

Abstract
Type III effectors from phytopathogenic bacteria exhibit a high degree of functional redundancy, hampering the evaluation of their precise contribution to pathogenicity. This is illustrated by the GALA type III effectors from Ralstonia solanacearum, which have been shown to be collectively, but not individually, required for disease on Arabidopsis thaliana and tomato. We investigated evolution, redundancy and diversification of this family in order to understand the individual contribution of the GALA effectors to pathogenicity.
From sequences available, we reconstructed GALA phylogeny and performed selection studies. We then focused on the GALAs from the reference strain GMI1000 to examine their ability to suppress plant defense responses and contribution to pathogenicity on three different host plants: A. thaliana, tomato (Lycopersicum esculentum) and eggplant (Solanum melongena).
The GALA family is well conserved within R. solanacearum species. Patterns of selection detected on some GALA family members, together with experimental results, show that GALAs underwent functional diversification.
We conclude that functional divergence of the GALA family likely accounts for its remarkable conservation during R. solanacearum evolution and could contribute to R. solanacearum’s adaptation on several host plants.
Keywords: evolution, functional divergence, host range, Ralstonia solanacearum, redundancy, type III effector
Introduction
Ralstonia solanacearum is a soilborne plant pathogenic bacterium which is able to cause ‘bacterial wilt’ disease on > 200 plant species (Denny, 2006). Soil living bacteria enter the root, colonize xylem vessels and cause wilting of the plant. R. solanacearum relies on a type III secretion system (T3SS) and a large repertoire of effectors (T3Es), 74 in the reference strain GMI1000 (Poueymiro & Genin, 2009), to promote pathogenicity.
Although it is frequent among phytopathogenic bacteria that single T3Es are individually dispensable for pathogenicity (Cunnac et al., 2004b; Castaneda et al., 2005; Kvitko et al., 2009), only a few examples of functional redundancy were documented. The best-studied cases include HopM1 and AvrE, located on the CEL locus in Pseudomonas syringae pv. tomato (Pst). Both are able to suppress callose deposition induced after perception of the bacteria by the plant (DebRoy et al., 2004). The double mutant ΔAvrEΔHopM1 is severely altered in its ability to grow in inoculated plants, while single mutants are not (Badel et al., 2006). Similar functional redundancy was described for AvrPto1 (previously AvrPto) and HopAB2 (previously AvrPtoB) (Lin & Martin, 2005), which target the same receptor-like kinase FLS2 (Gohre et al., 2008; Xiang et al., 2008). Genetic redundancy, through multigene families, can also contribute to functional redundancy. In bacteria, both gene duplication events and lateral gene transfers (LGTs) were shown to drive the appearance of multigene families (Lerat et al., 2005; Treangen & Rocha, 2011). A small number of T3Es are present in a given strain as multimember families. These include PopP (three members), AWR (five), SKWP (six), HLK (three) and GALA (seven) in R. solanacearum and TALs, with up to 28 members present in one Xanthomonas sp. strain (Poueymiro & Genin, 2009; Scholze & Boch, 2011). Large effector gene families appear to be more common among the predicted effectors from plant pathogenic fungi and oomycetes (Haas et al., 2009; Stergiopoulos & de Wit, 2009).
As originally stated by Ohno (1970) and later reviewed (Hahn, 2009; Innan & Kondrashov, 2010), when duplicates are retained during evolution, there are three major possible outcomes of gene duplication: neofunctionalization, the evolution of a new function through relaxed selection pressure in one of the two copies; subfunctionalization, the division of ancestral functions among duplicates; and redundancy, the conservation of all functions in both duplicates. The latter is generally explained by the benefit of increased dosage. In functional divergence models (neo- and subfunctionalization), the presence of both copies becomes indispensable because of their new specific functions. To our knowledge, the contribution of these different models to the evolution of effector gene families has never been addressed in plant pathogens.
GALAs are a family of seven effectors in GMI1000 which were initially identified based on their homology with plant F-box proteins (Angot et al., 2006). In eukaryotes, F-box proteins are components of E3-ubiquitin ligases complexes called SCF (SKP1–Cullin1–Fbox) (Hua & Vierstra, 2011). These enzymes target proteins for ubiquitination, leading either to their degradation by the proteasome or to modification of their activity by ubiquitination. The F-box domain mediates the interaction with the SKP1 subunit of the SCF complexes. A protein–protein interaction domain (leucin-rich repeats (LRRs) in the GALAs) enables specific interaction with proteins targeted for ubiquitination. Since bacteria do not possess a proteasome system, it is thought that GALAs could enable R. solanacearum to manipulate their host ubiquitin-proteasome system. GALA7 is a host-specificity factor on Medicago truncatula, and the F-box domain is required for its function (Angot et al., 2006). On other host plants (A. thaliana and tomato), none of the seven single gala mutants are affected in their pathogenicity (Cunnac et al., 2004b), whereas the septuple mutant is less pathogenic (Angot et al., 2006). This suggests that, on these plants, at least two different GALAs are redundant or have overlapping functions. In this work, we first show that the GALA family is well conserved within R. solanacearum species, indicating that the maintenance of GALAs could be important for bacterial fitness. Then, by comparing synonymous vs nonsynonymous substitution rates in GALA coding sequences, we show that GALAs are exposed to different patterns of selection. Finally, by combining sequence analysis with experimental data, we provide evidence of functional diversification among GALAs. Differential contribution of individual GALAs to pathogenicity on several hosts is likely to account for the conservation of this gene family.
Materials and Methods
Bacterial strains, growth conditions and plant material
The bacterial strains and plasmids used for this study are described in Supporting Information Table S1. Escherichia coli cells were grown in Luria-Bertani (LB) medium at 37°C. P. syringae was grown in King's B (KB) medium at 28°C. R. solanacearum was grown in complete medium B at 28°C (Plener et al., 2010). Antibiotics were used at the following concentrations (mg l−1): rifampicin, 50; spectinomycin, 40; gentamycin, 10; and kanamycin, 50. Plants used in this study were Arabidopsis thaliana ecotype Col0, tomato (Lycopersicum esculentum cv Marmande VR) and eggplant (Solanum melongena cv Zebrina).
DNA manipulation and genetic constructions
Standard methods were used unless otherwise stated. GMI1000 GALA open reading frames (ORFs) were cloned with stop codons using the Gateway system (Invitrogen). GALA4, GALA5, GALA6 and GALA7 were amplified by PCR with Phusion DNA polymerase (Finnzymes, Vantaa, Finland) and BP, gateway-cloned into pDON207 (Invitrogen). The first amplification was performed with specific primers, followed by a second amplification with adaptors oNP291/oNP292 to insert attB1 and attB2 sites. Despite several attempts, GALA2 could not be cloned, probably as a result of intramolecular recombinations. Stop codons were inserted into GALA1 and GALA3 pENTRY vectors (Angot et al., 2006) by site-directed mutagenesis, using the QuickChange II XL site-directed mutagenesis kit (Agilent, Santa Clara, CA, USA). GMI1000 GALA ORFs were recombined into pEDV6 destination vector (Sohn et al., 2007) using LRII Clonase (Invitrogen). R. solanacearum gala mutants were generated by natural transformation using genetic constructions described previously (Cunnac et al., 2004b; Angot et al., 2006). All oligonucleotides used in this study are listed in Table S2. Considering that plasmid-based expression only yields imperfect complementation of mutants in R. solanacearum (Angot et al., 2006), we decided not to generate plasmid complementation constructs for the different gala mutants.
