

Abstract
Structural studies of UV-induced lesions and their complexes with repair proteins reveal an intrinsic flexibility of DNA at lesion sites. Reduced DNA rigidity stems primarily from the loss of base stacking, which may manifest as bending, unwinding, base unstacking, or flipping out. The intrinsic flexibility at UV lesions allows efficient initial lesion recognition within a pool of millions to billions of normal DNA base pairs. To bypass the damaged site by translesion synthesis, the specialized DNA polymerase η acts like a molecular “splint” and reinforces B-form DNA by numerous protein–phosphate interactions. Photolyases and glycosylases that specifically repair UV lesions interact directly with UV lesions in bent DNA via surface complementation. UvrA and UvrB, which recognize a variety of lesions in the bacterial nucleotide excision repair pathway, appear to exploit hysteresis exhibited by DNA lesions and conduct an ATP-dependent stress test to distort and separate DNA strands. Similar stress tests are likely conducted in eukaryotic nucleotide excision repair.
Keywords: CPD, 6-4 PP, unstacking, hysteresis, bending, ATPase, NER, TLS, photolyase, endonuclease
Introduction
Ultraviolet radiation (UV) in the form of sunlight has bombarded the earth since long before life began. For nucleic acids to serve as the genetic material and blue-print of living beings, cells had to first repair base lesions induced by UV in nucleic acids. UV catalyzes covalent bond formation between adjacent pyrimidine bases, thymine or cytosine.1 A majority of UV lesions are cyclobutane purimidine dimers (CPDs, 80–90%) and 6-4 pyrimidinepyrimidone photoproduct (6-4 PPs, 10–20%). Dewar photoproducts (Dewar PPs) are valence isomers of 6-4 PPs [Fig. 1(A)]. Multiple repair pathways have been found from bacteria, bacterial phage, archaeal, and plants to humans, and they vary from specific repair of UV lesions by a single enzyme to repair of a broad range of damaged bases by multiprotein complexes [Fig. 1(A)].
Figure 1.
UV lesions and their repair. A: Diagrams of CPD, 6-4 and dewar photoproduct and three different repair pathways. B: An outline of multiple steps in NER. Each step is labeled in bold font and dark blue. UV lesions are represented by red TT with an inverted V above. The NER pathway is divided into global genome (GGR, highlighted in dark red) or transcription coupled repair (TCR, in pale green) based on how lesions are initially recognized. The two branches converge afterwards. Proteins responsible for each step are shown above (bacterial) or below (eukaryotic) each arrow. In the TCR branch RNA polymerase and co-factors are omitted for clarity. C: Bypass of UV lesions during replication is achieved by either translesion synthesis (TLS) or homologous recombination (HR). The template strands are shown in black and daughter strands in blue.
The simplest repair of UV lesions is catalyzed by photolyases, which use visible blue-light in sunlight to cleave the covalent bonds between pyrimidine bases.2 This pathway is also known as direct reversal. Photolyases specific for CPD and 6-4 photoproducts are found in bacteria, plants, and lower vertebrates. Marsupials and placental mammals, whose embryos develop inside a uterus without light exposure, have lost the capacity for blue-light-dependent UV-lesion repair by photolyases. Alternatively, in some bacteria and bacterial phages, a DNA glycosylase represented by T4 endonuclease V can specifically cleave off the 5′ crosslinked CPD base and the DNA backbone by β-elimination.3 Nicking at the 5′ to UV lesions may also be conducted by a DNA endonuclease (UVDE) in bacteria and fungi.4,5 Enzymes in the Base Excision Repair pathway (BER) are then recruited to completely remove the lesion and restore native DNA.6,7
The major repair pathway for UV lesions, however, is Nucleotide Excision Repair (NER).8–10 Unlike the highly specific photolyases or BER, which use different glycosylases to recognize and excise different base lesions, the same NER proteins repair many types of DNA lesions. NER is found in all organisms and can be divided into global genome-wide repair (GGR or GG-NER) or transcription-coupled repair (TCR) depending on how lesions are initially recognized.11,12 In bacteria, GG-NER requires 4 conserved proteins, UvrA, UvrB, UvrC, and UvrD13; TCR requires transcription and the Mfd protein in addition.14,15 In eukaryotes, seven XP (Xeroderma Pigmentosum) proteins (XPA to XPG) and trichothiodystrophy (TTD) protein are required for GG-NER16; 5 XP proteins (minus XPC and XPE) plus Cockayne Syndrome protein A and B are required for TCR.12 Except for the common SFII helicase motifs shared by UvrB, XPB, and XPD, no structural or functional homology can be detected between bacterial and eukaryotic NER proteins. Similar to all DNA repair pathways, NER initiates with lesion recognition, which is particularly difficult because the types of lesions to be repaired are unlimited. Lesion recognition is followed by DNA strand separation and incision on both sides of the lesion.17,18 NER finishes with lesion removal, DNA resynthesis, and ligation [Fig. 1(B)].13
