

Abstract
We have demonstrated that it is possible to radically change the specificity of maltose binding protein by converting it into a zinc sensor using a rational design approach. In this new molecular sensor, zinc binding is transduced into a readily detected fluorescence signal by use of an engineered conformational coupling mechanism linking ligand binding to reporter group response. An iterative progressive design strategy led to the construction of variants with increased zinc affinity by combining binding sites, optimizing the primary coordination sphere, and exploiting conformational equilibria. Intermediates in the design series show that the adaptive process involves both introduction and optimization of new functions and removal of adverse vestigial interactions. The latter demonstrates the importance of the rational design approach in uncovering cryptic phenomena in protein function, which cannot be revealed by the study of naturally evolved systems.
Furthering our understanding of substrate specificity redesign is essential not only for technological applications (1, 2), but also for characterizing the mechanisms leading to adaptation of protein function in biological evolution (3). Despite recent progress (4–6), the rational, structure-based redesign of substrate binding specificity in proteins and enzymes remains a major challenge in protein chemistry. Here we demonstrate that it is possible to change radically the specificity of maltose binding protein (MBP) by using automated computational techniques (7) to design tetrahedral zinc primary coordination spheres in the place of maltose binding residues.
MBP is one of the periplasmic binding proteins from Escherichia coli that are involved in chemotaxis and ligand transport (8). These soluble receptors are members of a protein superfamily (9) consisting of a single polypeptide chain that folds into two domains connected by a hinge region (10). Ligand binding sites are located at the interface between the two domains. The proteins adopt two conformations: a ligand-free open form and a ligand-bound closed form that interconvert through a combined bending–twisting motion around the hinge. This structural motif and binding mechanism is found not only in the E. coli periplasmic binding proteins (9), but also in DNA repressors (11, 12) and complex eukaryotic receptors (13, 14). This family exhibits large diversity in molecular recognition, binding chemically disparate species such as carbohydrates (15, 16), amino acids (17–20), neurotransmitters (14), oligopeptides (21, 22), anions (23, 24), and cations (25, 26).
The remarkable adaptability of this superfamily is likely to be the consequence of locating ligand binding sites at the interface between two domains. In this arrangement, the residues forming the binding site are placed at the surface of their respective domains; yet in the complex the ligand is placed within an environment that resembles the interior of a protein. This combines the plasticity of evolving sites on protein surfaces (27) with the desirable chemical characteristics of placing ligands within solvent-excluding regions. Furthermore, the ligand-mediated conformational change allows the development of linked functions by using a simple conformational coupling mechanism (28). In this way allosteric control and cooperative ligand binding have evolved in some of the members of this protein superfamily (29). Conformational coupling has also been exploited to design biosensors (1) in which ligand binding is linked to an optical or electrochemical response in several bacterial periplasmic binding proteins (30, 31), including MBP (32, 33).
Materials and Methods
Design.
The coordinates of the closed structure of MBP [PDB ID code 1anf (34)] were used in the calculations. Searches for His3⋅Zn sites by the DEZYMER program (7) were limited to residues located at the interface of the two domains. The geometry around the zinc center was described as an ideal tetrahedron with metal allowed to coordinate with the Nδ or Nɛ of histidine and within the plane of the imidazole ring. A subset of designs that have primary coordination spheres shared by the two domains were selected from the total set of predictions by visual inspection.
Mutagenesis, Protein Expression, and Purification.
The cloning vector used for expressing MBP is a variant of the described vector (32), lacking a C-terminal oligohistidine peptide. Oligonucleotide-directed mutagenesis was carried out as described (32). Proteins were produced by induction with isopropyl β-D-thiogalactoside (IPTG) as described (32). Mutants that retained maltose binding were purified on an amylose affinity column (35). Metal-binding mutants were purified by immobilized metal ion affinity chromatography and ion-exchange chromatography: cleared lysate (in 100 mM NaCl/20 mM Tris⋅HCl, pH 7.5) was loaded on 30 ml Chelating Fast Flow Sepharose (Amersham Pharmacia), eluted with a 0–100 mM imidazole gradient, exchanged into 20 mM Tris⋅HCl, pH 8, loaded on 10 ml DEAE Sepharose (Amersham Pharmacia), and eluted with a 0–100 mM NaCl gradient. Purified proteins were coupled to the fluorescent reporter group N-(((2-iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole (IANBD) as described (32). Conjugates were treated with 50 mM EDTA and dialyzed extensively to remove adventitious metal [10 mM Mops/10 mM NaCl, pH 7.5, which was rendered metal-free by extraction with dithizonechloroform (36)].
