

Abstract
M1 protein, a major virulence factor of the leading invasive strain of group A Streptococcus, is sufficient to induce toxic shock-like vascular leakage and tissue injury. These events are triggered by the formation of a complex between M1 and fibrinogen (Fg) that, unlike M1 or Fg alone, leads to neutrophil activation. Here we provide a structural explanation for the pathological properties of the M1-Fg complex. A conformationally dynamic coiled-coil dimer of M1 was found to organize four Fg molecules into a specific cross-like pattern. This pattern supported the construction of a supramolecular network that was required for neutrophil activation but was distinct from a fibrin clot. Disruption of this network into other supramolecular assemblies was not tolerated. These results have bearing on the pathophysiology of streptococcal toxic shock.
The M protein1 is the major surface-associated virulence factor of Streptococcus pyogenes (group A Streptococcus, GAS), a widespread bacterial pathogen that causes both mild infections and severe invasive diseases with high mortality rates (~30%), such as streptococcal toxic shock syndrome (STSS)2. Antigenic variation has resulted in >100 M protein types3 but only a few are frequently associated with invasive disease, with type M1 being the most prevalent4. Strains belonging to a globally disseminated subclone of the M1T1serotype have been the leading cause of severe invasive GAS infection worldwide for the past 30 years5. The M1 protein itself has proinflammatory properties and in animal models is sufficient to trigger vascular leakage and tissue injury similar to that observed in STSS6,7,8,9. These pathological properties of M1 require its interaction with fibrinogen (Fg). The M1-Fg complex binds β2 integrins on neutrophils and triggers the release of heparin binding protein (HBP), a potent vasodilator10 and a strong indicator of sepsis and circulatory failure in patients11. The M1-Fg complex also activates platelets in an integrin-dependent manner12, leading to further activation of neutrophils as well as monocytes.
How the M1-Fg complex causes neutrophil activation, when neither protein alone does6,8, is not known. To address this issue, we determined the ~3.3 Å resolution crystal structure of an M1-Fg complex containing the M1 fragment M1BC1 (residues 132-263, ~17 kDa) and fibrinogen fragment D (FgD, ~86 kDa)13,14 (Supplementary Table 1 and Supplementary Figs 1 and 2). The M1BC1 fragment contains the B-repeats, which are sufficient to bind Fg8,15, and the S-region, to which immunoglobulin G’s (IgG) bind and enhance the release of HBP through FcγRII7; it also contains the first C-repeat of M1. FgD, which comprises the majority of Fg, is necessary and sufficient to bind M116. The final model consists of M1BC1 residues 132-238, whose register was separately determined using anomalous scattering (see Methods); all but the first five residues of the C-repeats were apparently removed by proteolysis. The entirety of FgD, as seen in the crystal structure of the unbound form13,14, was visible except for several residues at either end. While residues of M1BC1 distal to the interface with FgD were often in incomplete electron density, residues at the interface had well defined electron density, enabling specific intermolecular contacts to be discerned.
Cross-like pattern
The most striking aspect of the structure is the fact that M1BC1 is surrounded by four FgD molecules in a cross-like pattern (Fig. 1). Two pairs of FgD molecules (each ~130 Å long) lie roughly perpendicular to one other as well as to M1BC1 (~160 Å long), which runs through the center of the cross. FgD, as previously described13, consists of a parallel heterotrimeric (αβγ) α-helical coiled coil connected to globular heads. M1BC1 forms a parallel homodimeric α-helical coiled coil throughout most of its length, including the B-repeats that bind FgD. There are four B-repeats in all, two per M1 chain, explaining the 2:4 M1BC1:FgD stoichiometry of this ~380 kDa complex. The upstream B-repeats (B1) bind two FgD molecules that are oriented ~180° to one another due to the dyad symmetry of the M1 coiled coil, as do the downstream B-repeats (B2). B1 and B2 are separated by 28 residues, roughly a quarter turn of the coiled-coil superhelix, meaning that the two pairs of FgD molecules are oriented ~90° to one another, giving rise to the cross-like pattern.
Figure 1. M1 assembles Fg into a cross-like pattern.
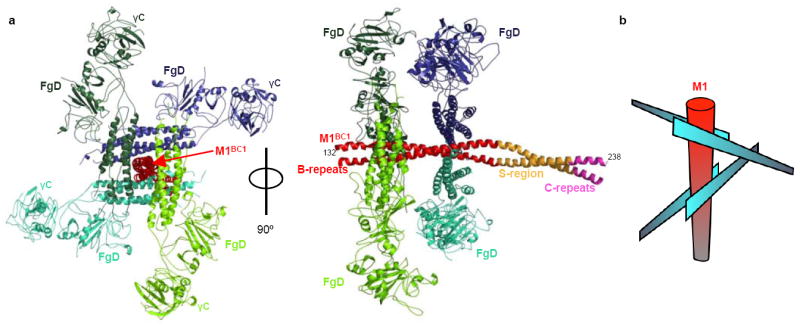
a, The M1BC1-FgD structure in ribbon representation. The M1BC1 B-repeats are in red, S-region in gold, and C-repeats in purple. The four FgD molecules bound by M1BC1 are in shades of blue or green, and the β2 integrin-binding γC domains are indicated. b, Schematic of the cross-like pattern of FgD (blue blades) surrounding M1BC1 (red cylinder).