Unrepaired UV lesions, whether CPDs or 6-4 PPs, hinder DNA replication and block normal progression of replication forks. Cells depend on two different mechanisms to bypass UV lesions during replication [Fig. 1(C)]. One is by homologous recombination (HR), which contributes both to template switching between sister chromatids at a replication fork and postreplication DNA break repair.19 The other is by specialized DNA polymerases, for example UmuCD' in bacteria and Pol η in eukaryotes,20,21 to conduct translesion DNA synthesis (TLS). Yeast cells without DNA pol η survive well under chronic low dose UV.22 But in humans failure to carry out translesion synthesis bypass of CPDs, which are more abundant and more difficult to recognize and repair than 6-4 PPs, leads to a variant form of the cancer predisposition disorder Xeroderma Pigmentosum (XPV) and extreme sensitivity to sunlight.23,24
Molecular mechanisms for coping with UV lesions by different repair pathways and translesion DNA synthesis have been extensively studied. In the last few years the structures of UvrA, UvrB, XPC, and XPE, which carry out the first step of UV-lesion recognition in bacteria and eukaryotes, have been determined alone or complexed with DNA substrates,25–36 as have yeast and human DNA pol η complexed with CPDs.37,38 DNA base lesions, which include mismatched basepairs, modified bases due to oxidation, deamination, or alkylation, losses of bases (abasic sites) and large base adducts like cisplatin and polyaromatic hydrocarbons, exhibit a general feature of reduced base stacking and reduced DNA persistence length (see the review39 and references therein). Changes in DNA rigidity due to base lesions have been hypothesized to be the key for lesion recognition and repair. In this brief review, I will focus on the mechanism of UV-lesion recognition by repair proteins and TLS polymerases. To learn more about the complete processes of repair and translesion synthesis, please refer to additional review articles including those mentioned above.
DNA Flexibility Due to UV Lesions
Crosslinking of adjacent pyrimidine bases in DNA, whether a CPD or 6-4 PP, leads to distortion of the double helix. The two bases in a CPD, although remaining roughly parallel, lose the planar π electron, the aromaticity of normal pyrimidines and the 36° relative rotation [Fig. 1(A)]. In a 6-4 PP, the crosslinked bases are nearly perpendicular to each other and thus bear no resemblance to adjacent bases in normal DNA.36,40,41 Both types of UV lesions eliminate base stacking and thus reduce the persistent length (rigidity) of DNA.
The reduced base stacking and increased backbone flexibility around a UV lesion is evident in the atomic-resolution crystal structures as well as in silico and solution studies.42–44 In the absence of any protein, a DNA decamer harboring a CPD is under-wound and bent by 30° at the lesion.42 The crosslinked bases of a thymine dimer are buckled relative to the adjacent bases [Fig. 2(A)]. DNA containing 6-4 PPs is distorted more severely as expected. Segmentation of DNA by bending and unwinding can be further exacerbated by the presence of a mismatched base opposite UV-crosslinked bases, which could result from mis-incorporation during DNA synthesis.43,45 Interestingly, the severity of DNA distortions correlates with the repair efficiency. 6-4 PPs are more destabilizing than CPDs and are also more efficiently removed and repaired in vivo.46 This observation agrees with in vitro data47 that bacterial NER proteins depend on DNA distortion for efficient lesion recognition.
Figure 2.
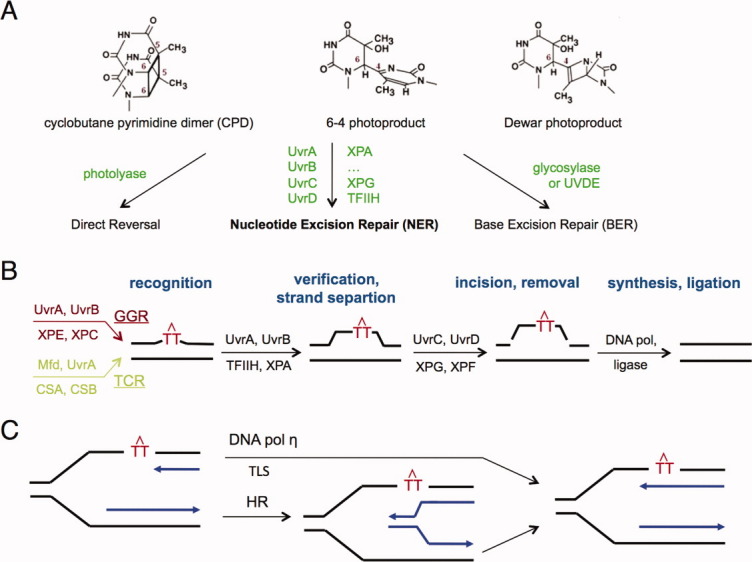
Structures of DNA with UV lesions. A: Cartoon diagram of a naked DNA with a CPD (PDB: 1SM5). The strand with the lesion is shown in orange, and the partner strand in yellow. The CPD is highlighted in red and bases opposite it in magenta. B: The structure of T4 endonuclease V complexed with CPD DNA (PDB: 1VAS). The protein is shown as blue ribbons and the DNA in the same color scheme as in A. C: The structure of photolyase-CPD complex (PDB: 1TEZ).
