

Abstract
The matricellular protein periostin is expressed in the skin. Although periostin has been hypothesized to contribute to dermal homeostasis and repair, this has not been directly tested. To assess the contribution of periostin to dermal healing, 6 mm full-thickness excisional wounds were created in the skin of periostin-knockout and wild-type, sex-matched control mice. In wild-type mice, periostin was potently induced 5–7 days after wounding. In the absence of periostin, day 7 wounds showed a significant reduction in myofibroblasts, as visualized by expression of α-smooth muscle actin (α-SMA) within the granulation tissue. Delivery of recombinant human periostin by electrospun collagen scaffolds restored α-SMA expression. Isolated wild-type and knockout dermal fibroblasts did not differ in in vitro assays of adhesion or migration; however, in 3D culture, periostin-knockout fibroblasts showed a significantly reduced ability to contract a collagen matrix, and adopted a dendritic phenotype. Recombinant periostin restored the defects in cell morphology and matrix contraction displayed by periostin-deficient fibroblasts in a manner that was sensitive to a neutralizing anti-β1-integrin and to the FAK and Src inhibitor PP2. We propose that periostin promotes wound contraction by facilitating myofibroblast differentiation and contraction.
Key words: α-Smooth muscle actin, Contraction, Matricellular protein, Myofibroblast, Periostin, Wound healing
Introduction
Periostin is a secreted 90 kDa, disulfide-linked protein, which has structural similarity with the insect adhesion protein, fasciclin-1 (Takeshita et al., 1993). During development, periostin is expressed by cardiac fibroblasts in the embryonic heart (Kruzynska-Frejtag et al., 2001), where it facilitates organization of the extracellular matrix (ECM) (Snider et al., 2009) and differentiation of mesenchymal cushion progenitor cells into contractile myofibroblasts (Conway and Molkentin, 2008). Recent research has now highlighted increased periostin expression in various disease states, including myocardial infarction (Iekushi et al., 2007; Oka et al., 2007; Shimazaki et al., 2008) and cancer (Baril et al., 2007; Erkan et al., 2007; Gillan et al., 2002). In particular, periostin is heavily implicated in fibrosis including bone marrow (Oku et al., 2008), subepithelial fibrosis of bronchial asthma (Takayama et al., 2006), fibrous dysplasia (Kashima et al., 2009), and keloid and hypertrophic scarring of the skin (Naitoh et al., 2005; Wang et al., 2007a). Additionally, induction of periostin expression has been described following acute injury to numerous tissues (Goetsch et al., 2003; Li et al., 2006; Lindner et al., 2005; Nakazawa et al., 2004), including skin (Jackson-Boeters et al., 2009; Lindner et al., 2005; Zhou et al., 2010), where expression peaks at 7–8 days and returns to basal levels by 4 weeks. Our previous reports on skin repair show that periostin expression is absent during inflammation, but instead corresponds with the proliferative and remodeling phases of repair (Jackson-Boeters et al., 2009; Zhou et al., 2010), suggesting a role for periostin during these later phases.
Fibroblasts are central to the proliferative and remodeling phases of skin (Ross, 1968). Dysregulation of fibroblast function in skin can result in inadequate repair (Blumbach et al., 2010; Shimazaki et al., 2008) or excessive matrix production (fibrosis) (Babu et al., 1992; Leask, 2010). Activated fibroblasts, or myofibroblasts, are fibroblasts that have differentiated into a contractile phenotype, characterized by the expression of α-smooth muscle actin (α-SMA) (Gabbiani et al., 1972). During wound repair, they serve to expedite wound closure by drawing the edges of the wound together through generation of contractile forces (Gabbiani et al., 1972; Gabbiani et al., 1971; Majno et al., 1971), which are transmitted to the ECM through integrin-containing adhesion complexes known as focal adhesions (Burridge and Chrzanowska-Wodnicka, 1996). Numerous studies have implicated periostin with expression of α-SMA (Jackson-Boeters et al., 2009; Shimazaki et al., 2008; Vi et al., 2009). In skin repair, expression of periostin coincides with expression of α-SMA (Jackson-Boeters et al., 2009).