The four separate M1-FgD contact sites are nearly identical in structure, being predominantly polar and each having a small buried surface area of ~645 Å2 (Fig. 2a). Residues in the B-repeats from both helices of the coiled coil and every heptad position (i.e., a-g) contribute to FgD binding (Fig. 2b). Although the two B-repeats are imperfect in sequence, the FgD-binding residues are identical between the two (Fig. 3). For FgD, the coiled coils in the β and γ chains are involved in binding M1, and contribute residues from the exposed b, c, and e positions to the interface. The site at which M1 binds FgD is notably quite distant (~90 Å) from the Fg γC globular head, which has been shown to bind β2 integrins17. The M1 S-region is freely available to bind IgG’s and thereby enhance HBP release.
Figure 2. M1-Fg interface.
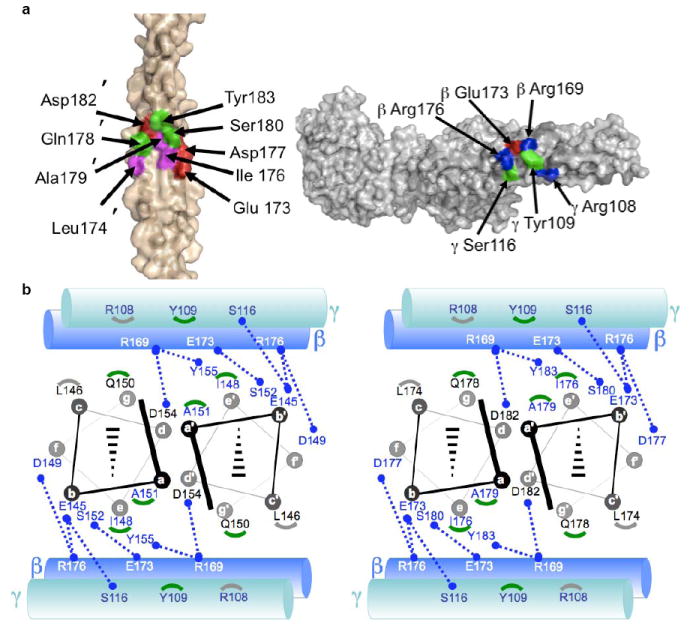
a, Interface between M1BC1 B2 (left, primed numbers refer to one helix and non-primed the opposing helix) and FgD (right) in surface representation (basic residues blue, acidic red, polar green, and nonpolar magenta). b, Schematic of the interface between M1BC1 B1 (left) and B2 (right) in helical projection and FgD (cylinders, β chain in blue and γ in blue-green). Blue dotted lines connect residues making polar contacts, and gray and green arcs correspond to M1 residues making van der Waals contacts to Fg γ 108 and 109, respectively.
Figure 3. Conformational Dynamics.
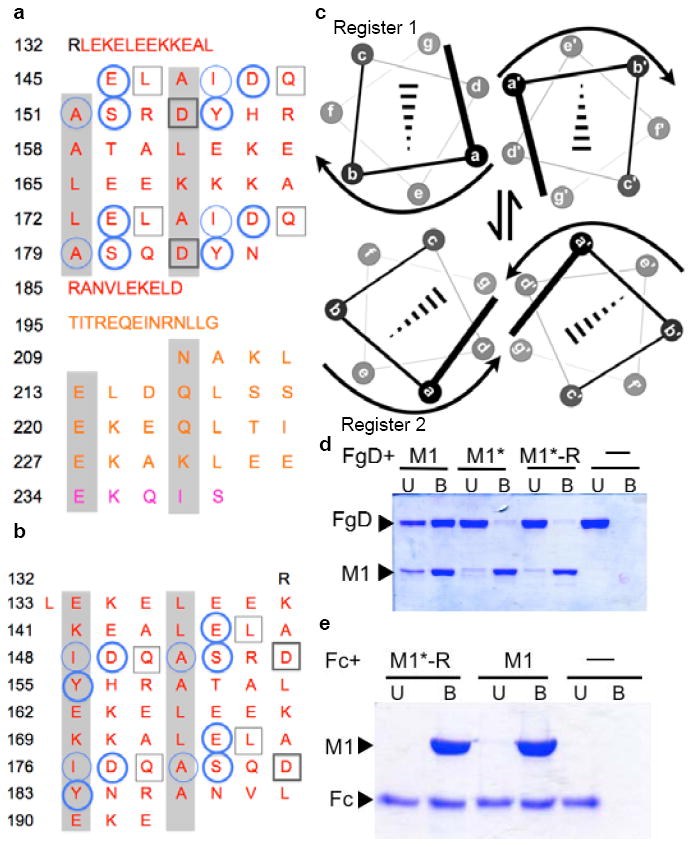
a, Heptad register of M1BC1 bound to FgD (register 2; a and d residues shaded gray). Residues not assigned a heptad position (132-144, 185-208) form an α-helical dimer but do not have coiled-coil “knobs-into-holes” packing. FgD-contacting residues are in circles for one M1 helix and in boxes for the other. Heavy lines denote polar contacts, and light lines vdW contacts. The B-repeats are in red, the S-region in gold, and C-repeat in purple. b, Heptad register of unbound M1AB (register 1; ref. 8). c, Relationship between registers 1 and 2. d, Association of His-tagged M1, M1*, and M1*-R with FgD at 37 °C, as assessed by a Ni2+-nitrilotriacetic acid (NTA) agarose coprecipitation assay and visualized by non-reducing, Coomassie-stained SDS-PAGE. U, unbound fraction; B, bound fraction. e, Association of His-tagged M1 and M1*-R with IgG Fc at 37 °C, as in panel d.
