

Abstract
Introduction
Stroke related tissue pressure increase in the core (Pcore) and penumbra (Ppen) determines regional cerebral perfusion pressure (rCPP) defined as a difference between local inflow pressure (Pi) and venous (Pv) or tissue pressure, whichever is higher. We previously showed that venous pressure reduction below the Pcore causes blood flow diversion - cerebral venous steal. Now we investigated how transition to collateral circulation after complete arterial occlusion affects rCPP distribution.
Methods
We modified two parallel Starling resistor model to simulate transition to collateral inflow after complete main stem occlusion. We decreased Pv from the arterial pressure (Pa) to zero, and investigated how arterial and venous pressure elevation augments rCPP.
Results
When core pressure exceeded venous (Pcore>Pv), rCPP=Pi−Pcore. Venous pressure (Pv) decrease from Pa to Pcore caused smaller Pi to drop augmenting rCPP. Further drop of Pv to Ppen decreased rCPP in the core but augmented rCPP in penumbra. After transition to collateral circulation, lowering Pv below Ppen further decreased rCPP and collaterals themselves became pathway for steal. Venous pressure level at which rCPP in the core becomes zero we termed the “point of no reflow” (PONR). Transition from direct to collateral circulation resulted in decreased Pi, decreased rCPP, and a shift of PONR to higher venous loading values. Arterial pressure augmentation increased rCPP, but only after venous pressure exceeded PONR.
Conclusion
In the presence of tissue pressure gradients, transition to collateral flow predisposes to venous steal (collateral failure) which may be reversed by venous pressure augmentation.
Keywords: Stroke, no reflow, hemodynamics, collaterals, venous, ischemia zero flow, perfusion pressure
INTRODUCTION
Extensive efforts have been devoted to understanding the basis of collateral failure following acute ischemic stroke. Collaterals should intuitively counteract the effects of cerebral arterial occlusion if they function properly 1. Yet, after ischemic stroke there is a tendency for collateral failure. 1. Collateral circulation may be defined as the blood flow that is supplied via secondary channels after arterial obstruction in a principal channel supplying downstream reaches of the brain. This article investigated the basis of collateral failure by investigating how continuation of flow in the non-obstructed, parallel venous outflow pathways leads to a drop in the collateral perfusion pressure and, at the extreme, to a zero, manifesting clinically as the phenomenon of no reflow when inflow pressure becomes equal to the external tissue pressure (zero flow pressure). Through our analysis we demonstrate that transition to collateral flow after arterial occlusion does not necessarily reestablish flow, but on the contrary, predisposes to venous pressure dependent flow diversion, known as cerebral venous steal 2, with collaterals themselves often serving as the pathways for steal.
METHODS
We modified two parallel Starling resistors with a common inflow model (Pranevicius, 2002) to simulate progression of partial inflow occlusion to the complete inflow occlusion with residual collateral flow (Fig. 1). Ischemic region was represented by two Starling resistors (Rcore and Rpen with external pressures Pcore and Ppen) and common inflow pressure Pi. Loading was defined by arterial pressure, Pa and venous pressure, Pv. The model assumed that regional cerebral blood flow is determined by the regional cerebral perfusion pressure (rCPP), defined as a Starling resistor: rCPPcore=0 if Pcore>Pi; rCPPcore=Pi−Pcore if Pi>Pcore>Pv; rCPPcore=Pi−Pv if Pcore<Pv. Equations defining rCPP are presented in the appendix.
Fig. 1.
