

Abstract
One of the main challenges for protein redesign is the efficient evaluation of a combinatorial number of candidate structures. The modeling of protein flexibility, typically by using a rotamer library of commonly-observed low-energy side-chain conformations, further increases the complexity of the redesign problem. A dominant algorithm for protein redesign is Dead-End Elimination (DEE), which prunes the majority of candidate conformations by eliminating rigid rotamers that provably are not part of the Global Minimum Energy Conformation (GMEC). The identified GMEC consists of rigid rotamers (i.e., rotamers that have not been energy-minimized) and is thus referred to as the rigid-GMEC. As a post-processing step, the conformations that survive DEE may be energy-minimized. When energy minimization is performed after pruning with DEE, the combined protein design process becomes heuristic, and is no longer provably accurate: a conformation that is pruned using rigid-rotamer energies may subsequently minimize to a lower energy than the rigid-GMEC. That is, the rigid-GMEC and the conformation with the lowest energy among all energy-minimized conformations (the minimized-GMEC) are likely to be different. While the traditional DEE algorithm succeeds in not pruning rotamers that are part of the rigid-GMEC, it makes no guarantees regarding the identification of the minimized-GMEC. In this paper we derive a novel, provable, and efficient DEE-like algorithm, called minimized-DEE (MinDEE), that guarantees that rotamers belonging to the minimized-GMEC will not be pruned, while still pruning a combinatorial number of conformations. We show that MinDEE is useful not only in identifying the minimized-GMEC, but also as a filter in an ensemble-based scoring and search algorithm for protein redesign that exploits energy-minimized conformations. We compare our results both to our previous computational predictions of protein designs and to biological activity assays of predicted protein mutants. Our provable and efficient minimized-DEE algorithm is applicable in protein redesign, protein-ligand binding prediction, and computer-aided drug design.
1 Introduction
Computational Protein Design
The ability to engineer proteins has many biomedical applications. A number of computational approaches to the protein redesign problem have been reported. To improve the accuracy of the redesign, protein flexibility has been incorporated into most previous structure-based algorithms for protein redesign [42, 20, 19, 18, 1, 26, 21]. A study of bound and unbound structures found that most structural changes involve only a small number of residues and that these changes are primarily side-chains, and not backbone [31]. Hence, many protein redesign algorithms use a rigid backbone and model side-chain flexibility with a rotamer library that consists of a discrete set of low-energy commonly-observed side-chain conformations [28, 34]. The major challenge for redesign algorithms is the efficient evaluation of the exponential number of candidate conformations, resulting not only from mutating residues along the peptide chain, but also by employing rotamer libraries. The development of pruning conditions capable of eliminating the majority of mutation sequences and conformations in the early, and less costly, redesign stages has been crucial.
GMEC-based algorithms for protein redesign are based on the assumption that protein folding and binding can be accurately predicted by examining the Global Minimum Energy Conformation (GMEC). Since identifying the GMEC using a model with a rigid backbone, a rotamer library, and a pairwise energy function is known to be NP-hard [33, 4], different heuristic approaches have been proposed [42, 20, 19, 18, 29, 9, 38]. A provable and efficient deterministic algorithm, which has become the dominant choice for GMEC-based protein design, is Dead-End Elimination (DEE) [8]. DEE reduces the size of the conformational search space by eliminating rigid rotamers that provably are not part of the GMEC. Most important, since no protein conformation containing a dead-ending rotamer is generated, DEE provides a combinatorial factor reduction in computational complexity.
When energy minimization is performed after pruning with DEE, the process becomes heuristic, and is no longer provably accurate: a conformation that is pruned using rigid-rotamer energies may subsequently minimize to a structure with lower energy than the rigid-GMEC. Therefore, the traditional DEE conditions are not valid for pruning rotamers when searching for the lowest-energy conformation among all energy-minimized rotameric conformations (the minimized-GMEC, or minGMEC).
NRPS Redesign and K*
Traditional ribosomal peptide synthesis is complemented by non-ribosomal peptide synthetase (NRPS) enzymes in some bacteria and fungi. NRPS enzymes consist of several domains, each of which has a separate function. Substrate specificity is generally determined by the adenylation (A) domain [39, 3, 37]. Among the products of NRPS enzymes are natural antibiotics (penicillin, vancomycin), antifungals, antivirals, immunosuppressants, and antineo-plastics. The main techniques for NRPS enzyme redesign are domain-swapping [40, 36, 10, 30], signature sequences [39, 12, 3], and active site manipulation from a structure-based mutation search utilizing ensemble docking (the K* method [25]).
The NRPS system discussed in this paper is the phenylalanine adenylation domain of Gramicidin Synthetase A (GrsA-PheA), which, together with Gramicidin Synthetase B (GrsB), produces the natural antibiotic gramicidin S. The K* algorithm has recently been used to gain new insights into the enzyme’s mechanism and selectivity [41]. Redesigning GrsA-PheA to switch its specificity from the wildtype phenylalanine to a different substrate (e.g., Leu or Tyr) may produce a modified version of gramicidin. Thus, structure-based computational protein redesign can play a role in engineering combinatorial biosynthesis for small-molecule diversity. The redesign of NRPS enzymes can lead to the synthesis of novel NRPS products, such as new libraries of antibiotics [2]. More generally, novel molecular function can be achieved by redesigning an enzyme’s active site so that it will perform its chemical reaction on a novel substrate.
The K* algorithm [25] has been demonstrated for NRPS redesign, but is a general algorithm that is, in principle, capable of redesigning any protein. K* is an ensemble-based scoring technique that uses a Boltzmann distribution to compute partition functions for the bound and unbound states of a protein. The ratio of the bound to the unbound partition function is used to compute a provably-good approximation (K*) to the binding constant for a given design sequence. A volume and a steric filter are applied in the initial stages of a redesign search to prune the majority of the conformations from more expensive evaluation. The number of evaluated conformations is further reduced by a provable ε-approximation algorithm. Protein flexibility is modeled for both the protein and the ligand using energy-minimization and rotamers [25]. In a recent study by Stevens et al. [41], the K* software was successfully applied in a redesign of GrsA-PheA: in vitro experiments showed that the top K*-predicted mutations improved the enzyme’s specificity for a novel substrate.
Contributions of the Paper
Boltzmann probability implies that low-energy conformations are more likely to be assumed than high-energy conformations. The motivation behind energy minimization is therefore well-established and algorithms that incorporate energy minimization often lead to more accurate results. However, if energy minimization is performed after pruning with DEE, then the combined protein design process is heuristic, and not provable. We show that a conformation pruned using rigid-rotamer energies may subsequently minimize to surpass the putative rigid-GMEC.
We derive a novel, provable, and efficient DEE-like algorithm, called minimized-DEE (Min-DEE), that guarantees that no rotamers belonging to the minGMEC will be pruned. We show that our method is useful not only in (a) identifying the minGMEC (a GMEC-based method), but also (b) as a filter in an ensemble-based scoring and search algorithm for protein redesign that exploits energy-minimized conformations. We achieve (a) by implementing a MinDEE/A* algorithm in a search to switch the binding affinity of the Phe-specific adenylation domain of the NRPS Gramicidin Synthetase A (GrsA-PheA) towards Leu. The latter goal (b) is achieved by implementing MinDEE as a combinatorial filter in a hybrid algorithm,1 combining A* search and our previous work on K* [25]. The experimental results, based on a 2-point mutation search on the 9-residue active site of the GrsA-PheA enzyme, confirm that the new Hybrid MinDEE-K* algorithm has a much higher pruning efficiency than the original K* algorithm. Moreover, it takes only 30 seconds for MinDEE to determine which rotamers can be provably pruned. We make the following contributions in this paper:
Derivation of MinDEE, a novel, provable, and efficient DEE-like algorithm that incorporates energy minimization, with applications in both GMEC- and ensemble-based protein design.
Introduction of a MinDEE/A* algorithm that identifies the minGMEC and returns a set of low-energy conformations;
Introduction of a Hybrid MinDEE-K* ensemble-based scoring and search algorithm, improving on our previous work on K* [25] by replacing a constant-factor with a combinatorial-factor provable pruning condition; and
The use of our novel algorithms in a redesign mutation search for switching the substrate specificity of the NRPS enzyme GrsA-PheA; we compare our results to previous computational predictions of protein designs and to biological activity assays of predicted protein mutants.
A preliminary version of this work was presented at a conference [15]. In [14], non-overlapping improvements to the current work and other algorithmic DEE enhancements are presented.
