

Abstract
Methods for rapid and reliable design and structure prediction of linker loops would facilitate a variety of protein engineering applications. Circular permutation, in which the existing termini of a protein are linked by the polypeptide chain and new termini are created, is one such application that has been employed for decreasing proteolytic susceptibility and other functional purposes. The length and sequence of the linker can impact the expression level, solubility, structure and function of the permuted variants. Hence it is desirable to achieve atomic-level accuracy in linker design. Here, we describe the use of RosettaRemodel for design and structure prediction of circular permutation linkers on a model protein. A crystal structure of one of the permuted variants confirmed the accuracy of the computational prediction, where the all-atom rmsd of the linker region was 0.89 Å between the model and the crystal structure. This result suggests that RosettaRemodel may be generally useful for the design and structure prediction of protein loop regions for circular permutations or other structure-function manipulations.
Keywords: circular permutation, RosettaRemodel, computational protein design, protein structure prediction
Introduction
Computational protein design is an increasingly efficient and powerful tool to manipulate protein structure and function.1–7 However, most computational design has focused on optimizing amino acid sequences on static backbone structures. One of the essential challenges of more aggressive protein remodeling is the de novo design and structure prediction of individual protein segments within a rigid protein. RosettaRemodel is a generalized method for protein design and structure prediction in which backbone conformational freedom and sequence variation can be restricted to particular protein segments.8 Here we have studied a case of circular permutation as one example of a common structural manipulation requiring design of a single protein segment.9 The starting molecule to be circularly permuted was an epitope-scaffold onto which the 4E10 HIV neutralization epitope had been transplanted, as previously described by Correia et al.10 The epitope-scaffold, which had high affinity towards the 4E10 antibody (Fig. 1), was used as a model system to assess the linker design and structure prediction capabilities of RosettaRemodel.
Figure 1.
Circular permutation of a 4E10 epitope-scaffold. The original termini of the epitope-scaffold are labeled in the left panel. The cutting point to generate the novel termini and the designed linker to join the original termini are schematically represented in the middle panel. The final permuted epitope-scaffold is shown in the right panel. The spatial proximity of the his tag (H) to the 4E10 antibody is also illustrated on the left panel.
Using RosettaRemodel we computationally modeled linker loops of different lengths to evaluate a variety of solutions to join the original termini. Most of the experimentally tested variants yielded soluble proteins and a crystal structure solved for one of the permuted designs showed very close agreement with the low-energy models generated by Rosetta.
Results
Computational prediction of the linker used in the circular permutaton
To accomplish the circular permutation and relocate the termini to a region distal from the transplanted epitope, a loop modeling protocol was used to build several linkers to join the original termini, and new termini were created in a loop located on the opposite side of the protein relative to the epitope (Fig. 1). Because of the spatial proximity of the original termini (Cα-Cα distance = 8.7 Å), we started by modeling linkers with lengths ranging from five to seven residues; the linker sequences were composed of different combinations of alanine and glycine (Table I). RosettaRemodel was used to perform the structure prediction calculations. The first stage was carried out at low resolution in “centroid mode”, with side-chains represented by spheres located at each side-chain center-of-mass, and the new backbone conformations were built based on a fragment insertion protocol together with cyclic coordinate descent (CCD) to maintain proper chain connectivity.11,12 The second stage of structure prediction was carried out with all-atom detail, and both backbone and side-chains were refined and minimized for accurate energy evaluation. For each of the different designed linkers, 2500 models were generated in which sampling of conformational degrees of freedom was restricted to the linker region. The 2500 models were clustered according to the Cα root mean square deviation (rmsd) in the loop region, and the three largest clusters included 303, 36, and 15 models, respectively [Fig. 2(A)]. The lowest energy model in the largest cluster was a logical selection as the top-ranked RosettaRemodel prediction for the loop conformation. The CPU (Intel 2GHz quad-core) time needed for RosettaRemodel to generate 2500 models was ∼ 2500 min.
Table I.
Sequence Features and Experimental Characterization of the Designed Permuted Variants
| Design | Loop Length | Sequence | Secondary Structure | Oligomeric State | Tm (°C) |
|---|---|---|---|---|---|
| Nonpermuted | — | — | — | Dimer | 56 |
| 003 | 6 | EAGGAM | HLLLLL | Dimer | 51 |
| 004 | 5 | EAGGM | HLLLL | Dimer | 49 |
| 005 | 6 | EAAGGM | HLLLLL | Multimer | NT |
| 006 | 5 | EAAGM | HLLLL | Dimer | 48 |
| 007 | 5 | EGGGM | HLLLL | Dimer | NT |
| 008 | 7 | EAGGAAM | HLLLLLL | Insoluble | - |
The solution oligomeric state was characterized by SEC/SLS. The melting temperatures were determined by circular dichroism spectroscopy.