Induction of the myofibroblast phenotype depends on a combination of three major factors (Tomasek et al., 2002): engagement of integrin cell surface receptors with the ED-A splice variant of fibronectin (Serini et al., 1998), stimulation with transforming growth factor β (TGFβ) (Desmouliere et al., 1993) and mechanical tension (Cevallos et al., 2006; Hinz et al., 2001). Recent work, however, suggests that fine-tuning of myofibroblast differentiation involves a complex interaction of many other factors (Blumbach et al., 2010; Liu et al., 2010; Shi-Wen et al., 2004), including the matricellular proteins, which include connective tissue growth factor (Liu et al., 2011), osteopontin (Lenga et al., 2008) and tenascin-C (Tamaoki et al., 2005). Matricellular proteins are a functionally related group of secreted proteins that have important roles in all phases of skin repair (Bornstein, 1995; Midwood et al., 2004). Often these proteins influence cell behavior through adhesive signaling, typically through integrin binding and activation of focal adhesion kinase (FAK) (Jun and Lau, 2010; Shi-wen et al., 2006; Sodek et al., 2000). Periostin is a matricellular protein that is known to modulate adhesive signaling through various integrins and FAK (Bao et al., 2004; Baril et al., 2007; Butcher et al., 2007; Gillan et al., 2002; Shao et al., 2004). Furthermore, periostin has been correlated with α-SMA expression in skin repair (Jackson-Boeters et al., 2009). However, the use of genetic deletion to determine the specific role of periostin in skin repair has not been attempted.
We therefore hypothesized that periostin facilitates myofibroblast differentiation during dermal wound repair and that the loss of periostin would impede wound resolution. Using a periostin-knockout (Postn−/−) mouse (Rios et al., 2005), we show that the loss of periostin results in altered wound closure kinetics, corresponding to the time of peak periostin expression. Histological analysis of wound tissue reveals that the granulation tissue of Postn−/− wounds is deficient in α-SMA. Furthermore, dermal fibroblasts isolated from Postn−/− mice are unable to contract a collagen matrix. This deficiency is corrected by addition of exogenous periostin, by a mechanism that is dependent on β1 integrin, Src and FAK.
Results
Loss of periostin results in altered wound-closure kinetics
Periostin is upregulated following acute injury to skin in wild-type mice (Jackson-Boeters et al., 2009; Lindner et al., 2005; Zhou et al., 2010). To investigate the contribution of periostin to the dermal wound repair process, full-thickness excisional wounds were created in Postn−/− mice and their sex- and age-matched Postn+/+ littermates. At 5 and 7 days post wounding, Postn+/+ wound size had reduced to 30% and 17%, respectively, of their initial wound area (Fig. 1). In Postn−/− mice, however, wounds were significantly larger than those in Postn+/+ mice (64% at day 5 and 41% at day 7, P<0.001) (Fig. 1). Histological analysis of sections from the centre of day 7 wounds confirmed that Postn−/− wounds were significant larger than those of Postn+/+ littermates (P<0.05) (supplementary material Table S1). Epithelial migration distance was not significantly different in day 7 wounds (P=0.25) (supplementary material Table S1). Wounds in both wild-type and knockout animals had closed by day 11.
Fig. 1.

Loss of periostin results in altered wound-closure kinetics. Full-thickness excisional punch wounds were created in the skin of Postn+/+ and Postn−/− mice using a 6 mm biopsy tool. (A) Wounds were photographed at 0, 3, 5, 7 and 11 days after wounding. Four pairs of mice (four wounds per mouse) were followed for 11 days. (B) Quantification of wound area was from photographs. Postn−/− (KO) wounds are delayed in closure at days 5 and 7 (P<0.01). Data are expressed as a fraction of the initial wound area; error bars represent s.d. (*P<0.01; two-way ANOVA).
We have previously reported that periostin protein is evident in granulation tissue of excisional wounds by day 3, peaking at day 7 (Jackson-Boeters et al., 2009), which we have confirmed in this study in Postn+/+ mice (Fig. 2A,B). Analysis of in vivo Postn gene expression by in situ hybridization and real-time quantitative polymerase chain reaction (RT-qPCR) confirmed that Postn mRNA is significantly, and selectively, increased during cutaneous wound repair at day 7 (P<0.01) (Fig. 2A,C). Inflammatory cell infiltration was similar between day 5 Postn+/+ and Postn−/− wounds (supplementary material Fig. S1), suggesting that periostin has no influence on the immune response.
Fig. 2.