Fluorescent Allosteric Signal Transduction (FAST) Assay.
Small amounts of fluorescently labeled protein (20–100 nM) were added to 3 ml of metal-free buffer (10 mM Mops/10 mM NaCl, pH 7.5) stirred at 25°C. The emission intensity of the fluorophore (λem = 538 nm, λex = 470 nm, emission bandpass = 16 nm) was measured as ligand was added incrementally.
Isothermal Titration Calorimetry (ITC).
Experiments were performed with a VP-ITC Microcalorimeter (Microcal, Amherst, MA; ref. 37). Ligand (maltose or zinc) was titrated into 1.5 ml of a 200 μM solution of protein in 10 mM Mops and 10 mM NaCl, pH 7.5. The heats of dilution of zinc and maltose are negligible.
Stopped-Flow Kinetics.
Experiments were performed with a SFM-400 stopped-flow machine (Biologic, Grenoble, France). A 20 μM solution of protein was mixed with an equal volume of buffer with varying concentration of ligand. The rate of formation of the closed state was monitored by the change in fluorescence intensity of IANBD (λex = 470 nm, λem > 515 nm). Each data point is the average of at least 18 individual mixing reactions, performed on at least two different days.
Results and Discussion
Design.
We have designed a small family of zinc binding sites at various locations near or within the maltose binding interface, using the diversity generated in this way to establish trends between location and ligand binding (38, 39). The initial designs were generated by using the DEZYMER program (7) to build idealized tetrahedral zinc coordination sites comprised of three histidines and a water molecule. These sites span the interdomain interface region such that at least one histidine is contributed by each domain. This arrangement is predicted to result in zinc-mediated formation of the closed state, analogous to maltose binding, and allows zinc binding to be monitored by using a fluorescent probe that is allosterically coupled to the conformational transition (32).
The DEZYMER program predicted 20 sites within the interdomain interface, four of which were subsequently constructed by oligonucleotide directed mutagenesis (Fig. 1). Two of these sites replace three maltose-binding residues [A1: A63IIH, R66IIH, W340IH; A2: A63IIH, R66IIH, Y155IH (subscripts refer to the domain in which the residue is placed; see Fig. 1)]; two are located at the rim of the maltose binding site, leaving the original binding site intact (B1: K42IIH, E44IIH, Y341IH; B2: P48IIH, G69IIH, S337IH). To maintain steric compatibility with the surrounding protein matrix, additional mutations needed to be introduced into the B sites (B1: E45IIA, R344IA; B2: R66IIS, Y70IIA), but not the A sites. Mutation R66IIS in the B2 site removes a hydrogen bond to maltose in the closed state. The structures of all four sites in the open state are sufficiently different that zinc cannot form a tetrahedral binding site in this conformation (models not shown).
Figure 1.

Location of the His3 sites as designed in the closed structure of MBP [PDB ID code 1anf (34)]. The two domains (Domain I, yellow; Domain II, blue) of MBP are connected by a β-strand hinge (green). The positioning of the zinc atom is the same for the A1 and A2 sites (red), and is located within the original maltose-binding pocket, near the hinge. The zinc centers for the B1 (orange) and B2 (pink) sites are located outside of the original maltose-binding pocket. Relative to the plane of the figure, the B2 site is closest to the viewer, followed by the B1 site and then the A sites. The large blue sphere represents the attachment position of the fluorophore IANBD at D95C. Detailed description of designs available in the supplemental data, which is published on the PNAS web site, www.pnas.org.
Binding of zinc in the presence and absence of maltose and the binding of maltose in the presence and absence of zinc were determined by measuring changes in the fluorescence intensity of IANBD conjugated to D95C, which monitors ligand-mediated formation of the closed state (Table 1). All four sites bind zinc, although two classes of behavior are observed. The A sites have lost the ability to bind maltose, whereas the B sites still exhibit maltose-mediated formation of the closed state. This is consistent with the mutation of three of the maltose-binding residues in the A site, and retention of the majority of these residues in the B sites. The A sites exhibit zinc-mediated formation of the closed state (Fig. 2), whereas the B sites do not, although the latter do bind zinc, as indicated by the observed zinc dependence of maltose affinity (Fig. 3). All four sites show the same magnitude in fluorescence change under saturating ligand concentrations (zinc or maltose), indicating that conformational coupling between the binding site and reporter group has been retained, and that the protein structure has not been greatly perturbed.
