

Abstract
Activation of the mTOR pathway subsequent to phosphatase and tensin homolog (PTEN) mutation may be associated with glucocorticoid (GC) resistance in acute lymphoblastic leukemia (ALL). The combination activity of rapamycin and dexamethasone in cell lines and xenograft models of ALL was determined. Compared with either drug alone, dexamethasone + rapamycin showed significantly greater apoptosis and cell cycle arrest in some cell lines, which was more frequently seen in T-lineage cell lines with PTEN mutation. The combination significantly extended the event-free survival of mice carrying PTEN mutated xenografts. Our data suggest that PI3K/mTOR pathway inhibitors could benefit patients with PTEN mutated T-ALL.
Keywords: dexamethasone, rapamycin, acute lymphoblastic leukemia
Introduction
Due to the potent pro-apoptotic properties of glucocorticoids (GC) against lymphoid cells, GCs are commonly employed in the therapy of lymphoid malignancies. The cell-death effect of GC is strictly dependent upon the interaction of GC with the cytosolic GC receptor (GR), a ligand-activated transcription factor of the nuclear receptor family [1], which mediates induction of apoptosis [1, 2]. However, mutations of the GR are infrequently detected in primary leukemia samples from GC-resistant patients, indicating that GC resistance is unlikely to be GR–mediated in lymphoid leukemia [3]. In spite of the poor understanding of GC resistance, enhancing GC-induced cytotoxicity remains important as the response to GC therapy is a solid prognostic factor for pediatric ALL.
The PI3K/Akt/mTOR signaling pathway is inappropriately activated in many cancers, which may result in uncontrolled growth and survival of cancer cells [4]. PTEN (phosphatase and tensin homolog) is a tumor suppressor that acts as a phosphatase for the lipid signaling intermediate phosphatidylinositol-3,4,5-triphosphate (PIP3), which turns off PI3K(phosphoinositide 3-kinase)/Akt signaling [5]. Genetic abnormalities, such as the loss of PTEN, can activate PI3K/Akt signaling [6]. The phosphorylation of Akt by mTOR complex 2 (mTORC2) results in full activation of this kinase [7], which then activates the mTOR complex 1 (mTORC1) to phosphorylate S6K1 and dissociate 4EBP1 (eukaryotic translation initiation factor 4E-binding protein 1) from the initiation factor [7]. S6K1 and 4EBP1 play major roles in the growth and proliferation of cancer cells [8]. Rapamycin, an mTOR inhibitor [9], dephosphorylates 4EBP1 and S6K1 by inhibiting mTORC1 phosphorylation of rictor, one of the proteins in the mTORC2 [10]. Rapamycin and its analogs showed anti-cancer activity in preclinical models [11] as well as modest clinical benefits in several cancers [12–17]. However, the clinical activity of rapamycin analogs used for anti-neoplastic therapy as single agents has been disappointing [18], suggesting that combining such agents with other drugs will be necessary to achieve substantial clinical activity.
The mTOR signaling pathway is essential for cell growth and survival in lymphoid malignancies [19, 20]. In addition, activation of mTOR has been suggested to one mechanism of GC resistance [21]. A gene expression-based screening study identified rapamycin as a sensitizer for leukemia cells to GC by inhibiting Mcl-1 translation in a cell culture model [22], while another study reported that RM reversed GC resistance in ALK+ lymphoid tumor cells [23]. PTEN, a negative regulator of the PI3K/mTOR pathway, is frequently mutated in T-lineage ALL (T-ALL), and the deletions of PTEN are a distinguishing feature between hematopoietic stem cells and leukemia initiating cells. [24, 25]. Therefore, inhibition of the mTOR pathway may sensitize ALL cells, especially T-ALL to chemotherapeutic agents. We assessed combination activity of rapamycin and dexamethasone in a panel of in vitro and in vivo models of pediatric cancers [26]. In this study, we evaluate the combination activity of rapamycin and dexamethasone and characterize a molecular mechanism of synergy between those agents using a larger panel of in vitro and in vivo models of lymphoid malignancies.