2 Derivation of the Minimized-DEE Criterion
2.1 The Original DEE Criterion
In this section we briefly review the traditional-DEE theorem [8, 32, 17, 22]. Traditional-DEE refers to the original DEE, which is not provably correct when used in a search for the minimized-GMEC. Our notation is chosen to remain consistent with previous work. The total energy, ET, of a given rotameric-based conformation can be written as ET = Et′ + Σi E(ir) + Σi Σj>i E(ir, js), where Et′ is the template self-energy (i.e., backbone energies or energies of rigid regions of the protein not subject to rotamer-based modeling), ir denotes rotamer r at position i, E(ir) is the self energy of rotamer ir (the intra-residue and residue-to-template energies), and E(ir, js) is the non-bonded pairwise interaction energy between rotamers ir and js. The rotamers assumed in the rigid-GMEC are written with a subscript g. Therefore ig is the rotamer assumed in the rigid-GMEC at position i. The following two bounds are then noted: for all i, j (i ≠ j), and , where Rj is the set of allowed rotamers for residue j. For clarity, we will not include Rj in the limits of the max and min terms, since it will be clear from the notation from which set s must be drawn. The DEE criterion for rotamer ir is defined as:
| (1) |
Any rotamer ir satisfying the DEE criterion (Eq. 1) is provably not part of the rigid-GMEC (ir ≠ ig), and is considered ‘dead-ending’ (Fig. 1). Extensions to this initial DEE criterion allow for additional pruning while maintaining correctness with respect to identifying the rigid-GMEC [8, 22, 16, 17, 32].
Figure 1. Rotamer Pruning by Dead-End Elimination.

A cartoon of the protein’s conformational energy for all conformations of residues j (j ≠ i) assuming the presence of rotamer r (orange) and rotamer t (blue) at position i. In this example, the lowest (best) conformational energy achievable with rotamer ir is indicated by the dotted line and the highest (worst) conformational energy achievable with rotamer it is indicated by the dashed line. Since the energy of all conformations is reduced in switching from ir to it, rotamer ir can be pruned as dead-ending. In practice, the use of Eq. (1) avoids the requirement of having to enumerate the exponential number of possible conformations for all residues j (j ≠ i).
2.2 DEE with Energy Minimization: MinDEE
We now derive generalized DEE pruning conditions which can be used when searching for the minimized-GMEC. The fundamental difference between traditional-DEE and MinDEE is that the former enjoys significant independence among multiple energy terms during a rotamer swap. For example, when conformations are not energy-minimized, changing rotamer ir to it does not affect the energy term E(js); however, when energy minimization is allowed, the value of this energy term may change as the rotameric conformations ir and js minimize from their initial rotameric conformations (Fig. 2). Therefore, to be provably correct, one must account for a range of possible energies. The conformation of a residue may change during energy minimization, however we constrain this movement to a region of conformation space called a voxel [43, 35] to keep one rotamer from minimizing into another. In this framework, the voxel
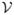 (ir) for rotamer ir is simply all conformations of residue i within a ±θ range around each rotamer dihedral when starting from the rotamer2 ir. We similarly define the voxel
(ir, js) for the pair of rotamers ir and js to be the region of conformation space
(ir) ×
(js). Next, we can define the maximum, minimum, and range of voxel energies:
(ir) for rotamer ir is simply all conformations of residue i within a ±θ range around each rotamer dihedral when starting from the rotamer2 ir. We similarly define the voxel
(ir, js) for the pair of rotamers ir and js to be the region of conformation space
(ir) ×
(js). Next, we can define the maximum, minimum, and range of voxel energies:
Figure 2. Energy-Minimized DEE.

Without energy minimization the swapping of rotamer ir for it (Panel A to Panel B) leaves unchanged the conformations and self and pairwise energies of residues j and k. When energy minimization is allowed, the swapping of rotamer ir for rotamer it (Panel C to Panel D) may cause the conformations of residues j and k to minimize (i.e., move) to form more energetically favorable interactions (from the faded to the solid conformations in Panels C and D).
Analogous definitions exist for pairwise terms:
We now define the MinDEE criterion for rotamer ir to be:
| (2) |
Proposition 1
When Eq. (2) holds, rotamer ir is provably not part of the minimized-GMEC.
The proof of Proposition 1 is given in Appendix A.
The most significant difference between traditional-DEE and MinDEE is the accounting for possible energy changes during minimization, which are incorporated through the introduction of the terms and . Using precomputed energy bounds, the MinDEE pruning condition (Eq. 2) can be computed as efficiently as the traditional-DEE pruning condition (Eq. 1). The complexity of deciding Eq. (2) is O(q2n2), where n is the number of residue positions and q is the maximum number of rotamers per residue position. The MinDEE framework can be used whenever a bound on a pairwise energy function can be obtained and is therefore not critically dependent upon the particular energy function or type of minimization employed.
In this section, we presented a generalization of traditional-DEE, to obtain an initial pruning criterion for MinDEE. Extensions to the traditional-DEE pruning conditions have made them more efficient [8, 22, 16, 17, 32]. An excellent review of these advanced pruning techniques appears in [32]. These methods allow more individual rotamers to be pruned during DEE and extend the DEE criterion to identify dead-ending rotamer pairs. Analogously to Sec 2.2, we have derived MinDEE equivalents to four extensions to traditional DEE for increased pruning efficiency [14].
2.3 Two Applications of MinDEE
The MinDEE criterion can efficiently reduce the search space for a given protein design problem by pruning rotamers that are provably not part of the minimized-GMEC. We have applied MinDEE as a pruning filter in two different protein design algorithms. The details of these algorithms are described in the following two sections. In Sec. 3, we use MinDEE as a part of MinDEE/A*, a provably-accurate GMEC-based protein design algorithm. In the MinDEE/A* algorithm, MinDEE is first used to provably prune the majority of the candidate conformations; the minimized-GMEC and all low-energy conformations (and thus sequences) within a specified threshold from the minimized-GMEC energy are then generated and energy-minimized using the A* search technique [24]. MinDEE/A* ranks mutation sequences3 based on the energy of the single best conformation for each mutation sequence (hence, the term GMEC-based algorithm). In Sec. 3, we first review the traditional-DEE/A* GMEC-based algorithm proposed in [24] (Sec. 3.1); we then derive the MinDEE/A* algorithm that, in contrast to traditional-DEE/A*, is provably-accurate with rotameric energy minimization (Sec. 3.2).
In Sec. 4, we describe how MinDEE can be used as a pruning filter in Hybrid MinDEE-K*, an ensemble-based protein design algorithm. For a given protein-ligand complex, Hybrid MinDEE-K* computes a provably-accurate approximation, K*, to the association binding constant by computing Boltzmann-weighted partition functions over rotameric ensembles of conformations. Given a set of candidate mutation sequences and a target ligand, Hybrid MinDEE-K* computes the K* scores for each sequence and ranks sequences in order of their computed scores (higher scores imply better binding). In the beginning of Sec. 4, we discuss the general motivation behind the Hybrid MinDEE-K* algorithm. In Sec. 4.1, we derive a provably-accurate algorithm for partition function computation over conformational ensembles that also exploits MinDEE pruning and the A* search; in Appendix B, we present an improvement to the partition function computation algorithm of Sec. 4.1. In particular, the efficient partition function computation is generalized to prune rotamers and sequences, so that in protein redesign the optimal sequences (in terms of K* score) are computed. Finally, in Sec. 4.2, we describe the application of the partition function computation algorithms in Hybrid MinDEE-K*, as well as the complete sequence of Hybrid MinDEE-K* algorithmic steps.
3 MinDEE/A* Search Algorithm (GMEC-Based Redesign)
3.1 Traditional-DEE with A*
In [24], an A* branch-and-bound algorithm was developed to compute a number of low-energy conformations for a single mutation sequence (i.e., a single protein). In this algorithm, traditional-DEE was first used to reduce the number of side-chain conformations, and then surviving conformations were enumerated in order of conformation energy by expanding sorted nodes of a conformation tree (Fig. 3).
Figure 3. A Sample Conformation Tree.

In a conformation tree, the rotamers of flexible residue i are represented by the branches at depth i. Internal nodes of a conformation tree represent partially-assigned conformations and each leaf node represents a fully-assigned conformation. Nodes marked with ×s have been pruned from further consideration.
The following derivation of the DEE/A* combined search closely follows [24]. The A* algorithm scores each node in a conformation tree using a scoring function f = g+h, where g is the cost of the path from the root to that node (the energy of all self and pairwise terms assigned through depth d) and h is an estimate (lower bound) of the path cost to a leaf node (a lower bound on the sum of energy terms involving unassigned residues). The value of g (at depth d) can be expressed as . The lower bound h can be written as , where n is the total number of flexible residues and . The A* algorithm maintains a list of nodes (sorted by f) and in each iteration replaces the node with the smallest f value by an expansion of the children of that node. This process of expansion is continued until the node with the smallest f value is a leaf node. This leaf node corresponds to a fully-assigned conformation and is returned by the algorithm. To reduce the branching factor of the conformation tree, the DEE algorithm is used to preprocess the set of allowed rotamers. If more than one low-energy conformation is to be extracted from the A* search, the DEE criterion must be modified. If low-energy conformations within Ew of the GMEC are to be returned by the DEE/A* search, then the DEE criterion must be modified to only eliminate rotamers that are provably not part of any conformation within Ew of the GMEC. The original DEE criterion (Eq. 1) is thus changed to: .