Table 1.
Ligand binding affinities of the designs
| Site | Kd Zn − Malt. | Kd Zn + Malt. | Kd Malt. − Zn | Kd Malt. + Zn |
|---|---|---|---|---|
| A1 | 56 ± 5 | 56 ± 5 | nb | nb |
| A2 | 135 ± 4 | 135 ± 4 | nb | nb |
| B1 | nb | 5 ± 1 | 0.9 ± 0.05 | 5 ± 0.3 |
| B2 | nb | 1.4 ± 0.3 | 500 ± 12 | 150 ± 7 |
| WT | nb | nb | 0.8 ± 0.03 | 0.8 ± 0.03 |
The A sites show changes in fluorescence in response to zinc but not maltose (1 mM). The B sites show changes in fluorescence in response to maltose, but not zinc (10 mM). The zinc-mediated change in fluorescence of the A sites is not modulated by the presence of maltose. The maltose-mediated changes in fluorescence in the B sites are modulated by the presence of zinc. All Kd values are reported in μM. WT, wild-type MBP; nb, no binding; Malt., maltose.
Figure 2.

Zinc binding curves for A1 (Kd = 56 μM) and A2 (Kd = 135 μM). Each data point represents the average of three separate titrations. (Inset) the goodness of fit at low zinc concentrations, with error bars removed for clarity. The data are fit to a hyperbolic binding isotherm (32).
Figure 3.

Dependence of maltose affinity on zinc concentration for B2. (Inset) Binding curves for maltose in the presence of five different concentrations of zinc, with error bars removed for clarity (all curves are fit to standard hyperbolic binding isotherms): 0 μM (●), 1 μM (■), 2 μM (⧫), 5 μM (▴), 10 μM (▾). The change in affinity for maltose as a function of zinc concentration can be fit to a hyperbolic binding isotherm, indicating a Kd for zinc of 1.4 ± 0.3 μM.
The binding patterns can be accounted for in a simple qualitative model in which zinc-mediated formation of the closed state is opposed by the burial of polar maltose-binding residues and the trapping of residual water molecules that would have been displaced in the maltose complex. These factors are likely to be more extensive in the B sites than the A sites because in the former the majority of maltose binding residues are left largely intact, whereas in the latter the maltose binding site is partially substituted by the zinc primary coordination sphere. This suggests that successful adaptation requires both the introduction of desired interactions (“target state optimization”), as well as removal of interfering vestigial interactions (“competing state destabilization”), which has also been observed to be a dominant factor in the design of other systems (38, 40). Next, we describe the use of progressive iterative design cycles (40) to test mechanisms by which binding behavior can be modulated and improved.
Design Iteration 1: Combining Zinc Binding Sites.
As a first approach to increasing zinc affinity, the A and B sites were combined to construct a receptor filled with two zinc centers. Of the four possibilities, molecular modeling revealed that only B1 combinations could be considered because B2 is sterically incompatible with both the A1 and A2 sites (B2 requires a small side chain at residue 66II; both A designs require H66II). The only B1 combination that is sterically allowed is with A2 (H341I of B1 interferes with the coordination sphere of A1). The resulting A2B1 construct exhibits sigmoidal zinc binding with a half-saturation point of 103 μM (Fig. 4), indicating that zinc-mediated formation of the closed state is improved as expected. Binding involves an almost perfectly cooperative interaction between the two sites, because the observed Hill coefficient (41) is 2.0 (Fig. 4 Inset). Unlike most of the other mutations (see above), the A2B1 mutations greatly alter the maximal zinc-mediated fluorescence change. The A2B1 mutant shows a 17.7-fold fluorescence increase upon zinc binding, whereas either parent shows only a 3.3-fold increase (the same as observed in the maltose-dependent response of the wild-type receptor). Furthermore, the fluorophore in the zinc-saturated, closed state of this mutant is much more sensitive to external quenchers such as iodide than the equivalent state in any of the other mutants (data not shown). Taken together these results suggest that the structure of the closed state of the A2B1 mutant protein has been perturbed, unlike either single site.
Figure 4.