Materials and Methods
In vitro Cell Culture
Cell lines from human T-cell leukemia established from children at diagnosis (COG-LL-329h) or at relapse (COG-LL-317h, COG-LL-332h, COG-LL-384h) and human pre-B leukemia cells established at diagnosis from children prior to therapy (COG-LL-319h) or at relapse (COG-LL-355h) were obtained from the Children’s Oncology Group (COG) Cell Line and Xenograft Repository (www.cogcell.org) approximately one month prior to each experiment. COG leukemia lines were cultured in Iscove’s modified Dulbecco medium (IMDM; Cambrex, Walkersville, MD) supplemented with 3 mM L-glutamine, 5 μg/mL insulin, and 20% heat-inactivated fetal bovine serum (FBS). NALM-6 (pre-B ALL, obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany), and RS4-11 (pre-B ALL), T-cell ALL cell lines (CCRF-CEM, MOLT-3, MOLT-4) from American Type Culture Collection (Manassas, VA) were maintained in RPMI-1640 medium (Mediatech, Herndon, VA) supplemented with 10% heat-inactivated FBS. All cell lines used were identified as mycoplasma free, and were cultured and treated with drugs in a 37°C incubator with 5% O2 (bone marrow–level hypoxia) [27], 5% CO2, and 90% N2. Cell line identities were confirmed after each expansion but prior to freezing by short tandem repeat (STR) profiling [28]; STR’s were unique for all cell lines except the ones established from the same patients at different stages of the disease (MOLT-3 and MOLT-4, and COG-LL-329 and COG-LL-332). Studies using human specimens were approved by the Investigational Review Board of Texas Tech University Health Sciences Center.
Cytotoxicity assay
The in vitro activities of dexamethasone (Sigma-Aldrich, St. Louis, MO), rapamycin (LC Laboratories, Woburn, MA) and their combination were determined using the DIMSCAN digital imaging microscopy cytotoxicity system in 11 ALL cell lines as previously described [29].
Cell lysates and immunoblot analysis
Whole-cell extracts were prepared by lysis of cells in radioimmunoprecipitation (RIPA) lysis buffer (Upstate, Lake Placid, NY) with 1 mM phenylmethanesulphonylfluoride (PMSF) and protease inhibitor cocktail (Sigma-Aldrich) for 30 minutes on ice. To analyze cytochrome c and Smac release from mitochondria, cytosol was extracted using Mitochondria/Cytosol Fractionation Kit (Biovision, Mountain View, CA). Immunoblotting was performed as previously described.[29] The following antibodies were used: Rabbit antihuman caspase-3 (8G10), caspase-9, E2F1, Rb, phospho-Rb (Ser807/811), phospho-Rb (Ser795), phospho-Rb (Ser780), Akt, phospho-Akt, S6K1, phospho-S6K1, S6, phospho-S6, phospho-4EBP1, XIAP antibodies from Cell Signaling Technology (Danvers, MA); PTEN, phospho-PTEN, cytochrome c, 4EBP1 from Santa Cruz Biotechnology (Santa Cruz, CA); antihuman Smac antibody from CalBiochem (Darmstadt, Germany); horseradish peroxidase (HRP) – conjugated rabbit anti-mouse IgG (Sigma) and donkey anti-rabbit/goat IgG (Santa Cruz).
Gene transfer by electroporation
We transfected CCRF-CEM cells with a small interfering RNA (siRNA) targeted against the S6K1 gene (Accession: NM_003161) from Integrated DNA Technologies (Skokie, IL) as previously described [29]. The sequences of the siRNAs used are
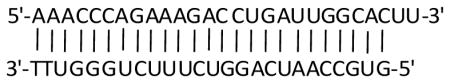 and
and
 Transfection conditions were optimized using Cy3TM DS Tranfection Control (Integrated DNA Technologies) at a final concentration of 10 nM. A non-targeting sequence was used as a negative control (DS scrambled negative control). Knock-down efficiency was assessed by measuring the amount of S6K1 protein by immunoblotting in cells transfected with siRNA against S6K1 relative to cells transfected with scrambled siRNA. The cytotoxicity effect was measured by DIMSCAN.
Transfection conditions were optimized using Cy3TM DS Tranfection Control (Integrated DNA Technologies) at a final concentration of 10 nM. A non-targeting sequence was used as a negative control (DS scrambled negative control). Knock-down efficiency was assessed by measuring the amount of S6K1 protein by immunoblotting in cells transfected with siRNA against S6K1 relative to cells transfected with scrambled siRNA. The cytotoxicity effect was measured by DIMSCAN.
Apoptosis, mitochondrial membrane depolarization (Δψm) and cell cycle analysis by flow cytometry
Apoptosis was quantified by staining cells with annexin and propidium iodide (PI) using an Annexin V – FITC apoptosis detection kit (BD bioscience, San Jose, CA), and the changes in mitochondrial membrane potential were measured using JC-1 as described previously [29]. The effect of the agents on cell cycle arrest was assessed as previously described [30]. Apoptosis, mitochondrial depolarization and cell cycle analysis were assessed using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA), operated with DiVA software (version 6.11). Bandpass filters were 525 ± 25 nm for FITC or JC-1 green and 610 ± 10 nm for PI or JC-1 red.
cDNA PCR assay with direct sequencing for PTEN mutation analysis
RNA was extracted from 7 cell lines using the Trizol reagent (Invitrogen, Carlsbad, CA) and reverse-transcribed to synthesize single - stranded cDNA with the use of the high capacity cDNA reverse transcription kit (Applied Biosytems, Foster City, CA). Primers were designed to amplify exon 1 to exon 8 of PTEN; Forward: 5’-CTTTTTCTTCAGCCACAGGC-3’ and Reverse: 5’-TGACGGCTCCTCTACTGTTTT-3’. The gene was polymerase chain reaction (PCR)–amplified and the PCR products (diluted to 100 ng/μl) were sequenced at the Biotechnology and Genomics Center, Texas Tech University.