α-SMA expression is reduced in the granulation tissue of Postn−/− mice. (A) Postn message detection in day 7 Postn+/+ wounds by in situ hybridization, showing periostin expression selectively in the wound. Arrowheads indicate the wound borders. (B) Histological analysis of day 7 wounds from Postn+/+ (WT) and Postn−/− (KO) animals. Sections were incubated with antibodies against periostin or α-SMA. Detection was with peroxidase-conjugated secondary antibodies and DAB. Day 7 Postn−/− wounds have reduced α-SMA staining (n=5). (C) Healthy skin or wound tissue biopsies were analyzed for Postn and (D) Acta2 mRNA by RT-qPCR. Target gene expression was normalized to 18S using the ΔΔCt method. Postn expression is significantly increased at day 7 (P<0.001, n=5). Postn mRNA was not detected in Postn−/− specimens. Increased Acta2 expression was observed at day 7 in Postn+/+ wounds, but not in Postn−/− wounds (P<0.001, n=5). Data are expressed relative to day 0 Postn+/+ expression; error bars represent s.e.m. (*P<0.001; two-way ANOVA).
α-SMA expression is reduced in the granulation tissue of Postn−/− mice
Our previous work showed that increased levels of periostin protein in the granulation tissue of excisional punch wounds is paralleled by an increase in α-SMA protein (Jackson-Boeters et al., 2009). It is not clear, however, whether periostin is required for α-SMA expression. Therefore, we assessed the level of α-SMA in Postn+/+ and Postn−/− wounds using immunohistochemistry (IHC). Immunoreactivity for α-SMA was evident at the wound edge and within the granulation tissue of Postn+/+ wounds at day 5 (supplementary material Fig. S2). At day 7, increased levels of α-SMA were detected at the wound border, throughout the granulation tissue and in blood vessel walls (Fig. 2B). In day 7 Postn−/− wounds, α-SMA immunoreactivity was significantly reduced when compared with that in sex-matched littermate controls (Fig. 2B). Reduced α-SMA gene (Acta2) expression in Postn−/− wounds was confirmed by RT-qPCR, where excised day 7 Postn−/− wound tissue contained significantly less Acta2 mRNA than day 7 Postn+/+ wounds (Fig. 2D) (P<0.01).
This deficit in α-SMA immunoreactivity was specific to the granulation tissue of Postn−/− wounds (Fig. 3A), with wound borders and vasculature positive for α-SMA in both Postn+/+ and Postn−/− wounds. To determine whether the reduction in α-SMA immunoreactivity was due to impaired fibroblast recruitment into the granulation tissue, sections were labeled for fibroblast-specific protein-1 (Fig. 3B). Indeed, fibroblasts dominated the granulation tissue of both Postn+/+ and Postn−/− day 7 wounds (Fig. 3B). Additionally, tissue sections were stained for nuclei with 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 3C). No significant differences in cell number were found between Postn+/+ and Postn−/− wounds at the wound borders or within the granulation tissue (Fig. 3C) (P=0.28). To further rule out a defect in fibroblast recruitment, we assessed fibroblast migration in vitro using a scratch wound assay. Scratch wounds in monolayers of Postn+/+ and Postn−/− were resolved by 20 hours. No difference in migratory ability was observed between cell types (supplementary material Fig. S3) (P=0.34).
Fig. 3.

Decreased α-SMA expression is restricted to the granulation tissue of Postn−/− mice. (A) Histological analysis of day 7 wounds from Postn+/+ and Postn−/− animals. Sections were incubated with an antibody for α-SMA (B) or fibroblast-specific protein-1. Detection was with peroxidase conjugated secondary antibodies and DAB. α-SMA is absent in the granulation tissue of Postn−/− wounds, but is present at the wound border (n=4). Granulation tissue is dominated by fibroblasts in both Postn+/+ and Postn−/− day 7 wounds. (C) Quantification of cell number at the wound border and within the granulation tissue in high magnification fields of view. Cell number was not different between Postn+/+ (WT) and Postn−/− (KO) wounds at either location (P=0.28). Data is expressed as mean number of cells per field of view; error bars represent s.d. (two-way ANOVA).
Canonical TGFβ signaling is not altered in Postn−/− fibroblasts
TGFβ is known to cause an increase in α-SMA expression through phosphorylation of Smad3 (Gu et al., 2007), and plays a major role in myofibroblast differentiation (Desmouliere et al., 1993). Therefore, to determine whether the reduction in α-SMA expression and immunoreactivity in Postn−/− granulation tissue was due to defective TGFβ–Smad3 signaling, we assessed the number of nuclei positive for phosphorylated Smad2 and Smad3 (Smad2/3–P) within the granulation tissue. The number of Smad2/3–P-positive nuclei was similar in both Postn+/+ and Postn−/− wounds (Fig. 4A,B) (P=0.25), suggesting that canonical TGFβ signaling is active in Postn−/− wounds. Furthermore, collagen production in the granulation tissue of both Postn+/+ and Postn−/− wounds, as evidenced by hydroxyproline content, was not significantly different (Fig. 4C,D) (P=0.69), a process for which TGFβ signaling is of great importance (Ignotz and Massague, 1986; Roberts et al., 1986; Wang et al., 2007b).