Phylogeny, synteny and promoter analysis
The GALA sequence dataset was collected from iANT (http://iant.toulouse.inra.fr/bacteria/annotation/cgi/ralso.cgi) and MaGe (http://www.genoscope.cns.fr/agc/microscope/mage/) (Vallenet et al., 2006) web resources. The strains used in this work and their associated phylotype (Fegan & Prior, 2005) are GMI1000 (phylotype I) (Salanoubat et al., 2002), RS1000 (phylotype I) (Mukaihara et al., 2004), IPO1609 (phylotype II) (Guidot et al., 2009), MolK2 (phylotype II) (C. Boucher & S. Genin, unpublished), CFBP2957 (phylotype II), CMR15 (phylotype III) and PSI07 (phylotype IV) (Remenant et al., 2010).
GALA sequences were aligned using the PRANK program, which implements the evolution-aware alignment algorithm (Loytynoja & Goldman, 2008), performing well with indel-rich data, as is our case. Phylogenies of all GALAs and of individual GALA genes were reconstructed using fast maximum likelihood (ML) heuristic search under model LG (Le & Gascuel, 2008) with Γ-rate variation among sites (Yang, 1994), as implemented in PhyMLv3.0 (Guindon et al., 2010). Branch supports were estimated using the new aBayes method, which is fast, accurate and has performance comparable with the Bayesian method (Anisimova et al., 2011).
The synteny analysis was based on data retrieved from MaGe website. The 500 nucleotides upstream of each GALA start codon were retrieved and submitted to an automatic search for the presence of hrpII box (Cunnac et al., 2004a).
Comparative genomic hybridization
Comparative genomic hybridization of 60 strains spanning the four R. solanacearum phylotypes was performed on a microarray under conditions described previously (Guidot et al., 2009). On this microarray, each GALA gene was represented by four to five specific 70-nucleotide-long probes (listed in Table S2). A GALA gene was considered absent in the tested strain if the base-2 logarithm of the ratio of the normalized hybridization signal with the tested strain over the normalized hybridization signal with the control DNA was lower than the cutoff value of −0.4 for every probe representative of the considered GALA gene. This cutoff value was increased compared with previous reports (Guidot et al., 2009) in order to efficiently remove false-positive signals.
Analyses of selection pressures
Selection pressures on GALA were analyzed using Markov models of codon substitution, and likelihood ratio tests (LRTs) were used to detect positive selection (for review see Anisimova & Kosiol, 2009). In these models, selection pressure on the protein is measured by ω = dN/dS– the ratio of nonsynonymous to synonymous substitution rates (Bielawski & Yang, 2001). Values of ω > 1 suggest an effect of positive selection on the protein, whereas values of ω < 1 suggest purifying selection of varying degree. For each orthologous GALA protein-coding alignment, the likelihood was optimized assuming inferred gene phylogenies. To ensure the robustness of our inferences, we used several models: one-ratio model M0, and site models M3, M1a, M2a, M7, M8 (Yang et al., 2000) and M8a (Swanson et al., 2003). Site models allow selection pressure to vary among sites and are used here to evaluate relative functional importance of different positions in the protein. Models M1a, M7 and M8a may represent the null hypothesis of no positive selection on the dataset, and were compared using LRTs to models M2a, M8 and M8, respectively, to test whether data supported the idea that a significant fraction of sites were under positive selection. The Bayes empirical Bayes (BEB) approach (Yang et al., 2005) was used for classifying sites in each alignment into site categories according to estimated ω ratios. The posterior probabilities (PPs) of a site to belong to a particular category were used to evaluate the support of BEB classification. A gene was classified to be under positive selection if at least two LRTs suggested the presence of positive selection.
Testing for recombination
For each GALA alignment package LDhat v2.1 (http://www.stats.ox.ac.uk/~mcvean/LDhat) was used to estimate population recombination rates using the approximate-likelihood coalescent method (Hudson, 2001), and performed recombination tests using the likelihood permutation test (McVean et al., 2002). In addition, Tajima's D values (Tajima, 1989) were computed by LDhat. In a classic neutrality test, the null hypothesis of neutral evolution corresponds to D = 0. Departures from neutrality may be indicated by significant deviations of D from 0. For example, D > 0 may be a result of positive (balancing or diversifying) selection or shrinking populations. Values |D| > 2 were considered as significant (Tajima, 1989). As standard neutrality tests assume a very simple population model, decoupling the effects of demography and selection is typically nontrivial (Nielsen, 2001).
Finally, for each GALA paralog we also estimated selection in the presence of recombination using an approximation to a population genetics coalescent (Wilson & McVean, 2006), as implemented in OmegaMap (http://www.danielwilson.me.uk). In this approach, inference is performed on both parameters simultaneously using the Bayesian method with reversible jump Markov chain Monte Carlo sampling (with 106 generations and discarding 2 × 105 burn-in). Variable models for both selection and recombination parameters were assumed.
Callose deposition assay
Pseudomonas syringae DC3000 overnight cultures were infiltrated in A. thaliana leaves at a concentration of 108 cfu ml−1. For each strain, three leaves were infiltrated on three plants. Leaves were harvested after 24 h and callose was stained with aniline blue as described (Adam & Somerville, 1996). Leaf samples were mounted in 50% glycerol and observed with a Zeiss Axiophot II epifluorescence microscope (365 nm BP excitation filter, 395 nm chromatic beam splitter, 397 nm LP emission filter). Between five and 15 independent images (c. 1 mm2) were taken for each strain and subsequently analyzed with ImageJ (Rasband, 1997). A two-tailed Mann–Whitney statistical test was performed with Prism, version 5.00 (GraphPad Software, La Jolla, CA, USA).
Ralstonia solanacearum pathogenicity assays, statistical analysis
Tomato and eggplant pathogenicity assays were performed by watering c. 5- to 6-wk-old plants with 50 ml of a bacterial suspension containing 107 cells ml–1. A. thaliana plants were inoculated with a bacterial suspension at the same concentration, as described by Deslandes et al. (1998). The plants were incubated in a growth chamber at 28°C for tomato and eggplant (14 h light : 10 h dark) or 16 h at 27°C (light) : 8 h at 26°C (dark) for A. thaliana. Disease development was scored daily, using a macroscopic scale describing the observed wilting: 1 for 25% of the leaves wilted; 2 for 50%; 3 for 75% and 4 for complete wilting. For subsequent analysis the data was transformed into a binary index: 0 for < 50% of wilted leaves and 1 for more or equal to 50% wilted leaves. To compare the disease development of two given strains, we used the Kaplan–Meier survival analysis (Bland & Altman, 1998) with the Gehan–Breslow–Wilcoxon method to compute the P-value to test the null hypothesis of identical survival experience of the two tested strains. A P-value smaller than 0.05 was considered significant. Statistical analyses were done with Prism version 5.00 (GraphPad Software).