Enzyme activity of p70S6 kinase
To measure S6K1 enzyme activity, cells were resuspended in PBS and lysed by sonication for 5–10 seconds. Cell lysates containing 100 μg of protein were immunoprecipitated with either S6K1 antibody or control non-immune rabbit immunoglobulin (RIgG). S6K1 enzyme activity was measured using an S6K1 assay kit (Upstate Biotechnology, Lake Placid, NY) containing a S6K1 peptide substrate (AKRRRLSSLRA) according to the manufacturer's instructions. Briefly, 10 μl of substrate cocktail, 10 μl of inhibitor cocktail and 10 μl of enzyme preparation were added to a microcentrifuge tube, and the reaction was started by adding 10 μl of the Mg2+/ATP cocktail containing [γ-32P]ATP. After 10 min of incubation at 30°C, 25 μl of sample mixture were spotted onto P81 phosphocellulose paper and incubated for 30s to allow the radiolabelled substrate to bind the filter paper. The papers were then washed three times with 0.75% phosphoric acid and once with acetone wash. Each square was transferred to a scintillation vial and the radioactivity was measured.
In vivo activity of dexamethasone and rapamycin
Systemic xenograft models
The in vivo activity of dexamethasone in combination with rapamycin was investigated in two ALL xenograft models, COG-LL-317x (T-ALL direct xenograft) and RS4;11 (Pre B-ALL). Female nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice aged 6 to 8 weeks were purchased from Charles River Laboratories (Wilmington, MA). On the day of inoculation, mice were irradiated (250 cGy of total-body irradiation), and injected via tail vein injection with 2 × 106 COG-LL-317x or 8 × 106 RS4;11 cells in 100 μL serum free medium. After two weeks, groups of 8 mice were injected intraperitoneally with dexamethasone (30mg/kg/day) and/or rapamycin (5mg/kg/day) daily (M-F) for 4 weeks. Event-free survival (EFS) was measured from the date of inoculation to the first observation of disease progression or drug-related toxicity (>20% weight loss, lethargy, ruffled fur).
Statistical analyses
For in vitro experiments, combination indices (CI) were calculated using Calcusyn (Biosoft, Cambridge, United Kingdom), which numerically quantifies drug synergism based on the multiple drug-effect equation of Chou-Talalay derived from enzyme kinetic methods [31]. A CI value < 0.9 indicates synergism; 0.9 to 1.10 additive; >1.1 antagonism. The CI values were calculated for each fixed-ratio concentration that was used for the cytotoxicity assays. Statistical evaluation of apoptosis, cell cycle progression, and S6K1 enzyme activity was performed using Student’s t-test. P-values were two sided and tests were considered significant at P < 0.05.
Mouse EFS was graphically represented by Kaplan-Meier analysis, and survival curves were compared by log-rank test where P values less than 0.05 were considered significant. The plotting and statistical analysis of the data was performed using GraphPad Prism (GraphPad Software, La Jolla, CA).
Results
Rapamycin enhanced dexamethasone cytotoxicity in some ALL and lymphoma cell lines
The sensitivities of 11 human ALL cell lines to dexamethasone, rapamycin, or dexamethasone + rapamtcin were determined at clinically achievable concentrations [32] using the DIMSCAN cytotoxicity assay. As shown in the dose-response curves (Fig. 1A and B), both rapamycin and dexamethasone were cytotoxic for < 90% of cells as single agents in all the cell lines tested except that dexamethasone achieved > 99% cell kill in RS4;11 cells. Moreover, the dose-response curves appeared to reach a plateau at higher concentrations of rapamycin when used as a single agent in the majority of the cell lines, which is commonly seen with cytostatic agents while dexamethasone cytotoxicity was concentration-dependent in cell lines relatively sensitive to rapamycin, e.g. CCRF-CEM, COG-LL-317h, RS4;11, COG-LL-319h. For dexamethasone + rapamtcin, the viabilities of four of the 11 ALL cell lines were affected more than 90%. Combination indices (CI) were calculated at the fixed-ratio drug concentrations to assess combination activity of dexamethasone and rapamycin as described under Methods. Strong synergy (CI <0.3) was observed in four of 11 cell lines: CCRF-CEM; COG-LL-317h; NALM-6; COG-LL-384.