3.2 MinDEE with A*
The traditional-DEE/A* algorithm [24] can be extended to include energy minimization by substituting our newly derived MinDEE (Sec. 2.2) for traditional-DEE. So that no conformations within Ew of the energy-minimized GMEC are pruned, the MinDEE equation (Eq. 2) becomes:
| (3) |
We modify the definition of the A* functions g and h to use the minimum energy terms E⊝(ir) and E⊝(ir, js) in place of E(ir) and E(ir, js). Thus, we have:
| (4) |
where
| (5) |
A lower bound on the minimized energy of the partially-assigned conformation is given by g, while a lower bound on the minimized energy for the unassigned portion of the conformation is given by h. Thus, the MinDEE/A* search generates conformations in order of increasing lower bounds on the conformation’s minimized energy.
We combine our modified MinDEE criterion (Eq. 3) with the modified A* functions (Eqs. 4–5) in a provable search algorithm for identifying the minimized-GMEC and obtaining a set of low-energy conformations. First, MinDEE prunes the majority of the conformations by eliminating rotamers that are provably not within Ew of the minimized-GMEC. The remaining conformations are then generated in order of increasing lower bounds on their minimized energies. The generated conformations are energy-minimized and ranked in terms of increasing actual minimized energies. The single best conformation for each unique mutation sequence is then used to rank the mutation sequence predictions.
The MinDEE/A* search must guarantee that upon completion all conformations within Ew of the minimized-GMEC are returned. Since in the A* algorithm conformations are returned in order of increasing lower bounds on the minimized energies, the minimized-GMEC may not be among the top conformations if the lower bound on its energy does not rank high. We therefore derive the following condition for halting the MinDEE/A* search. Let B(s) be the lower bound on the energy of conformation s (see Appendix C, which describes how lower energy bounds are precomputed for all rotamer pairs) and let Em be the current minimum energy among the minimized conformations returned so far in the A* search.
Proposition 2
The MinDEE/A* search can be halted once the lower bound B(c) on the energy of the next conformation c returned by A*, satisfies B(c) > Em + Ew. The set of returned conformations is guaranteed to contain every conformation whose energy is within Ew of the energy of the minimized-GMEC. Moreover, at that point in the search, the conformation with energy Em is the minimized-GMEC.
Proof
Let E(s) be the actual energy of a minimized conformation s. Let Y be the set containing conformation c (the next conformation returned by A*) and all conformations not yet returned. Since A* returns conformations in order of increasing lower bounds on the energy, we know that E(s) ≥ B(s) ≥ B(c) for any conformation s ∈ Y. Thus, if B(c) > Em + Ew holds, then E(s) > Em + Ew. Hence, no conformations in Y have energies within Ew of the energy of the minimized-GMEC, proving that all conformations within Ew of the minimized-GMEC energy have already been returned. Moreover, note that at that point in the search, the conformation with energy Em is actually the minimized-GMEC.
Using both MinDEE and A* search together, our algorithm obtains a combinatorial pruning factor by eliminating the majority of the conformations, which makes the search for the minimized-GMEC computationally feasible. The MinDEE/A* algorithm incorporates energy minimization with provable guarantees, and is thus more capable of returning conformations with lower energy states than traditional-DEE.
4 Hybrid MinDEE-K* Algorithm (Ensemble-Based Redesign)
We now present an extension and improvement to the original K* protein design algorithm [25] by using a version of the MinDEE criterion plus A* branch-and-bound search. The K* ensemble-based scoring function approximates the association binding constant for a given protein-ligand complex with the following quotient: , where qPL, qP, and qL are the partition functions for the protein-ligand complex, the free (unbound) protein, and the free ligand, respectively. For a given protein design problem, partition functions and K* scores are efficiently computed for all candidate mutation sequences with the target ligand; sequences are then ranked in order of their computed K* scores (higher scores imply better binding). In this section, we describe how our MinDEE pruning criterion and the A* search can be exploited for the partition function and K* computation.
A partition function q over a set (ensemble) of conformations S is defined as q = Σs∈S exp(−Es/RT ), where Es is the energy of conformation s, T is the temperature in Kelvin, and R is the gas constant. In a naive K* implementation, each partition function would be computed by a computationally-expensive energy minimization of all rotamer-based conformations. However, because the contribution to the partition function of each conformation is exponential in its energy, only a subset of the conformations significantly contribute to the partition function value. By identifying and energy-minimizing only the significantly-contributing conformations, a provably-accurate ε-approximation algorithm substantially improved the algorithm’s efficiency [25]. In this section we illustrate how the newly-derived MinDEE and A* algorithms (Sec. 3.2) can be used to generate and minimize only those conformations that contribute significantly to the partition function, and hence, for which energy minimization is required. The MinDEE criterion must be used in this algorithm because the K* scoring function is based on energy-minimized conformations. Since pruned conformations never have to be examined, the Hybrid MinDEE-K* algorithm provides a combinatorial improvement in runtime over the previously described constant-factor ε-approximation algorithm [25] (where a lower-bound on each conformation’s minimum energy was quickly examined to determine if full energy minimization was required).
4.1 Efficient Partition Function Computation Using A* Search
Here, we present an efficient algorithm for computing the qPL, qP, and qL partition functions used to compute a K* approximation score for a given mutation sequence. Using the A* algorithm with MinDEE, we can generate the conformations of a rotamerically-based ensemble in order of increasing lower bounds on the conformation’s minimized energy. We can efficiently compute the lower bound on a conformation’s energy as a sum of precomputed pairwise minimum energy terms (see Appendix C). As each conformation c is generated from the conformation tree, we compare its lower bound B(c) on the conformational energy to a moving stop-threshold and stop the A* search once B(c) becomes greater than the threshold. The A* algorithm guarantees that all remaining conformations will have minimized energies above the stop-threshold. We now prove that a partial partition function q* computed using only those conformations with energies below (i.e., better than) the stop-threshold will lie within a factor of ε of the true partition function q. Note that, by definition, q ≥ q*. Thus, q* is an ε-approximation to q, i.e., q* ≥ (1 − ε)q.
Since the application of the MinDEE criterion (Eq. 2) for each rotamer ir requires that the corresponding minimum energy terms be accessed, we can easily piggyback the computation of a lower bound Bir on the energy of all conformations that contain a pruned rotamer ir:
Let E0 be the minimum lower energy bound among all conformations containing at least one pruned rotamer, E0 = minir∈S Bir, where S is the set of pruned rotamers. E0 can be precomputed during the MinDEE stage and prior to the A* search. Let p* be the partition function computed over the set P of pruned conformations, so that p* ≤ k exp(−E0/RT ), where |P | = k. Also, let X be the set of conformations not pruned by MinDEE and let q* be the partition function for the top m conformations already returned by A*; let q′ be the partition function for the n conformations that have not yet been generated, all of which have energies above Et, so that q′ ≤ n exp(−Et/RT); note that |X| = m + n. Finally, let . We can then guarantee an ε-approximation to the full partition function q using:
Proposition 3
If the lower bound B(c) on the minimized energy of the (m + 1)st conformation returned by A* satisfies B(c) ≥ −RT (ln(q* ρ − k exp(−E0/RT)) − ln n), then the partition function computation can be halted, with q* guaranteed to be an ε-approximation to the true partition function q, that is, q* ≥ (1 − ε)q.
Proof
The full partition function q is computed using all conformations in both P and X:
| (6) |
Thus,
| (7) |
Hence, if
| (8) |
then q* ≥ (1 − ε)q. Solving Eq. (8) for Et, we obtain the desired stop-threshold:
| (9) |
We can halt the search once a conformation’s energy lower bound becomes greater than the stop-threshold (Eq. 9), since then q* is already an ε-approximation to q.