Fig. 4.
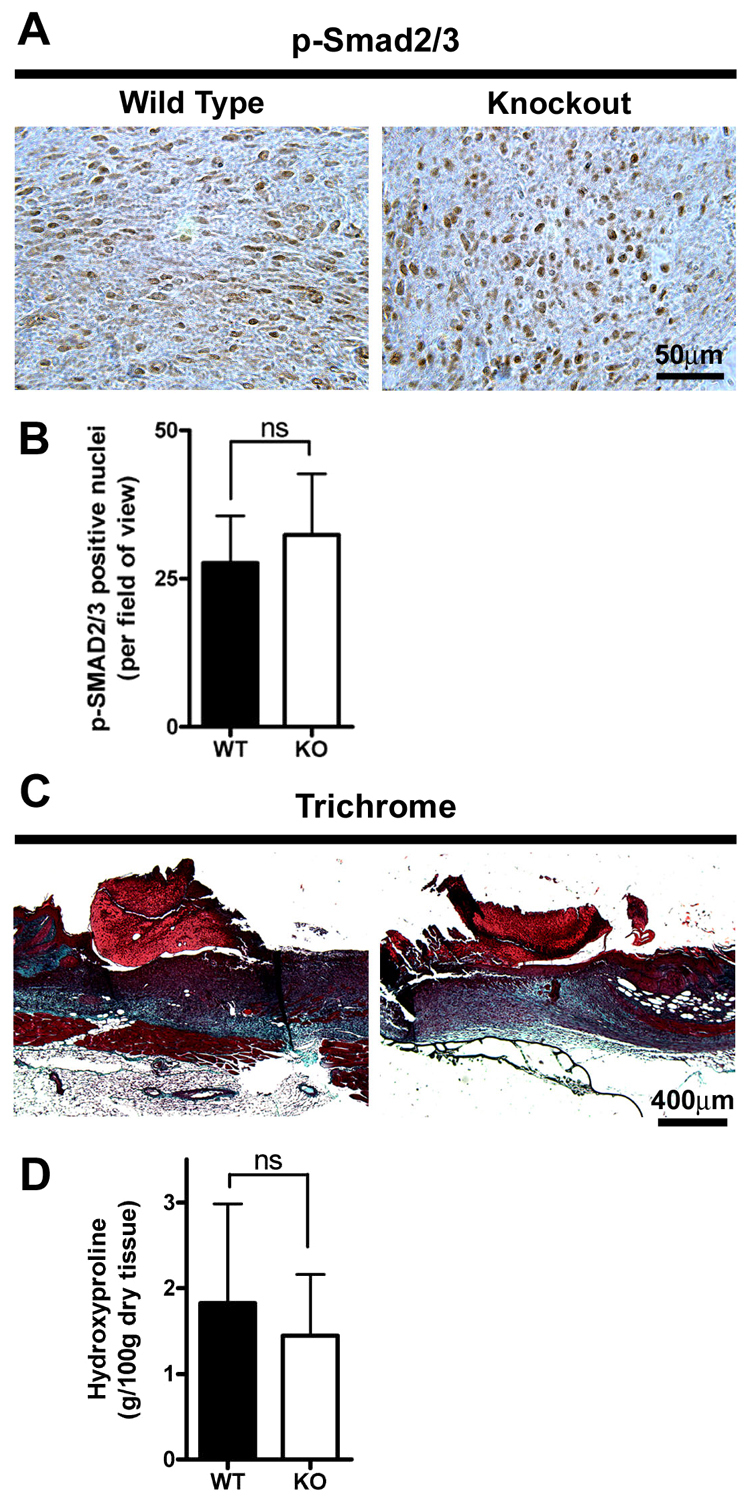
Canonical TGFβ signaling is not altered in Postn−/− fibroblasts. (A) Histological analysis of day 7 wounds from Postn+/+ and Postn−/− animals. Sections were incubated with an antibody for phosphorylated Smad2/3 (p-Smad2/3). Detection was with peroxidase conjugated secondary antibodies and DAB (n=3). (B) Numbers of positively stained nuclei per high power field of view were not significantly different between Postn+/+ (WT) and Postn−/− (KO) wounds (P=0.37; Student's t-test). (C) Masson's trichrome staining of day 7 wound sections. (D) Hydroxyproline content (g/100 g dry tissue) of excised day 7 wounds was not different between Postn+/+ and Postn−/− animals (P=0.60; Student's t-test). Data are expressed as means; error bars represent s.d.