Results
The GALA family is conserved in the R. solanacearum species complex
The R. solanacearum species complex exhibits a high degree of genetic diversity and is structured in four monophyletic groups, termed phylotypes. These groups correspond to specific geographic distributions (Fegan & Prior, 2005). Full genome sequences from seven strains, representative of the whole species diversity, are now available. These include the reference strain GMI1000 (Salanoubat et al., 2002) from phylotype I, draft sequences of phylotype II strains IPO1609 (Guidot et al., 2009) and MolK2 (C. Boucher & S. Genin, unpublished) and the newly sequenced strains CFBP2957, CMR15 and PSI07 from phylotypes II, III and IV, respectively (Remenant et al., 2010). GALA sequences were retrieved from these complete genomes (see Table S3 for accession numbers). A genome sequence from strain UW551 is also available (Gabriel et al., 2006) but we did not use it since its GALA sequences are exactly identical to IPO1609. Moreover, seven GALAs from RS1000, a phylotype I strain (Mukaihara et al., 2004), were included in the analysis. Between six (in MolK2, IPO1609) and nine (in PSI07) GALAs were detected in these strains. Using this dataset, we reconstructed the GALA phylogeny in the R. solanacearum species complex (Fig. 1). GALAs cluster by orthologous groups, indicating that the ancestral strain most likely had at least seven GALAs (namely GALA1, 2, 3, 4, 5, 6, 7), which subsequently evolved independently within each lineage. Two subclades of paralogs could be defined with high support: GALA2, 6, 7 and GALA1, 3, 4, 5. Using a large (60) and diverse set of R. solanacearum strains, we performed comparative genomic hybridization (CGH) on a dedicated microarray harboring T3Es probes designed on the sequenced GMI1000, IPO1609 and Molk2 strains (Guidot et al., 2009). We were able to detect the presence of GALA4 in all strains tested, the others, GALA2, 3, 5, 6 and 7, being present in most strains with some exceptions (Table S4). The four GALA1 probes do not hybridize with DNA extracted from any of the 35 phylotype II strains or any of the 13 phylotype III strains, indicating that GALA1 is either absent or highly divergent in these phylotypes. Based on the fact that GALA1 is present in CMR15, the sequenced phylotype III strain, and absent from the sequenced phylotype II strains IPO1609, Molk2 and CFBP2957, we hypothesize that GALA1 is indeed absent in phylotype II and present but divergent in the other phylotype III strains.
Fig. 1.
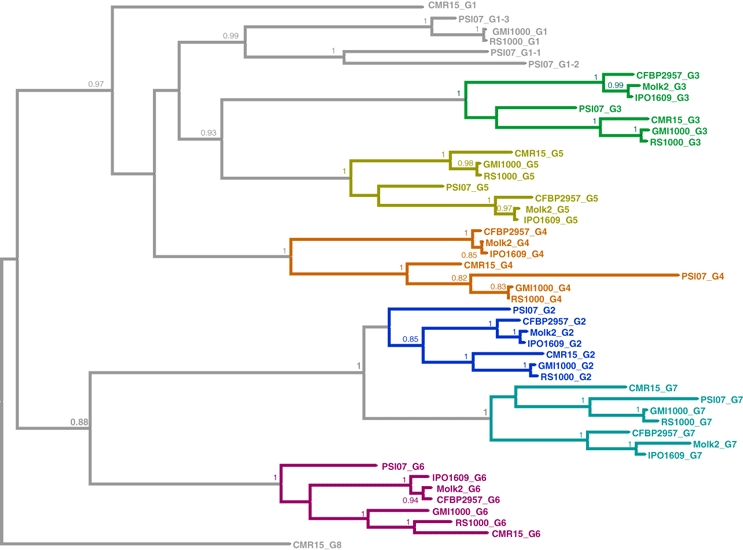
GALA phylogeny. Unrooted maximum-likelihood phylogenetic reconstruction of GALAs from strains GMI1000, RS1000, CMR15, IPO1609, Molk2, CFBP2957 and PSI07. Branch support values (aBayes) are shown only for clades with strong support (> 0.9).
Since GALAs are present in all sequenced strains, we scrutinized the loci of these different GALAs in the sequenced strains. GALA4 and GALA5 are in operon, while GALA6 and GALA7 are in direct tandem repeat. Together with GALA2, these three loci are well conserved in all sequenced strains (Fig. S1), strengthening the hypothesis that those genes were already present in the common ancestor of these strains. The GALA3 loci are more variable, with many ‘inserted genes’ in the phylotype II and IV strains compared with GMI1000. The strain PSI07 is somewhat peculiar since it possesses three copies of GALA1 (GALA1-1, GALA1-2 and GALA1-3) and GALA2 is found in the close vicinity of GALA1-3. Furthermore, only the 100 last amino acids of GALA4 from this strain are conserved and GALA1-3 is truncated after the first 200 amino acids.
In GMI1000, all GALAs but GALA1 are regulated by HrpB, the general regulator of T3SS and T3Es (Cunnac et al., 2004a). This regulation is associated with the presence of the hrpII box domain in the promoters of GALA2, GALA3, GALA4, GALA6 and GALA7 (Cunnac et al., 2004a). Promoter analysis of GALA genes from all sequenced strains indicates that GALAs (except for GALA1 and GALA5, which is in operon with GALA4) have a conserved hrpII box domain (Table S3). Unexpectedly, GALA1-1PSI07 also has a hrpII box sequence in its promoter. The distance between the position of the hrpII box and the start codon is well conserved in GMI1000 GALA promoters (c. 100 bp) but is greater, c. 400 bp, in GALA4IPO1609, GALA4CMR15, GALA6CFBP2957 and GALA3PSI07. This finding suggests that HrpB-regulation of most GALAs may be conserved in all R. solanacearum strains.
Differential selection pressures in GALA paralogs
We then investigated the patterns of selection acting on the GALAs. Positive selection was detected in GALA3, 4, 6 and 7 (Table 1) using at least two likelihood ratio tests (LRTs) for positive selection (see the Materials and Methods section). By contrast, no evidence of positive selection was found on GALA1, 2 and 5 (Table 1). The largest fraction of strictly conserved sites (57%) was found in GALA5, while the largest fraction of neutrally evolving sites (63%) was found in GALA2 (Table 1). GALA7 experienced most sites under positive selection (8%).
Table 1.