Recognition by T4 Endonuclease and Photolyases
The reduced DNA rigidity at a CPD is exploited by the UV repair enzyme T4 endonuclease V (endo V) and photolyases.48–50 Both repair proteins possess a concave surface, where the active site resides, and approach the minor groove of a DNA lesion that is bent away from the protein [Fig. 2(B–C)]. The lesion DNA is reminiscent of its structure without protein, but bending and unwinding are much more severe, and the conformations of the CPD and its partner bases are completely different (Fig. 2). In the T4 endoV complex, the thymine dimer remains in the duplex, while the adenine base opposite the 5′ thymine of the CPD is flipped out, thus leaving the CPD base exposed to the active site residues for cleavage [Fig. 2(B)]. In contrast, when complexed with the photolyase, it is the thymine dimer that is flipped out from the DNA duplex and into the active site of the protein [Fig. 2(C)]. The alternative flipping out of a CPD or the base opposite it illustrates two salient features. First, both DNA strands are affected by the loss of base stacking, unwinding and bending at the lesion site. Second, lesions cause plasticity in DNA and the exact structure surrounding the lesion is determined by and is complementary to the repair proteins.
The features of CPD recognition are shared by interactions between many base lesions and their cognate repair proteins.21 Similar to the CPD photolyase, a 6-4PP photolyase also binds the bent DNA and flips the 6-4 PP into its active site.40,41 Most strikingly, bacterial MutY and the human DNA glycosylase hOGG1, which recognize and remove an 8-oxo-G opposite C versus a misincorporated A opposite 8-oxo-G, both have a concave surface and bind the minor groove side of a bent and unwound DNA substrate.51,52 Yet depending on whether 8-oxo-G or the base opposite to it is to be cleaved, either the damaged 8-oxo-G or a normal but mispaired A is flipped out of the duplex and inserted into the active site of the repair protein, hOGG1 or MutY, respectively.
These observations lead to the conclusion that DNA double helices containing damaged or modified bases are destabilized due to poor base stacking and has the plasticity to adopt various bent, unwound and/or base flipped-out conformations in the absence of any repair proteins. All photolyases and glycosylases contain a concave DNA binding surface, presumably to bind an already bent or easy-to-bend damaged DNA,39 while normal DNA with a persistent length of ∼500 Å is more resistant to bending and other forms of deformation. The difference in the rigidity of normal versus damaged DNA allows repair proteins to reduce the sampling frequency of millions to billions of normal base pairs and “zoom in” on a pre-existing deformed substrate. After finding a bent lesion site, each repair enzyme needs to further examine damaged DNA by complementarity (base specificity) tests to reject non-substrates and cleave only cognate substrates. Coupling of complementarity tests and chemistry must be stringent. Forced binding of normal DNA or noncognate substrate to repair enzymes can bypass the first step of the DNA rigidity test, but usually cannot pass the second step of the base specificity test.53,54
Overcoming the Flexibility of CPD by TLS Polymerase η
Poor base stacking and the flexible DNA helix around UV lesions, which allow lesion recognition by repair enzymes, are detrimental to DNA polymerases. High-fidelity DNA synthesis depends upon normal base pairing and stacking and extends the primer strand one base at a time.21 Although most UV lesions are repaired before DNA replication initiates, a few are still present and require specialized polymerases to bypass. Because template-directed nucleotide incorporation depends on base pairing, a polymerase that can correctly synthesize DNA opposite a 6-4 or dewar photoproduct does not exist, perhaps owing to the grossly deformed shape and lack of hydrogen bonding potential of the lesion. After tens of millions of years of evolution, DNA polymerase η emerged to be able to incorporate correct incoming nucleotides opposite CPD lesions.23,24,37,38
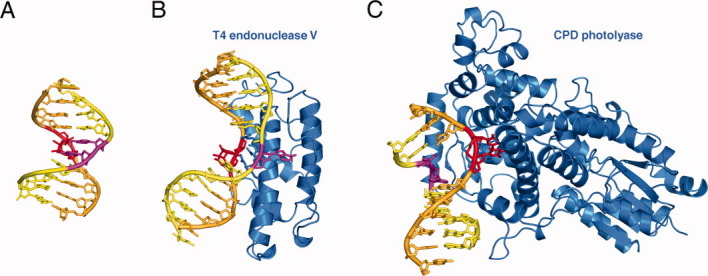
DNA polymerase η is a member of the Y-family of polymerases, which is generally characterized by an enlarged and solvent exposed active site such that these polymerases can accommodate base adducts and conduct TLS.21 The active site of Pol η readily accommodates a CPD or two normal pyrimidine bases simultaneously [Fig. 3(A)].37,38 This is unusual because the active site of a high-fidelity DNA polymerase and even many Y-family polymerases can accommodate only one template base at a time.21 The ability to bind a crosslinked CPD allows DNA pol η to use it as a template. To rigidify the lesion DNA and secure base pairing at a CPD site, Pol η has a positively charged phosphosugar-binding surface perfectly complementing DNA in the B-form conformation37 [Fig. 3(B)], and thus acts like a molecular splint to stiffen flexible CPD lesions. Facilitated by hydrogen bonding with the primer strand, Pol η is able to keep lesion-containing DNA in a normal helical structure even in the absence of proper base stacking.