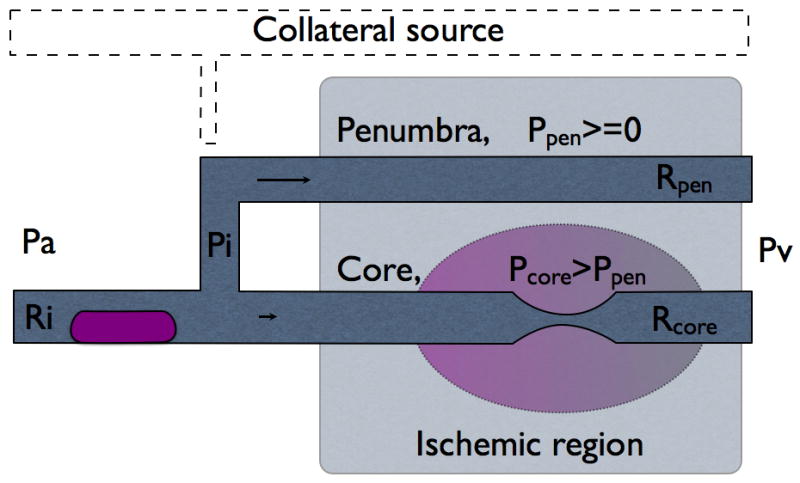
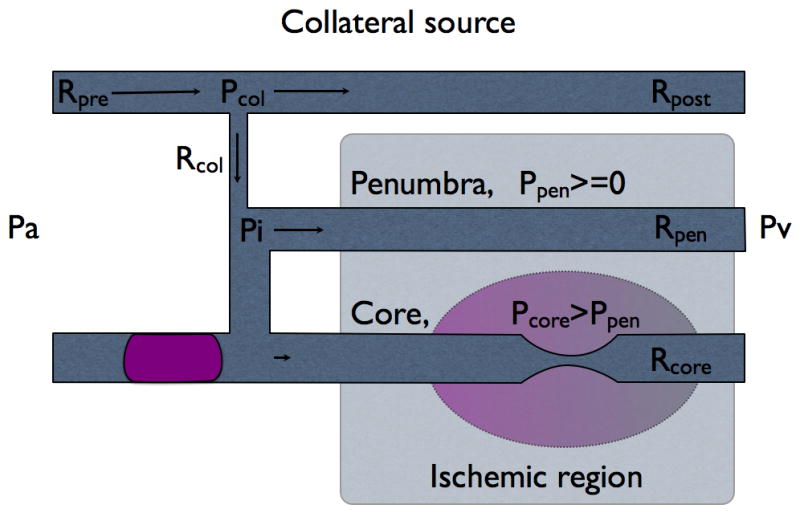
Fig. 1A Inflow through partially occluded direct connection.
Fig. 1B Inflow switched to collateral source when direct connection is occluded completely.
Collateral connection Rcol originates from the collateral source and supplies flow to the ischemic region. Starling resistors Rpen and Rcore represent penumbra and core of the ischemic region with corresponding external tissue pressures Ppen and Pcore. Pi is the common inflow pressure to the both, core and penumbra, distal to collateral connection. Pcol is pressure in the proximal end of collateral connection.
All resistors in series were described by the equivalent lumped resistor that already considers the effect of various sources of resistance including precapillary factors.
The model describes steady state rCPP distribution. The model assumes exhausted autoregulation as would be expected immediately after complete arterial occlusion and intraluminal pressure drop with transitioning to collateral inflow. Residual autoregulation, if present, would compensate for the reduced rCPP but autoregulation rarely, if ever, is present in the area of ischemia.
Assumed venous pressure drop from the level of arterial pressure Pa to zero is not a physiological phenomenon, but rather simulated example across a wide range of loading conditions to demonstrate the effect of venous pressure on the rCPP in both core and penumbra. In the situation of complete inflow occlusion (Fig. 1B) all flow to the ischemic region comes through collateral connections (Rcol) from the unaffected parallel vascular network- collateral source. Collateral pressure at the takeoff is Pcol and inflow pressure in the ischemic region at the distal end of collateral connection is Pi. Part of collateral source before collateral connection takeoff has resistance Rpre and part after takeoff- Rpost. We compared rCPPcore and rCPPpen while venous pressure was decreased from Pa to 0 in the event of complete and partial inflow obstruction.
Complete diversion of flow resulting in rCPPcore=0 we defined as the point of no reflow (PONR). External pressure Pcore at the point of no reflow is the minimal Pcore required to completely stop flow at the core. Venous pressure at the point of no reflow is the minimal venous pressure required to reestablish flow stopped by a given external pressure, Pcore.
RESULTS
Transition to the collateral circulation decreases inflow pressure Pi due to additional pressure drop at the collateral source (Pa-Pcol) (Fig. 2). Regional cerebral perfusion pressure of the ischemic core and penumbra is determined by the inflow pressure less venous pressure or tissue pressure, whichever is higher (Fig. 3). When venous pressure drops below the pressure level of the core (Pcore), rCPPcore decreases, yet at the same time rCPPpen increases (venous steal). Transition to collateral inflow decreases inflow pressure Pi and shifts the point of no reflow to the lower venous pressure values (Fig. 3). Additionally, when venous pressure drops below the pressure within the penumbra or Ppen, collaterals serve as a pathway for venous steal; whereas if inflow were to remain direct, rCPP would remain the same.
Fig. 2.