Selection pressures in GALA paralogs
| P-values for LRTs comparing codon modelsa | Proportions of sites in different selection regimesb | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GALA copy | Number of strains | Alignment length (nt) | Population recombination rate, Ner (PLPT)a | Tajima's Da | M0 vs M3 | M1a vs M2a | M7 vs M8 | M8a vs M8 | Strict negative (%) (ω < 0.15) | Relaxed negative (%) (0.15 < ω < 0.9) | Neutral (%) (0.9 < ω < 1) | Positive (%) (ω = 1) |
| GALA1 | 6 | 2046 | 0.00 (1.00) | 2.1 | 0.000 | 1.000 | 0.045 | 0.256 | 42 (ω = 0.08) | 25 (ω = 0.31) | 33 | 0 |
| GALA2 | 7 | 3237 | 0.00 (1.00) | 1.7 | 0.000 | 1.000 | 0.022 | 0.187 | 32 (ω = 0.04) | 4.5 (ω = 0.25) | 63 | 0 |
| GALA3c | 9 | 1971 | 12.25 (0.003) | 4.2 | 0.000 | 0.023 | 0.001 | 0.000 | 28 (ω = 0.05) | 57 (ω = 0.53) | 10 | 5 (ω = 3.28) |
| GALA4c | 8 | 1677 | 14.29 (0.001) | 3.5 | 0.000 | 0.040 | 0.002 | 0.002 | 39 (ω = 0.06) | 58 (ω = 0.49) | 0 | 3 (ω = 5.90) |
| GALA5 | 8 | 1683 | 0.00 (0.000) | 2.1 | 0.000 | 1.000 | 0.170 | 0.457 | 57 (ω = 0.06) | 19 (ω = 0.24) | 24 | 0 |
| GALA6c | 9 | 1866 | 6.12 (0.000) | 4.2 | 0.000 | 0.653 | 0.035 | 0.048 | 48 (ω = 0.06) | 48 (ω = 0.54) | 0 | 4 (ω = 2.58) |
| GALA7c | 9 | 2052 | 16.33 (0.000) | 4.3 | 0.000 | 0.000 | 0.000 | 0.000 | 46 (ω = 0.05) | 28 (ω = 0.51) | 18 | 8 (ω = 3.7) |
Significant values for selection, recombination and neutrality tests are shown in bold.
Estimates of selection regimes are reported according to the model M8 if the likelihood ratio test (LRT) comparing M8a and M8 was significant. Otherwise, selection regimes are reported according to model M8a. For strict and relaxed negative selection, the average ω -value over respective selection classes (with either ω < 0.15 or 0.15 < ω < 0.9) is shown. In each of four selection categories shown, the highest frequency value of sites is underlined.
GALAs where positive selection was detected using at least two LRTs for positive selection. Note that the LRT comparing M0 and M3 is not a test for positive selection but for variability of selection pressure among sites.
Given the inferred phylogeny of the GALAs, it is interesting to note that both GALA subclades (GALA2, 6, 7 and GALA1, 3, 4, 5) evolve in a similar fashion. Namely, two GALAs in each subclade (GALA6, 7 and GALA3, 4) are affected by positive selection, whereas at least one GALA in each clade (GALA2 and GALA1, 5) is mostly conserved. Of particular interest are the closely related GALA3 and GALA5, which originated from a recent duplication event, as shown in Fig. 1. GALA3 contains sites under positive selection and is generally evolving under relaxed purifying selection, but in GALA5 the majority of sites are very conserved and there are no sites under positive selection (Table 1). Similarly, the closely related GALA2 and GALA7 have very different patterns of selection: while GALA7 has a relatively large fraction of sites (8%) affected by strong positive selection, GALA2 mostly evolves neutrally (63%) and has no site under positive selection. In addition, compared with other GALA genes, GALA2 and GALA5 have a much larger fraction of sites (22 and 23%, respectively) that are very strictly conserved, with the ML estimates of ω < 0.1 and the posterior estimate of ω < 0.1. By contrast, GALA3, GALA4, GALA6 have none and GALA7 has only very few such sites (< 1%). Overall, such striking differences in patterns of selection between GALAs are indicative of functional divergence.
Interplay between selection and recombination
The presence of a high degree of recombination can hamper LRTs for positive diversifying selection, leading to elevated rates of false positives (Anisimova et al., 2003). However, inference of recombination may also be affected by selection forces (Reed & Tishkoff, 2006; O'Reilly et al., 2008).
In an attempt to disentangle the signatures of selection and recombination in our data, we performed tests for recombination and simultaneous inference of selection and recombination parameters. The population recombination rate Ner was estimated to be > 0 for GALA3, 4, 6, and 7, with significant likelihood permutation tests (PLPT < 0.05; Table 1). It is exactly for these GALA paralogs that strong positive selection was detected. However, our estimates of population recombination rates appear to be very low, and would be unlikely to affect our inferences of selection (Anisimova et al., 2003). Yet, judging by estimates from Table 1 and the occasional deviations from R. solanacearum phylogeny within each GALA clade (Fig. 1), it is more likely that a combination of both recombination and different types of selection (diversifying and directional) may be shaping GALA proteins. Indeed, inference of selection in the presence of recombination using OmegaMap confirmed that both positive selection and a low degree of recombination operate on GALA proteins. The amount of selection inferred by our codon models was consistent with estimates from OmegaMap, and with Tajima's D (Table 1).
GALA4, but not GALA5, interferes with callose deposition
An increasing number of studies show that many, if not most, T3Es are able to suppress PAMP (Pathogen-associated molecular pattern)- or Effector-Trigerred Immunity (PTI and ETI, respectively) (Hann et al., 2010; Block & Alfano, 2011). We could not detect any effect of GALA expression on AvrRpm1-triggered immunity (result not shown). In order to test if GALAs are able to interfere with PTI, we took advantage of the pEDV6 vector (Sohn et al., 2007) to express GALAs from P. syringae DC3000 strain ΔCEL (Alfano et al., 2000). This strain, with deleted HopM1 and AvrE T3Es, is no longer able to suppress callose papillae at the cell wall, a basic defence mechanism elicited during PTI. The pEDV6 vector enables expression of GALAs fused to the N-terminus of the P. syringae effector AvrRPS4 (Sohn et al., 2007). This N-terminal extension allows effective translocation of the chimeric T3E into plant cells and later cleavage to release the effective full-length GALA effectors in the plant cell. We monitored callose deposition in A. thaliana leaves after infiltration with P. syringae strain ΔCEL expressing each GALA. P. syringae-dependent delivery of GALA1, GALA3, GALA6 and GALA7 leads to a weak decrease in callose deposition but was not statistically significant (data not shown). P. syringae-dependent delivery of GALA4 reproducibly decreased the median number of callose spots per field of view, whereas this effect was not observed with GALA5 (Fig. 2). This latter observation is not because of the absence of GALA5 translocation since we could detect the cleaved full-length GALA5 by western blot (Sohn et al., 2007) after infiltration into A. thaliana leaves (data not shown). Surprisingly, the expression of GALA4 in P. syringae strain ΔCEL did not increase bacterial multiplication in A. thaliana leaves (data not shown), as is usually the case with PTI-suppressing effectors (Nomura et al., 2006; Sohn et al., 2007). The fact that callose suppression mediated by GALA4 is weaker than observed in the aforementioned studies may explain this result.