Figure 3.
Human DNA pol η during translesion synthesis. A: A close-up view of the active site with the 3′base of the CPD in the templating position (PDB: 3MR3). The CPD lesion is shown in red; the protein is represented by a silver semitransparent molecular surface. The template strand is colored orange, and the primer yellow. The incoming nucleotide is shown in multicolor and the two Mg2+ essential for the catalysis as purple spheres. Hydrogen bonds between the replicating base pair are represented by gray dotted lines. B: DNA pol η holds the lesion DNA in a straight B-form conformation. DNA is shown in the same color scheme as in A. The molecular surface of Pol η is shown with positive (blue) and negative (red) electrostatic potential.
Lesion Recognition in NER
NER primarily repairs UV lesions, but it is versatile and also repairs bulky polycyclic aromatic hydrocarbon adducts, abasic sites, and many other helix-distorting lesions. Because of its broad substrate range, lesion recognition cannot be based on specific shape complementarity. Instead NER proteins have to recognize a common feature of damaged DNA that is absent in a normal double helix. To achieve high specificity with a broad range of substrates, multiple ATPases are involved in NER, and ATP is thought to provide energy in addition to the direct binding to enhance recognition specificity.55 TCR takes advantage of transcription blockage by the presence of DNA lesions and recognizes lesions on the transcribed strand. For GG-NER, UvrA and UvrB, which carry out lesion recognition in bacteria, are both ATPases.13 In eukaryotes, initial recognition of UV lesions, particularly CPDs, is carried out by XPE in vivo and passed onto XPC.12,56–58 ATP-dependent proofreading and lesion-dependent strand separation in eukaryotes are likely performed by XPB and XPD in a 10-protein complex called TFIIH.18,59 Only after confirmation of the presence of a lesion do UvrB in bacteria and TFIIH and XPA in eukaryotes recruit specific nucleases for DNA cleavage and lesion removal.
Crystal structures of UvrA, UvrB, XPC-HR23B, and XPE (also known as DDB1-DDB2 complex) complexed with DNA lesions (Fig. 4) reveal interesting properties of how they interact with damaged DNA and initiate lesion recognition. Only DDB2 of XPE, which specifically enhances recognition of UV lesions in GG-NER, interacts directly with a UV lesion.36 UvrA, UvrB, and XPC make no direct contact with DNA lesions,28,34,35 which is consistent with the fact that they have no special preference for a particular lesion type. The lesions are disordered in cocrystal structures with UvrA, UvrB, and XPC-HR23B. XPC and UvrA instead interact extensively with the surrounding normal DNA duplex60,61 (Fig. 4). Another striking features is that the UvrB and XPC, which are the second players in UV-lesion recognition and take damaged DNA from UvrA and XPE, respectively, both thread a beta-hairpin loop through DNA at the lesion site leading to strand separation [Fig. 4(C–D)]. It is not clear whether UvrB pins down the DNA strand with a lesion or the one opposite it.62 Nevertheless it prefers DNA with an unpaired bubble structure and directly interacts with UvrA for DNA handover.63 On the other hand, XPC clearly recognizes and binds the undamaged strand opposite a lesion.35,61
Figure 4.
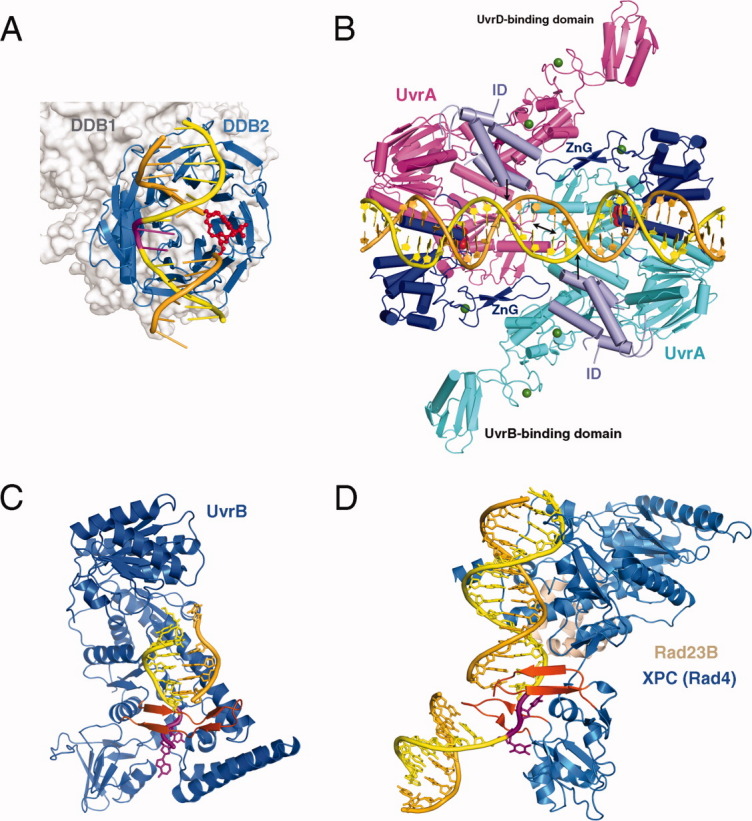
Proteins involved in NER. A: XPE complexed with 6-4 PP (PDB: 3EI1). The 6-4 PP is highlighted in red and the bases opposite it shown in magenta. DDB2 is a β-propeller protein and shown as blue ribbons. DDB1 binds DDB2 only and is shown in silver molecular surface in the background. B: UvrA-DNA complex (PDB: 3PIH). The bulk of two UvrA subunits are shown in cyan and pink ribbons diagrams except for the ATPase domains that directly contact DNA and contain the C-terminal Zinc-finger domain (dark blue) and the ID domains (pale blue). The UvrB-binding domains are labeled. Pyrophosphates marking the nucleotide-binding site are shown as red