Zinc binding in the A2B1 protein. Zinc shows sigmoidal binding. Data fit well to a binding isotherm that models perfect cooperativity between the two sites (41). (Inset) Hill plot transformation of the data (41). The slope of 2.0 in the linear region underscores the extreme cooperative linkage between the two sites. Error bars are smaller than the symbols used.
Design Iteration 2: Optimization of a Primary Coordination Sphere.
Another approach to enhance zinc affinities is to improve the intrinsic coordination geometry of the initial designs by constructing saturated primary coordination spheres, instead of His3⋅Zn sites. We limited these experiments to the A sites, because these initial designs exhibit metal-mediated formation of the closed state. As a first step, the contributions of the histidines in the A1 and A2 designs were quantified by alanine scanning mutagenesis (Table 2). The locations of the primary coordination spheres in these two sites overlap, having H63II and H66II in common. In both sites, the H63IIA mutation destroys binding. The mutations in the nonoverlapping residues (A1: H340IA; A2: H155IF) were also found to be important. Unexpectedly, in both cases H66IIA did not fully abolish binding, and it was found that nearby D65II plays in important role in coordinating zinc (Fig. 5).
Table 2.
Alanine scanning mutagenesis of the primary coordination sphere in designs A1 and A2
| Design | Variant | Kd Zn, μM |
|---|---|---|
| A1 | 56 ± 5 | |
| H63IIA | nb | |
| H66IIA | 400 ± 23 | |
| H340IA | ≈1200 | |
| D65IIA | nb | |
| A2 | 135 ± 4 | |
| H63IIA | nb | |
| H66IIA | 89 ± 13 | |
| H155IF | nb | |
| D65IIA | nb |
Subscript indicates the domain in which a residue is located (see Fig. 1). nb, no binding.
Figure 5.

Stereo image showing the residues of the A sites (clockwise, from top left: H155II, H340II, D65I, H66I, H63I). The image is presented in the same orientation as Fig. 1. The primary coordination spheres of A1 (H63I, H66I, H340II) and A2 (H63I, H66I, H155II) overlap, and are distinguished by the third histidine (positions 340II and 155II, respectively). Mutagenesis studies revealed that D65I is a fortuitous member of the metal coordination sphere in both sites (Table 2).
Molecular modeling revealed that completion of the primary coordination sphere at the A1 site requires placement of the fourth ligand at residue 155I, and at residue 340I for the A2 site. By happenstance, both positions have wild-type residues that can potentially coordinate metals. Mutations were therefore constructed to test the extent of potential, serendipitous coordination by these residues. In the A1 site, the Y155IF mutation significantly reduces zinc binding, indicating that the tyrosine hydroxyl completes the coordination sphere in this center (Table 3). Molecular modeling suggests, however, that tyrosine is nonoptimal, and that glutamate has better coordination geometry. As expected, the Y155IE mutant has higher affinity for zinc than the original A1 design.
Table 3.
Optimization of primary coordination spheres in the A1 and A2 designs
| Design | Variant | Kd Zn, μM |
|---|---|---|
| A1 | 56 ± 5 | |
| Y155F | 325 ± 37 | |
| Y155H | 292 ± 15 | |
| Y155A | 148 ± 15 | |
| Y155E | 18 ± 0.8 | |
| A2 | 135 ± 4 | |
| W340H | 292 ± 15 | |
| W340L | 920 ± 95 | |
| W340A | 115 ± 9 | |
| W340S | 57 ± 3 | |
| W340N | 462 ± 52 | |
| W340D | 58 ± 4 | |
| W340Q | 58 ± 3 | |
| W340E | 22 ± 1 | |
| A* | 155E + 340E | 5.1 ± 0.2 |
Both residues 155 and 340 are located in domain II.
In the A2 site the serendipitous wild-type coordinating group is the Nɛ of W340I. Construction of a series of mutations (Table 3) at this position is consistent with this interpretation, bearing in mind that mutants can either destroy coordination (W340IL, W340IN), isosterically substitute for Nɛ (W340IE, W340IQ, W340IH), or allow access of coordinating water (W340IA, W340IS, W340ID). The W340IE mutant in the A2 site has higher affinity for zinc than the original A2 design, presumably because in addition to satisfying coordination geometry, the charge balance is improved. The predicted fourth vertex in the A1 and A2 sites corresponds to the vertices that are not shared between these two designs, suggesting that a new site can be designed that is a fusion of these two original designs (Fig. 5). A simple His4 fusion of the two sites is not optimal, however, because histidines are too short to form a geometrically perfect tetrahedron. Inspection of the molecular model showed that a site combining the glutamate substitutions at each fourth vertex yields the geometrically best design. The corresponding His2Glu2 site that combines both optimized A1 and A2 coordination spheres yielded site A* (H63II, H66II, E155I, E340I) that has the best observed affinity for zinc (Kd = 5.1 μM).
