected significantly by PE exposure. Because dose-response gene data were not available for DEHP, DIHP, DCHP, and BBP in the Harlan SD rat, the ED50 values for these four PEs were assumed equivalent to the average of the ED50s for DBP and DIBP. This assumption was based upon the observation that these PEs all appear to induce malformations at about the same dosage levels in postnatal studies. Predictions were calculated using the DA and RA models for comparison. Hill slope and ED50 values for DBP were obtained from a similarly conducted dose-response study (Howdeshell, Lambright, Furr, Evans, Foster, Wilson, and Gray, unpublished data).
Two separate approaches were taken to compare the DA and RA model predictions to the observed mixture effects. First, the ED50 values derived from the DA and RA model predictions were compared with the ED50 value derived from the observed data by determining if the predicted ED50 fell within the 99% confidence interval (CI) of the observed ED50. Any model prediction ED50 falling outside of the observed ED50 99% CI was considered significantly different from observed. The second approach for comparison was to force fit the observed data (means, SEs, and sample sizes) to the DA or RA models by constraining the top, bottom, Hill slope, and ED50 parameters to that of the DA or RA model parameters. A model that perfectly predicted the observed results would produce an R2 value for the force fit that would be equal to the R2 value obtained from the observed data best-fit model. Therefore, the greater the DA or RA predicted models deviated from the observed effects, the greater the R2 of the force-fit model differed from the best-fit R2 value.
Data analysis and statistics.
Litter mean values were analyzed for T production, based on individual testis incubations. T production data were log10-transformed to correct for heterogeneity of variance and then analyzed with a one-way ANOVA model to detect significant differences in responses to DHeP, DHP, DIDP, or Wy-14,643 using PROC GLM in SAS, version 9.1 (SAS Institute, Cary, NC). Post hoc t-tests were performed with the LSMEANS procedure to compare individual doses when a significant overall dose effect was determined by the ANOVA model.
Gene expression was analyzed based on pooled testes samples from each litter. As seen in our previous study (Hannas et al., 2011b) where the two DINP formulations did not differ significantly in their ability to reduce fetal testicular T production, DINP 1 was not more or less potent than DINP 2 in reducing testis gene expression. Therefore, in the current study, litter means are presented by combining the values from both formulations of DINP. Fold change for each gene following exposure to DPeP, DIBP, DINP, DHeP, DHP, DIDP, and Wy-14,643 was considered to be statistically significant different if the F value for the overall ANOVA model was p < 0.01, followed by t-test (p < 0.01) as above. Effects with a p value between p < 0.05 and p > 0.01 were considered as “equivocal” changes. Genes with a mean control CT value > 33 were considered undetected.
Dose-response T production data were converted to percent of control and analyzed using a nonlinear four parameter regression analysis (sigmoidal fit with variable slope using Prism GraphPad 5.01 software; GraphPad Software, Inc., La Jolla, CA). The top and bottom parameters were constrained to 100 and 0% of control, respectively. Dose-response gene expression data were modeled as fold change using the sigmoidal fit with variable slope model, with the top constrained to 1 and the bottom constrained to 0. Potency comparisons between phthalates for individual genes and T production were made using the ED50 values derived from dose-response curves.
RESULTS
Maternal Body Weight and Litter Effects of DHP, DHeP, and DIDP
Maternal body weight gain and fetal mortality were not significantly affected by 5-day (GD 14–18) DHP, DHeP, or DIDP in utero exposure at any dose tested (Table 1). However, unlike the power to detect effects on T production and testis gene expression, which have power > 0.90 for the high-dose effects, the statistical power to detect reductions in litter size with an N of three dams per group is less than 0.80. As reported previously, DPeP, DIBP, and DINP, similarly did not induce maternal or fetal overt toxicity as defined by mortality, reduced maternal body weight or reduced litter size at any dose administered following GD 14–18 dosing (Hannas et al., 2011a,b).
TABLE 1.
Maternal Weight Gain and Fetus Survival Following Gestational Exposure From Day 14–18 to Increasing Doses of DHP, DHeP, DIDP, or Wy-14,643
| DHP | ||||||
| Control | 100 | 300 | 600 | 900 | ||
| Weight gaina | 47.7 ± 6.1 | 53.2 ± 8.1 | 38.8 ± 8.9 | 46.7 ± 4.3 | 32.5 ± 3.2 | |
| Number of live fetuses | 11.7 ± 1.5 | 14.3 ± 2.2 | 8.7 ± 3.4 | 13.0 ± 1.0 | 7.7 ± 1.8 | |
| Fetal survivalb | 89.7 ± 2.2 | 97.0 ± 3.0 | 73.3 ± 26.7 | 100.0 ± 0.0 | 53.9 ± 14.2 | |
| DHeP | ||||||
| Control | 100 | 300 | 600 | 900 | ||
| Weight gain | 53.2 ± 12.0 | 34.6 ± 16.9 | 25.2 ± 2.1 | 43.8 ± 4.2 | 56.2 ± 2.8 | |
| Number of live fetuses | 12.3 ± 0.3 | 10.5 ± 1.5 | 3.7 ± 0.9 | 12.0 ± 1.5 | 15.7 ± 1.2 | |
| Fetal survival | 97.6 ± 2.4 | 90.9 ± 9.1 | 77.8 ± 14.7 | 91.7 ± 4.8 | 100.0 ± 0.0 | |
| DIDP | ||||||
| Control | 100 | 300 | 600 | 900 | ||
| Weight gain | 48.7 ± 2.5 | 41.9 ± 1.4 | 37.2 ± 3.9 | 38.6 ± 2.0 | 41.5 ± 1.2 | |
| Number of live fetuses | 12.3 ± 2.2 | 13.7 ± 0.3 | 12.0 ± 2.1 | 13.3 ± 0.9 | 14.0 ± 0.6 | |
| Fetal survival | 93.3 ± 6.7 | 97.8 ± 2.2 | 100.0 ± 0.0 | 95.1 ± 2.5 | 100.0 ± 0.0 | |
| Wy-14,643 | ||||||
| Control | 50 Wy | 100 Wy | 200 Wy | 500 DIBP | ||
| Weight gain | 43.2 ± 3.1 | 55.1 ± 2.7 | 46.0 ± 3.7 | 46.0 ± 3.5 | 48.6 ± 2.8 | |
| Number of live fetuses | 12.3 ± 0.3 | 14.0 ± 1.0 | 11.0 ± 1.0 | 11.3 ± 1.2 | 14.7 ± 0.7 | |
| Fetal survival | 100.0 ± 0.0 | 97.6 ± 2.4 | 97.0 ± 3.0 | 100.0 ± 0.0 | 100.0 ± 0.0 | |
| 9-PE Mixture | ||||||
| Control | 8% | 17% | 33% | 67% | 100% | |
| Weight gain | 43.8 ± 2.0 | 51.8 ± 1.3 | 4.4 ± 6.6 | 45.6 ± 5.6 | 36.1 ± 7.0 | 50.1 ± 5.5 |
| Number of live fetuses | 11.0 ± 0.9 | 13.0 ± 1.0 | 8.5 ± 2.5 | 12.7 ± 1.5 | 8.0 ± 2.6 | 13.3 ± 0.9 |
| Fetal survival | 88.4 ± 6.1 | 95.2 ± 4.8 | 77.3 ± 16.0 | 100.0 ± 0.0 | 85.0 ± 15.0 | 95.6 ± 4.4 |
Note. Values are means ± SEM.