The application of the MinDEE criterion gives a combinatorial-factor speedup by caching the minimum lower energy bound for the set of all pruned conformations. Since the conformations pruned by MinDEE can potentially contribute significantly to the partition function, we bound their contribution, thus guaranteeing a provably-accurate approximation to the full partition function. The conformation tree could, in principle, be reduced by pruning an arbitrary subset of the rotamers, so long as a guarantee on the accuracy is still maintained through a bound on the contribution of the pruned conformations. However, in practice, the amount of pruning and the resulting approximation accuracy depend on which rotamers are chosen for pruning. Using MinDEE to determine the set of pruned rotamers guarantees that the pruned conformations will have high lower energy bounds by requiring that no conformations within Ew of the minimized-GMEC energy are pruned (Eq. 3), whereas an arbitrary rotameric set could easily contain conformations with very good (i.e., low) energies. Proposition 3 turns pruning with MinDEE into a provable heuristic. Note that: 1) the magnitude of p* is determined by the lower energy bounds of the pruned conformations, and 2) the number of conformations that A* must extract to guarantee a provably-accurate approximation to the partition function depends on the magnitude of p*. By using MinDEE pruning instead of an arbitrary set of rotamers, we increase the pruning efficiency. Since conformations that contain steric clashes do not contribute to the partition function for the given mutation sequence, we can further reduce p* by including in P only the pruned conformations whose lower energy bound does not contain a rotamer that always clashes sterically (such a reduction in P, and hence, k, can be computed during the MinDEE phase, since rotamers whose precomputed minimum-energy bounds indicate steric clashes, necessarily imply that all conformations containing these rotamers are also steric clashes).
If at some point in the search, the stop-threshold condition has not been reached and there are no remaining conformations for A* to extract (n = 0), then q′ = 0 by definition, and q = q* + p*. Hence, if q* ρ ≥ k exp(−E0/RT), then q* ≥ (1 − ε)(q* + k exp(−E0/RT)), so q* ≥ (1 − ε)q is already an ε-approximation to q; otherwise, we have
| (10) |
for some approximation accuracy δ > ε. Thus, the set of pruned rotamers must be reduced to guarantee the desired approximation accuracy. To assure that an ε-approximation is achieved when the search is repeated, a subset of the k pruned conformations in P must be re-introduced into the computation. Let l be the number of conformations from P (the set of pruned conformations) that are not to be pruned, such that p* ≤ (k − l) exp(−E0/RT). We will conservatively assume that the l conformations do not contribute to q*, although they no longer contribute to p* either. At the end of the second mutation search, we must have
| (11) |
Solving for l, we obtain the following condition, which guarantees the desired ε-approximation accuracy:
| (12) |
where again . Note that an ε-approximation may be achieved before all conformations have been extracted; Eq. (12) guarantees such an accuracy when all non-pruned conformations have been extracted by A*. To guarantee that at least l out of the k pruned conformations will be allowed during the repeated computation, we can choose a subset Q of the rotamers pruned by MinDEE, such that not pruning Q keeps at least l additional conformations.
In the algorithm for partition function computation described in this section, conformation pruning is performed only within a mutation sequence. In Appendix B, we derive an improvement to this partition function algorithm that further improves the efficiency of the partition function computation by allowing conformation pruning across mutation sequences. The improved algorithm in Appendix B also yields a provably-good approximation (see Proposition 4 therein).
4.2 Algorithm
We now have all the necessary tools for our ensemble-based Hybrid MinDEE-K* algorithm. The volume filter (see Sec. 5) in the original K* is applied first to eliminate under- and over-packed mutation sequences. For each of the remaining unpruned sequences, the scores are computed, using the partition function algorithms of Sec. 4.1 and Appendix B to efficiently compute the qPL, qP, and qL partition functions. The application of the MinDEE and A* algorithms in the partition function computation improves on the mere constant-factor speedup provided by the energy filter in the original K* algorithm [25]. By implementing a steric filter (see Sec. 5), similar to the one in [25], as a part of the A* search, we prevent some high-energy conformations (corresponding to steric clashes) with good lower bounds from being returned by A*, gaining an additional combinatorial speedup. Only the conformations that pass all of these filters are energy-minimized and used in the computation of the partition function for the conformational ensemble. In contrast to the original K* algorithm [25] where, for a given mutation sequence, pruning was performed during the (worst-case exponential) conformation enumeration, Hybrid MinDEE-K* uses the polynomial-time MinDEE criterion before the enumeration occurs. Our Hybrid MinDEE-K* algorithm efficiently prunes the majority of the mutation sequences and conformations from more expensive evaluation, while still giving provable guarantees about the accuracy of its score predictions (Eq. 33). Finally, the unpruned mutation sequences are ranked in order of their computed K* scores.
5 Methods
Structural Model
Our structural model is the same as the one used in the original K* [25]. In our experiments, the structural model consists of nine active site residues (D235, A236, W239, T278, I299, A301, A322, I330, C331) of GrsA-PheA (PDB id: 1AMU) [5], a steric shell (30 residues with at least one atom within 8 Å from the substrate), the amino acid substrate, and the AMP cofactor. The steric shell facilitates the computation of the energy between the active site residues and neighboring regions of the protein (the residue-to-template energy) and constrains the movement of the active site residues to only sterically-allowable conformations relative to the body of the GrsA-PheA protein. All nine active site residues are modeled as flexible using rotamers and are subject to energy minimization. The steric shell includes residues 186Y, 188I, 190T, 210L, 213F, 214F, 230A, 234F, 237S, 238V, 240E, 243M, 279L, 300T, 302G, 303S, 320I, 321N, 323Y, 324G, 325P, 326T, 327E, 328T, 329T, 332A, 333T, 334T, 515N, and 517K. In 1AMU [5], and also in [25], residues 235D and 517K make H-bonds to the amino acid backbone of the ligand, thereby stabilizing the substrate in a productive orientation for catalysis. Flexible residues are represented by rotamers from the Richardsons’ rotamer library [28]. The energy function consists of the AMBER electrostatic, vdW, and dihedral energy terms [44, 6], and the EEF1 pairwise solvation energy term [23]. A dielectric of 20 and a solvation energy scaling factor of 0.05 was used for the computational experiments. Each rotameric-based conformation is minimized using steepest-descent minimization (see Appendix C).
Energy Precomputation for Lower Bounds, B(·)
The MinDEE criterion (Eq. 2) uses both min and max precomputed energy terms to determine which rotamers are not part of the minimized-GMEC. There is no need to re-compute the min and max energies every time Eq. (2) is evaluated. See Appendix C for a detailed discussion.
Approximation Accuracy
We use an ε-value of 0.03, thus guaranteeing that the computed partial partition functions will be not less than 97% of the corresponding full partition functions. We use a value of 0.01 for γ, which requires that correct K* scores be computed for all mutation sequences whose score is at most two orders of magnitude less than the best score.
Filters
Volume filter: Mutation sequences that are over- or under-packed by more than 30Å3 compared to the wildtype PheA are pruned; Steric filter: Conformations in which a pair of atoms’ vdW radii overlap by more than 1.5Å prior to minimization are pruned; Sequence-space filter: The active site residues are allowed to mutate to the set (GAVLIFYWM) of hydrophobic amino acids; MinDEE: We use an implementation of the MinDEE analog to the simple coupled Goldstein criterion ([16] and [14]).
6 Results and Discussion
In this section, we compare the results of GMEC-based protein redesign without (traditional-DEE/A*) and with (MinDEE/A*) energy minimization. We also compare the redesign results when energy minimization is used without (MinDEE/A*) and with (Hybrid MinDEE-K*) conformational ensembles. We further compare our ensemble-based redesign results both to our previous computational predictions of protein designs and to biological activity assays of predicted protein mutants.
6.1 Comparison to Biological Activity Assays
Similarly to [25], we simulated the biological activity assays of L-Phe and L-Leu against the wild-type PheA enzyme and the double mutant T278M/A301G [39]. In [39], T278M/A301G was shown to have decreased specificity for Phe and increased specificity for Leu, as compared to the wild-type enzyme. The computed Hybrid MinDEE-K* scores qualitatively agreed with these results: the Hybrid MinDEE-K* score for wildtype with Phe was 17-fold higher than T278M/A301G with Phe; the Hybrid MinDEE-K* score for wildtype with Leu was 12-fold lower than T278M/A301G with Leu.
6.2 Comparison to Traditional-DEE
For comparison, the simple coupled Goldstein traditional-DEE criterion [16] was used in a redesign search for changing the specificity of the wildtype PheA enzyme from Phe to Leu, using the experimental setup in Sec. 5. A comparison to the rotamer assignments in the minimized-GMEC A236M/A322M (Sec. 6.3.2) revealed that A301, the minimized-GMEC identity at residue position 301, was in fact pruned by traditional-DEE. We then energy-minimized A236M/A301G, the rigid-GMEC obtained by traditional-DEE/A* and determined that its energy was higher (by appx. 6 kcal/mol) than the energy for the minimized-GMEC obtained by MinDEE/A*. Moreover, a total of 396 different conformations minimized to an energy lower than the minimized rigid-GMEC energy (see Fig. 6). These results confirm our claim that traditional-DEE is not provably-accurate with energy-minimization; they also show that conformations pruned by traditional-DEE may minimize to a lower energy state than the rigid-GMEC.
Figure 6. Energies of all conformations within 12.5 kcal/mol of the minimized–GMEC energy.