Exogenous periostin is sufficient to induce a contractile phenotype
The reduced expression of α-SMA observed in vivo indicated that a defect might exist in differentiation of fibroblasts to myofibroblasts and induction of contraction in Postn−/− wounds. Postn+/+ and Postn−/− primary dermal fibroblasts were isolated and assayed for their ability to contract a floating collagen gel, as well as exert contractile forces across a collagen gel matrix (Fig. 5A). Postn+/+ cells were able to generate contractile forces across the collagen gel lattice, but Postn−/− fibroblasts showed a significant reduction in ability to contract the lattice. The anchored matrix gel contraction assay was then used, because it more closely resembles the mechanical environment of granulation tissue (Grinnell, 1994). Quantification of contraction through measurement of gel weight confirmed that Postn+/+ fibroblasts were able to significantly contract the collagen matrix (Fig. 5B,C), in comparison with Postn−/− fibroblasts (Fig. 5B), indicating that periostin is required for contraction of a collagen matrix by dermal fibroblasts (P<0.01). To further investigate this finding, recombinant human periostin (rhPN) was added to the collagen matrix and was sufficient to induce contraction of the gels by Postn−/− dermal fibroblasts. Addition of 5 μg/ml rhPN fully recovered the contractile ability of Postn−/− cells (Fig. 5C), supporting the notion that periostin facilitates wound repair by promoting wound contraction.
Fig. 5.

Exogenous periostin is sufficient to induce a contractile phenotype. (A) The effect of periostin (PN) deletion on the ability of dermal fibroblasts to exert contractile force in a fixed, tethered floating collagen gel lattice was investigated using a culture force monitor. Forces generated by fibroblasts were measured over 24 hours; a representative trace is shown (n=3). (B) Cells contracted collagen gels over an additional 24 hours at 37°C, 5% CO2. (C) Gel contraction was quantified by loss of gel weight, compared with gels lacking cells. Postn−/− (KO) fibroblasts were unable to significantly contract collagen gels. Note that Postn+/+ (WT) fibroblasts were able to contract collagen gels. Addition of 5 μg/ml rhPN to the collagen gels rescued the contractile ability of Postn−/− fibroblasts (n=3). Data is expressed as a fraction of the initial gel weight; error bars represent s.d. (*P<0.05; two-way ANOVA). (D) Gels were treated with 10 μM PP2 (or DMSO vehicle) or 10 μg/ml β1-integrin blocking antibody (mouse IgG for controls). Data are expressed as a fraction of the initial gel weight; error bars represent s.d. (*P<0.05; one-way ANOVA, n=3). A Dunnett's multiple comparison test was used where Postn−/−, rhPN and DMSO were used as the reference group. Extracellular periostin influences contractility through a β1-integrin- and FAK-dependent mechanism in vitro. (E) Fluorescent labeling of fibroblast populated collagen gels for α-SMA (green) and nuclei (blue). α-SMA-positive cells were counted in high magnification fields of view. Percentage of α-SMA-positive cells was significantly reduced in Postn−/− fibroblast populated gels (P<0.01; n=3). Addition of 5 μg/ml rhPN to the collagen gels restored the percentage of α-SMA-positive cells. Data are expressed as mean; error bars represent s.d. (#P<0.01; one-way ANOVA).
To understand the mechanism by which periostin induces contraction, we attempted to reverse the effect of rhPN on Postn−/− fibroblast contraction with various inhibitors of signal transduction. As in Fig. 5C, Postn−/− fibroblasts were unable to contract the collagen matrix. With the addition of rhPN, however, these cells contracted gels to the same extent as Postn+/+ fibroblasts (Fig. 5D). Blockade of β1-integrin ligation by incorporation of a β1-integrin neutralizing antibody (10 μg/ml) completely reversed the rhPN-induced gel contraction (Fig. 5D) (P<0.01). As a negative control, non-specific mouse IgG was instead incorporated into gels at the same concentration. Gels containing IgG contracted to the same degree as Postn−/− fibroblast populated gels with rhPN. Additionally, inhibition of Src and FAK phosphorylation with PP2 completely reversed the influence of rhPN on contraction of collagen gels populated with Postn−/− fibroblasts (P<0.01).