Maternal weight gain = Body weight GD 18 − Body weight GD 14.
Fetal survival (%) = Number of live fetuses/total fetuses.
Ex vivo Fetal Testicular T Production Dose-Response for DHP, DHeP, and DIDP
Dose-response studies were performed to assess the relationship between in utero exposure (GD 14–18) to DHeP, DHP, and DIDP and fetal testicular ex vivo T production. Both DHeP and DHP reduced T production in a dose-responsive manner, whereas DIDP had no effect (Fig. 1). T Production was reduced significantly at doses of 600 mg DHeP/kg/day and greater and 100 mg DHP/kg/day and greater (p < 0.05). The ED50s derived from regression analysis of the dose-response data for DHP and DHeP were 75.25 and 444.2 mg/kg/day, respectively (Table 2).
FIG. 1.

Fetal testicular ex vivo T production following 3-h incubation from SD rats exposed in utero to DHeP, DHP, or DIDP during sexual differentiation on GD 14–18. Each data point represents the mean (± SEM) of 2–3 pooled litter values (n= 2 for 300 mg DHeP/kg/day group and 100 mg DHP mg/kg/day, n = 3 for all other groups).
TABLE 2.
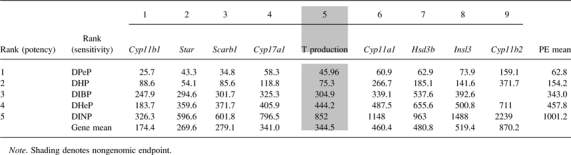
Potency Ranking of the Five Tested “Positive” PEs (first column) and the Nine Fetal Endpoints (first row), Based on the Mean ED50 Following 5-day in utero Exposure to Increasing Doses of 1 of 6 Phthalates (GD 14–18)
 |
Wy-14,643 Maternal Liver Weight and Fetal Endocrine Effects
Overall maternal body weight gain was not affected by the DIBP or Wy-14,643 5-day (GD 14–18) dosing regimen (Table 1). Maternal liver weight was increased in all Wy-14,643 dosage groups but not by DIBP treatment (positive control for fetal endocrine effects; data not shown). DIBP decreased fetal testicular ex vivo T production at a dose of 500 mg/kg/day, whereas Wy-14,643 had no effect on T production (Fig. 2).
FIG. 2.

Fetal testicular ex vivo T production following 3-h incubation from SD rats exposed in utero to Wy-14,643 during sexual differentiation on GD 14–18. Each data point represents the mean (± SEM) of three pooled litter values. An asterisk (*) denotes statistically different from control (p < 0.01).
Gene Array Screening of DHP, DIBP, DHeP, DPeP, DINP, and DIDP Fetal Testes
The effect of DHeP, DIBP, DHP, DPeP, DINP, and DIDP on gene expression following 5-day in utero exposure was assessed using a PCR array containing 89 individual candidate target genes (Fig. 3). All tested phthalates with the exception of DIDP were previously ranked (DPeP, DIBP, and DINP in Hannas et al., 2011a,b) or ranked in the current study (DHeP and DHP) as positive for antiandrogenic activity based on the ability to reduce ex vivo T production in the fetal testes. These PEs consistently reduced gene expression levels of Cyp11b1, Scarb1, Star, Cyp11a1, Cyp17a1, Insl3, and Hsd3b in a dose-responsive manner (Fig. 4, see Fig. 3 for lowest dose at which gene expression level reduction is significant). Comparison of the ED50 values for each of these genes across the positive phthalates demonstrated that DPeP was the most potent phthalate for reducing each gene and DINP was the least potent for most of the affected genes (Table 2). DHP, DHeP, and DIBP all had potencies in between that of DPeP and DINP for the affected genes. We ranked the overall sensitivity of each gene endpoint and T production for the six phthalates and determined that the order of sensitivity from most to least was Cyp11b1 > Star =Scarb1 > Cyp17a1 = T production > Cyp11a1= Hsd3b = Insl3 > Cyp11b2 (Table 2).
FIG. 4.

Dose-response curves for fold inhibition of fetal testis RNA expression levels for (A) Cyp11b1, (B) Scarb1, (C) Star, (D) Cyp11a1, (E) Hsd3b, (F) Insl3, (G) Cyp17a1, and (H) Cyp11b2 on GD 18 following in utero exposure (GD 14–18) to DPeP, DIBP, DHP, DHeP, or DINP. Each data point represents the mean (± SEM) of pooled litters (n = 3–4 l). The 2−▵▵CT method was used to analyze data and change in gene expression levels are reported as fold change.
Some mRNA levels for additional genes were occasionally affected by one or two PEs but not consistently affected across all PEs and generally not in a dose-related manner (Fig. 3). Additional research would be required to determine if these “changes” were actually treatment-related or statistical vagaries.