Fig. 2.
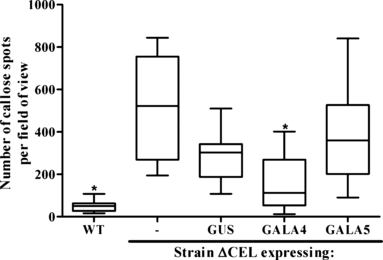
GALA4 but not GALA5 can interfere with callose deposition. Arabidopsis thaliana leaves were inoculated with Pseudomonas syringae strains and stained for callose 24 h after infection. A box plot diagram of papillae number from at least five independent fields of view is shown for each strain. The horizontal black bar is the median and the boxes indicate lower and upper quartiles. This experiment was repeated four times with similar results. *, median number of callose spots per field of view in the tested strain is statistically different from the strain Pst DC3000 ΔCEL (Mann–Whitney test, P < 0.05). WT, wild-type.
Differential GALA requirement for pathogenicity on A. thaliana, tomato and eggplant
We previously showed that GALAs are collectively required for pathogenicity on tomato and A. thaliana, and that GALA7 is a host-specificity factor on M. truncatula (Angot et al., 2006). In order to identify the contribution of each GALA to the pathogenicity of R. solanacearum GMI1000, we generated strains carrying various combinations of GALA gene disruptions and assayed them for their pathogenicity on A. thaliana, tomato and eggplant. We identified strain GRS460 (gala2, 3, 6, 7) as phenocopying the septuple mutant GRS447 on A. thaliana and tomato (Fig. 3a,b). This strain has an intermediate phenotype on eggplant, significantly different from both GMI1000 and GRS447 (Fig. 3c). To identify which of GALA2, 3, 6 or 7 were required for pathogenicity on A. thaliana and tomato, we generated the four triple mutants, GRS536 (gala 2, 3, 7), GRS537 (gala 2, 3, 6), GRS538 (gala 3, 6, 7) and GRS539 (gala 2, 6, 7). On A. thaliana, these four triple mutants have a wild-type phenotype, indicating that GALA2, 3, 6 and 7 are all functional and redundant on A. thaliana (Fig. 4a,b,c). Indeed, the presence of any of them, in conjunction with the still present GALA1, 4, and 5, is sufficient to promote pathogenicity at a wild-type level. On tomato, GRS536 (gala2, 3, 7) phenocopies GRS460, indicating that GALA6 has no significant virulence effect in a GALA1, 4, 5 background (Fig. 4d). Conversely, GRS537 (gala2, 3, 6) and GRS539 (gala2, 6, 7) have a wild-type phenotype, indicating that GALA7 and GALA3 are functional on tomato (Fig. 4d,e). The phenotype of strain GRS538 (gala3, 6, 7) is intermediate and disease kinetics induced by this strain is not statistically different from either GRS460 or GMI1000 (Fig. 4d). GALA2 may therefore have a weak virulence effect on tomato, not sufficient to restore full pathogenicity in a GALA1, 4, 5 background.
Fig. 3.
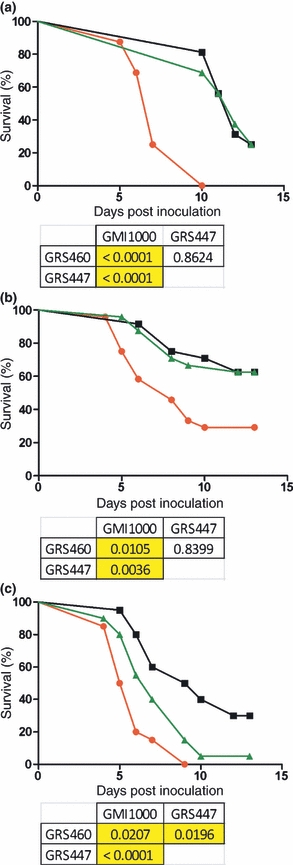
gala2, 3, 6, 7 mutant (strain GRS460, triangles) phenocopies the septuple gala mutant (strain GRS447, squares) on Arabidopsis thaliana and Lycopersicon esculentum (tomato) but not on Solanum melongena (eggplant). Kaplan–Meier survival analysis of A. thaliana (a), tomato (b) and eggplant (c) plants inoculated with Ralstonia solanacearum. GMI1000, wild-type, circles. Each strain was inoculated on 16 A. thaliana plants (a), 24 tomato plants (b) and 20 eggplant plants (c). Correspondence between strains and color code is conserved in the three graphs. P-values from Gehan–Breslow–Wilcoxon tests are associated with each graph. Yellow boxes indicate a P-value of <0.05. These experiments were performed at three times with similar results.
Fig. 4.
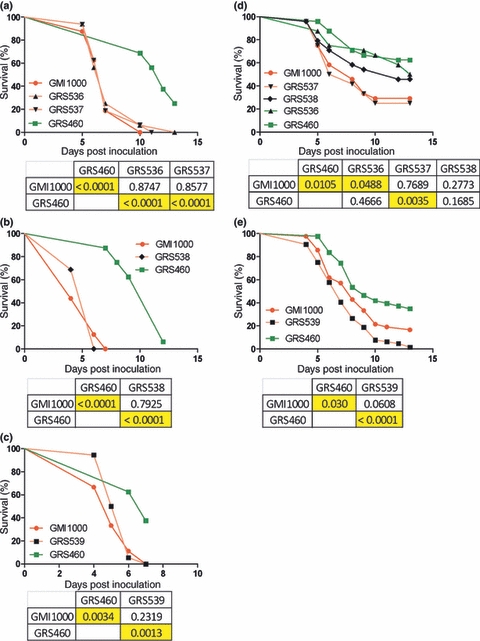
Pathogenicity assays of Ralstonia solanacearum triple gala mutants on Arabidopsis thaliana and Lycopersicon esculentum (tomato). Kaplan–Meier survival analysis of A. thaliana (a, b, c) and tomato (d, e) plants inoculated with R. solanacearum mutants. Genotypes of the tested strains are the following: GMI1000, wild-type; GRS536, gala2, 3, 7; GRS537, gala2, 3, 6; GRS538, gala3, 6, 7; GRS539, gala2, 6, 7; GRS460, gala2, 3, 6, 7. Each strain was inoculated on 16 A. thaliana plants (a, b, c) and 24 tomato plants (d, e). P-values from Gehan–Breslow–Wilcoxon tests are associated with each graph. Yellow boxes indicate a P-value of <0.05. These experiments were performed three times with similar results.
Altogether, our data show that different subsets of GALAs promote disease on different hosts, providing additional evidence of functional diversification of the GALA family.