To assess whether the differences in collagen matrix contraction were the result of differences in α-SMA levels, fibroblast-populated collagen gels were immunolabeled for α-SMA (Fig. 5E). Postn+/+ fibroblasts were generally more spread and displayed strong α-SMA labeling. In comparison, the number of Postn−/− fibroblasts positive for α-SMA was significantly reduced (89% positive in Postn+/+, 40% in Postn−/−; P<0.01) (Fig. 5E). In line with gel contraction data, addition of 5 μg/ml rhPN to the collagen matrix resulted in an increase in the number of α-SMA-positive Postn−/− cells (93%; P<0.01) (Fig. 5E).
Periostin is known to influence cell adhesion (Takeshita et al., 1993). Therefore, it was conceivable that the lack of matrix contraction by Postn−/− fibroblasts was simply due to a defect in adhesion. To determine the influence of periostin on dermal fibroblast adhesion, we conducted adhesion assays with tissue culture plates coated with fibronectin, collagen-1 and periostin. Postn+/+ and Postn−/− cells adhered to all matrix molecules tested equally, following 2 hours of incubation (supplementary material Fig. S4). Adhesion was higher on fibronectin than on collagen-1, as anticipated (P<0.01). Adhesion on periostin was significantly lower than on either fibronectin or collagen-1 (P<0.01). Coating wells with a combination of rhPN and fibronectin had no effect on dermal fibroblast adhesion compared with fibronectin alone. Coating with rhPN and collagen-1, however, greatly reduced adhesion of dermal fibroblasts (supplementary material Fig. S4) (P<0.001). This effect was consistent between Postn+/+ and Postn−/− cells. Therefore, it is unlikely that the influence of periostin on contractility is simply due to cell adhesion.
Periostin influences fibroblast morphology in 3D, but not 2D, culture
The presence of α-SMA immunoreactivity in the borders of day 7 Postn−/− wounds, but not within the granulation tissue, implies that alternative signals for myofibroblast differentiation are at work in these regions. Differentiation of fibroblasts into myofibroblasts requires TGFβ-dependent signalling, but also depends heavily on matrix stiffness (Tomasek et al., 2002). To explore the contribution of matrix stiffness on myofibroblast differentiation, dermal fibroblasts were seeded onto either collagen-1-coated tissue culture plates or anchored collagen gels. Fibroblasts seeded on collagen-coated tissue culture plates adopted a planar, well-spread morphology typical of fibroblasts in culture (Fig. 6A). These cells developed very distinct stress fibers, which often incorporated α-SMA, indicating myofibroblast differentiation. Postn−/− fibroblasts grown on collagen-coated tissue culture plates were indistinguishable from Postn+/+ fibroblasts. Moreover, α-SMA expression, as assessed by western blot, was not significantly different between Postn+/+ and Postn−/− cells. Addition of 5 ng/ml recombinant human TGFβ1 did not further increase α-SMA levels (Fig. 6B), indicating that the cultures might have already become maximally differentiated.
Fig. 6.

Periostin influences fibroblast morphology in 3D culture but not 2D culture. Isolated dermal fibroblasts were seeded on collagen-coated tissue culture plates or onto precast anchored collagen gels. Cells were incubated for 48 hours before fixation or harvesting lysates. (A) Filamentous actin was visualized with Rhodamine-conjugated Phalloidin. Distinct stress fibers were observed in both Postn+/+ and Postn−/− fibroblasts (n=3). (B) Western blot analysis of lysates was carried out to quantify the level of α-SMA. Equal loading was confirmed by blotting for GAPDH. No difference was detected between Postn+/+ (WT) and Postn−/− (KO) fibroblasts. (C) Fibroblasts seeded on 3D collagen gels were labeled with Rhodamine-conjugated phalloidin and assessed for cell morphology (n=3). Postn−/− fibroblasts were more likely to adopt a dendritic phenotype, characterized by a lack of stress fibers and extension of thin branching cytoplasmic extensions (P<0.01). Addition of 5 μg/ml rhPN to the gels restored the percentage of dendritic fibroblasts to Postn+/+ levels. Data is expressed as mean; error bars represent s.d. (*P<0.01; one-way ANOVA). (D) 3D collagen gels were homogenized and cells were lysed by sonication. α-SMA levels were assessed by western blot.