The energies (after minimization) of the minimized–GMEC (red cross) and the rigid–GMEC (yellow circle) are shown. The minimized–GMEC A236M/A322M is (by definition) the lowest–energy conformation, while the rigid–GMEC is ranked 397th.
6.3 Redesign for Leu
6.3.1 Hybrid MinDEE-K*
The experimental setup for Leu redesign with Hybrid MinDEE-K* is as described in Sec 5. The 2-point mutation search took approximately 9 hours on a cluster of 24 processors. Only 30% of the mutation sequences passed the volume filter, while MinDEE pruned 98% of the remaining conformations. The use of the ε-approximation algorithms reduced the number of conformations that had to be subsequently generated and energy-minimized by an additional factor of fifty (see Table I). A brute-force version of Hybrid MinDEE-K* that did not utilize any of the filters, would take approximately 8,700 times longer (appx. 3,262 days) for the same experimental setup for redesign.
Table I. Conformational Pruning with Hybrid MinDEE-K*.
The initial number of conformations for the GrsA-PheA 2-residue Leu mutation search is shown with the number of conformations remaining after the application of volume, MinDEE, steric, and energy (with A*) pruning. The A* energy filter is based on the ε-approximation algorithms in Secs. 4.1 and Appendix B. The pruning factor represents the ratio of the number of conformations present before and after the given pruning stage. The pruning-% (in parentheses) represents the percentage of remaining conformations eliminated by the given pruning stage.
| Conf. Remaining | Pruning Factor (%) | |
|---|---|---|
| Initial | 6.8 × 108 | - |
| Volume Filter | 2.04 × 108 | 3.33 (70.0) |
| MinDEE Filter | 4.13 × 106 | 49.43 (98.0) |
| Steric Filter | 3.86 × 106 | 1.07 (6.5) |
| A* Energy Filter | 7.82 × 104 | 49.41 (98.0) |
To determine the per-sequence pruning efficiency of Hybrid MinDEE-K*, we further computed the fraction of fully-evaluated conformations (the number of conformations that pass all of the Hybrid MinDEE-K* filters, divided by the total number of conformations) separately for each sequence. Fig. 5 shows the fraction of fully-evaluated conformations vs. the computed log K* scores for each of the unpruned sequences, for the protein-ligand bound-state partition function computation. As expected, the fraction of fully-evaluated conformations that contribute significantly to the computation of the provably-accurate ε-approximation to the partition function is very small (less than 0.5%) for all sequences, confirming again the efficiency of Hybrid MinDEE-K*. However, there is no correlation between the magnitude of the sequence scores and the fraction of fully-evaluated conformations.
Figure 5. Fraction of fully-evaluated conformations for the Hybrid MinDEE-K* bound-state ensembles (GrsA-PheA active site redesign).

For each of the unpruned mutation sequences, the log of the computed K* score is shown vs. the fraction of fully-evaluated conformations used to compute an ε-approximation to the partition function for the bound protein-ligand complex. The fraction of fully-evaluated conformations for a given sequence is the ratio of the number of conformations that pass all of the Hybrid MinDEE-K* pruning filters (see Table I) divided by the total number of conformations for that sequence.
The two top-scoring sequences are A301G/I330W and A301G/I330F for both Hybrid MinDEE-K* and the original-K*. These novel mutation sequences were tested in the wetlab and were shown to have the desired switch of specificity from Phe to Leu (for details of the wetlab experiments, see [25]). Moreover, the other known successful redesign T278M/A301G [39] is ranked 3rd by Hybrid MinDEE-K* (this sequence was ranked 12th by the original-K* in [25]). Furthermore, all of the top 13 Hybrid MinDEE-K* sequences contain the mutation A301G, which is found in all known native Leu adenylation domains [3]. These results show that our algorithms can give reasonable predictions for redesign.
Comparison to Original-K*
An initial comparison to the original-K* results showed only a small overlap between the top-ranking mutations for Hybrid MinDEE-K* and the original-K* [25]. To facilitate a fair comparison between the two algorithms, we applied the same energy function (as described in Sec. 5, but without solvation energies) and energy-minimization module (see Appendix C) for both Hybrid MinDEE-K* and the original-K*. This comparison revealed that both the mutation-sequence rankings and the scores for a given mutation sequence are very similar for the two algorithms: the top 19 sequences are identical, while all of the top 40 sequences for Hybrid MinDEE-K* can be found in the top 40 sequences for K*, and vice versa; the trend is similar for the remaining sequences, as well. This fact shows that, all other factors being equal, both algorithms converge to very similar results, despite the different (but still provably-accurate) filters used. To compare the efficiency of the two algorithms, we measured the number of fully-evaluated conformations, since the full energy minimization of the conformations is the most computationally-expensive part of both algorithms. The original-K* algorithm fully-evaluated approximately 30% more conformations than Hybrid MinDEE-K*. Thus, Hybrid MinDEE-K* is much more efficient at obtaining the desired results.
6.3.2 MinDEE/A*
We now discuss results from our GMEC-based experiments using MinDEE/A*. To redesign the wildtype PheA enzyme so that its substrate specificity is switched towards Leu, we used the experimental setup described in Sec. 5. The MinDEE filter on the bound protein:ligand complex pruned 206 out of the 421 possible rotamers for the active site residues, reducing the number of conformations that were subsequently supplied to A* by a factor of 2,330. We then extracted and minimized all conformations over the 2-point mutation sequences using the A* search until the halting condition defined in Proposition 2 was reached, for Ew = 12.5 kcal/mol. A total of 7261 conformations, representing 221 unique mutation sequences, had actual minimized energies within 12.5 kcal/mol of the minimized-GMEC energy (see Fig. 6), which confirms that a mutation sequence can be found in multiple low-energy states. The top-ranked MinDEE/A* mutation sequence is A236M/A322M; the minimized-GMEC is obtained from this sequence. The entire redesign process took approximately 4 days on a single processor (the MinDEE pruning stage took less than a minute, and the remainder of the time was spent in the A* enumeration stage), with more than 60000 extracted conformations before the search could be provably halted. Thus, the provable accuracy of the results comes at the cost of this computational overhead, since the number of extracted conformations is much larger than the actual number of conformations within Ew of the minimized-GMEC energy. Note, however, that a redesign effort without a MinDEE filter and a provably-accurate halting condition would be computationally infeasible.
Since a mutation sequence can be found in multiple low-energy states (see above), it is interesting to determine how similar these states are. We therefore selected the set of conformations generated by MinDEE/A* for the minimized-GMEC sequence A236M/A322M for further analysis. For this sequence, Fig. 7 shows the all-atom RMSD (active site residues only) for the minimized-GMEC with each of the 337 conformations within 12.5 kcal/mol of the minimized-GMEC energy. As Fig. 7 shows, the similarity of the structures varies significantly, with 75% of the structures clustered within the range 0.6 – 1.1 RMSD (average of 0.83). Although the correlation between the RMSD values and the conformational energies is weak (R2 of 0.24), there is a general trend for conformations with a larger deviation from the minimized-GMEC structure to also have higher energies.
Figure 7. All-atom RMSD (active site residues only) vs. energy for all A236M/A322M conformations generated by MinDEE/A*.

A total of 337 conformations for the A236M/A322M sequence have energies within 12.5 kcal/mol of the MinDEE/A* minimized-GMEC. The all-atom RMSD with the minimized-GMEC (red cross) for each of these conformations is shown vs. the corresponding computed conformational energy.
As another measure of similarity between the low-energy conformations for the A236M/A322M sequence, we computed the frequency for each observed rotamer identity at each active site residue position (Fig. 8). As Fig. 8 shows, with the exception of T278 and C331 which assume all allowed rotamers for the corresponding amino acid types from the Richardsons’ rotamer library, all other residues preferentially assume only a small subset of the possible rotamers (cf. [28]), thus indicating some (though not high) rotamer diversity between the different structures. This rotamer diversity, in combination with the rotameric energy minimization allowed in our model, are the reasons for the structure variability observed in Fig. 7.
Figure 8. Rotamer diversity for the A236M/A322M conformations generated by MinDEE/A*.

For each active site residue, the normalized frequency for each observed rotamer (number of occurrences divided by the total number of structures) is shown: the highest-occurring rotamer is in blue, the second-highest is in red, followed by yellow, green, and light blue. For clarity, A301 is not shown here since Ala has only one rotamer.
Only 2 of the top 40 MinDEE/A* mutation sequences can be found in the top 40 Hybrid MinDEE-K* sequences, and vice versa, indicating that ensemble-scoring yields substantially different predictions from single-structure scoring using the minimized-GMEC, where only the minimized bound state of a single conformation is considered (see Fig. 9).
Figure 9. Distribution of Mutations.