Figure 1.

Combination cytotoxicity of DEX and RAP in T-ALL (A) and B-ALL cell lines (B). Cytotoxicity was evaluated after treating the cells with DEX, RAP, or DEX+RAP (D+P) for 72h by DIMSCAN system. The fractional survival was determined by mean fluorescence of the treated cells/mean fluorescence of vehicle control cells. Each point represents the mean value for 12 replicates, error bars correspond to standard deviations. * and †: Cell lines with the same symbol were established from the same patient. For B-C the points/columns are the mean and error/vertical bars are standard deviation from experiments performed twice (n=12).
Rapamycin plus dexamethasone induced apoptosis via mitochondrial pathway
We investigated the effect of dexamethasone, rapamycin, and dexamethasone + rapamycin on a mitochondrial apoptosis in COG-LL-317h cells. In COG-LL-317h cells, the percentages of apoptotic cells by Annexin V positivity were vehicle control = 3.9% ± 0.5%, dexamethasone = 32.8% ± 0.9% (P = 0.012), rapamycin = 2.4% ± 0.1% (P < 0.01), and rapamycin + dexamethasone = 81.1 ± 10.6% (vs dexamethasone, P=0.012; vs rapamycin, P < 0.01) (Fig. 2A). In addition, dexamethasone + rapamycin resulted in greater mitochondrial membrane depolarization than either drug alone (Fig. 2B) in COG-LL-317h cells (P < 0.001 compared with dexamethasone or rapamycin). To further confirm the combination effect of dexamethasone and rapamycin on induction of the mitochondrial apoptosis pathway, we also examined cytochrome c and Smac release from mitochondria to cytosol. The levels of cytosolic cytochrome c and Smac were greater in cells treated with dexamethasone + rapamycin than cells treated with dexamethasone or rapamycin as single agents (Fig. 2C). Also, the decrease in procaspase-9 and the increase in cleaved caspase-3 were enhanced by dexamethasone + rapamycin compared with that found in cells treated with single agents (Fig. 2D).
Figure 2.

DEX + RAP on apoptosis. (A) COG-LL-317h cells were incubated with vehicle control, DEX, RAP (10 ng/ml), or DEX+RAP for 36 hours, and subjected to flow cytometry after staining. (B) The percentages of cells undergoing apoptosis (black bars) and the cells with mitochondrial depolarization (white bar) were measured by flow cytometry after treating the cells with vehicle control, DEX, RAP, or DEX+RAP for 36h. (C) Cytochrome c and Smac release into cytosol was assessed at 36h of incubation with the agents. (D) Whole-cell extracts from cells incubated with DEX, RAP, or DEX+RAP for 48h were immunoblotted with caspase-9 and caspase-3 antibodies. β-actin was used for loading control. Experiments were performed twice and were consistently repeatable; for simplicity, one representative experiment for each condition is shown.
Effect of dexamethasone plus rapamycin on cell cycle
As both rapamycin or dexamethasone are suggested to induce G0/G1 arrest in leukemia cells, we investigated the effect of dexamethasone + rapamycin on cell cycle arrest by assessing DNA content in COG-LL-317h and CCRF-CEM cell lines. Cell cycle was analyzed after the cells were treated with dexamethasone, rapamycin, or dexamethasone + rapamycin for 12, 24, or 36h. Dexamethasone + rapamycin treated COG-LL-317h cells displayed a significantly greater G0/G1 arrest (86.2 ± 4.0%) compared with vehicle control (57.3 ± 1.6%, P < 0.01), dexamethasone (74.1 ± 3.0%, P < 0.05), or rapamycin (74.4 ± 4.7% P < 0.05) after 24 hours of treatment. The differences in cell cycle distribution among groups were even greater by 36 h, leading to greater G0/G1 arrest (Fig. 3A). Similar changes in cell cycle distribution were observed in CCRF-CEM cells, another synergistic cell line (data not shown). In COG-LL-317h cells, changes in proteins that contribute to G0/G1 arrest were observed to a greater extent by the dexamethasone + rapamycin combination at 36h as compared with single agents in a time-dependent manner. Changes observed in COG-LL-317h include induction of p27, and decreased retinoblastoma protein (Rb), phospho-Rb (p-Rb), E2F1, cyclin E, and cyclin D2 (Fig. 3B).
Figure 3.