Tissue culture plastic provides an exceedingly stiff environment, whereas 3D collagen gels provide a much more compliant matrix, reminiscent of granulation tissue (Grinnell, 1994). The surface of the anchored collagen gels used in this study provides a mechanically intermediate environment (Arora et al., 1999), which produced mainly planar cells (Fig. 6C). 21% of Postn+/+ cells, however, adopted a dendritic phenotype, characterized by poor spreading, lack of stress fibers and extension of numerous thin branching processes (Grinnell, 2003) (Fig. 6C). Interestingly, the proportion of Postn−/− cells adopting the dendritic phenotype was significantly higher at 39% (P<0.01). Furthermore, addition of 5 μg/ml rhPN to the collagen gels decreased the proportion of dendritic cells to the level of Postn+/+ cultures. α-SMA protein was reduced in Postn−/− fibroblasts cultured on compliant collagen gels, but increased with the addition of rhPN to the gel (Fig. 6D).
Periostin facilitates α-SMA expression on compliant substrates, but is not required with increasing substrate stiffness
Because Postn−/− fibroblasts showed altered spreading and a deficit in α-SMA protein when cultured on compliant collagen gels but not on collagen-coated plastic, we sought to further assess periostin-induced myofibroblast differentiation in the context of matrix stiffness. Postn+/+ and Postn−/− fibroblasts were seeded on collagen-coated flexible polyacrylamide gels of varying stiffness. On soft substrates (Young's modulus of 4800 Pa), 64% of Postn+/+ fibroblasts were positive for α-SMA (Fig. 7B). The proportion of α-SMA-positive Postn+/+ fibroblasts peaked on 19200 Pa substrates at 90%, with no additional increase on the stiff 50,000 Pa substrates. The percentage of α-SMA-positive Postn−/− fibroblasts increased with an increase in substrate stiffness (P<0.01). Compared with Postn+/+ fibroblasts, however, the proportion of α-SMA-positive Postn−/− fibroblasts was significantly lower on 4800 Pa and 19,200 Pa substrates at 29% and 57%, respectively (P<0.05) (Fig. 7B). On 50,000 Pa substrates, the proportion of α-SMA-positive Postn−/− fibroblasts was equivalent to that of Postn+/+ fibroblasts (Postn+/+, 87%; Postn−/−, 84%). To further investigate the mechanism by which periostin facilitates α-SMA expression we focused on the soft 4800 Pa substrates. Postn−/− fibroblasts showed an increase in α-SMA protein and phosphorylated FAK(Y397) when polyacrylamide gels were coated with 100 μg/ml collagen and 5 μg/ml rhPN, compared with gels coated with 100 μg/ml collagen alone (Fig. 7C). This increase was attenuated by 10 μM PP2.
Fig. 7.

Periostin facilitates α-SMA expression on a soft substrate, but is compensated for by increased substrate stiffness. Isolated dermal fibroblasts were seeded on collagen-coated flexible polyacrylamide substrates with Young's moduli of 4800, 19,200 or 50,000 Pa. Cells were incubated for 48 hours before fixation. (A) Fibroblasts were fluorescently labeled for α-SMA (green), filamentous actin (red) and nuclei (blue). α-SMA positive cells were counted from high magnification fields of view. (B) Percentage of α-SMA-positive cells was significantly reduced in Postn−/− (KO) fibroblasts grown on the soft 4800 and 19,200 Pa substrates (P<0.05, n=3). On the stiff 50,000 Pa substrates, however, the proportion of α-SMA-positive Postn−/− fibroblasts was equivalent to that of Postn+/+ (WT) fibroblasts. Data are expressed as means; error bars represent s.d. (*P<0.05; two-way ANOVA). (C) Western blot analysis of lysates from Postn−/− fibroblasts grown on 4800 Pa substrates was carried out to quantify the level of FAK-P [at Y397; pFAK(Y397)] and α-SMA. Loading was corrected by blotting for total FAK and GAPDH. pFAKY397/FAK and α-SMA/GAPDH indicate results of densitometry, relative to control. Incorporation of rhPN on 4800 Pa substrates resulted in an increase in phosphorylated FAK(Y397) and α-SMA protein. Increased α-SMA was attenuated by 10 μM PP2.
Delivery of recombinant periostin via electrospun collagen scaffolds stimulates α-SMA expression
Since rhPN was sufficient to restore α-SMA expression and contractility in Postn−/− fibroblasts in vitro, we attemped to reintroduce periostin into Postn−/− wounds, in vivo. We incorporated rhPN into electrospun collagen scaffolds (Fig. 8A,B) and these scaffolds (or control scaffolds lacking periostin) were laid into Postn−/− wounds immediately following wounding. Immunohistochemical analysis of day 7 wounds revealed a marked increase in α-SMA immunoreactivity within the wounds that received collagen and periostin scaffolds, compared with collagen-only controls (Fig. 8C). Moreover, α-SMA immunoreactivity was detected throughout the granulation tissue of wounds treated with collagen and periostin, mimicking the wild-type expression pattern.