Conformational Dynamics
The heptad register in the B-repeats observed here differs from that observed previously in the crystal structure of the M1AB fragment (ref. 8), which includes the A-region and B-repeats but not the S-region and C-repeats. Residues that were in the d-g heptad face of M1AB (register 1) occupy the a-d face of M1BC1 bound to FgD (register 2) (Figs 3a and 3b). These two registers are related by a rotation of one helical face, or ~51.4° (Fig. 3c). The ability of the B-repeats to adopt these two competing registers is supported by coiled coil propensity analysis18, which indicates that both registers 1 and 2 are embedded within the B-repeats as short interspersed stretches (Supplementary Fig. 3). Surprisingly, residues that bind Fg have a preference for register 1, which is incapable of binding Fg, but are surrounded by residues that have a preference for register 2, the FgD-binding register. In addition to these two registers being alternately sampled by the B-repeats, a splayed conformation is likely to exist, as suggested by the dynamic dissociation and reassociation of M1 chains8. Presumably the splayed conformation enables transitions between registers 1 and 2, the latter being stabilized by Fg binding.
To experimentally test for the presence of conformational dynamics, we stabilized register 1 in the B-repeats without altering Fg-binding residues. We hypothesized that Fg binding should be decreased through this process if register 1 were sampled, because M1 would be ‘locked’ in the nonbinding register. The ideal coiled-coil residues Val and Leu were substituted at a and d positions, respectively, of register 1 in the B-repeats19,20, except at the six a and d positions involved in FgD binding (Supplementary Fig. 4). Most notable among these six were Tyr155 and Tyr183, which are at core a positions in register 1 but at exposed e positions in register 2, from which they make π-cation interactions with FgD β169. This variant of M1, called M1*-Revertant (M1*-R), is equivalent to the previously characterized M1* except with all the Fg-binding residues present8. M1* was shown to be more stable than wild-type M1 but substantially diminished in FgD binding. We found that M1*-R, like M1*, was also greatly attenuated for FgD binding (Fig. 3d), and that this attenuation was specific as M1*-R maintained wild-type levels of interaction with IgG Fc fragments (Fig. 3e). This latter interaction occurs through M1 protein regions outside the B-repeats8. These mutational results support the conclusion that register 1, along with register 2, is sampled in the B-repeats. Altogether our observations provide evidence for large conformational dynamics in the B-repeats. What purpose these dynamics serve is unknown, but one possibility is that they are advantageous for GAS immune evasion, in effect providing a ‘moving target’ for antibody recognition.
M1-Fg network
To address the mechanism of neutrophil activation, we modeled Fg and intact M1 in place of FgD and M1BC1 (Fig. 4, Supplementary Movie 1), respectively. Importantly, because Fg is a dimer (of αβγ heterotrimers), a Fg molecule has two M1-binding sites as opposed to the single site in FgD. From this modeling emerged a non-clashing M1-Fg network with Fg acting as struts and M1 acting as joints. The two M1 molecules that bound an individual Fg were tilted with respect to each other due to the inherent flexibility of Fg21. This tilt gave the network three-dimensional character and meant that the network incorporated M1 molecules pointing in opposite directions. The variation in M1 directionality suggests that the network is formed by free M1 released from the bacterial surface by neutrophil proteases6, as opposed to M1 anchored unidirectionally by its C-terminus to the bacterial cell wall22. Consistent with this notion, the greater proportion of M1 in samples from STSS patients occurs as free released protein7.
Figure 4. M1-Fg network.
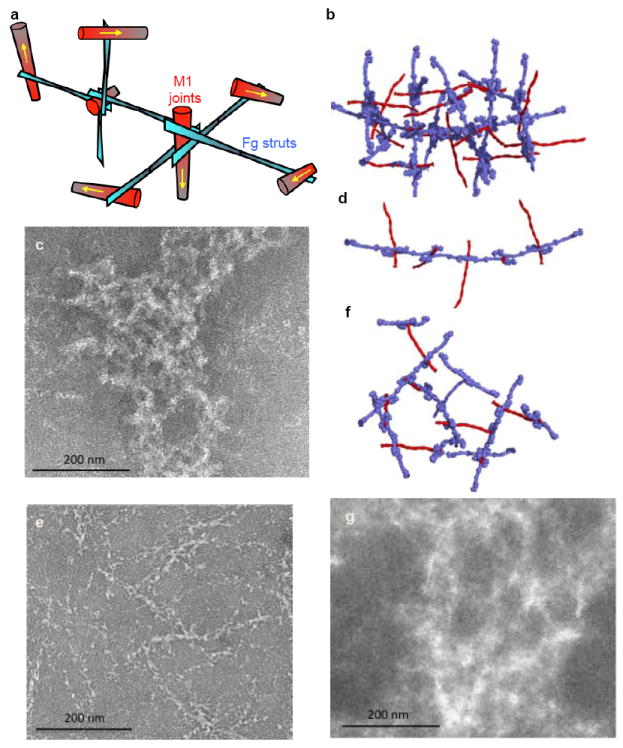
a, Schematic of the M1-Fg network. The yellow arrows specify the direction of M1. b, Model of the M1-Fg network, with M1 (red) and Fg (blue) in surface representation, and (c) negative-stained electron micrograph of M1 co-incubated with Fg. Following panels are identical, except with (d, e) ΔB2 and (f, g) B2C modeled and co-incubated with Fg.