Figure 2.
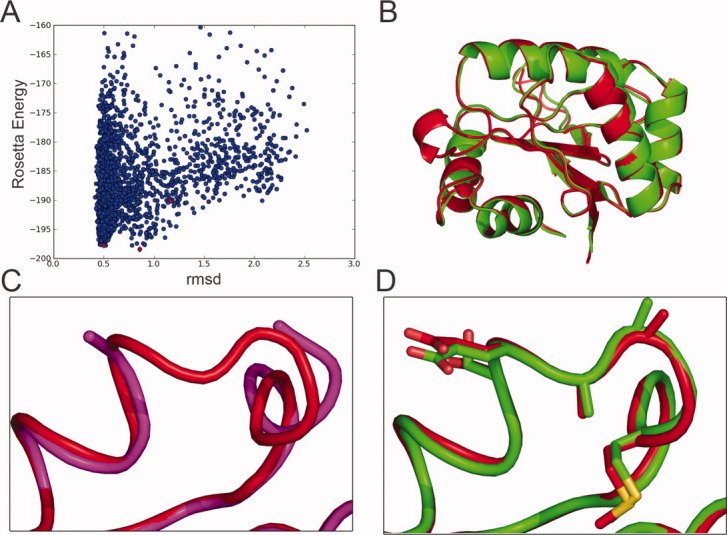
Identification of a low Rosetta energy model which closely mimics the crystal structure of a designed permuted variant. A: Plot of Rosetta energy versus rmsd to the crystal structure of 006, for the 2500 models with different loop conformations (blue circles) generated by RosettaRemodel. The 2500 models were clustered by all-against-all rmsd of the designed linkers, and red diamonds show the lowest Rosetta energy models from the three largest clusters. B: The crystal structure of the 006 permuted variant (red) was in close agreement with the lowest energy model from the largest cluster (green). No major changes were observed in terms of the overall fold relative to the original epitope-scaffold. C: Comparison of the linker region in the original epitope-scaffold (magenta) and the permuted version (red). D: Comparison of the linker region in the lowest energy model from the largest cluster (green) and the crystal structure (red). Side-chains are shown in sticks.
The sequences of the native protein and the permuted variants were tested for their similarity to known proteins by performing a Blast13 search against the nonredundant protein sequence database. While multiple full length matches were found for the native protein, none were found for the permuted variants. Instead, searches with the permuted variants recovered only discontinuous matches in which the N-terminus of the permuted variant matched to the C-terminus of the hits and the C-terminus of the permuted variant matched to the N-terminus of the hits (Fig. S1). Hence, circular permutation in this case created novel proteins.
Stability and solution behavior
To assess the solution behavior and thermal stabilities, all 6 permuted constructs were expressed in E. coli and experimentally characterized. Five of the six designs were purifiable and soluble. The solution oligomeric state was assessed by static light scattering (SLS) in-line with size exclusion chromatography (SEC). Four of five designs formed dimers in solution like the parent molecule, while one design formed a higher order multimer. The thermal stability of the designs was assessed using circular dichroism temperature melt analysis. Three of the designs had Tms ranging from 48°C to 51°C (Table I) where two other variants showed no transition. The permuted variants were prone to aggregation, as many 4E10 scaffolds have been,10 and this prevented quantitative assessment of binding affinities for the 4E10 antibody.
Structural characterization and modeling accuracy
To evaluate the accuracy of the computational modeling, crystal structures of the designs were pursued. Crystallization trials were conducted for all purifiable designs. One design (006) formed diffraction-quality crystals and a structure was determined (Table II). The overall fold of the parent protein was maintained in the permuted variant, with a backbone (N, Cα, C, O) rmsd of 0.4 Å between permuted variant and nonpermuted parent [Fig. 2(B)]. Upon the circular permutation, some of the residues included in the original termini underwent subtle conformational rearrangements [Fig. 2(C)]. The backbone and all-atom rmsd values in the designed loop region between the crystal structure of 006 and the lowest energy model in the largest cluster were 0.5 Å and 0.89 Å, respectively [Fig. 2(D)].