DEX+RAP on cell cycle progression. (A) Cells (COG-LL-317h) were treated with DEX, RAP or DEX+RAP for 24h and then fixed in 70% ethanol overnight and stained with PI for subsequent analysis by flow cytometry (n=3). Experiments were performed twice and were consistently repeatable; one representative experiment for each condition is shown. (B) COG-LL-317h cells were treated with DEX, RAP, and DEX+RAP for 12, 24 and 36h. Cells were lysed and the lysates were immunobloted with antibodies to CDK2, 4, 6, cyclin D2, cyclin E, p27, retinoblastoma (Rb), p-Rb, and E2F1.
Dexamethasone and rapamycin combination on PI3K pathway
Changes in the expression of proteins in the PI3K pathway were analyzed in COG-LL-317h and CCRF-CEM cell lines after treating the cells with dexamethasone, rapamycin, or dexamethasone + rapamycin. As shown in Figure 4, PTEN expression and phosphorylation were minimal in COG-LL-317h and were relatively low in CCRF-CEM cells. Subsequent sequencing experiments show that all T-cell ALL cell lines (COG-LL-317h, CCRF-CEM, MOLT-3, MOLT-4, COG-LL-329h, COG-LL-332h) have mutations on exon 5 and/or exon 7, but not B-lineage ALL cell lines (RS4;11 and COG-LL-319h). Interestingly, phospho-S6K1 (p-S6K1) was inhibited by dexamethasone + rapamycin, and the phosphorylation of the S6K1 substrate, S6 was inhibited by rapamycin in COG-LL-317h and CCRF-CEM (Fig. 4A).
Figure 4.

Changes in PI3K pathway proteins by DEX + RAP. Cells were treated with DEX (50 nM), RAP (10 ng/ml), or DEX+RAP for 24 and 36h. Cells were lysed and the extracts were subjected to analysis by western blotting using antibodies specific for p85, AKT, p-AKT, PTEN, p-PTEN, S6K1, p-S6K1, S6, and p-S6 (A). Changes in S6K1 enzyme activity after the treatment (B) were assessed in COG-LL-317 cell line as described under Methods (*, p < 0.05; **, p < 0.01). The data represent the mean values for triplicate measurements from two independent experiments, and the values are normalized to vehicle control; error bars represent standard deviation (n=3). The knockdown efficiency (C) was assessed. Although S6K1 protein level was low up to 72 hours of transfection, p-S6 levels increased by 72 hours. Therefore, the cytotoxicity assay after the transfection of siRNAs against S6K1 was performed at 48h after the treatment (D). The cytotoxicity of DEX+RAP after transfecting scrambled siRNA (●) or siRNAs against S6K1 (○) was assessed at 48h after the treatment. Bars represent standard deviation of 12 samples. Experiments were performed twice and were consistently repeatable; one representative experiment for each condition is shown.
To further investigate the relevance of S6K1 in the synergy of dexamethasone + rapamycin, we assessed S6K1 enzyme activity after treating cells with dexamethasone, rapamycin, or the combination of the two drugs in CCRF-CEM cells. Although S6K1 activity was not affected by dexamethasone or rapamycin as single agents compared with controls (P > 0.13), the combination of dexamethasone + rapamycin significantly suppressed the S6K1 activity (P < 0.02, Fig. 4B).
Subsequently, we reduced S6K1 protein levels using siRNA (transfection efficiency 86%, Fig. 4C). The suppression of S6K1 protein expression and the inhibition of S6 phosphorylation were not apparent until 48h after transfection, and the inhibitory effect on S6 phosphorylation was diminished at 72h (Fig. 4D). S6K1 knock-down using siRNA did not affect single drug cytotoxicity of dexamethasone or rapamycin. In contrast, the dexamethasone + rapamycin-induced cytotoxicity was significantly enhanced in cells transfected with siRNA against S6K1 compared with scrambled siRNA-transfected cells (Fig. 4D).
Activity of dexamethasone and rapamycin against ALL and lymphoma xenografts
The in vivo activity of dexamethasone, rapamycin and dexamethasone + rapamycin was assessed in a model that showed synergy in cell culture experiments (COG-LL-317x: direct xenograft of COG-LL-317h) and one without synergy (RS4;11) in mouse xenografts. In COG-LL-317x systemic xenograft model, rapamycin as a single agent extended event-free survival (EFS) of leukemia-bearing mice compared with control, and dexamethasone + rapamycin significantly prolonged EFS of the xenograft mice compared with control or either single agent (Fig. 5A). In the RS4;11 systemic xenograft model, consistent with the high sensitivity of the cell line in vitro to dexamethasone, the EFS was extended in mice treated with dexamethasone (P < 0.001 ) or dexamethasone + rapamycin (P = 0.001) compared with the mice treated with vehicle control or rapamycin (Fig. 5B). However, also consistent with the lack of cytotoxic synergy for RS4;11 in vitro, rapamycin did not enhance EFS compared with dexamethasone alone in RS4;11 xenograft mice.