Design Iteration 3: Manipulation of Intrinsic Conformational Equilibria.
Thermodynamic linkage between the intrinsic equilibrium of the open and closed states in the absence of ligand (ΔAGoc) and the intrinsic affinity of the closed state for zinc (ΔCGb) dictates that the overall affinity for zinc (ΔAppGb) is the sum of these two components (Scheme 1 and Eq. 1):
 |
 |
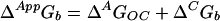 |
1 |
Consequently, zinc binding can also be improved by manipulating the intrinsic equilibrium between the open and closed states. We have explored two avenues to achieve this: replacement of vestigial maltose binding residues with hydrophobic residues within the ligand-binding pocket to stabilize formation of the closed state, and constructing a mutant in the hinge region at a location that is conformationally distinct in the two states [“protoallosteric” site (32)] to destabilize the open state.
E111I and K15II form hydrogen bonds with maltose in the closed state (8) that are not satisfied in the zinc design. These two residues were changed to Met and Ala, respectively, within the A* background, and showed significant improvement in zinc affinity over A*, both singly and in a double-mutant combination (Table 4).
Table 4.
Manipulation of conformational equilibria
| Design | Variant | Kd Zn, μM |
|---|---|---|
| A* | 5.1 ± 0.2 | |
| K15IIA | 1.0 ± 0.1 | |
| E111IM | 1.0 ± 0.1 | |
| K15IIA + E111IM | 1.0 ± 0.2 | |
| A* | I329F | 0.35 ± 0.04 |
Residues K15 and E111 are residues that form hydrogen bonds with maltose in the wild-type receptor. Removal of these vestigial interactions is intended to favor formation of the closed state. Residue 329 is located in the hinge region. Mutation I329F is intended to destabilize the open state. Subscript indicates the domain in which a residue is located (see Fig. 1). Residue 329 is located in the hinge region.
Residue I329 is located in a hinge-region crevice that opens and closes as the protein changes conformations. It is tightly packed in the open state but not the closed state, and thus is predicted to be a determinant of the relative stability of each state. Substitution with an I329F mutation—which is predicted to be sterically less compatible with the open state, but does not pack badly in the closed state—resulted in a significantly increased affinity for zinc within the A* background (Kd = 350 nM). This mutant also resulted in a significant decrease in the maximum, zinc-mediated fluorescence change. Because such a change in fluorescence was not observed in the E111IM and K15IIA mutants, this effect is most likely to be due to a change in the environment of the fluorophore, which is attached in close proximity to residue 329, rather than reflecting an observable change in the equilibrium between the open and closed states.
Analysis of Ligand-Binding Mechanisms.
To further determine whether the properties of the redesigned receptors are consistent with the intent of the designs, we compared the thermodynamics and kinetics of ligand binding of the wild-type and engineered proteins. Isothermal titration calorimetry (ITC) experiments (37) show that for wild-type MBP, the enthalpy of maltose binding (ΔAppHb) is endothermic and strongly temperature dependent, with a large, negative ΔCp (42; Fig. 6). This indicates that ΔAppGb is dominated by entropic effects, presumably resulting from desolvation of the nonpolar surfaces of both maltose and the maltose-binding pocket (43). In contrast, zinc binding is exothermic and has significantly smaller ΔCp values (Fig. 6), consistent with a process that is dominated by enthalpic contributions. This is further underscored by the observation that, as vestigial maltose binding residues are removed, the zinc binding mutants show decreased ΔCp values (A* > A*K15A+E111M > A*I329F > A2B1). Indeed, in the limit, zinc binding may be enthalpically determined, analogous to the formation of the Zn⋅EDTA complex.
Figure 6.

Temperature dependence of the enthalpy of zinc binding for A* (■), A*K15A+E111 M (▴), A*I329F (▾), A2B1 (⊙), and EDTA (♦) and of maltose binding for MBP (●) as determined by isothermal titration calorimetry. The slope of these lines gives the observed heat capacities, ΔCp, (cal mol−1⋅K−1): MBP, −297; A*, −194; A*K15A+E111M, −184; A*I329F, −99; A2B1, −10; EDTA, −6.