spheres, and Zn2+ as green spheres. The unstacked central G:C base pairs are indicated by a black double-headed arrow and the location of fluorescein labeled bases are marked by black arrows. C: UvrB – DNA complex (PDB: 2FDC). The protein is shown as a blue ribbon diagram, and the β hairpin that threads through DNA duplex and clamps down one strand is highlighted in orange-red. The flipped-out bases are highlighted in magenta. They are inserted into a conserved binding pocket. D: The structure of yeast homolog of XPC (Rad4) bound to a UV-damaged DNA. XPC is shown in blue and the partner protein Rad23 in light orange. Two β hairpins (in orange red) “hug” the un-damaged strand, while the damaged bases are flipped out and disordered.
DNA at the lesion site is severely distorted in all cases. DDB2 (or the p48 subunit) of XPE, which is responsible for DNA binding, approaches UV lesions with a concave surface on the minor groove of a bent and unwound DNA36 just like T4 endonuclease V or photolyase, although they share no structural similarity. The 6-4 PP cocrystalized with XPE is flipped out from the duplex into a shallow pocket in the protein [Fig. 4(A)]. Thus UV-lesion recognition by XPE is based on both the flexibility of DNA and complementarity between the lesion and protein. With specific recognition of UV lesions, XPE then passes the lesion DNA to XPC, which in turn recruits TFIIH (XPB and XPD) and XPA for lesion verification and strand separation in GGR.10,18 Consistent with their lesion recognition roles, XPC and DDB2 are not required in TCR.12
In the UvrA-DNA complex (without an ATP or ADP analog) structure, which contains two fluorescein labeled thymines as mimics of damaged bases, the DNA appears to be fully base paired and nearly straight34 [Fig. 4(B)]. The plasticity of the modified DNA is manifested in severe unwinding (20°) and base unstacking. The adjacent two central GC base pairs are separated by 6.5 Å, nearly double of the usual 3.4 Å. But UvrA has no direct contact with the DNA region containing the modified bases. Two domains of UvrA (the insertion domain (ID) and the C-terminal Zn finger (ZnG), which contribute to lesion-dependent DNA binding,27,64,65 are not in contact with the DNA.
A Stress-Test Model For Lesion Recognition by UvrA and UvrB
In bacteria, UvrA binds a lesion first and passes it onto UvrB, which then recruits UvrC nuclease to remove the lesion.1,9 The fully base-paired DNA in the UvrA-DNA complex, although unstacked and under-wound, cannot be easily bound by UvrB, which appears to prefer unpaired DNA strands. Some conformational changes must take place in the DNA. UvrA is an ABC-family ATPase66 and a functional dimer67 (Fig. 5). Its ATP binding domains are in direct contact with DNA duplex surrounding the lesion [Fig. 4(B)].34 ATP binding and hydrolysis likely results in domain movement in UvrA and may be correlated with DNA binding.26,64,65,68 One possibility is that in the absence of ATP UvrA unwinds and stretches the DNA as observed in the crystal structure [Fig. 4(B)]; upon binding and hydrolysis of ATP UvrA may continue to untwist the DNA but compress rather than stretching it (Fig. 5). Normal DNA with strong base stacking is resistant to such deformation. If forced to bend or unwound, normal DNA can readily reverse to the B-form when the external forces are removed. For example, normal DNA can bind UvrA presumably in a distorted form, but it dissociate from the protein upon ATP hydrolysis.68 In contrast, damaged DNA without proper base stacking is prone to deformation and does not return to normal B-form in a reversible path. Hence it can be considered “hysteresis”. Under the probing of unwinding and stretching/compressing by UvrA, the region of DNA with poor base stacking is likely to bend and buckle, which leads to strand separation (Fig. 5). The ID and ZnG domains of UvrA may interact with the severely deformed DNA and enhance both UvrA binding to lesion DNA and handing over lesion DNA to UvrB.
Figure 5.