Figure 5.

In vivo activity of DEX, RAP, DEX+RAP with ALL and lymphoma xenograft models. NOD/SCID mice (n=8 per group) were inoculated with COG-LL-317x (A) or RS4;11 (B) through tail vein injection within 8h of irradiation. Mice were treated with vehicle control (Con, black), DEX (green), RAP (blue), or DEX+RAP (red) by intraperitoneal injection. The rapamycin dose was 5mg/kg/day, and dexamethasone 30mg/kg/day as a single agent or 7.5mg/kg/day in combination with rapamycin. The EFS of mice was quantified as the time from the initiation of treatment until mice were killed due to treatment-related toxicity or disease. Each line represents the proportion of mice remaining event-free over time.
Discussion
Pediatric ALL is the classic example of successful application of combination chemotherapy. The cure rate of pediatric ALL, which was less than 50% up until the 1970’s, is now over 80% with appropriate treatment [33]. However, the T-cell phenotype is one of the negative prognostic factors for pediatric ALL patients [34]. Patients with T-cell ALL have worse outcome when T-lineage and B-lineage ALL patients get the same age/WBC-stratified therapy [35, 36]. Ultimate outcomes are similar when T-ALL patients receive optimal therapy (high-risk therapy), but T-ALL patients who relapse tend to relapse earlier and more often in the CNS [37]. Initial response to a glucocorticoid is one of the clinical prognostic factors in T-cell ALL [38], suggesting that understanding the overcoming GC resistance in T-cell ALL provides a route toward improving clinical outcome. Activation of mTOR is suggested to be related to glucocorticoid resistance [39]. Here we have examined the in vitro cytotoxicity of dexamethasone, rapamycin, and their combination in 11 different cell culture models of acute lymphoblastic leukemia, identified mechanisms of synergistic activity with dexamethasone + rapamycin, and we have confirmed those observations in xenograft models. In several cell lines, dexamethasone + rapamycin was synergistic although the synergism was not limited to the cell lines that are resistant to dexamethasone, suggesting that the synergism of dexamethasone + rapamycin may not be directly associated with dexamethasone resistance. Further, the synergistic cytotoxicity of the dexamethasone + rapamycin combination was more frequent in T-lineage than in B-lineage in ALL cell lines. In fact, NALM-6 was the only B-lineage ALL cell line that showed synergism between dexamethasone and rapamycin.
Our data suggest that both apoptosis and cell cycle arrest by either agents are enhanced by combining two agents together in the cell lines that were synergistically affected by by the dexamethasone + rapamycin although the observations need to be confirmed in a larger panel of preclinical models and in primary samples from future clinical trials. Rapamycin and other mTOR inhibitors are cytostatic via interference with the activation of S6K1 by Akt [40]. S6K1 is a downstream effector of mTOR that can be activated by phosphorylation through PI3K-dependent effectors. S6K1 plays a key role in cell cycle progression through the G1 phase [41, 42]. Therefore, mTOR inhibition results in decreased S6K1 activity which subsequently enhances dexamethasone cytotoxicity. Considering that PTEN is a negative regulator of the PI3K/Akt pathway [6], which converts the Akt activator, PIP3 (active) to phosphatidylinositol-4,5-bisphosphate (PIP2, inactive), we speculated that PTEN mutations might have resulted in abnormal S6K1 activation in the synergistic cell lines. PTEN mutations or deletions are frequent in T-lineage ALL and PTEN deletions are associated with less favorable outcome in T-cell ALL [24, 43]. We observed that synergistic activity of rapamycin + dexamethasone was more frequent in lines from T-cell relative to B-lineage ALL. To investigate the association between the synergistic activity of rapamycin + dexamethasone and PTEN mutations, we sequenced DNA in the cell lines, and the data revealed that COG-LL-317h (T-lineage ALL, a cell line with synergism) have deletions in exon 7 of PTEN and do not express PTEN protein (Fig. 4). This may have resulted in high activity of S6K1, which is inhibited by rapamycin and further by the combination with dexamethasone (Fig. 4) in COG-LL-317h cell line. In the isogenic direct xenograft model (COG-LL-317x), rapamycin as a single agent enhanced EFS of leukemia-bearing mice compared with control, and dexamethasone + rapamycin further extended the survival. Our data suggest that the synergy between dexamethasone and rapamycin might be associated with PTEN mutations that result in high S6K1 activity.