The kinetic mechanistic consequences of binding were investigated by stopped-flow studies. In MBP (Fig. 7a) and other sugar-binding proteins (44), ligand binding and formation of the closed state are concerted, exhibiting simple pseudo-first-order kinetics (Scheme 2 and Eq. 2):
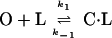 |
 |
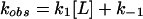 |
2 |
where k1 and k−1 are the on and off rates of binding (45). In contrast, we observe a two-step mechanism for zinc binding in the A* protein (Fig. 7b) in which an encounter complex (O⋅L) is formed before the closed ligand-bound state (C⋅L) (Scheme 3 and Eq. 3):
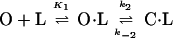 |
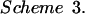 |
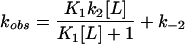 |
3 |
where K1 is the equilibrium of formation of an open state encounter complex (k1/k−1) and k2 and k−2 are the rate constants for closing and opening of the encounter complex (45).
Figure 7.

Kinetics of ligand binding, as monitored by changes in fluorescence of IANBD in a stopped-flow apparatus, at 25°C. (a) MBP binds maltose with classic pseudo-first-order dependence on maltose concentration. (b) The binding of zinc to A* shows more complex behavior that is consistent with a two-state model in which formation of the closed state is rate-limiting (see text, Scheme 3, and Eq. 3).
The degree of linkage between formation of the ligand complex and the closed state is indicative of the differences between the fundamental mechanisms of ligand binding. Maltose binding requires desolvation of the entire ligand binding pocket and the nonpolar sugar surface. Because formation of the closed state also requires desolvation of the binding pocket, the two processes are tightly coupled. Zinc binding, however, requires only a subset of the primary coordination sphere to form an intermediate zinc complex, albeit with weaker affinity than the final state. Binding and closure are therefore not obligately linked. A similar two-step mechanism is also observed for the periplasmic phosphate binding protein (46). The thermodynamic and kinetic changes observed in the A* design are, therefore, consistent with the nature of the new receptor ligand.
Conclusions
We have demonstrated that it is possible to radically change the specificity of the maltose binding protein by converting it into a zinc sensor using a rational design approach. An iterative progressive design strategy led to variants with increased zinc affinity by combining binding sites, optimizing the primary coordination sphere, and exploiting conformational equilibria. As anticipated, the placement of the binding site at the interface of the two domains provides a great adaptive potential in receptors such as MBP that can be exploited in the redesign of specificity.
Rational protein design can be viewed as an evolutionary pathway that can be experimentally observed, allowing explicit analysis of partially successful intermediates, which are usually not observed. We find that the adaptive process involves not only the introduction and optimization of new functions, but also the removal of vestigial interactions. The latter can be considered a cryptic adaptive process because we can typically only identify interactions that are present in the final result of adaptation, and not those that have been removed. It is becoming increasingly clear that rational protein design is an important tool by which such cryptic processes in proteins can be uncovered. For instance, the importance of destabilizing competing interactions, another cryptic protein function, in determining specificity in proteins has also been demonstrated by design approaches (40, 47–49).
The parental MBP protein contains a rationally designed link between ligand binding and fluorescent response of a reporter group (32). This was originally established by exploiting a conformational coupling mechanism in which the reporter group, placed at some distance from the binding site, is sensitive to changes in local environment that occur as a consequence of ligand-mediated domain closure, rather than by direct steric interactions with the ligand itself (32). The absence of direct interactions between the reporter group and ligand binding site allows ligand specificity to be manipulated while retaining fluorescent signal transduction, as we show here. As predicted (1), it is therefore possible to develop families of biosensors with different specificities by engineering receptor binding surfaces. The next challenge is to redesign receptors by using ligands that are both sterically more complex than metals and whose thermodynamics are not dominated by enthalpic interactions alone.
Supplementary Material
Acknowledgments
This work was supported by grants from the Office of Naval Research and the National Institutes of Health.