The distribution of the mutation types for the top 40 mutation sequences for (A) MinDEE/A* and (B) Hybrid MinDEE-K* algorithms is shown as the fraction of each mutating type for each active site residue. The types and frequencies for the mutations are quite different for the two methods, which indicates that the difference in the information content for GMEC- and ensemble-based algorithms can be substantial.
7 Limitations and Extensions
The MinDEE criterion can efficiently prune a large number of the possible conformations (see Sec. 6.3). However, because of the use of min and max energy terms, the pruning efficiency of MinDEE cannot be as high as that of traditional-DEE. This trade-off in efficiency results from the provable guarantees that MinDEE can (while traditional-DEE cannot) make when energy minimization is employed. An increase of the pruning capabilities of MinDEE would require the derivation and computation of tighter upper and lower energy bounds. Since (with a rigid backbone) the conformational changes due to switching the identity of a single rotamer should decrease in magnitude as the proximity to the modified rotamer decreases, it may also be possible to increase the pruning factor by scaling the terms in the MinDEE condition (Eq. 2), depending on the proximity of the residues involved.
The goal of our ensemble-based Hybrid MinDEE-K* algorithm is to find mutation sequences with better binding constants for the novel substrate than the wildtype enzyme. An assessment of catalytic activity is not explicitly included in the algorithm. In general, it would be interesting to generalize K* to stabilize the transition state. Since the transition state is not known structurally, K* maintains backbone contacts of the substrate in proximity to the nucleotide cofactor. As was shown in [41], the top K*-predicted mutations in a GrsA-PheA redesign improved the catalytic specificity (kcat/KM ) as well.
Several limitations of our computational model warrant a discussion. Since using a continuous representation for the partition functions is currently not feasible, our algorithm discretizes the conformational space. Rotamer discretization has been shown to work well in practice [7, 21, 27, 9, 26, 11]. A further limitation of our model is the use of a rigid backbone. However, our algorithm aims to simultaneously find the best mutations and to stabilize the sidechain placements for the given backbone, rather than assuming the backbone will remain rigid. All dead-end elimination algorithms, and the majority of structure-based protein design algorithms in general, use a model with a rigid backbone. The incorporation of backbone flexibility, however, will likely improve the computational predictions, and thus represents interesting future work.
8 Conclusions
When energy-minimization is required, the traditional-DEE criterion makes no guarantees about pruning rotamers belonging to the minimized-GMEC. In contrast, a rotamer is only pruned by MinDEE if it is provably not part of the minimized-GMEC. We showed experimentally that the minimized-GMEC can minimize to lower energy states than the rigid-GMEC, confirming the feasibility and significance of our novel MinDEE criterion. When used as a filter in ensemble-based redesign, MinDEE efficiently reduced the conformational and sequence search spaces, leading both to predictions consistent with previous redesign efforts and novel sequences that are unknown in nature. Our Hybrid MinDEE-K* algorithm showed a significant improvement in pruning efficiency, as compared to the original K* algorithm. Redesign searches for two other substrates, Val and Tyr, have also been performed, confirming the generality of our algorithms.
Protein design using traditional-DEE uses neither ensembles nor rotamer minimization. In our experiments, we reported the relative benefits of incorporating ensembles and energy-minimization into a provable redesign algorithm. A major challenge for protein redesign algorithms is the balance between the efficiency and accuracy with which redesign is performed. While the ability to prune the majority of mutation/conformation search space is extremely important, increasing the accuracy of the model is a prerequisite for successful redesign. It would be interesting to implement finer rotamer sampling and more accurate (and hence more expensive) energy functions, and remove bias in the rotamer library by factoring the Jacobian into the partition function over torsion-angle space. MinDEE can also be generalized to incorporate backbone flexibility [13]. An accurate and efficient algorithm for redesigning the enzymes that synthesize natural products should prove useful as a technique for drug design.
Figure 4. Efficient Partition Function Computation with Energy Minimization Using the A* Search.

q* is the running approximation to the partition function. The function B(·) computes the energy lower bound for the given conformation (see Appendix C). The function ComputeMinEnergy(·) returns a conformation’s energy after energy minimization. The function GetNextAStarConf() returns the next conformation from the A* search. The function RepeatSearch(·) sets up and repeats the mutation search if an ε-approximation is not achieved after the generation of all A* conformations; the search is repeated at most once. Upon completion, q* represents an ε-approximation to the true partition function q, such that q ≥ q* ≥ (1 − ε)q.
Acknowledgments
We thank Prof. A. Anderson, Dr. S. Apaydin, Mr. J. MacMaster, Mr. A. Yan, Mr. B. Stevens, and all members of the Donald Lab for helpful discussions and comments on drafts. This work is supported by grants to B.R.D. from the National Institutes of Health (R01 GM-65982 and R01 GM-078031), and the National Science Foundation (EIA-0305444).
APPENDIX
In Appendix A, we present a detailed proof of Proposition 1 from Sec. 2.2. Appendix B presents an improvement to the algorithm of Sec. 4.1 for more efficient partition function computation. Appendix C provides details on the energy precomputation for computing the lower energy bounds B(·).
A MinDEE Derivation
In this section, we present a detailed proof of Proposition 1. For clarity, we restate Proposition 1 here:
Proposition 1
When Eq. (2) holds, rotamer ir is provably not part of the minimized-GMEC.
Proof
For a given protein, we define a rotamer vector A = (A1, A2, ···, An) to specify the rotamer at each of the n residue positions; Ai = r when rotamer r is assumed by residue i. We then define the conformation vector such that is the conformation of residue i in the voxel-constrained minimized conformation, i.e., and
| (13) |
where E(B) is the energy of the system specified by conformation vector B. For the energy-minimized conformation starting from rotamer vector A, we define the self-energy of rotamer ir as and the pairwise interaction energy of the rotamer pair ir, js as where is the self-energy of residue i in conformation and is the pairwise energy between residues i and j in conformations and . We can then express the minimized energy of A, ET (A) as:
| (14) |
Let G represent the rotamer vector that minimizes into the minimized-GMEC and ET (G) be the energy of the minimized-GMEC. Let Gig→it be the rotamer vector G where rotamer ig is replaced with it. We know that ET (Gig→it) ≥ ET (G), so we can pull residue i out of the two summations, obtaining:
| (15) |
The Et′ terms (Sec. 2.1) correspond to the rigid portion of the molecule; they are independent of rotamer choice, are equal, and can be canceled. We make the following trivial upper and lower-bound observations (the E⊝(·), E⊕(·), and E⊘(·) terms are as defined in Sec. 2.2):
| (16) |
| (17) |
| (18) |
| (19) |
Substituting Eqs. (16–19) into Eq. (15), we obtain:
| (20) |
When the MinDEE pruning condition Eq. (2) holds, we can substitute the left-hand side of Eq. (2) for the first two terms of Eq. (20), and simplify the resulting equation to:
| (21) |
We then substitute the following two bounds and into Eq. (21) and reduce:
| (22) |
Thus, when the MinDEE pruning condition Eq. (2) holds, ir ≠ ig and we can provably eliminate rotamer ir as not being part of the energy-minimized GMEC.
B Improved Partition Function Computation
We now describe an improvement to the algorithm of Sec. 4.1 for more efficient partition function computation. In Sec. 4.1, provably-accurate K* scores are computed for all mutation sequences. However, since we are only interested in mutation sequences with high K* scores (i.e., sequences that are good binders), we need only require that a provably-accurate score be computed only for the top fraction of the mutation sequences. To achieve this, we will allow conformational pruning across mutation sequences. Hence, for clarity, we will refer to the partition function computation described in this Appendix as inter-mutation, while the computation described in Sec. 4.1 (where conformational pruning could be performed only within a sequence) will be referred to as intra-mutation. Below, we use the following idea (cf. [25]). When using K* to perform a mutation search, we can bootstrap the pruning condition for improved efficiency (by caching partition functions, we can exploit K* bounds from other mutations in the same search). Our search algorithm has the desirable property that provably-accurate ε-approximations are computed for top-ranking mutations, while the bounds we can prove on the quickly-computed K* values for lower-ranked mutations do not enjoy the same degree of accuracy. This idea is briefly formulated and then exploited below.
We first review some of the definitions from [25]. We let γ ∈ [0, 1] be a parameter that defines the set of mutation sequences for which an ε-approximation is to be computed. We require that an ε-approximation be guaranteed for a mutation sequence i only when , where is the score for sequence i and is the best score observed so far in the search. When γ = 1.0, an ε-approximation is guaranteed only for the best-scoring K* mutation sequence; γ = 0.0 computes an ε-approximation for all K* mutation sequences. Let us assume that A* has already generated the first m conformations and that there are n remaining conformations that have not been generated yet. We use the definitions for q′, p*, E0, and k from Proposition 3 above. We assume that we have already computed qP using the intra-mutation filter only (Proposition 3), and now describe how to efficiently compute qPL.