The structure of the M1-Fg network suggested a mechanism for neutrophil activation. Prior work had shown that antibody crosslinking of β2 integrin had the same effect on neutrophil activation as the M1-Fg complex6,9,23, indicating that β2 integrin clustering and avidity are involved in signaling by M1-Fg. Based on these data, we surmised that the Fg density induced by the M1-Fg network was likely to be a critical factor for neutrophil activation. To test this model, we compared HBP release by neutrophils stimulated by various M1 deletion constructs. M1 in which either the upstream or downstream B-repeat was deleted, ΔB1 and ΔB2, respectively, retained Fg binding due to the continued presence of one of the B-repeats (Fig. 5a). However, as the modeling predicted, ΔB1 and ΔB2 formed fibers (Figs 4d, e and Supplementary Fig. 5a) rather than the networks formed by wild-type M1 (Figs 4b, c). Despite being able to bind Fg, neither ΔB1 nor ΔB2 triggered release of HBP from neutrophils, which was in sharp contrast to wild-type M1 (Fig. 5b). This result indicates that the M1-Fg network rather than Fg-binding itself is required for neutrophil activation. We also demonstrated that the addition of FgD, which is unable to support network formation because it has only one M1-binding site, blocks M1-mediated neutrophil activation (Fig. 5c). Excess FgD was necessary in this experiment as binding of M1 to FgD is weaker than it is to Fg24, the difference being explained by the high avidity between M1 and Fg as compared to the weaker affinity between M1 and FgD.
Figure 5. Fg binding and neutrophil activation.
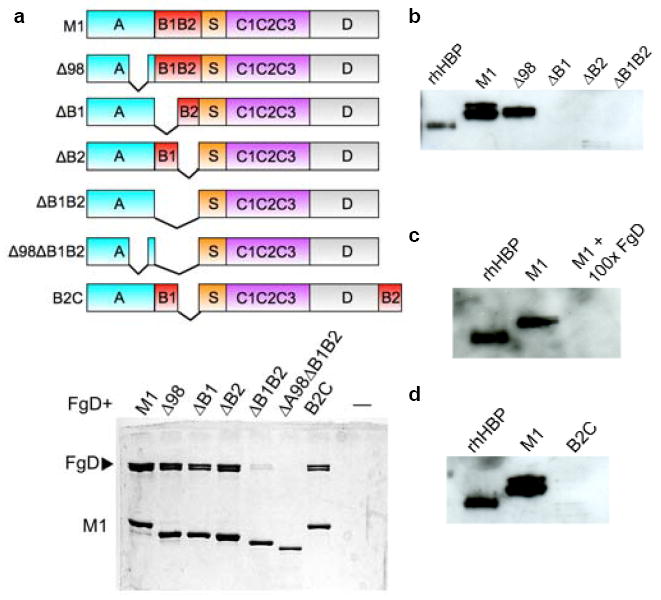
a, Schematic of M1 constructs (top), with domains denoted. Bottom, association of His-tagged M1, Δ98 (Δ98-125), ΔB1 (Δ133-161), ΔB2 (Δ162-189), ΔB1B2 (Δ133-189), Δ98ΔB1B2 (Δ98-125, Δ133-189), and B2C (residues 162-189 deleted and inserted after C-terminal residue 453) with FgD as assessed by a Ni2+-NTA agarose coprecipitation assay and visualized by non-reducing Coomassie-stained SDS-PAGE. Only bound fractions are shown. b, Release of HBP by human neutrophils incubated with M1, Δ98, ΔB1, ΔB2, or ΔB1B2, as assayed by an anti-HBP western blot. The leftmost lane contains recombinant HBP, rHBP, as a positive control. The difference between this and other HBP samples is due to glycosylation. c, Release of HBP inhibited by a 100-fold excess of FgD, and (d) elicited by B2C, both visualized as in panel b.
As expected, deletion of both B-repeats (ΔB1B2) resulted in no networks, no fibers, and no induction of HBP release (Fig. 5b and Supplementary Fig. 5b). However, ΔB1B2 retained a low level of Fg binding (Fig. 5a). Based on this and other evidence, we uncovered a cryptic Fg-binding site in the A-region. A molecular replacement solution of a low-resolution crystal (7.5 Å resolution limit) of the M1 A-region bound to FgD (M1A-FgD) confirmed the existence of this site. This solution revealed two molecules of FgD oriented 180° to one another and arranged perpendicularly to the A-region, similar to the binding mode observed for each of the B-repeats (Supplementary Fig. 6). Although the low resolution limited our abilities to discern A-region residues, it was apparent that the same FgD residues bound by the B-repeats were bound by the A-region. This suggested A-region residues 106-119 to be the likely Fg-binding site (Supplementary Fig. 7), as this region has some sequence similarity to the B-repeats, including a Tyr capable of forming a π-cation bond to FgD β169; tyrosines are otherwise rare in the M1 sequence. In line with observations for the B-repeats, the A-region site would also require a ~51.4° rotation in helical register from the conformation observed in M1AB (ref. 8) to bind Fg. Deletion of this putative Fg-binding site in the A-region along with both B-repeats completely abrogated Fg binding (Fig. 5a, Δ98ΔB1B2). While we found that the A-region site was not required for network formation or for HBP release (Fig. 5b, Δ98; Supplementary Fig. 5c), the possibility that this cryptic site has other functions related to Fg binding (e.g., evasion of phagocytosis) merits future exploration.