Abbreviations
- MBP
maltose binding protein
- IANBD
N-(((2-iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole
References
- 1.Hellinga H W, Marvin J S. Trends Biotechnol. 1998;16:183–189. doi: 10.1016/s0167-7799(98)01174-3. [DOI] [PubMed] [Google Scholar]
- 2.Rubingh D N. Curr Opin Biotechnol. 1997;8:417–422. doi: 10.1016/s0958-1669(97)80062-6. [DOI] [PubMed] [Google Scholar]
- 3.Babbitt P C, Gerlt J A. Adv Protein Chem. 2000;55:1–28. doi: 10.1016/s0065-3233(01)55001-9. [DOI] [PubMed] [Google Scholar]
- 4.Bone R, Silen J L, Agard D A. Nature (London) 1989;339:191–195. doi: 10.1038/339191a0. [DOI] [PubMed] [Google Scholar]
- 5.Wilks H M, Cortes A, Emery D C, Halsall D J, Clarke A R, Holbrook J J. Ann NY Acad Sci. 1992;672:80–93. doi: 10.1111/j.1749-6632.1992.tb32662.x. [DOI] [PubMed] [Google Scholar]
- 6.Cedrone F, Menez A, Quemeneur E. Curr Opin Struct Biol. 2000;10:405–410. doi: 10.1016/s0959-440x(00)00106-8. [DOI] [PubMed] [Google Scholar]
- 7.Hellinga H W, Richards F M. J Mol Biol. 1991;222:763–785. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 8.Quiocho F A, Ledvina P S. Mol Microbiol. 1996;20:17–25. doi: 10.1111/j.1365-2958.1996.tb02484.x. [DOI] [PubMed] [Google Scholar]
- 9.Tam R, Saier M H., Jr Microbiol Rev. 1993;57:320–346. doi: 10.1128/mr.57.2.320-346.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quiocho F A. Philos Trans R Soc London B. 1990;326:341–351. doi: 10.1098/rstb.1990.0016. [DOI] [PubMed] [Google Scholar]
- 11.Lewis M, Chang G, Horton N G, Kercher M A, Pace H C, Schumacher M A, Brennan R G, Lu P. Science. 1996;271:1247–1254. doi: 10.1126/science.271.5253.1247. [DOI] [PubMed] [Google Scholar]
- 12.Mauzy C A, Hermodson M A. Protein Sci. 1992;1:843–849. doi: 10.1002/pro.5560010702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armstrong N, Sun Y, Chen G Q, Gouaux E. Nature (London) 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- 14.Slotboom D J, Konings W N, Lolkema J S. Microbiol Mol Biol Rev. 1999;63:293–307. doi: 10.1128/mmbr.63.2.293-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quiocho F A. Biochem Soc Trans. 1993;21:442–448. doi: 10.1042/bst0210442. [DOI] [PubMed] [Google Scholar]
- 16.Quiocho F A. Annu Rev Biochem. 1986;55:287–315. doi: 10.1146/annurev.bi.55.070186.001443. [DOI] [PubMed] [Google Scholar]
- 17.Oh B H, Pandit J, Kang C H, Nikaido K, Gokcen S, Ames G F, Kim S H. J Biol Chem. 1993;268:11348–11355. [PubMed] [Google Scholar]
- 18.Sack J S, Saper M A, Quiocho F A. J Mol Biol. 1989;206:171–191. doi: 10.1016/0022-2836(89)90531-7. [DOI] [PubMed] [Google Scholar]
- 19.Yao N, Trakhanov S, Quiocho F A. Biochemistry. 1994;33:4769–4779. doi: 10.1021/bi00182a004. [DOI] [PubMed] [Google Scholar]
- 20.Kang C H, Shin W C, Yamagata Y, Gokcen S, Ames G F, Kim S H. J Biol Chem. 1991;266:23893–23899. [PubMed] [Google Scholar]
- 21.Nickitenko A V, Trakhanov S, Quiocho F A. Biochemistry. 1995;34:16585–16595. doi: 10.1021/bi00051a006. [DOI] [PubMed] [Google Scholar]
- 22.Sleigh S H, Tame J R, Dodson E J, Wilkinson A J. Biochemistry. 1997;36:9747–9758. doi: 10.1021/bi970457u. [DOI] [PubMed] [Google Scholar]
- 23.Luecke H, Quiocho F A. Nature (London) 1990;347:402–406. doi: 10.1038/347402a0. [DOI] [PubMed] [Google Scholar]
- 24.Pflugrath J W, Quiocho F A. J Mol Biol. 1988;200:163–180. doi: 10.1016/0022-2836(88)90341-5. [DOI] [PubMed] [Google Scholar]