Perfusion pressure drop along the segments of cerebral circulation. Pressure progressively drops along the vascular network and determines regional cerebral perfusion pressure (rCPP) at the point of common inflow Pi. If inflow switches to the collateral source, Pi drops further due to pressure drop at the collateral source (Pa-Pcol). If venous pressure drops to Ppen, Pi drops below the pressure of the core, thus making rCPPcore equal zero, with only penumbra remaining perfused. Scale is for illustrative purposes.
Fig. 3. Inflow transition from direct to collateral decreases rCPP and induces collateral steal.

When direct inflow switches to the collateral source, inflow pressure Pi drops, decreasing rCPP, and moving PONR to the left. When venous pressure drops below Pcore, core rCPP drops to zero (core steal). When venous pressure decreases below Ppen, rCPPpen is not affected with residual direct inflow. (no steal with direct inflow) but slightly decreases when inflow switches to the collateral source (collateral steal). Pressures are scaled up for illustrative purposes.
Alternatively, if venous pressure is raised from zero to Ppen, the outflow pressure is not affected, while at same time inflow pressure increases, but only when inflow is supplied by the collateral connection (Fig. 3). Fig. 4 shows the combined effects of both arterial and venous pressure on rCPP: in the presence of the cerebral venous steal, both venous and arterial pressures can augment rCPP, yet the venous augmentation effect is an order of magnitude higher
Fig. 4. Arterial and venous augmentation of core perfusion pressure.

(Pcore =10; Ppen=0). Pa is increased from 0 to 150mmHg, while Pv from 0 to 20. Regional cerebral perfusion pressure decreases to zero when Pv<=5 and Pa<=80. If venous pressure is zero, Pa has to exceed 130mmHg to increase rCPP above zero. Note the scale difference for the arterial and venous pressures.
Fig. 5 shows that moving the collateral takeoff position from arterial to venous end decreases inflow pressure and rCPP, which is partially reversed by increased venous pressure. Fig. 6 shows that increasing collateral resistance decreases rCPP in the core which is further compromised by even relatively small tissue pressure increase. Increasing venous pressure largely reverses this steal.
Fig. 5. Variable collateral takeoff.

If collateral connection is moved from the arterial to venous end at collateral source (Rpre increases from 0- to 100%), that drops the collateral inflow pressure Pcol from Pa to Pv. Venous pressure drop from Pv=Pcore to Pv′=0 causes Pcol to drop to Pcol′ and Pi to Pi′, reducing rCPP. Decreasing venous pressure only at the collateral source causes collateral “steal” while reducing it at the penumbra ads core steal. Residual rCPP becomes zero at PONR. Pcore for illustrative purposes was chosen to be 20% of Pa.
Fig. 6.

Effect of collateral resistance on rCPP. Collateral resistance Rcol is expressed as percentage of total resistance in the ischaemic region. As Rcol increases to 100%, rCPP drops to zero. When core tissue pressure is 10% of rCPP drops to zero at lower collateral resistances (63%). Increase of Pv from 0 to Pcore recruits most of the rCPP drop. Rpre was chosen to be 50% of collateral source resistance.
Discussion
A two parallel Starling resistor model may be used to explain the evolution of hemodynamic events in the brain following acute ischemic stroke. Our simulations revealed that under conditions of local tissue pressure increases due to early ischemic infarction apparent as cytotoxic edema, a steal-like phenomenon can be triggered immediately after stroke. The degree of steal is higher if there is a persistent tissue pressure gradient between the ischemic core and the penumbra, and even higher if the blood flow supply comes solely from the collaterals, which as a rule typically have higher resistance to flow. These events provide a logical explanation for collateral failure following stroke onset 3.
However, once the stroke is established, steal phenomenon may become beneficial, maintaining perifocal flow.
Even with current stroke therapies, no reflow after revascularization remains problematic. Recanalization without effective reperfusion is an enigma and is considered to be a principal reason for failure of current reperfusion strategies. Reperfusion is often incomplete after reopening on the occluded arterial lumen 4. This condition has been termed “no reflow” phenomenon and describes failure of microvasculature perfusion after blood flow is restored in the principal arteries. No reflow phenomenon was first observed in rabbit brain after the transient interruption of cerebral blood flow 4, but since then it has been viewed as the primary culprit for reperfusion failures in stroke. Explanations for this phenomenon have been numerous: blockage of the vascular lumen by platelets and red cell aggregates 5; change in blood viscosity 6; local intravascular coagulation; and direct capillary compression from edematous endothelial and glial cells 7. Similarly, an alternative pattern of post-ischemic circulatory disturbance was discovered where an initial increase in cerebral blood flow, ‘reactive hyperemia’, is followed by a reduction in flow, ‘delayed hypoperfusion’. This secondary hypoperfusion has been demonstrated in the isolated canine brain 8; following global cerebral ischemia in the cat 9, dog 10, monkey 11 and rat 12. Yet up to this day these explanations have failed to find their way into routine clinical practice and management of stroke patients.