The PPAR-related genes and target genes included on the plate were Ppara, Pparb, Pparg, Rxra, Rxrb, Rxrg, Acox1, Cyp4a1, Fabp1, and Apoa1. Of this gene set, DIBP downregulated Rxrg at the highest dosage level of 900 mg/kg/day, and DINP downregulated Acox1 at the highest dose level of 1500 mg/kg/day. Pparg, Cyp4a1, and Fabp1 were not detected in the fetal testes, based on a mean control CT values > 33 (Fig. 3).
The PPARα agonist, Wy-14,643, induced several PPAR-regulated genes, as expected, including Rxrg, Acox1, Cyp4a1, Fabp1, Adh1, and Aldh1a1 in the maternal livers following 5-day dosing at 50 mg/kg/day and greater (Fig. 5). Wy-14,643 induced Rxrb at 100 mg/kg/day and greater and Rxra at the highest dose of 200 mg/kg/day. Cyp17a1, Tspo, Dixdc1, and Nr3c1 were also upregulated by this compound, whereas Tle1 and Inhba were downregulated.
FIG. 5.

Gene expression changes in maternal livers and fetal testes on GD 18 following 5-day in utero exposure to increasing doses of Wy-14,643 (GD 14–18). Control CT refers to the mean (± SEM) of control litter values (n = 3) for comparison of relative expression. Control CT values ≥ 33 were considered nondetectable and represented by black boxes. Red and yellow boxes indicate downregulation with p < 0.01 and p < 0.05, respectively, and green boxes represent upregulation of the gene with p < 0.01. The number inside the boxes indicates the lowest dose at which a significant change from control was detected.
Phthalate Mixture Gene Expression Results
A dose-response study was conducted using dilutions of the top dose of a 9-PE mixture to determine if response of the affected genes was more accurately predicted by a dose-additive or response-additive model. The mixture significantly reduced expression of (ED50 in mg/kg/day PE mixture shown in parentheses following each gene) Star (25.64), Cyp11a (34.61), Hsd3b (35.7), Cyp11b2 (77.32), Cyp17a1 (27.22), Scarb1 (19.96), Insl3 (46.23), Cyp11b1 (14.21), Lhcr7 (43.65), Dhcr7 (103.7), Inha (75.64), and Nr5a1 (170.8) in a dose-responsive manner (Fig. 6, data shown for seven of the genes). The mixture also upregulated several genes, including Sry, Sox9, Ptgds2, Pdgfa, Fgf8, Inhbb, Dvl3, Rara, and Rarb at 8 or 17% of the top dose, but these effects were generally not dose related (Fig. 3).
FIG. 6.

Gene expression changes in fetal testes on GD 18 following 5-day in utero exposure to a dose range of a 9-PE mixture including DEHP, DIHP, DIBP, DBP, BBP, DNCP, DHeP, DHP, and DPeP. The top dose contained a total of 650 mg phthalate/kg/day including 10 mg DPeP/kg/day and 80 mg/kg/day of all other phthalates. Observed (OBS) values are litter means (± SEM) of 3–4 l. RA and DA models are shown for each gene. The ED50 values are derived from the logistic regression model for each fit of the data. The R2 values describe the percentage of variance described when the OBS data were force fit to each model parameters, using the respective ED50 and Hill slope values for the RA or DA models.
For three of the genes modeled for DA and RA predictions (Cyp11b1, Cyp11a, and Cyp17a1), the DA model was slightly more accurate in predicting the ED50 of the mixture of nine PEs than the RA model. DA provided a better fit to the mixture data than did the RA model with the ED50s being OBS < DA < RA, based upon lack of overlap of the observed data 99% CIs with the model ED50s (Fig. 6). Neither model was superior to the other in predicting the ED50 of the mixture for the following genes: Scarb1 (the observed ED50 of the mixture was slightly and significantly lower than those predicted by both DA and RA models, with OBS < DA = RA), Star (RA = OBS = DA), Hsd3b (both model ED50 predictions differed slightly from the observed ED50 with RA < OBS < DA), and Insl3 (both model predictions were lower than the observed ED50 with RA = DA < OBS). When the observed data were force fit by constraining all four parameters from the DA and RA logistic regression models, the R2 values for DA exceeded that of the RA fit for two of seven genes (Cyp11b1 and Cyp11a), whereas fitting the observed data to DA and RA model parameters provided similar R2 values for the other five genes (Fig. 6).
DISCUSSION
In the current study, we evaluated the dose-related effects of several PEs on fetal testis gene expression using custom-designed 96-gene real-time (RT) PCR arrays. We compared the sensitivity of the affected genes to T production to identify potential genomic biomarkers of effect and exposure. These data indicate that the PEs which reduce T production act through a similar mode of action in the fetal testis, due to the consistency in reducing expression of a subset of genes involved in steroid transport and synthesis. The order of sensitivity from most to least affected genes was Cyp11b1 > Star = Scarb1> Cyp17a1 =T production > Cyp11a1 = Hsd3b = Insl3 > Cyp11b2. Interestingly, two of these consistently downregulated fetal testis genes, Cyp11b1 and Cyp11b2, code for adrenal enzymes in the adult and are not present in adult LCs. Although several genes involved in androgen synthesis and steroid transport are dramatically downregulated in the fetal testis, genes in the PPARα pathway were not induced by any PE treatment, suggesting that this pathway is not involved in PE-induced fetal testis toxicity.
We determined that the order of potency for reducing expression of the affected genes was generally consistent across most of the genes with the order of potency for reducing T production (DPeP > DHP > DIBP ≥ DHeP > DINP; Table 2). We also demonstrated that DIDP was not an active antiandrogenic phthalate, as defined by the lack of an effect on fetal T production.
The relative potency of each phthalate for reducing gene expression was used to conduct a mixture study with nine phthalates in which we found that dose-addition models adequately predicted the mixture effects on the genes (being slightly and statistically superior to RA predictions for two of seven modeled genes and equal to RA predictions for the other five).