We define the score for the ith mutation sequence to be , while . We let be the partial partition function for the bound protein-ligand state, computed from the m already-generated conformations. We define . Finally, let and .
Proposition 4
If the lower bound B(c) on the minimized energy of the (m + 1)st conformation returned by A* satisfies B(c) ≥ −RT(ln(ψ −k exp(−E0/RT)) − ln n), then the partition function computation can be halted, with guaranteed to be an ε-approximation to the true partition function qPL for a mutation sequence whose score satisfies .
Proof
Since the ligand is invariant throughout the search, qL = oqL. Let us assume that we have a sequence for which holds. Thus,
| (23) |
First, we note again that
| (24) |
| (25) |
From the definition of qPL, we obtain
| (26) |
Now, if
| (27) |
then by Eqs. (24) and (25) we have
| (28) |
and by Eq. (23),
| (29) |
and finally, by Eq. (26), we obtain
| (30) |
which is the definition of the partition function ε-approximation. Thus, if Eq. (27) holds, then we will have an ε-approximation to the true partition function qPL. Solving Eq. (27) for Et, we obtain the stop-threshold:
| (31) |
The first conformation that has an energy above the stop-threshold (Eq. 31) halts the partition function computation, since we already have an ε-approximation. Thus, combining Eq. (31) and the intra-mutation stop-threshold (Eq. 9), our stopping condition for the computation of qPL becomes
| (32) |
where and B(·) is the lower bound on the minimized energy of a conformation.
If the desired approximation accuracy is not achieved at the end of the mutation search, after all conformations have been extracted by A*, we can modify Eq. (12) to incorporate the inter-mutation filter, obtaining the number of conformations l from P (the set of pruned conformations) that must be allowed in the repeated search:
We have derived the stop-threshold that guarantees an ε-approximation to the partition function when conformations are generated in order of increasing lower bounds on the conformation’s energy. This generalizes the inter-mutation proof in [25] which is valid when the energy lower bounds for all of the conformations are evaluated. We should note that Eq. (32) was derived assuming holds, so we can guarantee an ε-approximation to qPL only for this case. When , then we might not obtain an ε-approximation for the given mutation sequence, but we do not require a provably-good approximation for such low-scoring sequences.
Similarly to [25], we define to be an ε-approximation to the full score of a mutation sequence (the score if the full partition functions are used, instead of the partial ones) when . If holds for a mutation sequence i, then by Proposition 4, . Also, since qP is already computed using Proposition 3, . Since , we have
| (33) |
Thus, the algorithm guarantees that an ε-approximation to the full score is computed when .
C Energy Precomputation for Lower Bounds
We first derive a lower bound for the energy of a minimized conformation, closely following [25]. We then present improvements on the energy precomputation algorithm, as compared to [25].
C.1 Computing a Lower Bound on Minimized Energies
In our structural model, (Sec. 5), some residues are treated as rigid, while others have a rigid backbone but flexible side-chains. Let h be the number of flexible residues in our system. Let A be a (h+1) × (h+1) precomputed residue-indexed energy matrix that describes the energy interactions of a given residue i within itself (Ai0), with the backbone (A0i), and with other residues (Aij); the matrix element A00 is reserved for the energy interactions between the atoms of the backbone only. We term A00 to be the template energy, A0i is the residue-to-template energy, Ai0 is the intra-residue energy, and Aij is the pairwise energy for residue i. The energy of the system can be computed as
| (34) |
To compute the energy of a minimized conformation, we use a matrix M, whose elements are analogous to the elements of A, but the precomputed energies correspond to the energy-minimized structure. If we obtain the lower bounds on the energy terms in M and store these bounds in a matrix D, then we can define the lower bound Emin on the energy of a minimized system as
| (35) |
The computation of Emin can be done in time O(h2) with a precomputed pairwise energy matrix. The use of a precomputed residue-indexed lower-bound pairwise energy matrix avoids the computation of O(a2) energy terms, where a ≫ h is the total number of atoms in the system.
The precomputed energy matrix in the original K* is indexed over all residues and over all rotamers for each residue, since the same rotamer can be in several different conformations, depending on the type of the neighboring residues (see Sec. 2.2). Thus, for a system with h flexible residues and m rotamers for each residue, we precompute a (hm + 1) × (hm + 1) residue-indexed lower-bound pairwise energy matrix V whose elements V00, V0i, Vi0, and Vij are analogous to the elements of D.
To compute the lower bounds on the minimized template, intra-residue, residue-to-template, and pairwise energy terms, we allow rotamers to assume the best possible conformation for the given relative system (template, self-, or pairwise). However, the movement of the rotamer dihedrals is constrained to a hypercuboid region of conformation space, called a voxel [43, 35], so that one rotamer will not minimize into another. We use a voxel of ±9° for each χ angle.
C.2 Application of the Pairwise Energy Matrix
Energy precomputation is employed both for pruning with MinDEE (Sec. 2.2) and for the ε-approximation algorithms (Secs. 4.1 and Appendix B). The MinDEE criterion (Eq. 2) uses both the lower- and the upper-bound (Appendix C.3) precomputed energy terms to determine which rotamers are not part of the energy-minimized GMEC. Thus, there is no need to re-compute the minimum and maximum energies every time Eq. (2) is evaluated.
Both the intra- and inter-mutation filters (Propositions 3 and 4, respectively) require that a lower bound on the energy-minimized conformation be computed. For this purpose, a lookup in the lower-bound pairwise energy matrix is performed and the terms involved in the given conformation are added, analogously to Eq. (35). The computation of a lower bound on the energy of a conformation permits a subset of the conformations to be pruned before the computationally-expensive full energy-minimization stage. The full energy minimization of a given system requires the simultaneous minimization of all of the flexible residues for the system, a much more costly process than the pairwise minimization performed for the precomputations. Moreover, once the pairwise matrices are precomputed, they can be used in any mutation search that involves the same residues. Thus, in a protein-ligand system, a redesign for a different ligand requires the re-computation only of the terms involving the ligand.
C.3 Improved Energy Bounds Computation
Analogously to the definition of matrix D in Appendix C.1, we define the matrix F to be the residue-indexed upper-bound pairwise energy matrix, which facilitates the computation of the upper-bound Emax on the maximized energy of a system:
| (36) |
Analogously to the definition of V (see Appendix C.1), when we index over all rotamers for all residues, we can define the (hm + 1) × (hm + 1) residue-indexed upper-bound pairwise energy matrix U, whose elements U00, U0i, Ui0, and Uij are upper-bounds on the corresponding energy terms.
The original K* algorithm [25] used a steepest-descent minimization scheme to precompute lower-bound energy matrices. To improve the minimization results, we 1) refined the implementation of the steepest-descent algorithm, and 2) implemented a random sampling with steepest descent algorithm that explores the energy landscape within a voxel better than the local steepest-descent algorithm. Empirically, however, the computed minimum energy bounds using multiple random-sampling starting points appear to be over-optimistic and present a worse approximation to the actual conformation energies. The resulting lower bounds lm from multiple minimization starting points are necessarily at least as low as the corresponding lower bounds ls computed by minimizing only from the center of the voxels, lm ≤ ls. Choosing a good starting point for the energy minimization of a full conformation that could use the additional information of the pair-wise lm bounds is a difficult task, since the different addends involved in the computation of lm (analogous to Eq. 35) may actually result from incompatible starting points. Moreover, using multiple starting points for full energy-minimization is computationally infeasible (see Appendix C.2). Thus, using multiple minimization starting points for lower-bounds computation in fact increases the gap between lower bounds and actual energies (i.e., the lower bounds are less achievable). As a result, the ε-approximation algorithms (Secs. 4.1 and Appendix B) require the full minimization of a larger number of conformations before the provable halting conditions (Propositions 3 and 4) are reached. Hence, we chose to compute the pairwise minimum energy bounds using steepest-descent minimization starting at the center of the voxel space.
While min energies may appear as a natural concept, the computation of max energies (pairwise-computed maximum energy bounds) presents both conceptual and practical challenges. A simple maximization algorithm cannot be used, since most rotamer systems will maximize into a steric clash, which would make max bounds biophysically inapplicable. Moreover, energy functions, such as AMBER [44, 6], are not well-defined for high energies. However, max bounds are used only in the MinDEE framework, where, indirectly, minimized conformations are compared to determine which ones are provably not the minimized-GMEC. We can thus think of the max energy for a given rotamer system as the worst minimization this system can achieve. Hence, we chose to compute max energies as max(M), where M is the set of energies obtained by steepest-descent minimization from multiple starting points (max of mins). In all our experiments we used 200 randomly-chosen starting points per voxel.
Footnotes
For brevity, we will henceforth refer to this algorithm as the Hybrid MinDEE-K* algorithm.
The voxel space for each rotamer can be multi-dimensional, depending on the number of dihedrals. The largest number of dihedrals for a single rotamer is 4 (Arg and Lys).