Recanalization without reperfusion in stroke remains an influential and recognized challenge in routine clinical practice, yet strict definitions of such revascularization results are often variable unless documented in systematic prospective studies. Detailed imaging protocols with noninvasive and conventional angiography measures have previously noted discordance between recanalization and reperfusion 13. Distinguishing no reflow from other causes of neurological worsening requires such detailed imaging. No reflow is suspected in cases when clinical improvement is lacking despite blood flow restoration in the major arteries. One study noted the lack of such improvement in one third of stroke victims with early recanalization, while four-fifths of these nonresponders had persistent severe neurological deficits at 24 hours after stroke 14.
The model simulates platelet aggregation as inflow occlusion and/or collateral connection resistance increases. Mechanical compression of the capillary bed is simulated using Starling resistor with external pressure Pcore. Increased inflow resistanse and focal compression creates conditions for venous steal and rCPP drops to zero even if arterial pressure remains higher than focal tissue pressure.
Our analyses provide compelling evidence that implicate the transition to collateral circulation after ischemic stroke as a trigger for cerebral venous steal and consequentially, to collateral failure, evolution of ischemia, and no reflow phenomenona. We applied a “global Starling resistor” equation, used to define cerebral perfusion pressure: CPP=MAP−ICP (when ICP>CVP) 15,16, for the purposes to study regional cerebral blood flow, when tissue pressure is locally increased. The diversion of blood flow occurs under these conditions, but it can be reversed by the elevation of venous pressure 2.
Core-to penumbra steal phenomenon may help preserve penumbra’s flow in case of established stroke, while during stroke progression it may expand the core; additionally it is important to keep in mind that our model simulated rCPP, not rCBF, distribution. rCBF distribution was implicitly assumed to follow rCPP distribution but in this model it was not studied explicitly as that would require knowledge of the absolute resistor values. Striking experimental findings indicate a close relationship between collateral flow and systemic venous pressures. Most illustrative of these findings were obtained from the experiments in cats, when immediately after MCA occlusion, pial artery pressure dropped from 56.2+/−1.6 to 7.8+/−0.4 mmHg and exhibited respiratory pressure variations 17, passively following respiratory variations of venous pressure, pointing to the conjecture, that venous pressure and local tissue pressure can be the primary determinants of collateral flow and collateral flow distribution after arterial stroke.
Through our analyses we were able to show that arterial pressure augmentation increases collateral flow, but only if venous pressure exceeds the critical zero flow threshold -PONR. The point of no reflow defines the very essence of complete flow diversion: arterial pressure augmentation has no effect on ischemic flow if Pv<PONR. Augmentation of venous pressure to the level of PONR increases inflow pressure into ischemic core, and reestablishes flow at the point when inflow pressure exceeds external tissue pressure.
Although the models presented in this paper are deductive in nature, meaning that the conclusions should be correct, if the assumptions are correct, the model assumptions are very intuitive - that rCPP is determined by the local tissue pressure and that rCBF passively follows rCPP when autoregulation is exhausted. Both these assumptions have to be verified experimentally. Future studies utilizing imaging and supportive translational research paradigms will facilitate our growing understanding of such complex phenomena in acute ischemic stroke. Limitations of our analyses also included the fact that we only considered steady-state, without any reference to the transitional states, and that we did not incorporate three-dimensional geometry of the realistic cerebrovascular tree.
Future research efforts should incorporate three-dimensional cerebral blood flow models, incorporating the principles outlined in this article by adding additional Starling resistors in both parallel and serial arrangements.
Such consideration will allow virtual manipulation of CBV-CBF distribution and correlation of this distribution with imaging that may provide a framework not only for explanation of evolution of hemodynamic events after ischemic stroke due to vascular occlusion, but also to disclose novel therapeutic strategies.
Summary
A schematic physiological framework for hemodynamics after onset of ischemic stroke, incorporating seemingly unrelated phenomena such as no reflow, postischemic luxury perfusion, collateral failure, and brain edema, may expand current clinical strategies. Consideration of established cerebral blood flow physiology, in terms of cerebral vessel configuration, inflow/outflow pressures, and cerebrovascular resistance, reveal that the transition from direct to collateral supply predisposes to cerebral venous steal.