We previously evaluated the potencies of DPeP, DINP, DIBP, DEHP, and DIHP for reducing fetal testicular T production (Hannas et al., 2011a,b). DPeP is the most potent phthalate tested to date for reducing T production and inducing postnatal reproductive tract malformations (Hannas et al., 2011a) and testicular toxicity in the pubertal male model (Foster et al., 1980). Based on the results of the current study, DHP is the second most potent for reduction of fetal T production. DHP induces malformations of the reproductive tract in male rat offspring, reduces AGD and induces permanent female-like nipples (Saillenfait et al., 2009) at slightly higher dosage levels than does DPeP. When DHP is administered to pregnant SD rats (GD 12–21) at doses of 0, 50, 125, 250, or 500 mg/kg/day, AGD was reduced and areola/nipples were retained (in infants and adults) at 250 and 500 mg DHP/kg/day. In addition, low incidences of severe reproductive malformations were observed in young adult males at 125 and 250 mg DnHP/kg/day, whereas most males were malformed at 500 mg/kg/day. We previously determined that the ED50 for reduction of male AGD on PND 2 by DPeP was 252.3 mg/kg/day (Hannas et al., 2011a) with severe malformations occurring at lower doses (Gray, personal communication). Together, these data indicate that the potency of DHP appears to verge upon that of DPeP for inhibiting differentiation of the reproductive tract of the fetal male rat.
DHeP, which was less potent than both DPeP and DHP for reducing T production and testis gene expression, has one and two additional carbons in the ester side chain compared with DHP and DPeP, respectively. Saillenfait et al. (2011) demonstrated that DHeP only reduced AGD in male fetuses exposed in utero (GD 6–20) at doses of 500–1000 mg/kg/day administered orally to the dam, whereas testis descent was normal for this stage of development in all dosage groups.
Our observations that DIDP was negative and DINP was the least potent active PE for reducing T production and testis gene expression are consistent with published data on the ability of these PEs to induce the Phthalate Syndrome in male rats. For example, DINP only reduces AGD (Boberg et al., 2011) and induces reproductive tract malformations (Boberg et al., 2011; Gray et al., 2000) at dosage levels of 900 and 750 mg/kg/day, respectively. DIDP, which is a mixture of isomers containing two carbon chains with 9–11 carbons, was inactive in the current fetal screening protocol. This result was expected based on both its structure and the lack of effect on reproductive endpoints examined in two separate two-generation studies (Hushka et al., 2001). In those studies, the male offspring (F1) of Sprague-Dawley rats (F0) administered DIDP in feed from 10 weeks prior to mating through female lactation, displayed no reproductive abnormalities.
Taken together, these results demonstrate that our fetal T production and gene expression findings are predictive of postnatal androgen- and INSL3-dependent tissue malformations. Numerous studies have demonstrated the ability of PE’s to reduce testicular T levels (Howdeshell et al., 2008; Lehmann et al., 2004; Shultz et al., 2001), which occurs through a nonandrogen receptor (AR)–mediated mechanism (Parks et al., 2000). Additionally, expression of Insl3 is downregulated by antiandrogenic PEs (Howdeshell et al., 2008; Wilson et al., 2004). In the current study, the tested PE’s that were classified as positive for reducing fetal testicular T production were all consistent in downregulating gene expression of steroidogenic enzymes, steroid regulatory, and transport proteins (Fig. 7) and Insl3. Some of these effects on T production and gene expression were expected based on previous work demonstrating that DBP downregulated many of these genes including Star, Cyp11a1, Insl3, Scarb1, Hsd3b, and Cyp17a1 (Barlow et al., 2003; Howdeshell et al., 2008; Johnson et al., 2007; Lahousse et al., 2006; Lehmann et al., 2004; Plummer et al., 2007; Shultz et al., 2001; Wilson et al., 2004). Nevertheless, only a few of these studies have included PEs other than DBP (Howdeshell et al., 2008) or included a sufficient range of doses to be able to determine ED50s or relative potency values for these genomic endpoints. Results of the current study also clearly indicate that all PEs that induce the phthalate syndrome alter fetal LC function via a common endocrine and genomic mode of action and that the overall potency in inducing fetal testis alterations is predictive of their potency to induce reproductive tract malformations.
FIG. 7.

Steroid biosynthesis pathways of the testes (pathway outside dashed circle) and adrenals (pathway inside dashed circle). Steroidogenesis-related enzymes and transport proteins (noted by parentheses) affected by in utero phthalate exposure are circled. Dashed lines around CYP17A1 signify this enzyme acting on multiple steps.
We derived two important comparisons from the dose-response data generated using the PCR gene array: (1) comparison of the individual phthalate potency for reducing expression of a particular gene and (2) comparison of the sensitivity of different genes to phthalate exposure. In comparing phthalate potency, we previously demonstrated a link between the reduction of fetal T production and early postnatal male reproductive tract malformations following dosing during the sexual differentiation period by determining that the potency of DPeP for the fetal endocrine endpoints was predictive of the postnatal endocrine endpoints (Hannas et al., 2011a). Here, we further demonstrate that phthalate effects on expression of androgen-related genes are linked to reduced T production, based not only on the biological relevancy but also on the congruency in phthalate potency between the endpoints. We thereby further assume that these gene endpoints are also predictive of the postnatal malformations.
The second comparison we made as mentioned above was between the sensitivity of the different affected endpoints to each phthalate. In general, the order of most sensitive to least was consistent for all phthalates. A few of the genomic endpoints ranked more sensitive to PE disruption than T production, including Cyp11b1, Scarb1, and Star, although the difference among Scarb1, Star, and T production was not great. Cyp17a1 was as sensitive to PE disruption as was T production.