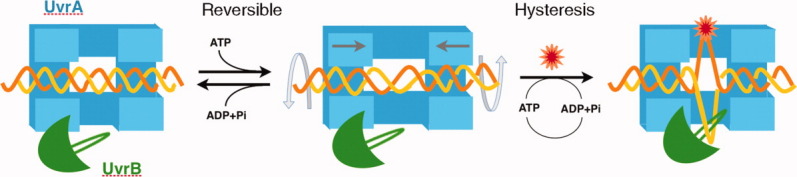
A diagram of lesion recognition and kinetic proofreading by UvrA. UvrA dimer is shown as a symmetric blue open box, and UvrB is shown in green with a β-hairpin. DNA helix (in yellow and orange) binds to UvrA and may be unwound and stretched somewhat, but once released from UvrA (e.g., during UvrA ATP-hydrolysis) it returns to normal B form. But damaged DNA is likely to become bent and unwound under such stress test and undergoes strand separation. UvrB, which is physically attached to UvrA, can then grab the ssDNA and complete lesion recognition.
Concluding Remarks
UV damage causes the most prevalent lesions in DNA. All living organisms employ multiple repair pathways to remove, repair, and bypass UV lesions. The absence of base stacking and reduced DNA rigidity are intrinsic features of UV lesions, which are actively exploited by repair proteins for lesion recognition. UV lesion-specific repair proteins, for example T4 endonuclease V, photolyase, and XPE, use a direct readout mechanism and interact with both the bent DNA and the lesion or its base pairing partners. On the other hand, DNA polymerase η, which is specialized in translesion synthesis to bypass CPD lesions and has an extensive and complementary binding surface for B-form DNA, acts like a molecular splint to fortify the backbone at a lesion site and allow it to template new DNA synthesis. In contrast, repair proteins with a broad substrate range, such as those in the NER pathway, use an indirect readout mechanism exemplified by UvrA. UvrA binds normal and damaged DNA alike and conducts a “stress resistance” test using its intrinsic ATPase activity. Only DNA with a lesion may undergo strand separation due to hysteresis and can thus be passed onto UvrB. Therefore UvrA achieves high binding specificity for different types of damaged DNA with the help of ATPase and a partner protein UvrB.
Acknowledgments
The author thanks Drs. B. Craigie and D. Leahy for critical reading of the manuscript, and M. Gellert for suggesting the term “hysteresis” to describe damaged DNA.
REFERENCES
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2nd ed. Washington: DC: ASM Press; 2006. [Google Scholar]
- 2.Sancar A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev. 2003;103:2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 3.Latham KA, Lloyd RS. T4 endonuclease V. Perspectives on catalysis. Ann NY Acad Sci. 1994;726:181–196. doi: 10.1111/j.1749-6632.1994.tb52813.x. discussion 196–187. [DOI] [PubMed] [Google Scholar]
- 4.Goosen N, Moolenaar GF. Repair of UV damage in bacteria. DNA Repair (Amst) 2008;7:353–379. doi: 10.1016/j.dnarep.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Kaur B, Doetsch PW. Ultraviolet damage endonuclease (Uve1p): a structure and strand-specific DNA endonuclease. Biochemistry. 2000;39:5788–5796. doi: 10.1021/bi000189r. [DOI] [PubMed] [Google Scholar]
- 6.Lindahl T. Keynote: past, present, and future aspects of base excision repair. Prog Nucleic Acid Res Mol Biol. 2001;68:xvii–xxx. doi: 10.1016/s0079-6603(01)68084-x. [DOI] [PubMed] [Google Scholar]
- 7.Fromme JC, Verdine GL. Base excision repair. Adv Prot Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 8.Friedberg EC. Nucleotide excision repair of DNA: the very early history. DNA Repair (Amst) 2011;10:668–672. doi: 10.1016/j.dnarep.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 9.Truglio JJ, Croteau DL, Van Houten B, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem Rev. 2006;106:233–252. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- 10.Naegeli H, Sugasawa K. The xeroderma pigmentosum pathway: decision tree analysis of DNA quality. DNA Repair (Amst) 2011;10:673–683. doi: 10.1016/j.dnarep.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 11.de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 12.Hanawalt PC, Spivak G. Transcription-coupled DNA repair two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 13.Sancar A, Reardon JT. Nucleotide excision repair in IE. coli Iand man. Adv Prot Chem. 2004;69:43–71. doi: 10.1016/S0065-3233(04)69002-4. [DOI] [PubMed] [Google Scholar]
- 14.Savery NJ. The molecular mechanism of transcription-coupled DNA repair. Trends Microbiol. 2007;15:326–333. doi: 10.1016/j.tim.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Deaconescu AM, Chambers AL, Smith AJ, Nickels BE, Hochschild A, Savery NJ, Darst SA. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 16.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 17.Zou Y, Van Houten B. Strand opening by the UvrA(2)B complex allows dynamic recognition of DNA damage. EMBO J. 1999;18:4889–4901. doi: 10.1093/emboj/18.17.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oksenych V, Coin F. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle. 2010;9:90–96. doi: 10.4161/cc.9.1.10267. [DOI] [PubMed] [Google Scholar]