Discussion
Evolution of type III effectors is shaped by strong selection pressures, either to escape recognition by the plant immune system or to gain efficiency to promote bacterial multiplication within one or several host plants. T3Es are exchanged between strains by LGT at relatively high frequency or can adapt to optimize their function via pathoadaptation (Ma & Guttman, 2008; Arnold & Jackson, 2011). Those two mechanisms are thought to be responsible for much of the variation observed in T3Es complements from phytopathogenic bacteria, in terms of both presence/absence and allelic variations. Here we describe an unusual fate for T3Es genes, that is, creation of a multigene family via several rounds of duplications and subsequent retention of six to nine family members across the R. solanacearum species complex.
Since very few bacterial species have F-box proteins (Angot et al., 2007) and considering that GALA2, 3, 4, 5, 6 and 7 have a common ancestor, it is very likely that those GALAs are of monophyletic origin, as suggested previously (Kajava et al., 2008). The more variable presence of GALA1 could be explained by additional LGT events or by multiple independent losses. This could also apply to GALA8, a newly identified GALA in strain CMR15 (Remenant et al., 2010).
The inferred R. solanacearum phylogeny and good synteny conservation support vertical inheritance among GALA paralogs. From our phylogeny, synteny and CGH analyses, we conclude that at least seven GALA genes were already present in the ancestral strain and have subsequently evolved within R. solanacearum genome for a long time. The GALA T3E family, as a whole, can therefore be considered as a ‘core’ pathogenicity determinant in the R. solanacearum species complex. Conversely, the family of TAL T3Es expanded greatly in Xanthomonas sp. (up to 28 members per strain), but only in some lineages (Scholze & Boch, 2011). This suggests a more plant-specific requirement of these T3Es as opposed to the seemingly ubiquitous use of the GALAs.
In order to understand which selection forces were responsible for GALA conservation, we conducted statistical sequence analyses followed by functional description of these type III effectors. Our bioinformatics analyses suggest that both recombination and selection operate on GALA proteins. A low degree of recombination appears to be coupled with strong positive selection on GALA 3, 4, 6, and 7. Neither selection nor recombination can be detected on GALA 1, 2, and 5.
Two patterns of selection can be described. GALA3, GALA4, GALA6 and GALA7 show signs of diversifying selection while GALA1, GALA2 and GALA5 do not. The differences of selection pressures between GALAs are strongly indicative of a functional divergence evolutionary scenario. We therefore performed additional experiments to test this hypothesis. GALA4 significantly decreases callose deposition elicited by P. syringae DC3000 strain ΔCEL, while the closely related GALA5 did not. Putative E3-ubiquitine ligase function of GALA proteins is in part dependent on their ability to interact with SKP1-like proteins, which has previously been characterized (Angot et al., 2006). GALA4 is unable to interact, in yeast-two-hybrid, with any member of the Arabidopsis SKP1-like (ASK) family, probably because of a variation in a conserved residue in the F-box domain (Angot et al., 2006). GMI1000 GALA4’s function is therefore probably independent of any E3-ubiquitine ligase activity, as has already been reported for other F-box proteins (Galan et al., 2001). Other GALAs exhibited different interaction specificities for ASK and MSK (Medicago SKP1-like) proteins, in a way reminiscent of plant F-box proteins (Risseeuw et al., 2003; Angot et al., 2006). Although the significance of these interaction differences is not known, this suggests variations within F-box domains of ASK-binding GALAs. However, these variations are not thought to affect the general ability of the GALAs to be part of SCF complexes, because GALA1, 3, 5, 6 and 7 are all able to interact with some ASKs/MSKs. Finally, pathogenicity assays with gala polymutants enabled us to uncover plant-specific functions for some GALAs. Contrary to the situation on A. thaliana, GALA3 and GALA7 are not redundant with GALA6 on tomato. The presence of GALA1, 4 and 5 is sufficient to increase pathogenicity on eggplant, but not on tomato or A. thaliana. Differential GALA requirement on tested host plants indicates that GALAs contribute to pathogenesis on these hosts. This is further confirmed by GALA7 which is required to extend R. solanacearum’s host range to M. truncatula (Angot et al., 2006). Interestingly, among the positively selected sites of GALA7, 66% are in the LRR region and 92% of the sites present in an LRR are exposed to the solvent, according to the previously predicted structure (Kajava et al., 2008). This could suggest an adaptation of GALA7 to interact with specific M. truncatula target proteins. Divergence in substrate recognition during evolution of F-box proteins has been documented in the case of the TIR1/AFB family of auxin receptors (Parry et al., 2009). Hence, even if some GALAs perform partially redundant functions during infection, several lines of evidence clearly show that the GALA gene family underwent diversification at the protein-coding level.
Functional diversification occurs either by subfunctionalization or by neofunctionalization. During subfunctionalization, the rate of evolution of the two copies is expected to be symmetrical (Innan & Kondrashov, 2010). Inversely, during neofunctionalization, one copy evolves faster and acquires a new function while the other maintains its original function. We have shown that GALAs are organized into two subclades: GALA1, 3, 4, 5 and GALA2, 6, 7. In each of these subclades, at least one GALA experienced purifying and neutral selection (GALA2 and GALA1, GALA5) while the two others in each subclade underwent diversifying positive selection. This is consistent with a neofunctionalization model in which GALA2 and GALA5 (and/or GALA1) would have kept the ‘ancestral’ function, while GALA6 and GALA7, on one hand, and GALA3 and GALA4 on the other hand, could have acquired new functions through diversifying selection. The functional analysis provides data to support this model. Indeed, we show that GALA5 does not affect callose deposition. By parsimony principle, this may indicate that GALA4 acquired this ability in the course of divergence from the ancestral allele. Pathogenicity assays on tomato show that GALA7 is different from GALA6 by its requirement for full pathogenicity when associated with GALA1, 4 and 5. Furthermore, GALA7 evolved essential pathogenicity function on M. truncatula, not shared by any other GALAs (Angot et al., 2006).
Overall, our work shows that the GALA gene family underwent functional diversification, possibly via neofunctionalization, which resulted in specific adaptation of some GALAs on different host plants. Given R. solanacearum’s wide host range, these specificities likely provided the selective advantage responsible for the remarkable conservation of the GALAs in the entire species. A similar mode of evolution was recently described for the ROP5 family of injected pseudokinases from Toxoplasma gondii and was proposed to contribute to the ability of this parasite to infect a wide host range (Reese et al., 2011). Hence, duplications of important pathogenicity genes may represent a general mechanism providing pathogens with the potential to adapt to a large host spectrum by evolving new functions.