Currently, T production is being considered as a critical endpoint for some phthalate risk assessments. Nevertheless, the findings of this study could potentially support the use of genomic endpoints as the critical effect in future risk assessments, with a few caveats. When considering gene expression as the most sensitive endpoint of phthalate exposure, it is critically important to consider the biological role of the products of the genes. This point is illustrated when taking into account the role of the most sensitive genes detected in our assessment. The “most” sensitive gene detected in the fetal testis, Cyp11b1, does not appear to be biologically linked to the postnatal outcomes of concern. CYP11B1, also known as 11β-hydroxylase, is an enzyme responsible for conversion of 11-deoxycortisol to cortisol in the adrenal cortex. It is not expressed in adult testes. On the contrary, recent studies, which examined mouse fetal testes, have demonstrated that a subpopulation of steroidogenic cells that express Cyp11b1 occur in the fetal LC (Hu et al., 2007; Val et al., 2006). Hu et al. (2007) determined that although Cyp11b1 gene expression was detected, the protein product was not detected by immunohistochemistry, and enzyme activity was also not detected. The authors of this study suggest that translation of the enzyme product is suppressed to prevent high levels of corticosteroid production in the fetal testes. Although our results demonstrate that message of this gene in the fetal testes is highly susceptible to phthalate exposure, this vulnerability is not likely to translate into the postnatal phthalate effects, and therefore, Cyp11b1 gene expression would not be a suitable critical effect endpoint in risk assessment. Likewise, Cyp11b2, another adrenal enzyme gene detected in the fetal testes, is not likely linked to the postnatal reproductive tract toxicity. In contrast, SR-B1, StAR, and Cyp17A1 are all critical for normal testosterone synthesis. SR-B1 protein facilitates cholesterol uptake into steroidogenic cells, whereas StAR acts as the transport protein for cholesterol across mitochondrial membranes. Cyp17A1 is the steroidogenic enzyme that converts progesterone to the androgen androstenedione, which is then converted to testosterone by another enzyme (Fig. 7). It is reasonable to assume that the vulnerability of these genes is linked to the postnatal reproductive malformations we detect following in utero phthalate exposure because the products of these genes feed directly into production of T during the critical period for androgen-dependent tissue development. Furthermore, the ED50 values for the effects of PEs on Scarb1, Star, and Cyp17a1 gene expression do not differ greatly from the ED50 value for T production, suggesting that these genomic biomarkers could be considered additional sensitive critical or supportive endpoints for PE risk assessments.
In order to verify that our custom PCR arrays responded appropriately to activation of PPARα, we determined that the potent PPARα agonist, Wy-14,643, was effective in increasing liver weights of the exposed dams and upregulating genes in the PPARα pathway including: Rxra, Rxrb, Rxrg, Acox1, Cyp4a1, and Fabp1. In contrast, in the same experiment, DIBP had no effect on these genes. Furthermore, we confirmed that Wy-14,643 had no effect on fetal testicular T production, whereas DIBP significantly reduced T production. From this experiment, we conclude that insensitive methodology is not the cause for the lack of phthalate-induced activation of PPARα target genes in the fetal testis.
We did not detect any Pparg expression in the fetal testis, also eliminating this as a likely PE pathway for PE-induced reproductive toxicity. Furthermore, in utero administration of rosiglitazone, a potent PPARγ agonist, did not reduce fetal AGD, T levels, and Insl3 or steroidogenic gene expression or induce histopathological changes in the fetal testis, whereas DIBP administration induced all of these affects (Boberg et al., 2008). We did not specifically test the effects of a PPARβ agonist on fetal T production; however, there is currently no evidence to suggest that PPARβ is involved in testicular toxicity. The existing data do not support the hypothesis that activation of PPARα or PPARγ pathways is involved in the effects of PEs on sexual differentiation of the male rat.
Despite lack of effect on the PPARα pathway, the current study provides evidence supporting the hypothesis that phthalate exposure reduces T production by interfering with cholesterol regulation. This mode of action can be inferred based on the consistency of the antiandrogenic phthalates in reducing Star and Scarb1 gene expression as well as the reduction of Dhcr7 at relatively high doses of DPeP, DHP, and DINP. DHCR7 or 7-dehydrocholesterol is the enzyme, which mediates the final step in cholesterol production. As previously mentioned, SR-B1 and StAR are involved in transport of cholesterol into the cell and mitochondria, respectively, as the precursor to testosterone. Johnson et al. (2007) detected reduced Star and Dhcr7 expression following 3 h of in utero exposure (GD 19) to 10 or greater and 100 or greater mg DBP/kg, respectively. Plummer et al. (2007) also demonstrated downregulation of Scarb1 and Star expression by DBP in a time-dependent fashion between GD 15.5 and 19.5. In light of these collective results, upstream genes in this pathway and earlier time points and/or short phthalate exposure durations during the critical period warrant further investigation.
We previously determined that our 9-PE mixture reduces fetal testicular T production in a dose-additive manner (Hannas et al., 2011b). The impetus behind that study was to provide data to support the recommendation provided to the U.S. EPA by the National Academy of Science National Research Council committee that a cumulative assessment be conducted for antiandrogenic phthalates (National Academy of Sciences, 2008). In the current study, we further assessed fetal testicular samples for gene expression changes to determine if the effects could similarly be modeled using a DA mixture model. The mixture was designed so that each of the nine phthalates would contribute equally in terms of potency for reducing fetal T production if they acted in a dose-additive manner. The top dose was expected to dramatically reduce T production and gene expression if the effects of the nine PEs were dose additive. Based on the data from this mixture study, we can conclude that the DA model adequately predicted the observed values for all seven genes that showed consistent dose-related PE-induced downregulation. The DA model was slightly superior to the RA model for two of seven genes, whereas the models were roughly equivalent in their ability to predict the ED50 of the other five genes.
In conclusion, we used a targeted RT-PCR array approach of toxicity assessment in an attempt to address several of the challenges faced in the human health risk assessment process related to phthalate exposure. Based on the results, we confirmed that the antiandrogenic phthalates we assessed act through a similar mode of toxicity, despite not yet fully understanding the proximate molecular target. We additionally demonstrated that the rank of potency of the individual phthalates largely translates from reduction of T production to the downregulation of gene expression, suggesting that most of the consistently downregulated genes from our array plate are directly linked to the postnatal reproductive tract malformations. Finally, we demonstrated that the targeted genomic response of the fetal testis to a mixture of nine antiandrogenic phthalates was predicted using a DA mathematical model, supporting the notion that a cumulative risk assessment of the phthalates would be most protective of human health as compared with assessments of individual phthalates. Using this targeted gene array approach, we can continue to investigate the behavior of phthalates in male fetuses during a critical period of development by focusing on timing within the sexual differentiation period and additional target genes/pathways in future assessments.
FUNDING
This project was provided by an Interagency Agreement between the United States Environmental Protection Agency and the NTP at the National Institute of Environmental Health Sciences (IA no. 75-92285501-1) and B.R.H. was funded through a National Academy of Sciences postdoctoral fellowship.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Mary Cardon and Hunter Sampson for assistance during necropsies.