- 19.Budzowska M, Kanaar R. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem Biophys. 2009;53:17–31. doi: 10.1007/s12013-008-9039-y. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann AR. Replication of UV-damaged DNA: new insights into links between DNA polymerases, mutagenesis and human disease. Gene. 2000;253:1–12. doi: 10.1016/s0378-1119(00)00250-x. [DOI] [PubMed] [Google Scholar]
- 21.Yang W, Woodgate R. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci USA. 2007;104:15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hishida T, Kubota Y, Carr AM, Iwasaki H. RAD6-RAD18-RAD5-pathway-dependent tolerance to chronic low-dose ultraviolet light. Nature. 2009;457:612–615. doi: 10.1038/nature07580. [DOI] [PubMed] [Google Scholar]
- 23.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 24.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 25.Pakotiprapha D, Inuzuka Y, Bowman BR, Moolenaar GF, Goosen N, Jeruzalmi D, Verdine GL. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol Cell. 2008;29:122–133. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi F, Khanduja JS, Bortoluzzi A, Houghton J, Sander P, Guthlein C, Davis EO, Springer B, Bottger EC, Relini A, Penco A, Muniyappa K, Rizzi M. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 2011;39:7316–7328. doi: 10.1093/nar/gkr271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timmins J, Gordon E, Caria S, Leonard G, Acajjaoui S, Kuo MS, Monchois V, McSweeney S. Structural and mutational analyses of Deinococcus radiodurans UvrA2 provide insight into DNA binding and damage recognition by UvrAs. Structure. 2009;17:547–558. doi: 10.1016/j.str.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Truglio JJ, Karakas E, Rhau B, Wang H, DellaVecchia MJ, Van Houten B, Kisker C. Structural basis for DNA recognition and processing by UvrB. Nat Struct Mol Biol. 2006;13:360–364. doi: 10.1038/nsmb1072. [DOI] [PubMed] [Google Scholar]
- 29.Machius M, Henry L, Palnitkar M, Deisenhofer J. Crystal structure of the DNA nucleotide excision repair enzyme UvrB from Thermus thermophilus. Proc Natl Acad Sci USA. 1999;96:11717–11722. doi: 10.1073/pnas.96.21.11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Theis K, Chen PJ, Skorvaga M, Van Houten B, Kisker C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999;18:6899–6907. doi: 10.1093/emboj/18.24.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakagawa N, Sugahara M, Masui R, Kato R, Fukuyama K, Kuramitsu S. Crystal structure of Thermus thermophilus HB8 UvrB protein, a key enzyme of nucleotide excision repair. J Biochem. 1999;126:986–990. doi: 10.1093/oxfordjournals.jbchem.a022566. [DOI] [PubMed] [Google Scholar]
- 32.Eryilmaz J, Ceschini S, Ryan J, Geddes S, Waters TR, Barrett TE. Structural insights into the cryptic DNA-dependent ATPase activity of UvrB. J Mol Biol. 2006;357:62–72. doi: 10.1016/j.jmb.2005.12.059. [DOI] [PubMed] [Google Scholar]
- 33.Waters TR, Eryilmaz J, Geddes S, Barrett TE. Damage detection by the UvrABC pathway: crystal structure of UvrB bound to fluorescein-adducted DNA. FEBS Lett. 2006;580:6423–6427. doi: 10.1016/j.febslet.2006.10.051. [DOI] [PubMed] [Google Scholar]
- 34.Jaciuk M, Nowak E, Skowronek K, Tanska A, Nowotny M. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat Struct Mol Biol. 2011;18:191–197. doi: 10.1038/nsmb.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 36.Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biertumpfel C, Zhao Y, Kondo Y, Ramon-Maiques S, Gregory M, Lee JY, Masutani C, Lehmann AR, Hanaoka F, Yang W. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silverstein TD, Johnson RE, Jain R, Prakash L, Prakash S, Aggarwal AK. Structural basis for the suppression of skin cancers by DNA polymerase eta. Nature. 2010;465:1039–1043. doi: 10.1038/nature09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair (Amst) 2006;5:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Glas AF, Schneider S, Maul MJ, Hennecke U, Carell T. Crystal structure of the T(6-4)C lesion in complex with a (6-4) DNA photolyase and repair of UV-induced (6-4) and Dewar photolesions. Chemistry. 2009;15:10387–10396. doi: 10.1002/chem.200901004. [DOI] [PubMed] [Google Scholar]
- 41.Maul MJ, Barends TR, Glas AF, Cryle MJ, Domratcheva T, Schneider S, Schlichting I, Carell T. Crystal structure and mechanism of a DNA(6-4) photolyase. Angew Chem Int Ed Engl. 2008;47:10076–10080. doi: 10.1002/anie.200804268. [DOI] [PubMed] [Google Scholar]