There are challenges in targeting the PI3K/Akt/mTOR pathway in ALL patients. Testing novel agents in combination in recurrent ALL is challenging due to the baseline systemic toxicities from the disease process [44] and there is the potential of exacerbating toxicities of current ALL re-induction regimens due to the immunosuppressive effects of rapamycin [45]. In our in vivo experiments, the single agent dexamethasone dose was not tolerable when combined with rapamycin, requiring that dexamethasone dosing for the combination treatment to be reduced to one-fourth of a single dexamethasone dose. Yet, even using lower dosing, the dexamethasone + rapamycin prolonged EFS of mice bearing COG-LL-317x cells compared with the full dose dexamethasone single agent, suggesting that for patients with leukemias sensitive to this combination, rapamycin could improve responses to dexamethasone, even with lower doses of the latter drug being employed to decrease systemic toxicities. An inhibitor of the PI3K/Akt/mTOR pathway without apparent immunosuppression would be an option to investigate in combination with current therapies.
In summary, we showed that rapamycin enhanced the in vitro cytotoxicity and activity against xenografts of dexamethasone in some but not all preclinical models of lymphoid malignancies. We also showed that rapamycin + dexamethasone in combination enhanced apoptosis and cell cycle arrest effect relative to either agent alone, and knock-down of S6K1 resulted in greater in vitro activity for rapamycin and dexamethasone in combination. Our data suggest that a subset of acute lymphoblastic leukemia may be sensitive to dexamethasone + rapamycin and suggest that clinical trials testing this approach be undertaken in acute lymphoblastic leukemia.
Acknowledgments
COG cell lines were kindly provided by Children’s Oncology Group (COG) Cell Line and Xenograft Repository (www.COGcell.org), and were established by Dr. C. Patrick Reynolds, (School of Medicine, TTUHSC) from primary samples via a protocol, ABTR04B1. CZ’s current affiliation is College of Pharmaceutical Sciences, Zhejiang University, China.
Role of the funding source
This work was supported by the National Institutes of Health National Cancer Institute (Grant CA159308). CZ was partially supported by CSC Scholarship Program, The China Scholarship Council.
Footnotes
Authors’ Contributions
C.Z. and Y-K.R. contributed equally to this work; C.Z. and Y.K.R. designed, performed and analyzed the experiments; C.P.H. participated in the interpretation of the data; T.Z.C. provided critical techniques (kinase activity assay); D.R.W. revised the article for important content; M.H.K. designed, analyzed, interpreted the studies, and revised the manuscript.
Conflict of Interest
No potential conflicts of interest were disclosed. No commercial funds or input contributed to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helmberg A, Auphan N, Caelles C, Karin M. Glucocorticoid-induced apoptosis of human leukemic cells is caused by the repressive function of the glucocorticoid receptor. EMBO J. 1995;14:452–60. doi: 10.1002/j.1460-2075.1995.tb07021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA Binding of the Glucocorticoid Receptor Is Not Essential for Survival. Cell. 1998;93:531–41. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 3.Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 2005;65:9712–8. doi: 10.1158/0008-5472.CAN-05-1227. [DOI] [PubMed] [Google Scholar]
- 4.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 5.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118:3762–74. doi: 10.1172/JCI34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 8.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Mol Cell Biol. 2004;24:200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mills JR, Hippo Y, Robert F, Chen SMH, Malina A, Lin CJ, et al. mTORC1 promotes survival through translational control of Mcl-1. PNAS. 2008;105:10853–8. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Molecular Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 11.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 12.Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27:3822–9. doi: 10.1200/JCO.2008.20.7977. [DOI] [PubMed] [Google Scholar]
- 13.Ansell SM, Inwards DJ, Rowland KM, Jr, Flynn PJ, Morton RF, Moore DF, Jr, et al. Low-dose, single-agent temsirolimus for relapsed mantle cell lymphoma: a phase 2 trial in the North Central Cancer Treatment Group. Cancer. 2008;113:508–14. doi: 10.1002/cncr.23580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36:443–50. doi: 10.1016/j.exphem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23:5347–56. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- 16.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–22. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 17.Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, et al. Sirolimus for Kaposi's Sarcoma in Renal-Transplant Recipients. N Engl J Med. 2005;352:1317–23. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- 18.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR Signaling Network for Cancer Therapy. J Clin Oncol. 2009;27:2278–87. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 20.Wendel HG, De SE, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 21.Beesley AH, Firth MJ, Ford J, Weller RE, Freitas JR, Perera KU, et al. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. Br J Cancer. 2009;100:1926–36. doi: 10.1038/sj.bjc.6605072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10:331–42. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Gu L, Gao J, Li Q, Zhu YP, Jia CS, Fu RY, et al. Rapamycin reverses NPM-ALK-induced glucocorticoid resistance in lymphoid tumor cells by inhibiting mTOR signaling pathway, enhancing G1 cell cycle arrest and apoptosis. Leukemia. 2008;22:2091–6. doi: 10.1038/leu.2008.204. [DOI] [PubMed] [Google Scholar]