References
- Altenburger R, Backhaus T, Boedeker W, Faust M, Scholze M, Grimme LH. Predictability of the toxicity of multiple chemical mixtures to Vibrio fischeri: Mixtures composed of similarly acting chemicals. Environ. Toxicol. Chem. 2000;19:2341–2347. [Google Scholar]
- Andrade AJ, Grande SW, Talsness CE, Grote K, Golombiewski A, Sterner-Kock A, Chahoud I. A dose-response study following in utero and lactational exposure to di-(2-ethylhexyl) phthalate (DEHP): Effects on androgenic status, developmental landmarks and testicular histology in male offspring rats. Toxicology. 2006;225:64–74. doi: 10.1016/j.tox.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Arikawa E, Quelhorst G, Han Y, Pan H, Yang J. RT2 Profiler™ PCR Arrays: Pathway-focused gene expression profiling with qRT-PCR. SABiosciences. 2011 . Available at: http://www.sabiosciences.com/manuals/pcrarraywhitepaper.pdf. Accessed June 1, 2011. [Google Scholar]
- Barlow NJ, Foster PM. Pathogenesis of male reproductive tract lesions from gestation through adulthood following in utero exposure to di(n-butyl) phthalate. Toxicol. Pathol. 2003;31:397–410. doi: 10.1080/01926230390202335. [DOI] [PubMed] [Google Scholar]
- Barlow NJ, Phillips SL, Wallace DG, Sar M, Gaido KW, Foster PM. Quantitative changes in gene expression in fetal rat testes following exposure to di(n-butyl) phthalate. Toxicol. Sci. 2003;73:431–441. doi: 10.1093/toxsci/kfg087. [DOI] [PubMed] [Google Scholar]
- Bhattacharya N, Dufour JM, Vo MN, Okita J, Okita R, Kim KH. Differential effects of phthalates on the testis and the liver. Biol. Reprod. 2005;72:745–754. doi: 10.1095/biolreprod.104.031583. [DOI] [PubMed] [Google Scholar]
- Biegel LB, Liu RCM, Hurtt ME, Cook JC. Effects of ammonium perfluorooctanoate on Leydig-cell function: In vitro, in vivo, and ex vivo studies. Toxicol. Appl. Pharmacol. 1995;134:18–25. doi: 10.1006/taap.1995.1164. [DOI] [PubMed] [Google Scholar]
- Boberg J, Christiansen S, Axelstad M, Kledal TS, Vinggaard AM, Dalgaard M, Nellemann C, Hass U. Reproductive and behavioral effects of diisononyl phthalate (DINP) in perinatally exposed rats. Reprod. Toxicol. 2011;31:200–209. doi: 10.1016/j.reprotox.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Boberg J, Metzdorff S, Wortziger R, Axelstad M, Brokken L, Vinggaard AM, Dalgaard M, Nellemann C. Impact of diisobutyl phthalate and other PPAR agonists on steroidogenesis and plasma insulin and leptin levels in fetal rats. Toxicology. 2008;250:75–81. doi: 10.1016/j.tox.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Borch J, Metzdorff SB, Vinggaard AM, Brokken L, Dalgaard M. Mechanisms underlying the anti-androgenic effects of diethylhexyl phthalate in fetal rat testis. Toxicology. 2006;223:144–155. doi: 10.1016/j.tox.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Bowman CJ, Turner KJ, Sar M, Barlow NJ, Gaido KW, Foster PM. Altered gene expression during rat Wolffian duct development following di(n-butyl) phthalate exposure. Toxicol. Sci. 2005;86:161–174. doi: 10.1093/toxsci/kfi172. [DOI] [PubMed] [Google Scholar]
- Carruthers CM, Foster PMD. Critical window of male reproductive tract development in rats following gestational exposure to di-n-butyl phthalate. Birth Defects Res. B Dev. Reprod. Toxicol. 2005;74:277–285. doi: 10.1002/bdrb.20050. [DOI] [PubMed] [Google Scholar]
- Corton JC, Lapinskas PJ. Peroxisome proliferator-activated receptors: Mediators of phthalate ester-induced effects in the male reproductive tract? Toxicol. Sci. 2005;83:4–17. doi: 10.1093/toxsci/kfi011. [DOI] [PubMed] [Google Scholar]
- Foster PM. Disruption of reproductive development in male rat offspring following in utero exposure to phthalate esters. Int. J. Androl. 2006;29:140–147. doi: 10.1111/j.1365-2605.2005.00563.x. discussion 181–185. [DOI] [PubMed] [Google Scholar]
- Foster PM, Thomas LV, Cook MW, Gangolli SD. Study of the testicular effects and changes in zinc excretion produced by some n-alkyl phthalates in the rat. Toxicol. Appl. Pharmacol. 1980;54:392–398. doi: 10.1016/0041-008x(80)90165-9. [DOI] [PubMed] [Google Scholar]
- Foster PM, Thomas LV, Cook MW, Walters DG. Effect of di-n-pentyl phthalate treatment on testicular steroidogenic enzymes and cytochrome P-450 in the rat. Toxicol. Lett. 1983;15:265–271. doi: 10.1016/0378-4274(83)90226-6. [DOI] [PubMed] [Google Scholar]
- Gazouli M, Yao ZX, Boujrad N, Corton JC, Culty M, Papadopoulos V. Effect of peroxisome proliferators on Leydig cell peripheral-type benzodiazepine receptor gene expression, hormone-stimulated cholesterol transport, and steroidogenesis: Role of the peroxisome proliferator-activator receptor alpha. Endocrinology. 2002;143:2571–2583. doi: 10.1210/endo.143.7.8895. [DOI] [PubMed] [Google Scholar]
- Gray LE, Jr, Ostby J, Furr J, Price M, Veeramachaneni DN, Parks L. Perinatal exposure to the phthalates DEHP, BBP, and DINP, but not DEP, DMP, or DOTP, alters sexual differentiation of the male rat. Toxicol. Sci. 2000;58:350–365. doi: 10.1093/toxsci/58.2.350. [DOI] [PubMed] [Google Scholar]
- Greco W, Unkelbach H-D, Pöch G, Sühnel J, Kundi M, Boedeker W. Consensus on concepts and terminology for combined action assessment: The Saariselkä agreement. Arch. Complex Environ. Studies. 1992;4:65–69. [Google Scholar]