- 42.Park H, Zhang K, Ren Y, Nadji S, Sinha N, Taylor JS, Kang C. Crystal structure of a DNA decamer containing a cis-syn thymine dimer. Proc Natl Acad Sci USA. 2002;99:15965–15970. doi: 10.1073/pnas.242422699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JH, Choi YJ, Choi BS. Solution structure of the DNA decamer duplex containing a 3′-T x T basepair of the cis-syn cyclobutane pyrimidine dimer: implication for the mutagenic property of the cis-syn dimer. Nucleic Acids Res. 2000;28:1794–1801. doi: 10.1093/nar/28.8.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Bae SH, Choi BS. The Dewar photoproduct of thymidylyl(3′-->5′)-thymidine (Dewar product) exhibits mutagenic behavior in accordance with the “A rule”. Proc Natl Acad Sci USA. 2000;97:4591–4596. doi: 10.1073/pnas.080057097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bang J, Kang YM, Park CJ, Lee JH, Choi BS. Thermodynamics and kinetics for base pair opening in the DNA decamer duplexes containing cyclobutane pyrimidine dimer. FEBS Lett. 2009;583:2037–2041. doi: 10.1016/j.febslet.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 46.Chandrasekhar D, Van Houten B. In vivo formation and repair of cyclobutane pyrimidine dimers and 6-4 photoproducts measured at the gene and nucleotide level in Escherichia coli. Mutat Res. 2000;450:19–40. doi: 10.1016/s0027-5107(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 47.Luo C, Krishnasamy R, Basu AK, Zou Y. Recognition and incision of site-specifically modified C8 guanine adducts formed by 2-aminofluorene, N-acetyl-2-aminofluorene and 1-nitropyrene by UvrABC nuclease. Nucleic Acids Res. 2000;28:3719–3724. doi: 10.1093/nar/28.19.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vassylyev DG, Kashiwagi T, Mikami Y, Ariyoshi M, Iwai S, Ohtsuka E, Morikawa K. Atomic model of a pyrimidine dimer excision repair enzyme complexed with a DNA substrate: structural basis for damaged DNA recognition. Cell. 1995;83:773–782. doi: 10.1016/0092-8674(95)90190-6. [DOI] [PubMed] [Google Scholar]
- 49.Mees A, Klar T, Gnau P, Hennecke U, Eker AP, Carell T, Essen LO. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science. 2004;306:1789–1793. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]
- 50.Torizawa T, Ueda T, Kuramitsu S, Hitomi K, Todo T, Iwai S, Morikawa K, Shimada I. Investigation of the cyclobutane pyrimidine dimer (CPD) photolyase DNA recognition mechanism by NMR analyses. J Biol Chem. 2004;279:32950–32956. doi: 10.1074/jbc.M404536200. [DOI] [PubMed] [Google Scholar]
- 51.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 52.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 53.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 54.Qi Y, Spong MC, Nam K, Banerjee A, Jiralerspong S, Karplus M, Verdine GL. Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature. 2009;462:762–766. doi: 10.1038/nature08561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reardon JT, Sancar A. Thermodynamic cooperativity and kinetic proofreading in DNA damage recognition and repair. Cell Cycle. 2004;3:141–144. [PubMed] [Google Scholar]
- 56.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 57.Zolezzi F, Linn S. Studies of the murine DDB1 and DDB2 genes. Gene. 2000;245:151–159. doi: 10.1016/s0378-1119(00)00022-6. [DOI] [PubMed] [Google Scholar]
- 58.Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol. 2010;30:2708–2723. doi: 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 60.Van Houten B, Gamper H, Sancar A, Hearst JE. DNase I footprint of ABC excinuclease. J Biol Chem. 1987;262:13180–13187. [PubMed] [Google Scholar]
- 61.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 62.Jia L, Kropachev K, Ding S, Van Houten B, Geacintov NE, Broyde S. Exploring damage recognition models in prokaryotic nucleotide excision repair with a benzo[a]pyrene-derived lesion in UvrB. Biochemistry. 2009;48:8948–8957. doi: 10.1021/bi9010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pakotiprapha D, Liu Y, Verdine GL, Jeruzalmi D. A structural model for the damage-sensing complex in bacterial nucleotide excision repair. J Biol Chem. 2009;284:12837–12844. doi: 10.1074/jbc.M900571200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Croteau DL, DellaVecchia MJ, Wang H, Bienstock RJ, Melton MA, Van Houten B. The C-terminal zinc finger of UvrA does not bind DNA directly but regulates damage-specific DNA binding. J Biol Chem. 2006;281:26370–26381. doi: 10.1074/jbc.M603093200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagner K, Moolenaar GF, Goosen N. Role of the insertion domain and the zinc-finger motif of IEscherichia coli IUvrA in damage recognition and ATP hydrolysis. DNA Repair (Amst) 2011;10:483–496. doi: 10.1016/j.dnarep.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 66.Doolittle RF, Johnson MS, Husain I, Van Houten B, Thomas DC, Sancar A. Domainal evolution of a prokaryotic DNA repair protein and its relationship to active-transport proteins. Nature. 1986;323:451–453. doi: 10.1038/323451a0. [DOI] [PubMed] [Google Scholar]
- 67.Mazur SJ, Grossman L. Dimerization of IEscherichia coli IUvrA and its binding to undamaged and ultraviolet light damaged DNA. Biochemistry. 1991;30:4432–4443. doi: 10.1021/bi00232a009. [DOI] [PubMed] [Google Scholar]
- 68.Thiagalingam S, Grossman L. Both ATPase sites of IEscherichia coliI UvrA have functional roles in nucleotide excision repair. J Biol Chem. 1991;266:11395–11403. [PubMed] [Google Scholar]