- 25.de Pina K, Navarro C, McWalter L, Boxer D H, Price N C, Kelly S M, Mandrand-Berthelot M A, Wu L F. Eur J Biochem. 1995;227:857–865. doi: 10.1111/j.1432-1033.1995.tb20211.x. [DOI] [PubMed] [Google Scholar]
- 26.Bruns C M, Nowalk A J, Arvai A S, McTigue M A, Vaughan K G, Mietzner T A, McRee D E. Nat Struct Biol. 1997;4:919–924. doi: 10.1038/nsb1197-919. [DOI] [PubMed] [Google Scholar]
- 27.Murzin A G. Trends Biochem Sci. 1993;18:403–405. doi: 10.1016/0968-0004(93)90132-7. [DOI] [PubMed] [Google Scholar]
- 28.Perutz M F. Mechanisms of Cooperativity and Allosteric Regulation in Proteins. Cambridge, U.K.: Cambridge Univ. Press; 1990. [DOI] [PubMed] [Google Scholar]
- 29.Sauer R T. Structure (London) 1996;4:219–222. doi: 10.1016/s0969-2126(96)00025-1. [DOI] [PubMed] [Google Scholar]
- 30.Marvin J S, Hellinga H W. J Am Chem Soc. 1998;120:7–11. [Google Scholar]
- 31.Brune M, Hunter J L, Corrie J E, Webb M R. Biochemistry. 1994;33:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 32.Marvin J S, Corcoran E E, Hattangadi N A, Zhang J V, Gere S A, Hellinga H W. Proc Natl Acad Sci USA. 1997;94:4366–4371. doi: 10.1073/pnas.94.9.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilardi G, Zhou L Q, Hibbert L, Cass A E. Anal Chem. 1994;66:3840–3847. doi: 10.1021/ac00093a047. [DOI] [PubMed] [Google Scholar]
- 34.Quiocho F A, Spurlino J C, Rodseth L E. Structure (London) 1997;5:997–1015. doi: 10.1016/s0969-2126(97)00253-0. [DOI] [PubMed] [Google Scholar]
- 35.Kellermann O K, Ferenci T. Methods Enzymol. 1982;90:459–463. doi: 10.1016/s0076-6879(82)90171-9. [DOI] [PubMed] [Google Scholar]
- 36.Holmquist B. Methods Enzymol. 1988;158:6–12. doi: 10.1016/0076-6879(88)58042-4. [DOI] [PubMed] [Google Scholar]
- 37.Wiseman T, Williston S, Brandts J F, Lin L N. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 38.Benson D E, Wisz M S, Hellinga H W. Proc Natl Acad Sci USA. 2000;97:6292–6297. doi: 10.1073/pnas.97.12.6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wisz M S, Garrett C Z, Hellinga H W. Biochemistry. 1998;37:8269–8277. doi: 10.1021/bi980718f. [DOI] [PubMed] [Google Scholar]
- 40.Hellinga H W. J Am Chem Soc. 1998;120:10055–10066. [Google Scholar]
- 41.Segel I H. Enzyme Kinetics. New York: Wiley Interscience; 1975. [Google Scholar]
- 42.Thomson J, Liu Y, Sturtevant J M, Quiocho F A. Biophys Chem. 1998;70:101–108. doi: 10.1016/s0301-4622(97)00113-0. [DOI] [PubMed] [Google Scholar]
- 43.Makhatadze G I, Privalov P L. Adv Protein Chem. 1995;47:307–425. doi: 10.1016/s0065-3233(08)60548-3. [DOI] [PubMed] [Google Scholar]
- 44.Miller D M, III, Olson J S, Pflugrath J W, Quiocho F A. J Biol Chem. 1983;258:13665–13672. [PubMed] [Google Scholar]
- 45.Johnson K A. The Enzymes. New York: Academic; 1992. pp. 1–61. [Google Scholar]
- 46.Ledvina P S, Tsai A L, Wang Z, Koehl E, Quiocho F A. Protein Sci. 1998;7:2550–2559. doi: 10.1002/pro.5560071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O'Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 48.Hecht M H, Richardson J S, Richardson D C, Ogden R C. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 49.Quinn T P, Tweedy N B, Williams R W, Richardson J S, Richardson D C. Proc Natl Acad Sci USA. 1994;91:8747–8751. doi: 10.1073/pnas.91.19.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.