- 24.Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 26.Houghton PJ, Morton CL, Gorlick R, Lock RB, Carol H, Reynolds CP, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Mol Cancer Ther. 2010;9:101–12. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown JM, Lemmon MJ. Tumor hypoxia can be exploited to preferentially sensitize tumors to fractionated irradiation. International Journal of Radiation Oncology, Biology, Physics. 1991;20:457–61. doi: 10.1016/0360-3016(91)90057-b. [DOI] [PubMed] [Google Scholar]
- 28.Masters JR, Thomson JA, ly-Burns B, Reid YA, Dirks WG, Packer P, et al. Short tandem repeat profiling provides an international reference standard for human cell lines. PNAS. 2001;98:8012–7. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang MH, Wan Z, Kang Y, Sposto R, Reynolds CP. Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst. 2008;100:580–95. doi: 10.1093/jnci/djn076. [DOI] [PubMed] [Google Scholar]
- 30.Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Mills GB, Younes A. Inhibition of the phosphatidylinositol-3 kinase/Akt promotes G1 cell cycle arrest and apoptosis in Hodgkin lymphoma. Br J Haematol. 2006;132:503–11. doi: 10.1111/j.1365-2141.2005.05881.x. [DOI] [PubMed] [Google Scholar]
- 31.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 32.Jimeno A, Rudek MA, Kulesza P, Ma WW, Wheelhouse J, Howard A, et al. Pharmacodynamic-Guided Modified Continuous Reassessment Method-Based, Dose-Finding Study of Rapamycin in Adult Patients With Solid Tumors. J Clin Oncol. 2008;26:4172–9. doi: 10.1200/JCO.2008.16.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. New England Journal of Medicine. 2006;354:166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 34.Sen L, Borella L. Clinical importance of lymphoblasts with T markers in childhood acute leukemia. N Engl J Med. 1975;292:828–32. doi: 10.1056/NEJM197504172921604. [DOI] [PubMed] [Google Scholar]
- 35.Gaynon PS, Angiolillo AL, Carroll WL, Nachman JB, Trigg ME, Sather HN, et al. Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a Children's Oncology Group Report. Leukemia. 2010;24:285–97. doi: 10.1038/leu.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moricke A, Zimmermann M, Reiter A, Henze G, Schrauder A, Gadner H, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia. 2010;24:265–84. doi: 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg JM, Silverman LB, Levy DE, Dalton VK, Gelber RD, Lehmann L, et al. Childhood T-Cell Acute Lymphoblastic Leukemia: The Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Experience. J Clin Oncol. 2003;21:3616–22. doi: 10.1200/JCO.2003.10.116. [DOI] [PubMed] [Google Scholar]
- 38.Pieters R, den Boer ML, Durian M, Janka G, Schmiegelow K, Kaspers GJ, et al. Relation between age, immunophenotype and in vitro drug resistance in 395 children with acute lymphoblastic leukemia--implications for treatment of infants. Leukemia. 1998;12:1344–8. doi: 10.1038/sj.leu.2401129. [DOI] [PubMed] [Google Scholar]
- 39.Miller AL, Garza AS, Johnson BH, Thompson EB. Pathway interactions between MAPKs, mTOR, PKA, and the glucocorticoid receptor in lymphoid cells. Cancer Cell Int. 2007;7:3. doi: 10.1186/1475-2867-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsurutani J, West KA, Sayyah J, Gills JJ, Dennis PA. Inhibition of the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Pathway but not the MEK/ERK Pathway Attenuates Laminin-Mediated Small Cell Lung Cancer Cellular Survival and Resistance to Imatinib Mesylate or Chemotherapy. Cancer Res. 2005;65:8423–32. doi: 10.1158/0008-5472.CAN-05-0058. [DOI] [PubMed] [Google Scholar]
- 41.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–87. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, et al. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–10. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 43.Jotta PY, Ganazza MA, Silva A, Viana MB, da Silva MJ, Zambaldi LJ, et al. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia. 2010;24:239–42. doi: 10.1038/leu.2009.209. [DOI] [PubMed] [Google Scholar]
- 44.Horton TM, Sposto R, Brown P, Reynolds CP, Hunger SP, Winick NJ, et al. Toxicity assessment of molecularly targeted drugs incorporated into multiagent chemotherapy regimens for pediatric acute lymphocytic leukemia (ALL): review from an international consensus conference. Pediatr Blood Cancer. 2010;54:872–8. doi: 10.1002/pbc.22414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calne RY, Lim S, Samaan A, Collier DS, Pollard SG, White DJG, et al. Rapamycin for immunosuppression in organ allografting. The Lancet. 1989;334:227. doi: 10.1016/s0140-6736(89)90417-0. [DOI] [PubMed] [Google Scholar]