- Hannas BR, Furr J, Lambright CS, Wilson VS, Foster PMD, Gray LE. Dipentyl phthalate dosing during sexual differentiation disrupts fetal testis function and postnatal development of the male Sprague Dawley rat with greater relative potency than other phthalates. Toxicol. Sci. 2011a;120:184–193. doi: 10.1093/toxsci/kfq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannas BR, Lambright CS, Furr J, Howdeshell KL, Wilson VS, Gray LE., Jr Dose-response assessment of fetal testosterone production and gene expression levels in rat testes following in utero exposure to diethylhexyl phthalate, diisobutyl phthalate, diisoheptyl phthalate, and diisononyl phthalate. Toxicol. Sci. 2011b;123:206–216. doi: 10.1093/toxsci/kfr146. [DOI] [PubMed] [Google Scholar]
- Hotchkiss AK, Parks-Saldutti LG, Ostby JS, Lambright C, Furr J, Vandenbergh JG, Gray LE., Jr. A mixture of the “antiandrogens” linuron and butyl benzyl phthalate alters sexual differentiation of the male rat in a cumulative fashion. Biol. Reprod. 2004;71:1852–1861. doi: 10.1095/biolreprod.104.031674. [DOI] [PubMed] [Google Scholar]
- Howdeshell KL, Rider CV, Wilson VS, Gray LE. Mechanisms of action of phthalate esters, individually and in combination, to induce abnormal reproductive development in male laboratory rats. Environ. Res. 2008;108:168–176. doi: 10.1016/j.envres.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Hu L, Monteiro A, Johnston H, King P, O'Shaughnessy PJ. Expression of Cyp21a1 and Cyp11b1 in the fetal mouse testis. Reproduction. 2007;134:585–591. doi: 10.1530/REP-07-0133. [DOI] [PubMed] [Google Scholar]
- Hurst CH, Waxman DJ. Activation of PPAR alpha and PPAR gamma by environmental phthalate monoesters. Toxicol. Sci. 2003a;74:297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- Hurst CH, Waxman DJ. Trans-activation of PXR by phthalate monoesters. Toxicol. Sci. 2003b;72:176. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- Hushka LJ, Waterman SJ, Keller LH, Trimmer GW, Freeman JJ, Ambroso JL, Nicolich M, McKee RH. Two-generation reproduction studies in rats fed di-isodecyl phthalate. Reprod. Toxicol. 2001;15:153–169. doi: 10.1016/s0890-6238(01)00109-5. [DOI] [PubMed] [Google Scholar]
- Johnson KJ, Hensley JB, Kelso MD, Wallace DG, Gaido KW. Mapping gene expression changes in the fetal rat testis following acute dibutyl phthalate exposure defines a complex temporal cascade of responding cell types. Biol. Reprod. 2007;77:978–989. doi: 10.1095/biolreprod.107.062950. [DOI] [PubMed] [Google Scholar]
- Lahousse SA, Wallace DG, Liu DL, Gaido KW, Johnson KJ. Testicular gene expression profiling following prepubertal rat mono-(2-ethylhexyl) phthalate exposure suggests a common initial genetic response at fetal and prepubertal ages. Toxicol. Sci. 2006;93:369–381. doi: 10.1093/toxsci/kfl049. [DOI] [PubMed] [Google Scholar]
- Lampen A, Zimnik S, Nau H. Teratogenic phthalate esters and metabolites activate the nuclear receptors PPARs and induce differentiation of F9 cells. Toxicol. Appl. Pharmacol. 2003;188:14–23. doi: 10.1016/s0041-008x(03)00014-0. [DOI] [PubMed] [Google Scholar]
- Lehmann KP, Phillips S, Sar M, Foster PM, Gaido KW. Dose-dependent alterations in gene expression and testosterone synthesis in the fetal testes of male rats exposed to di(n-butyl) phthalate. Toxicol. Sci. 2004;81:60–68. doi: 10.1093/toxsci/kfh169. [DOI] [PubMed] [Google Scholar]
- Liu RCM, Hahn C, Hurtt ME. The direct effect of hepatic peroxisome proliferators on rat Leydig cell function in vitro. Fundam. Appl. Toxicol. 1996;30:102–108. doi: 10.1006/faat.1996.0047. [DOI] [PubMed] [Google Scholar]
- Mahood IK, Hallmark N, McKinnell C, Walker M, Fisher JS, Sharpe RM. Abnormal Leydig cell aggregation in the fetal testis of rats exposed to di(n-butyl) phthalate and its possible role in testicular dysgenesis. Endocrinology. 2005;146:613–623. doi: 10.1210/en.2004-0671. [DOI] [PubMed] [Google Scholar]
- Maloney EK, Waxman DJ. trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol. Appl. Pharmacol. 1999;161:209–218. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- McKinnell C, Sharpe RM, Mahood K, Hallmark N, Scott H, Ivell R, Staub C, Jegou B, Haag F, Koch-Nolte F, et al. Expression of insulin-like factor 3 protein in the rat testis during fetal and postnatal development and in relation to cryptorchidism induced by in utero exposure to di(n-butyl) phthalate. Endocrinology. 2005;146:4536–4544. doi: 10.1210/en.2005-0676. [DOI] [PubMed] [Google Scholar]
- Mylchreest E, Cattley RC, Foster PM. Male reproductive tract malformations in rats following gestational and lactational exposure to di(n-butyl) phthalate: An antiandrogenic mechanism? Toxicol. Sci. 1998;43:47–60. doi: 10.1006/toxs.1998.2436. [DOI] [PubMed] [Google Scholar]
- Mylchreest E, Sar M, Cattley RC, Foster PM. Disruption of androgen-regulated male reproductive development by di(n-butyl) phthalate during late gestation in rats is different from flutamide. Toxicol. Appl. Pharmacol. 1999;156:81–95. doi: 10.1006/taap.1999.8643. [DOI] [PubMed] [Google Scholar]
- Mylchreest E, Wallace DG, Cattley RC, Foster PMD. Dose-dependent alterations in androgen-regulated male reproductive development in rats exposed to di(n-butyl) phthalate during late gestation. Toxicol. Sci. 2000;55:143–151. doi: 10.1093/toxsci/55.1.143. [DOI] [PubMed] [Google Scholar]
- National Academy of Sciences. Phthalates and Cumulative Risk Assessment: The Tasks Ahead. 2008. Available at: http://dels.nas.edu/dels/rpt_briefs/phthalates_final.pdf. Accessed May 5, 2010. [PubMed] [Google Scholar]