Acknowledgments
We are grateful to Drs Kee Sohn and Jonathan Jones (JIC, Norwich, UK) for providing the pEDV6 vector before publication, and to Sebastien Carrere (LIPM) for help with bioinformatics. We also wish to thank Philippe Prior (Cirad, La Reunion) for access to sequence data before publication and selection of strains that were spotted on the microarray, and Christian Boucher and Fabienne Vailleau for critical review of the manuscript. We also would like to acknowledge the high quality and thoroughness of the critique provided by an anonymous reviewer. This work was supported by an ANR grant 07-JCJC-0133 to N.P., a grant from ‘Ministère de l'Enseignement Supérieur et de la Recherche’ to P.R., and a grant 31003A_127325 from the Swiss National Science Foundation to M.A.
Supporting Information
Additional supporting information may be found in the online version of this article.
Fig. S1 Synteny in GALAs' loci.
Table S1 List of strains and plasmids used in this study
Table S2 List of oligonucleotides used in this study
Table S3 GALA accession numbers andposition of hrpII box
Table S4 Comparative genomic hybridization analysis
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
References
- Adam L, Somerville SC. Genetic characterization of five powdery mildew disease resistance loci in Arabidopsis thaliana. Plant Journal. 1996;9:341–356. doi: 10.1046/j.1365-313x.1996.09030341.x. [DOI] [PubMed] [Google Scholar]
- Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, van Dijk K, Collmer A. The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proceedings of the National Academy of Sciences, USA. 2000;97:4856–4861. doi: 10.1073/pnas.97.9.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot A, Peeters N, Lechner E, Vailleau F, Baud C, Gentzbittel L, Sartorel E, Genschik P, Boucher C, Genin S. Ralstonia solanacearum requires F-box-like domain-containing type III effectors to promote disease on several host plants. Proceedings of the National Academy of Sciences, USA. 2006;103:14620–14625. doi: 10.1073/pnas.0509393103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot A, Vergunst A, Genin S, Peeters N. Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathogens. 2007;3:e3. doi: 10.1371/journal.ppat.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Gil M, Dufayard JF, Dessimoz C, Gascuel O. Survey of branch support methods demonstrates accuracy, power and robustness of fast-likelihood-based approximation schemes. Systematic Biology. 2011 doi: 10.1093/sysbio/syr041. doi: 10.1093/sysbio/syr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Molecular Biology and Evolution. 2009;26:255–271. doi: 10.1093/molbev/msn232. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Nielsen R, Yang Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics. 2003;164:1229–1236. doi: 10.1093/genetics/164.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DL, Jackson RW. Bacterial genomes: evolution of pathogenicity. Current Opinion in Plant Biology. 2011;4:385–391. doi: 10.1016/j.pbi.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Badel JL, Shimizu R, Oh HS, Collmer A. A Pseudomonas syringae pv. tomato avrE1/hopM1 mutant is severely reduced in growth and lesion formation in tomato. Molecular Plant-Microbe Interactions. 2006;19:99–111. doi: 10.1094/MPMI-19-0099. [DOI] [PubMed] [Google Scholar]
- Bielawski JP, Yang Z. Positive and negative selection in the DAZ gene family. Molecular Biology and Evolution. 2001;18:523–529. doi: 10.1093/oxfordjournals.molbev.a003831. [DOI] [PubMed] [Google Scholar]
- Bland JM, Altman DG. Survival probabilities (the Kaplan–Meier method) BMJ. 1998;317:1572. doi: 10.1136/bmj.317.7172.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block A, Alfano JR. Plant targets for Pseudomonas syringae type III effectors: virulence targets or guarded decoys? Current Opinion in Microbiology. 2011;14:39–46. doi: 10.1016/j.mib.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda A, Reddy JD, El-Yacoubi B, Gabriel DW. Mutagenesis of all eight avr genes in Xanthomonascampestris pv. campestris had no detected effect on pathogenicity, but one avr gene affected race specificity. Molecular Plant-Microbe Interactions. 2005;18:1306–1317. doi: 10.1094/MPMI-18-1306. [DOI] [PubMed] [Google Scholar]
- Cunnac S, Boucher C, Genin S. Characterization of the cis-acting regulatory element controlling HrpB-mediated activation of the type III secretion system and effector genes in Ralstonia solanacearum. Journal of Bacteriology. 2004a;186:2309–2318. doi: 10.1128/JB.186.8.2309-2318.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnac S, Occhialini A, Barberis P, Boucher C, Genin S. Inventory and functional analysis of the large Hrp regulon in Ralstonia solanacearum: identification of novel effector proteins translocated to plant host cells through the type III secretion system. Molecular Microbiology. 2004b;53:115–128. doi: 10.1111/j.1365-2958.2004.04118.x. [DOI] [PubMed] [Google Scholar]
- DebRoy S, Thilmony R, Kwack Y-B, Nomura K, He SY. A family of conserved bacterial effectors inhibits salicylic acid-mediated basal immunity and promotes disease necrosis in plants. Proceedings of the National Academy of Sciences, USA. 2004;101:9927–9932. doi: 10.1073/pnas.0401601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny TP. Plant pathogenic Ralstonia species. In: Gnanamanickam SS, editor. Plant-associated bacteria. Dordrecht, the Netherlands: Springer Publishing; 2006. pp. 573–644. [Google Scholar]
- Deslandes L, Pileur F, Liaubet L, Camut S, Can C, Williams K, Holub E, Beynon J, Arlat M, Marco Y. Genetic characterization of RRS1, a recessive locus in Arabidopsis thaliana that confers resistance to the bacterial soilborne pathogen Ralstonia solanacearum. Molecular Plant-Microbe Interactions. 1998;11:659–667. doi: 10.1094/MPMI.1998.11.7.659. [DOI] [PubMed] [Google Scholar]
- Fegan M, Prior P. How complex is the “Ralstonia solanacearum species complex”. In: Allen C, Prior P, Hayward C, editors. Bacterial wilt: the disease and the Ralstonia solanacearum species complex. St. Paul, MN, USA: APS Press; 2005. pp. 449–462. [Google Scholar]
- Gabriel DW, Allen C, Schell M, Denny TP, Greenberg JT, Duan YP, Flores-Cruz Z, Huang Q, Clifford JM, Presting G, et al. Identification of open reading frames unique to a select agent: Ralstonia solanacearum race 3 biovar 2. Molecular Plant-Microbe Interactions. 2006;19:69–79. doi: 10.1094/MPMI-19-0069. [DOI] [PubMed] [Google Scholar]
- Galan JM, Wiederkehr A, Seol JH, Haguenauer-Tsapis R, Deshaies RJ, Riezman H, Peter M. Skp1p and the F-box protein Rcy1p form a non-SCF complex involved in recycling of the SNARE Snc1p in yeast. Molecular and Cellular Biology. 2001;21:3105–3117. doi: 10.1128/MCB.21.9.3105-3117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohre V, Spallek T, Haweker H, Mersmann S, Mentzel T, Boller T, de Torres M, Mansfield JW, Robatzek S. Plant pattern-recognition receptor FLS2 is directed for degradation by the bacterial ubiquitin ligase AvrPtoB. Current Biology. 2008;18:1824–1832. doi: 10.1016/j.cub.2008.10.063. [DOI] [PubMed] [Google Scholar]