A mutation sequence specifies an assignment of amino-acid type to each residue position in a protein.
References
- 1.Bolon D, Mayo S. Enzyme-like proteins by computational design. PNAS USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cane D, Walsh C, Khosla C. Harnessing the biosynthetic code: combinations, permutations, and mutations. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 3.Challis G, Ravel J, Townsend C. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 4.Chazelle B, Kingsford C, Singh M. A semidefinite programming approach to side-chain positioning with new rounding strategies. INFORMS Journal on Computing, Computational Biology Special Issue. 2004;16(4):380–392. [Google Scholar]
- 5.Conti E, Stachelhaus T, Marahiel M, Brick P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of Gramicidin S. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornell W, Cieplak P, Bayly C, Gould I, Merz K, Ferguson D, Spellmeyer D, Fox T, Caldwell J, Kollman P. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 7.Dahiyat B, Mayo S. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 8.Desmet J, Maeyer M, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 9.Desmet J, Spriet J, Lasters I. Fast and accurate side-chain topology and energy refinement (FASTER) as a new method for protein structure optimization. Proteins. 2002;48:31–43. doi: 10.1002/prot.10131. [DOI] [PubMed] [Google Scholar]
- 10.Doekel S, Marahiel M. Dipeptide formation on engineered hybrid peptide synthetases. Chem Biol. 2000;7:373–384. doi: 10.1016/s1074-5521(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 11.Dwyer M, Looger L, Hellinga H. Computational design of a biologically active enzyme. Science. 2004;304:1967–1971. doi: 10.1126/science.1098432. [DOI] [PubMed] [Google Scholar]
- 12.Eppelmann K, Stachelhaus T, Marahiel M. Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry. 2002;41:9718–9726. doi: 10.1021/bi0259406. [DOI] [PubMed] [Google Scholar]
- 13.Georgiev I, Donald BR. Dead-end elimination with backbone flexibility. Bioinformatics. 2007;23(13):i185–94. doi: 10.1093/bioinformatics/btm197. [DOI] [PubMed] [Google Scholar]
- 14.Georgiev I, Lilien R, Donald BR. Improved pruning algorithms and divide-and-conquer strategies for dead-end elimination, with application to protein design. Bioinformatics; Proc. International Conference on Intelligent Systems for Molecular Biology (ISMB); Fortaleza, Brazil. 2006; 2006a. pp. e174–183. [DOI] [PubMed] [Google Scholar]
- 15.Georgiev I, Lilien R, Donald BR. A novel minimized dead-end elimination criterion and its application to protein redesign in a hybrid scoring and search algorithm for computing partition functions over molecular ensembles. International Conference on Research in Computational Molecular Biology (RECOMB); Venice, Italy. 2006b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein R. Efficient rotamer elimination applied to protein side-chains and related spin glasses. Biophys J. 1994;66:1335–1340. doi: 10.1016/S0006-3495(94)80923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon D, Mayo S. Radical performance enhancements for combinatorial optimization algorithms based on the dead-end elimination theorem. J Comput Chem. 1998;19:1505–1514. [Google Scholar]
- 18.Hellinga H, Richards F. Construction of new ligand binding sites in proteins of known structure: I. computer-aided modeling of sites with pre-defined geometry. J Mol Biol. 1991;222:763–785. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 19.Jaramillo A, Wernisch L, Héry S, Wodak S. Automatic procedures for protein design. Comb Chem High Throughput Screen. 2001;4:643–659. doi: 10.2174/1386207013330724. [DOI] [PubMed] [Google Scholar]
- 20.Jin W, Kambara O, Sasakawa H, Tamura A, Takada S. De novo design of foldable proteins with smooth folding funnel: Automated negative design and experimental verification. Structure. 2003;11:581–591. doi: 10.1016/s0969-2126(03)00075-3. [DOI] [PubMed] [Google Scholar]
- 21.Keating A, Malashkevich V, Tidor B, Kim P. Side-chain repacking calculations for predicting structures and stabilities of heterodimeric coiled coils. Proc Natl Acad Sci USA. 2001;98:14825–14830. doi: 10.1073/pnas.261563398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lasters I, Desmet J. The fuzzy-end elimination theorem: correctly implementing the side chain placement algorithm based on the dead-end elimination theorem. Protein Eng. 1993;6:717–722. doi: 10.1093/protein/6.7.717. [DOI] [PubMed] [Google Scholar]
- 23.Lazaridis T, Karplus M. Effective energy function for proteins in solution. PROTEINS: Structure, Function, and Genetics. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 24.Leach A, Lemon A. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins. 1998;33:227–239. doi: 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 25.Lilien R, Stevens B, Anderson A, Donald BR. A novel ensemble-based scoring and search algorithm for protein redesign, and its application to modify the substrate specificity of the Gramicidin Synthetase A phenylalanine adenylation enzyme. Journal of Computational Biology. 2005;12(6–7):740–761. doi: 10.1089/cmb.2005.12.740. [DOI] [PubMed] [Google Scholar]
- 26.Looger L, Dwyer M, Smith J, Hellinga H. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423:185–190. doi: 10.1038/nature01556. [DOI] [PubMed] [Google Scholar]
- 27.Looger L, Hellinga H. Generalized dead-end elimination algorithms make large-scale protein side-chain structure prediction tractable: Implications for protein design and structural genomics. J Mol Biol. 2001;307:429–445. doi: 10.1006/jmbi.2000.4424. [DOI] [PubMed] [Google Scholar]
- 28.Lovell S, Word J, Richardson J, Richardson D. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 29.Marvin J, Hellinga H. Conversion of a maltose receptor into a zinc biosensor by computational design. PNAS. 2001;98:4955–4960. doi: 10.1073/pnas.091083898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mootz H, Schwarzer D, Marahiel M. Construction of hybrid peptide syn-thetases by module and domain fusions. Proc Natl Acad Sci USA. 2000;97:5848–5853. doi: 10.1073/pnas.100075897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Najmanovich R, Kuttner J, Sobolev V, Edelman M. Side-chain flexibility in proteins upon ligand binding. Proteins. 2000;39(3):261–8. doi: 10.1002/(sici)1097-0134(20000515)39:3<261::aid-prot90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Pierce N, Spriet J, Desmet J, Mayo S. Conformational splitting: a more powerful criterion for dead-end elimination. J Comput Chem. 2000;21:999–1009. [Google Scholar]
- 33.Pierce N, Winfree E. Protein design is NP-hard. Protein Eng. 2002;15:779–782. doi: 10.1093/protein/15.10.779. [DOI] [PubMed] [Google Scholar]
- 34.Ponder J, Richards F. Tertiary templates for proteins: Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 35.Rienstra C, Tucker-Kellogg L, Jaroniec C, Hohwy M, Reif B, McMahon M, Tidor B, Lozano-Pérez T, Griffin R. De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proc Natl Acad Sci USA. 2002;99:10260–10265. doi: 10.1073/pnas.152346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider A, Stachelhaus T, Marahiel M. Targeted alteration of the substrate specificity of peptide synthetases by rational module swapping. Mol Gen Genet. 1998;257:308–318. doi: 10.1007/s004380050652. [DOI] [PubMed] [Google Scholar]
- 37.Schwarzer D, Finking R, Marahiel M. Nonribosomal peptides: from genes to products. Nat Prod Rep. 2003;20:275–287. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 38.Shah P, Hom G, Mayo S. Preprocessing of rotamers for protein design calculations. J Comput Chem. 2004;25:1797–1800. doi: 10.1002/jcc.20097. [DOI] [PubMed] [Google Scholar]
- 39.Stachelhaus T, Mootz H, Marahiel M. The specificiy-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- 40.Stachelhaus T, Schneider A, Marahiel M. Rational design of peptide antibiotics by targeted replacement of bacterial and fungal domains. Science. 1995;269:69–72. doi: 10.1126/science.7604280. [DOI] [PubMed] [Google Scholar]
- 41.Stevens B, Lilien R, Georgiev I, Donald BR, Anderson A. Redesigning the PheA domain of Gramicidin Synthetase leads to a new understanding of the enzyme’s mechanism and selectivity. Biochemistry. 2006;45(51):15495–15504. doi: 10.1021/bi061788m. [DOI] [PubMed] [Google Scholar]
- 42.Street A, Mayo S. Computational protein design. Structure. 1999;7:R105–R109. doi: 10.1016/s0969-2126(99)80062-8. [DOI] [PubMed] [Google Scholar]
- 43.Tucker-Kellogg L. PhD thesis. Massachusetts Institute of Technology; 2002. Systematic Conformational Search with Constraint Satisfaction. [Google Scholar]
- 44.Weiner S, Kollman P, Case D, Singh U, Ghio C, Alagona G, Profeta S, Weiner P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]