- Parks LG, Ostby JS, Lambright CR, Abbott BD, Klinefelter GR, Barlow NJ, Gray LE., Jr The plasticizer diethylhexyl phthalate induces malformations by decreasing fetal testosterone synthesis during sexual differentiation in the male rat. Toxicol. Sci. 2000;58:339–349. doi: 10.1093/toxsci/58.2.339. [DOI] [PubMed] [Google Scholar]
- Plummer S, Sharpe RM, Hallmark N, Mahood IK, Elcombe C. Time-dependent and compartment-specific effects of in utero exposure to di(n-butyl) phthalate on gene/protein expression in the fetal rat testis as revealed by transcription profiling and laser capture microdissection. Toxicol. Sci. 2007;97:520–532. doi: 10.1093/toxsci/kfm062. [DOI] [PubMed] [Google Scholar]
- Rider CV, Furr J, Wilson VS, Gray LE., Jr. A mixture of seven antiandrogens induces reproductive malformations in rats. Int. J. Androl. 2008;31:249–262. doi: 10.1111/j.1365-2605.2007.00859.x. [DOI] [PubMed] [Google Scholar]
- Rider CV, Furr JR, Wilson VS, Gray LE. Cumulative effects of in utero administration of mixtures of reproductive toxicants that disrupt common target tissues via diverse mechanisms of toxicity. Int. J. Androl. 2010;33:443–462. doi: 10.1111/j.1365-2605.2009.01049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saillenfait AM, Roudol AC, Gallissot F, Sabate JP. Prenatal developmental toxicity studies of di-n-heptyl and di-n-octyl phthalates in Sprague-Dawley rats. Reprod. Toxicol. 2011;32:268–276. doi: 10.1016/j.reprotox.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Saillenfait AM, Sabate JP, Gallissot F. Diisobutyl phthalate impairs the androgen-dependent reproductive development of the male rat. Reprod. Toxicol. 2008;26:107–115. doi: 10.1016/j.reprotox.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Saillenfait AM, Sabate JP, Gallissot F. Effects of in utero exposure to di-n-hexyl phthalate on the reproductive development of the male rat. Reprod. Toxicol. 2009;28:468–476. doi: 10.1016/j.reprotox.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Scott HM, Hutchison GR, Jobling MS, McKinnell C, Drake AJ, Sharpe RM. Relationship between androgen action in the “male programming window,” fetal Sertoli cell number, and adult testis size in the rat. Endocrinology. 2008;149:5280–5287. doi: 10.1210/en.2008-0413. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Waxman DJ. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicol. Appl. Pharmacol. 2004;199:275–284. doi: 10.1016/j.taap.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Shultz VD, Phillips S, Sar M, Foster PMD, Gaido KW. Altered gene profiles in fetal rat testes after in utero exposure to di(n-butyl) phthalate. Toxicol. Sci. 2001;64:233–242. doi: 10.1093/toxsci/64.2.233. [DOI] [PubMed] [Google Scholar]
- Silva E, Rajapakse N, Kortenkamp A. Something from “nothing”—Eight weak estrogenic chemicals combined at concentrations below NOECs produce significant mixture effects. Environ. Sci. Technol. 2002;36:1751–1756. doi: 10.1021/es0101227. [DOI] [PubMed] [Google Scholar]
- Silva MJ, Barr DB, Reidy JA, Malek NA, Hodge CC, Caudill SP, Brock JW, Needham LL, Calafat AM. Urinary levels of seven phthalate metabolites in the U.S. population from the National Health and Nutrition Examination Survey (NHANES) 1999-2000. Environ. Health Perspect. 2004;112:331–338. doi: 10.1289/ehp.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CJ, Ross SM, Gaido KW. Di(n-butyl) phthalate impairs cholesterol transport and steroidogenesis in the fetal rat testis through a rapid and reversible mechanism. Endocrinology. 2004;145:1227–1237. doi: 10.1210/en.2003-1475. [DOI] [PubMed] [Google Scholar]
- Thompson CJ, Ross SM, Hensley J, Liu K, Heinze SC, Young SS, Gaido KW. Differential steroidogenic gene expression in the fetal adrenal gland versus the testis and rapid and dynamic response of the fetal testis to di(n-butyl) phthalate. Biol. Reprod. 2005;73:908–917. doi: 10.1095/biolreprod.105.042382. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val P, Jeays-Ward K, Swain A. Identification of a novel population of adrenal-like cells in the mammalian testis. Dev. Biol. 2006;299:250–256. doi: 10.1016/j.ydbio.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Ward JM, Peters JM, Perella CM, Gonzalez FJ. Receptor and nonreceptor-mediated organ-specific toxicity of di(2-ethylhexyl)phthalate (DEHP) in peroxisome proliferator-activated receptor alpha-null mice. Toxicol. Pathol. 1998;26:240–246. doi: 10.1177/019262339802600208. [DOI] [PubMed] [Google Scholar]
- Welsh M, Saunders PT, Fisken M, Scott HM, Hutchison GR, Smith LB, Sharpe RM. Identification in rats of a programming window for reproductive tract masculinization, disruption of which leads to hypospadias and cryptorchidism. J. Clin. Invest. 2008;118:1479–1490. doi: 10.1172/JCI34241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson VS, Lambright C, Furr J, Ostby J, Wood C, Held G, Gray LE., Jr Phthalate ester-induced gubernacular lesions are associated with reduced insl3 gene expression in the fetal rat testis. Toxicol. Lett. 2004;146:207–215. doi: 10.1016/j.toxlet.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Wittassek M, Wiesmuller GA, Koch HM, Eckard R, Dobler L, Muller J, Angerer J, Schluter C. Internal phthalate exposure over the last two decades—A retrospective human biomonitoring study. Int. J. Hyg. Environ. Health. 2007;210:319–333. doi: 10.1016/j.ijheh.2007.01.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.