Table II.
Crystallographic Statistics
| Data Collection | |
|---|---|
| Space group | P21 |
| Lattice constants | 36.1, 65.3, 73.1 Å; β = 99.6° |
| Resolution (Å) | 30.31–1.95 (2.02–1.95) |
| Number observed reflections | 74,218 (4485) |
| Number unique reflections | 24,452 (2431) |
| Redundancy | 3.04 (1.84) |
| Completeness (%) | 99.7 (99.6) |
| Rmerge | 0.052 (0.273) |
| Average I/σ(I) | 12.5 (2.0) |
| Structure Refinement | |
| Resolution (Å) | 30.32–1.95 |
| Rwork/Rfree | 0.204/0.249 |
| Number of atoms | |
| Protein | 2430 |
| Water | 194 |
| r.m.s. deviations from ideal values | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.040 |
| Chiral volume (Å3) | 0.061 |
| Ramachandran plot statistics (Procheck) | |
| Residues in most favored regions (%) | 94.0 |
| Residues in additional allowed regions (%) | 6.0 |
| Residues in generously allowed regions (%) | 0.0 |
| Residues in disallowed regions (%) | 0.0 |
| Estimated coordinate error (maximum likelihood e.s.u.) (Å) | 0.115 |
| Average B factor (Å2) | |
| Protein | 43.8 |
| Water | 50.7 |
Statistics for the highest resolution shell are shown in parentheses.
Discussion
Circular permutation has been used for multiple purposes that span the optimization of solution behavior14 and function.15–17 Here we report a fast and accurate computational method that allows for the modeling of linkers to join the pre-existing termini, enabling the generation of the circular permuted variants in a controlled and rational fashion. The computational model and the solved crystal structure were in close agreement in terms of backbone and side-chain conformations. The computational model was selected based on cluster size and Rosetta full-atom energy, so the accuracy of the model supports the validity of both the conformational sampling and the energy function implemented in Rosetta.
Several computationally designed loops have been previously reported. Hu et al.18 accomplished the design of a 10 residue loop for which the conformation was predicted with subangstrom accuracy. In that work, several iterations of sequence-design and structural optimization were utilized to obtain the final sequence and structure. Correia et al.19 designed a 16 residue helix-loop segment that contributed to a protein core, also with subangstrom accuracy. That work followed a similar methodology, but unlike RosettaRemodel the conformational sampling and sequence design stages were not automated within a self-contained protocol. Here, for the design of a shorter five residue linker, the RosettaRemodel protocol achieved similarly accurate structure prediction with less sampling (2500 models). Hence, RosettaRemodel holds promise for more complex protein engineering tasks.
Methods
Computational Method
The RosettaRemodel protocol implemented in the software package Rosetta20 was used to sample low energy loop conformations with different predefined sequences (Table I). In the starting structure used in the computational protocol (PDB accession code 1xiz), new termini were imposed by removing two residues (K16 and E17 numbered as in 1xiz.pdb) and the original termini were joined by computationally modeled linkers. In the computational simulations, 2500 models were generated for each linker and only the side-chains included on the linker were allowed to sample different conformations, while the coordinates of the remainder of the structure were kept fixed. The conformational space for the newly designed linkers was sampled based on fragment insertion in conjunction with a Cyclic Coordinate Descendent step to guarantee proper polypeptide chain connectivity.11,12 The fragments used were collected from available crystal structures21 and selected according to secondary structure defined for the newly designed linkers.19 Initially, conformational sampling was carried with a low-resolution description of the side-chains, which were represented as centroids, and in the final stage the sampled conformations were refined and scored using a full atom description of the protein.8 The generated models were clustered according to the rmsd of the designed loop using a hierarchical cluster algorithm as implemented in Rosetta, with a cluster radius of 0.1 Å. The lowest full atom energy21 structure from the largest cluster was selected to establish structural comparisons with the solved crystal structure.
The following command line was used to run the RosettaRemodel protocol as implemented in the software package Rosetta version 2.0: rosetta.intel -pose1 -remodel -s input.pdb -blueprint input.blueprint -try 50 -save_top 50 -num_frag_moves 10 -use_non_monotone_line_search –paths paths.txt. The blueprint file is composed of lines with residue specific instructions. The following examples illustrate the lines necessary for the designs in this paper: “1 V.” - residue 1 with the native residue valine will be untouched; “137 G L PIKAA A” – residue 137 with native residue glycine where fragments with loop conformation (L) will be inserted and the sequence change allowed is to alanine (PIKAA A). The use of this simplified syntax enables the manipulation of sequence and structure using RosettaRemodel. The computational models of the variants described in Table I are available in the electronic Supporting Information.