Lastly, we asked whether the density of the M1-Fg network was consequential. To address this, we deleted the downstream B-repeat (B2) and inserted it at the C-terminus of M1, thereby increasing the spacing between the two B-repeats (Fig. 5a). Modeling predicted that a sparser network should be formed by this construct, called B2C. B2C bound FgD and formed networks, but significantly, did not trigger release of HBP (Figs 4f, g and 5d). We note that the resolution of the electron micrographs did not allow us to distinguish between M1-Fg and B2C-Fg networks, but our modeling strongly suggests that the difference in network density accounted for the lack of neutrophil activation.
Conclusions
In summary, we have shown that a key proinflammatory property of M1, as exemplified by the induction of neutrophil HBP release, is due to the organization of Fg into a specific cross-like pattern that supports the formation of an M1-Fg network. This process requires the presence of two appropriately spaced B-repeats. Repeats are a common feature of M protein sequences, but whether other Fg-binding M protein types with repeat sequences have similar proinflammatory capabilities is unknown and under investigation. Because disruption of the M1-Fg network into fibers or sparse networks resulted in loss of neutrophil activation, we conclude that the density of Fg in the network is the critical factor in neutrophil activation. An alternative model would require a conformational change in Fg upon M1 binding, as has been suggested for the unmasking of a β2 integrin-binding tail in the γC globular domain of Fg25. This possibility cannot be excluded as the tail is absent in FgD, but it seems unlikely to us as the tail would be quite distant from M1 and no large conformational changes were evident in FgD to transmit a binding signal to the tail. While the M1-Fg network is distinct from a fibrin clot, these supramolecular assemblies both present high densities of integrin-binding sites, suggesting that integrin clustering and avidity are conserved mechanisms for leukocyte activation. Interference with the M1-Fg interaction visualized here represents a potential therapeutic target to ameliorate the severe outcomes of STSS.
METHODS SUMMARY
Preparation of M1 and FgD, fibrinogen binding assays, and HBP release assays were carried out as previously described8. Diffraction data from crystals of M1BC1-FgD and M1A-FgD were processed using MOSFLM26 or HKL200027, and phases were determined by molecular replacement using the program Phaser28 and the structure of human FgD14 (PDB code 3E1I) as a search model. The register of M1BC1 was verified by an anomalous dispersion experiment using crystals of selenomethionine-substituted M1BC1(I148M)-FgD. Samples for electron microscopy were negatively stained with 0.2% uranyl acetate and imaged using a FEI Tecnai F20 Twin transmission electron microscope at an accelerating voltage of 120 kV.
METHODS
DNA manipulation
The DNA sequences of intact mature M1 protein (residues 42-453), M1BC1 (residues 128-263), and M1A (residues 42-132) were cloned as described previously8 from human GAS isolate 544829 into pET28b (Novagen). The M1 deletion mutants and B2C were generated using the QuickChange II Site-Directed mutagenesis kit (Stratagene), according to manufacturer’s instructions, or by the mega-primer method30. All M1 protein constructs had a C-terminal His-tag for purification purposes, except for M1BC1.
Protein expression and purification
M1 protein constructs were expressed in Escherichia coli BL21 (DE3), which were grown in LB containing 34 mg/mL kanamycin at 37 °C until mid-logarithmic phase and then induced at room temperature with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and grown further for 18 h. Bacteria were harvested by centrifugation and resuspended in either 100 mM NaCl, 50 mM sodium phosphate buffer, pH 8 (SP) or 100 mM NaCl, 50 mM Tris, pH 8 (ST), both with protease inhibitors (Complete tablet, Roche), for biochemical analysis or crystallization experiments, respectively. Bacteria were lysed using an EmulsiFlex-C5 (Avestin). His-tagged M1 constructs were then purified as previously reported8. For M1BC1, which lacks a His-tag, the lysate was heat denatured at 75 °C for 30 minutes, cooled on ice for 30 minutes, and clarified by centrifugation. Nucleic acids were removed by the addition of 0.5% polyethyleneimine and the resulting supernatant was then precipitated with 75% (NH4)2SO4. Precipitated protein was resuspended in either SP or ST and dialyzed overnight in the same buffer. Proteins were then purified on a Q-Sepharose anion exchange column (GE-Healthcare). Selenomethionine incorporation into M1BC1 was carried out as previously described31, and the purification was carried out as above except with the addition of 1 mM dithiothreitol throughout.
Fibrinogen fragment D (FgD) was purified from human fibrinogen (Fg) as described previously32. Briefly, human Fg (Calbiochem) was trypsinized overnight in 150 mM NaCl, 5 mM CaCl2, 50 mM imidazole, pH 7 and purified on a Gly-Pro-Arg column equilibrated with the same buffer. The protein was eluted from the column with 1 M NaBr, 0.05 M NaOAc, pH 5.3 and subsequently exchanged by ultrafiltration into ST.