Supplementary Material
Footnotes
Disclosures: None
References
- 1.Liebeskind DS. Collaterals in acute stroke: beyond the clot. Neuroimaging Clin N Am. 2005;15:553–73. x. doi: 10.1016/j.nic.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Pranevicius M, Pranevicius O. Cerebral venous steal: blood flow diversion with increased tissue pressure. Neurosurgery. 2002;51:1267–73. doi: 10.1097/00006123-200211000-00023. discussion 1273–4. [DOI] [PubMed] [Google Scholar]
- 3.Liebeskind DS. Reperfusion for acute ischemic stroke: arterial revascularization and collateral therapeutics. Curr Opin Neurol. 2010;23:36–45. doi: 10.1097/WCO.0b013e328334da32. [DOI] [PubMed] [Google Scholar]
- 4.Ames A, 3rd, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol. 1968;52:437–453. [PMC free article] [PubMed] [Google Scholar]
- 5.Cantu RC, Ames A, 3rd, Dixon J, DiGiacinto G. Reversibility of experimental cerebrovascular obstruction induced by complete ischemia. J Neurosurg. 1969;31:429–431. doi: 10.3171/jns.1969.31.4.0429. [DOI] [PubMed] [Google Scholar]
- 6.Fischer EG, Ames A, 3rd, Lorenzo AV. Cerebral blood flow immediately following brief circulatory stasis. Stroke. 1979;10:423–427. doi: 10.1161/01.str.10.4.423. [DOI] [PubMed] [Google Scholar]
- 7.Hossmann KA, Hossmann V. Coagulopathy following experimental cerebral ischemia. Stroke. 1977;8:249–254. doi: 10.1161/01.str.8.2.249. [DOI] [PubMed] [Google Scholar]
- 8.Zimmer R, Lang R, Oberdorster G. Post-ischemic reactive hyperemia of the isolated perfused brain of the dog. Pflugers Arch. 1971;328:332–343. doi: 10.1007/BF00586835. [DOI] [PubMed] [Google Scholar]
- 9.Tomita M, Gotoh F, Amano T, Tanahashi N, Tanaka K. “Low perfusion hyperemia” following middle cerebral arterial occlusion in cats of different age groups. Stroke. 1980;11:629–636. doi: 10.1161/01.str.11.6.629. [DOI] [PubMed] [Google Scholar]
- 10.Snyder JV, Nemoto EM, Carroll RG, Safar P. Global ischemia in dogs: intracranial pressures, brain blood flow and metabolism. Stroke. 1975;6:21–27. doi: 10.1161/01.str.6.1.21. [DOI] [PubMed] [Google Scholar]
- 11.Kofke WA, Nemoto EM, Hossmann KA, Taylor F, Kessler PD, Stezoski SW. Brain blood flow and metabolism after global ischemia and post-insult thiopental therapy in monkeys. Stroke. 1979;10:554–560. doi: 10.1161/01.str.10.5.554. [DOI] [PubMed] [Google Scholar]
- 12.Pulsinelli WA, Levy DE, Duffy TE. Regional cerebral blood flow and glucose metabolism following transient forebrain ischemia. Ann Neurol. 1982;11:499–502. doi: 10.1002/ana.410110510. [DOI] [PubMed] [Google Scholar]
- 13.Tomsick T. TIMI, TIBI, TICI: I came, I saw, I got confused. AJNR Am J Neuroradiol. 2007;28:382–384. [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov AV, Hall CE, Labiche LA, Wojner AW, Grotta JC. Ischemic stunning of the brain: early recanalization without immediate clinical improvement in acute ischemic stroke. Stroke. 2004;35:449–452. doi: 10.1161/01.STR.0000113737.58014.B4. [DOI] [PubMed] [Google Scholar]
- 15.Pranevicius M, Pranevicius O. Modified calculation of the cerebral perfusion pressure in a sitting position: jugular Starling resistor and related clinical implications. APSF Newsletter. 2008;23:05/05/2009–33. [Google Scholar]
- 16.Hallenbeck JM, Bradley ME. Experimental model for systematic study of impaired microvascular reperfusion. Stroke. 1977;8:238–243. doi: 10.1161/01.str.8.2.238. [DOI] [PubMed] [Google Scholar]
- 17.Shima T, Hossmann KA, Date H. Pial arterial pressure in cats following middle cerebral artery occlusion. 1. Relationship to blood flow, regulation of blood flow and electrophysiological function. Stroke. 1983;14:713–719. doi: 10.1161/01.str.14.5.713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.