Expression and purification
Protein expression, purification, and thermal stability measurements were carried out as described in Correia et al.10,19
Crystallization and crystallography
Crystals of T298 (12 mg/mL) were grown by vapor diffusion (well solution: 10% w/w PEG 4000, 20% v/v isopropanol) and cryo-protected with ethylene glycol. Diffraction data to 1.95 Å were collected at −170°C on a Saturn CCD detector with HF optic (Rigaku) and processed with d*TREK.22 Initial structure factor phases were determined by molecular replacement, using the program Phaser23 as implemented in the CCP4i graphical user interface,24 and a search model consisting of the partially refined model of a related epitope-scaffold with the epitope and several key residues removed. Successive rounds of modeling and positional and individual B factor refinement were carried out with the programs Coot25 and Refmac5.26 Structure validation was carried out with Procheck,27 the MolProbity server,28 and the RCSB ADIT validation server. The structure has been deposited in the RCSB PDB29 with PDB ID 3T43. Data collection and structure refinement statistics are shown in Table II.
Acknowledgments
The authors thank Colin Corrent for assistance with the structure deposition.
Supplementary material
REFERENCES
- 1.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 2.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 3.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 5.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 6.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang PS, Ban YEA, Richter F, Andre I, Vernon R, Schief WR, Baker D. RosettaRemodel: A generalized framework for flexible backbone protein design. PloSOne. 2011;6:e24109. doi: 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Y, Lutz S. Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol. 2011;29:18–25. doi: 10.1016/j.tibtech.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Correia BE, Ban YE, Holmes MA, Xu H, Ellingson K, Kraft Z, Carrico C, Boni E, Sather DN, Zenobia C, et al. Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure. 2010;18:1116–1126. doi: 10.1016/j.str.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Bradley P, Baker D. Protein-protein docking with backbone flexibility. J Mol Biol. 2007;373:503–519. doi: 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 12.Canutescu AA, Dunbrack RL., Jr Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitehead TA, Bergeron LM, Clark DS. Tying up the loose ends: circular permutation decreases the proteolytic susceptibility of recombinant proteins. Protein Eng Des Sel. 2009;22:607–613. doi: 10.1093/protein/gzp034. [DOI] [PubMed] [Google Scholar]
- 15.Reitinger S, Yu Y, Wicki J, Ludwiczek M, D'Angelo I, Baturin S, Okon M, Strynadka NC, Lutz S, Withers SG, et al. Circular permutation of Bacillus circulans xylanase: a kinetic and structural study. Biochemistry. 2010;49:2464–2474. doi: 10.1021/bi100036f. [DOI] [PubMed] [Google Scholar]
- 16.Qian Z, Lutz S. Improving the catalytic activity of Candida antarctica lipase B by circular permutation. J Am Chem Soc. 2005;127:13466–13467. doi: 10.1021/ja053932h. [DOI] [PubMed] [Google Scholar]
- 17.Cheltsov AV, Guida WC, Ferreira GC. Circular permutation of 5-aminolevulinate synthase: effect on folding, conformational stability, and structure. J Biol Chem. 2003;278:27945–27955. doi: 10.1074/jbc.M207011200. [DOI] [PubMed] [Google Scholar]
- 18.Hu X, Wang H, Ke H, Kuhlman B. High-resolution design of a protein loop. Proc Natl Acad Sci USA. 2007;104:17668–17673. doi: 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Correia BE, Ban YE, Friend DJ, Ellingson K, Xu H, Boni E, Bradley-Hewitt T, Bruhn-Johannsen JF, Stamatatos L, Strong RK, et al. Computational protein design using flexible backbone remodeling and resurfacing: case studies in structure-based antigen design. J Mol Biol. 2011;405:284–297. doi: 10.1016/j.jmb.2010.09.061. [DOI] [PubMed] [Google Scholar]
- 20.Das R, Baker D. Macromolecular modeling with rosetta. Ann Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 21.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 22.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Cryst. 1999;D55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 23.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Cryst. 2003;D59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 28.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.