Crystallization and data collection
For crystallization of M1BC1-FgD, M1BC1 was mixed with FgD at a 4:1 molar ratio, and the FgD-M1BC1 complex was purified on a Superdex 200 16/60 size exclusion column (Amersham) in 20 mM NaCl, 10 mM Tris, pH 8. Crystallization was performed by the vapor-diffusion method in two steps. The first step involved mixing of an equal volume of M1BC1-FgD at 8 mg/mL and precipitant solution containing 0.6 M K2/Na2 PO4, 0.12 M (NH4)2SO4, 0.1 M HEPES, pH 7.5. This produced crystals that did not diffract X-rays. These non-diffracting crystals were crushed and diluted in the precipitant solution before being used as a 0.3 μL seeding additive in a second round of crystallization performed at 4 °C with 1 μL M1BC1-FgD at 4 mg/mL and 1 μL of 16% PEG 3350 and 0.2 M sodium tartrate. Crystals were cryoprotected in the mother liquor solution supplemented with 25% ethylene glycol and flash cooled in liquid N2. A native FgD-M1BC1 data set was recorded at 1.033 Å wavelength to 3.3 Å resolution limit at the Advanced Photon Source (APS, 23-ID-B, Argonne, IL). Data were processed with the programs MOSFLM26 and SCALA33.
For crystallization of M1A-FgD, M1A was mixed with FgD and purified as described above. Crystals were grown using M1A-FgD at 10.7 mg/mL and 1.3 M ammonium tartrate, 0.1 M MES, pH 6.25 as a precipitant. Crystals were cryoprotected in the mother liquor solution supplemented with 25% glycerol and flash cooled in liquid N2. A native data set for crystals of M1A-FgD was recorded at 0.9800 Å wavelength to 7.5 Å resolution at APS (23-ID-B), and data were processed as described above.
The diffraction data were truncated at resolution limits of 3.3 Å (I/σI of 1.4) and 7.5 Å (I/σI of 1.8) for M1BC1-FgD and M1A-FgD, respectively, as suggested by an analysis of σA values as a function of resolution34.
Structure determination and refinement
Phases for M1BC1-FgD and M1A-FgD were determined by molecular replacement using the program Phaser28 and the structure of human FgD14 (PDB code 3E1I) as a search model. Four FgD molecules were identified through molecular replacement to occupy the asymmetric unit of M1BC1-FgD (final rotation function and translation function Z-scores of 12.2 and 69.7, and initial Rwork, Rfree of 41.6%, 41.2%). Two FgD molecules were identified through molecular replacement to occupy the asymmetric unit of M1A-FgD (final rotation function and translation function Z-scores of 5.2 and 25.5, and initial Rwork, Rfree of 44.1%, 42.1%). A single M1BC1-FgD complex occupied the asymmetric unit of its crystal, and a single M1A-FgD complex occupied the asymmetric unit of its crystal.
Continuous electron density corresponding to the backbone of M1BC1 in M1BC1-FgD was evident in initial electron density maps, with sufficient side chain density for a tentative register to be assigned. The register was verified by independent means as follows. Ile148 in M1BC1 was substituted with methionine, and the resulting mutant protein was biosynthetically labeled with selenomethionine and crystallized in complex with FgD, as above. A highly redundant (1080°) data set at the selenium anomalous absorption peak (0.9795 Å wavelength) was recorded at the APS (23-ID-D) in order to increase the signal to noise ratio. Data were processed using HKL200027. A difference anomalous map was calculated using FFT33, and two anomalous peaks were located, resulting in the unambiguous location of residue 148 on each chain of the M1BC1 coiled coil (Supplementary Figure 2). The entirety of M1BC1 visible in the crystal structure was α-helical, and thus specification of the position of residue 148 enabled assignment of the remaining residues.
Refinement of the M1BC1-FgD model was performed using CNS35 and Refmac33. All B-factors were initially set to 90 Å2 and subsequently refined as side chain and main chain groups using bgroup from CNS36. Four-fold noncrystallographic symmetry (NCS) restraints were applied to FgD (medium restraints for the main chain and loose restraints for the side chain); each of the three chains of the Fg αβγ heterotrimer formed a separate NCS group. The model was then refined using Refmac5, with five alternating macro-cycles of model building and refinement. Model building was guided by inspection of σA-weighted 2Fo-Fc and Fo-Fc omit maps37, and was carried using COOT38. Side chains were modeled as preferred rotamers. Each refinement macro-cycle consisted of 10 micro-cycles of maximum likelihood restrained refinement using standard parameters, except for the following adjustments. Due to the moderate resolution of the data, a weighting term of 0.01 was used to favor geometric restraints, and an overall temperature factor model and a Babinet scaling model were used34.
Structure validation was performed using Procheck39 and Molprobity40. In the final M1BC1-FgD model, 97.8% and 99.3% of residues were in allowed and generously allowed Ramachandran regions, respectively. The final map had correlation coefficients of 0.90 and 0.68 for the main chain and side chains, respectively, as calculated with OVERLAPMAP33. The Molprobity40 clash score was 24.63 (89th percentile) and overall score was 3.18 (77th percentile).
Molecular figures were generated with PyMol (http://pymol.sourceforge.net).