- Guidot A, Elbaz M, Carrere S, Siri MI, Pianzzola MJ, Prior P, Boucher C. Specific genes from the potato brown rot strains of Ralstonia solanacearum and their potential use for strain detection. Phytopathology. 2009;99:1105–1112. doi: 10.1094/PHYTO-99-9-1105. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Kamoun S, Zody MC, Jiang RH, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- Hahn MW. Distinguishing among evolutionary models for the maintenance of gene duplicates. Journal of Heredity. 2009;100:605–617. doi: 10.1093/jhered/esp047. [DOI] [PubMed] [Google Scholar]
- Hann DR, Gimenez-Ibanez S, Rathjen JP. Bacterial virulence effectors and their activities. Current Opinion in Plant Biology. 2010;13:388–393. doi: 10.1016/j.pbi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Hua Z, Vierstra RD. The cullin-RING ubiquitin-protein ligases. Annual Review of Plant Biology. 2011;62:299–334. doi: 10.1146/annurev-arplant-042809-112256. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Two-locus sampling distributions and their application. Genetics. 2001;159:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nature Reviews Genetics. 2010;11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- Kajava AV, Anisimova M, Peeters N. Origin and evolution of GALA-LRR, a new member of the CC-LRR subfamily: from plants to bacteria? PLoS ONE. 2008;3:e1694. doi: 10.1371/journal.pone.0001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitko BH, Park DH, Velasquez AC, Wei CF, Russell AB, Martin GB, Schneider DJ, Collmer A. Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathogens. 2009;5 doi: 10.1371/journal.ppat.1000388. e1000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Molecular Biology and Evolution. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Lerat E, Daubin V, Ochman H, Moran NA. Evolutionary origins of genomic repertoires in bacteria. PLoS Biology. 2005;3 doi: 10.1371/journal.pbio.0030130. e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin NC, Martin GB. An avrPto/avrPtoB mutant of Pseudomonas syringae pv. tomato DC3000 does not elicit Pto-mediated resistance and is less virulent on tomato. Molecular Plant-Microbe Interactions. 2005;18:43–51. doi: 10.1094/MPMI-18-0043. [DOI] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320:1632–1635. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- Ma W, Guttman DS. Evolution of prokaryotic and eukaryotic virulence effectors. Current Opinion in Plant Biology. 2008;11:412–419. doi: 10.1016/j.pbi.2008.05.001. [DOI] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaihara T, Tamura N, Murata Y, Iwabuchi M. Genetic screening of Hrp type III-related pathogenicity genes controlled by the HrpB transcriptional activator in Ralstonia solanacearum. Molecular Microbiology. 2004;54:863–875. doi: 10.1111/j.1365-2958.2004.04328.x. [DOI] [PubMed] [Google Scholar]
- Nielsen R. Statistical tests of selective neutrality in the age of genomics. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- Nomura K, Debroy S, Lee YH, Pumplin N, Jones J, He SY. A bacterial virulence protein suppresses host innate immunity to cause plant disease. Science. 2006;313:220–223. doi: 10.1126/science.1129523. [DOI] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. NewYork, NY, USA: Springer-Verlag; 1970. [Google Scholar]
- O'Reilly PF, Birney E, Balding DJ. Confounding between recombination and selection, and the Ped/Pop method for detecting selection. Genome Research. 2008;18:1304–1313. doi: 10.1101/gr.067181.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry G, Calderon-Villalobos LI, Prigge M, Peret B, Dharmasiri S, Itoh H, Lechner E, Gray WM, Bennett M, Estelle M. Complex regulation of the TIR1/AFB family of auxin receptors. Proceedings of the National Academy of Sciences, USA. 2009;106:22540–22545. doi: 10.1073/pnas.0911967106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plener L, Manfredi P, Valls M, Genin S. PrhG, a transcriptional regulator responding to growth conditions, is involved in the control of the type III secretion system regulon in Ralstonia solanacearum. Journal of Bacteriology. 2010;192:1011–1019. doi: 10.1128/JB.01189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poueymiro M, Genin S. Secreted proteins from Ralstonia solanacearum: a hundred tricks to kill a plant. Current Opinion in Microbiology. 2009;12:44–52. doi: 10.1016/j.mib.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Rasband W. ImageJ. Bethesda, MD, USA: U.S. National Institutes of Health; 1997. [Google Scholar]
- Reed FA, Tishkoff SA. Positive selection can create false hotspots of recombination. Genetics. 2006;172:2011–2014. doi: 10.1534/genetics.105.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, Boyle JP. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proceedings of the National Academy of Sciences, USA. 2011;108:9625–9630. doi: 10.1073/pnas.1015980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C, Fegan M, Pruvost O, Elbaz M, Calteau A, et al. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics. 2010;11:379. doi: 10.1186/1471-2164-11-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risseeuw EP, Daskalchuk TE, Banks TW, Liu E, Cotelesage J, Hellmann H, Estelle M, Somers DE, Crosby WL. Protein interaction analysis of SCF ubiquitin E3 ligase subunits from Arabidopsis. Plant Journal. 2003;34:753–767. doi: 10.1046/j.1365-313x.2003.01768.x. [DOI] [PubMed] [Google Scholar]
- Salanoubat M, Genin S, Artiguenave F, Gouzy J, Mangenot S, Arlat M, Billault A, Brottier P, Camus JC, Cattolico L, et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature. 2002;415:497–502. doi: 10.1038/415497a. [DOI] [PubMed] [Google Scholar]
- Scholze H, Boch J. TAL effectors are remote controls for gene activation. Current Opinion in Microbiology. 2011;14:47–53. doi: 10.1016/j.mib.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Sohn KH, Lei R, Nemri A, Jones JD. The downy mildew effector proteins ATR1 and ATR13 promote disease susceptibility in Arabidopsis thaliana. Plant Cell. 2007;19:4077–4090. doi: 10.1105/tpc.107.054262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergiopoulos I, de Wit PJ. Fungal effector proteins. Annual review of Phytopathology. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Nielsen R, Yang Q. Pervasive adaptive evolution in mammalian fertilization proteins. Molecular Biology and Evolution. 2003;20:18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Rocha EP. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genetics. 2011;7 doi: 10.1371/journal.pgen.1001284. e1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G, Scarpelli C, Medigue C. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Research. 2006;34:53–65. doi: 10.1093/nar/gkj406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DJ, McVean G. Estimating diversifying selection and functional constraint in the presence of recombination. Genetics. 2006;172:1411–1425. doi: 10.1534/genetics.105.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T, Zong N, Zou Y, Wu Y, Zhang J, Xing W, Li Y, Tang X, Zhu L, Chai J, et al. Pseudomonas syringae effector AvrPto blocks innate immunity by targeting receptor kinases. Current Biology. 2008;18:74–80. doi: 10.1016/j.cub.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. Journal of Molecular Evolution. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Molecular Biology and Evolution. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.