Modeling of M1-Fg
The A-region of intact M1 was modeled based on the structure of M1AB (ref. 8), and the B-repeats, S-region, and the initial few residues of the C-repeats were modeled based on the structure of M1BC1 from M1BC1-FgD. For C-terminal portions of M1 for which no structural information exists, an α-helical dimeric coiled coil was modeled. Fg was modeled using the structure of chicken Fg41, which has been determined to a higher resolution limit than human Fg21. The human and chicken Fg structures show a similar organization, with some flexibility between the FgD portions due to a bend in the central fragment E portion. The initial M1-Fg model, which has intact M1 at the center of a cross-like structure formed by four Fg molecules, was enlarged as follows. Since each of the four Fg molecules has a second binding site for M1, M1 was modeled at these second sites based on the structure of M1BC1-FgD. Further, since M1 binds four Fg molecules, three more Fg molecules were modeled at these second sites based on the structure of M1BC1-FgD. This procedure was carried on iteratively to yield the model of the M1-Fg network. Similar procedures were carried out for M1 mutants.
Coprecipitation assays
Ten μg of wild-type or variant M1 proteins were mixed with 20 μg of FgD in 50 μl of binding buffer (300mM NaCl, 50 mM NaPi, pH 8.0, 50 mM imidazole, 0.1% (v/v) Triton X-100) at 37 °C for 30 minutes. Twenty μL of Ni2+-NTA agarose beads were equilibrated in binding buffer and then added to the protein mix and incubated for 30 minutes at 37 °C under agitation. The beads were washed three times in 200 μL of binding buffer and eluted by boiling the beads for 5 min in non-reducing 5x SDS-PAGE sample loading buffer. Fractions corresponding to unbound and bound proteins were resolved by non-reducing SDS-PAGE.
A similar procedure was used for human IgG Fc fragment (Calbiochem), except for the following changes: 25 μg of M1 or M1*-R protein and 100 μg of Fc were mixed; this mixture was added to 50 μL of equilibrated Ni2+-NTA agarose beads; washes were 1 mL each; and reducing SDS-PAGE sample loading buffer was used.
Heparin binding protein immunoblot
Human neutrophils were purified from healthy donors blood using the PolymorphPrep™ system (Axis-Shield). Thirteen million neutrophils were incubated with 86 μg/mL wild-type or variant M1 protein at 37 °C for 30 minutes. After centrifugation, the supernatants were resolved on a 12% SDS-PAGE reducing gel and transferred to a PVDF membrane for immunoblotting. Recombinant human heparin binding protein (HBP) (R&D Systems) was used as a reference. The membrane was blocked with Tris buffered saline containing 5% nonfat milk or 5% bovine serum albumin (BSA). HBP was detected using either a primary rabbit anti-human-HBP polyclonal antibody (Sigma) or a mouse anti-human-HBP monoclonal antibody (R&D), peroxidase-conjugated secondary antibodies (Santa Cruz), and the ECL system (Pierce).
Electron microscopy
Mixtures containing 10 μg of M proteins and 100 μg of human fibrinogen (Calbiochem) were incubated at 37°C for 15 minutes before being deposited on glow-discharged copper grids coated with carbon film (Electron microscopy sciences, Cat #CF300-Cu) for 2 minutes. The grids were subsequently washed twice with water and negatively stained with 0.2% uranyl acetate for 45 seconds. Images were recorded using a FEI Tecnai F20 Twin transmission electron microscope at an accelerating voltage of 120 kV.
Supplementary Material
Acknowledgments
We thank the Argonne Photon Source Laboratory GM/CA CAT staff for help with data collection, Russell Doolittle and Antoine Royant for advice, and Stephanie Mel and Gouri Ghosh for comments on the manuscript. This work was supported by NIH R21 AI071167 (PG), T32 GM007240 (CB), R01 AI077780 (VN), R01 GM 54076 (JEJ), and a fellowship (JC) from the National Health and Medical Research Council of Australia (514639).
Footnotes
Author Contributions PM and PG designed the experiments. PM carried out the structure determination, modeling, and FgD coprecipitation assay. CB carried out the Fc coprecipitation assay. PM and CF carried out the electron microscopy under the supervision of JEJ. PM, ASZ, and JNC carried out the HBP release assays under the supervision of VN. PM and PG wrote the manuscript, and the other authors provided editorial advice.
Atomic coordinates and structure factors for M1BC1-FgD (2XNX) and M1A-FgD (2XNY) have been deposited with the Protein Data Bank.
The authors declare that they have no competing financial interests.
References
- 1.Fischetti VA. Streptococcal M protein: molecular design and biological behavior. Clin Microbiol Rev. 1989;2:285–314. doi: 10.1128/cmr.2.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Facklam RF, et al. Extension of the Lancefield classification for group A streptococci by addition of 22 new M protein gene sequence types from clinical isolates: emm103 to emm124. Clin Infect Dis. 2002;34:28–38. doi: 10.1086/324621. [DOI] [PubMed] [Google Scholar]
- 4.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9:611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 5.Aziz RK, Kotb M. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis. 2008;14:1511–1517. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herwald H, et al. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 2004;116:367–379. doi: 10.1016/s0092-8674(04)00057-1. [DOI] [PubMed] [Google Scholar]
- 7.Kahn F, et al. Antibodies against a surface protein of Streptococcus pyogenes promote a pathological inflammatory response. PLoS Pathog. 2008;4:e1000149. doi: 10.1371/journal.ppat.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNamara C, et al. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science. 2008;319:1405–1408. doi: 10.1126/science.1154470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soehnlein O, et al. Neutrophil degranulation mediates severe lung damage triggered by streptococcal M1 protein. Eur Respir J. 2008;32:405–412. doi: 10.1183/09031936.00173207. [DOI] [PubMed] [Google Scholar]
- 10.Gautam N, et al. Heparin-binding protein (HBP/CAP37): a missing link in neutrophil-evoked alteration of vascular permeability. Nat Med. 2001;7:1123–1127. doi: 10.1038/nm1001-1123. [DOI] [PubMed] [Google Scholar]
- 11.Linder A, Christensson B, Herwald H, Bjorck L, Akesson P. Heparin-binding protein: an early marker of circulatory failure in sepsis. Clin Infect Dis. 2009;49:1044–1050. doi: 10.1086/605563. [DOI] [PubMed] [Google Scholar]
- 12.Shannon O, et al. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol. 2007;65:1147–1157. doi: 10.1111/j.1365-2958.2007.05841.x. [DOI] [PubMed] [Google Scholar]
- 13.Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–462. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 14.Bowley SR, Lord ST. Fibrinogen variant BβD432A has normal polymerization but does not bind knob “B”. Blood. 2009;113:4425–4430. doi: 10.1182/blood-2008-09-178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ringdahl U, et al. A role for the fibrinogen-binding regions of streptococcal M proteins in phagocytosis resistance. Mol Microbiol. 2000;37:1318–1326. doi: 10.1046/j.1365-2958.2000.02062.x. [DOI] [PubMed] [Google Scholar]
- 16.Akesson P, Schmidt KH, Cooney J, Bjorck L. M1 protein and protein H: IgGFc- and albumin-binding streptococcal surface proteins encoded by adjacent genes. Biochem J. 1994;300(Pt 3):877–886. doi: 10.1042/bj3000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medved L, Litvinovich S, Ugarova T, Matsuka Y, Ingham K. Domain structure and functional activity of the recombinant human fibrinogen gamma-module (gamma148-411) Biochemistry. 1997;36:4685–4693. doi: 10.1021/bi962795l. [DOI] [PubMed] [Google Scholar]
- 18.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 19.Tripet B, Wagschal K, Lavigne P, Mant CT, Hodges RS. Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil: 20 amino acid substitutions in position “d”. J Mol Biol. 2000;300:377–402. doi: 10.1006/jmbi.2000.3866. [DOI] [PubMed] [Google Scholar]
- 20.Wagschal K, Tripet B, Lavigne P, Mant C, Hodges RS. The role of position a in determining the stability and oligomerization state of alpha-helical coiled coils: 20 amino acid stability coefficients in the hydrophobic core of proteins. Protein Sci. 1999;8:2312–2329. doi: 10.1110/ps.8.11.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kollman JM, Pandi L, Sawaya MR, Riley M, Doolittle RF. Crystal structure of human fibrinogen. Biochemistry. 2009;48:3877–3886. doi: 10.1021/bi802205g. [DOI] [PubMed] [Google Scholar]
- 22.Navarre WW, Schneewind O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautam N, Herwald H, Hedqvist P, Lindbom L. Signaling via beta(2) integrins triggers neutrophil-dependent alteration in endothelial barrier function. J Exp Med. 2000;191:1829–1839. doi: 10.1084/jem.191.11.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitnack E, Beachey EH. Inhibition of complement-mediated opsonization and phagocytosis of Streptococcus pyogenes by D fragments of fibrinogen and fibrin bound to cell surface M protein. J Exp Med. 1985;162:1983–1997. doi: 10.1084/jem.162.6.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated unmasking of the cryptic binding site for integrin αMβ2 in the γC-domain of fibrinogen. Biochemistry. 2002;41:12942–12951. doi: 10.1021/bi026324c. [DOI] [PubMed] [Google Scholar]
- 26.Leslie A. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. 1997;276 doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.McCoy AJ, et al. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun. 2000;68:6362–6369. doi: 10.1128/iai.68.11.6362-6369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geiser M, Cebe R, Drewello D, Schmitz R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;31:88–90, 92. doi: 10.2144/01311st05. [DOI] [PubMed] [Google Scholar]
- 31.Doublié S. Methods in Enzymology. Vol. 276. Academic Press; 1997. pp. 523–530. [PubMed] [Google Scholar]
- 32.Everse SJ, Pelletier H, Doolittle RF. Crystallization of fragment D from human fibrinogen. Protein Sci. 1995;4:1013–1016. doi: 10.1002/pro.5560040523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.DeLaBarre B, Brunger AT. Considerations for the refinement of low-resolution crystal structures. Acta Crystallogr D Biol Crystallogr. 2006;62:923–932. doi: 10.1107/S0907444906012650. [DOI] [PubMed] [Google Scholar]
- 35.Brünger A. Crystallography & NMR system: a new software for macromolecular structure determination. Acta Crystallogr sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 37.Brunger AT, Adams PD, Rice LM. New applications of simulated annealing in X-ray crystallography and solution NMR. Structure. 1997;5:325–336. doi: 10.1016/s0969-2126(97)00190-1. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 40.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Z, Kollman JM, Pandi L, Doolittle RF. Crystal structure of native chicken fibrinogen at 2.7 A resolution. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.