Fig. 8.

Delivery of recombinant periostin via electrospun collagen scaffolds recovers α-SMA expression in Postn−/− mice. (A) Collagen type 1 electrospun scaffolds, with and without rhPN, were cut into 6 mm disks to match the size of punch wounds. (B) Periostin (green) protein was detected throughout the collagen + periostin scaffolds, but not the control collagen scaffolds. (C) Punch wounds were created in Postn−/− animals and scaffolds were immediately laid into the wounds. Half of the wounds received two control (collagen only) scaffolds and the other half of the wounds received two collagen and periostin scaffolds. Wounds were harvested for IHC at day 7. Addition of periostin to Postn−/− wounds by electrospun collagen scaffolds resulted in a marked increase in α-SMA immunoreactivity throughout the granulation tissue, when compared with that in wounds receiving control collagen scaffolds (n=3).
Discussion
In this report we show that the loss of periostin by use of the periostin knockout mouse (Rios et al., 2005) results in altered dermal wound-closure kinetics, specifically during the pro-fibrotic phase of wound repair. The alteration in wound closure corresponds with the onset and peak of periostin expression in Postn+/+ animals. Immunohistochemistry and RT-qPCR reveal that α-SMA is strikingly reduced in the granulation tissue of day 7 Postn−/− wounds. Altered wound-closure kinetics in Postn−/− mice might therefore be due to a reduction in α-SMA-positive myofibroblasts within the granulation tissue, and thus a reduction in wound contraction. Other phases of wounds repair appeared to be unaffected by the loss of periostin. Infiltration of inflammatory cells (macrophages and neutrophils) was similar between Postn+/+ and Postn−/− wounds (supplementary material Fig. S1). The eventual closure of both Postn+/+ and Postn−/− wounds by day 11 indicates that Postn−/− animals close dermal wounds by a method other than contraction, possibly re-epithelialization. Recently, Nishiyama and colleagues reported a reduced percentage re-epithelialization following dermal wounding in an independently derived Postn−/− mouse (Nishiyama et al., 2011). We observed no significant difference in re-epithelialization of Postn−/− wounds, when compared with Postn+/+ controls (supplementary material Table S1), but wound size was greater in Postn−/− animals. Owing to the difference in wound size, the percentage of epithelialization appeared to be lower in Postn−/− wounds (not significant), yet the epithelial migration distance was actually higher. Therefore we feel that percentage epithelialization is an inappropriate measurement for wounds of different sizes (Gorin et al., 1996) and epithelial migration distance is the more reliable method.
Reduced levels of α-SMA, specifically within the granulation tissue of Postn−/− wounds, in which wound borders and vasculature are positive for α-SMA, raised the question of whether fibroblast recruitment was deficient in Postn−/− wounds. In fact, by immunohistochemical detection of fibroblast-specific protein-1, fibroblasts were observed to dominate the newly formed granulation tissue of both Postn+/+ and Postn−/− wounds. Cell number was also similar for Postn+/+ and Postn−/− wounds in the granulation tissue and at wound borders. Moreover, fibroblast migration, as assessed by scratch wound assays in vitro, revealed no difference in migration between Postn+/+ and Postn−/− fibroblasts. Together, these results suggest that fibroblast recruitment is not the underlying cause for the pattern of α-SMA expression in Postn−/− wounds. Collagen content from day 7 wounds, as assessed by hydroxyproline content, was not significantly different between Postn+/+ and Postn−/− wounds. Therefore, we suggest that the granulation tissue of Postn−/− wounds harbors a synthetic fibroblast population, which is, however, deficient in contractile machinery.
Because fibroblast recruitment did not appear to be altered in Postn−/− wounds, we sought to determine whether reduced α-SMA within Postn−/− granulation tissue was the result of a defect in differentiation of fibroblasts into myofibroblasts. Supporting this hypothesis, isolated dermal fibroblasts from Postn−/− animals were unable to significantly contract anchored collagen gels, which assess contractility rather than tractional forces of migration (Grinnell, 1994). Addition of exogenous rhPN, however, fully rescued the phenotype of the Postn−/− fibroblasts. Immunocytochemistry revealed that the degree of gel contraction corresponded to the number of α-SMA-positive cells within the gel. Therefore, we conclude that periostin is required for gel contraction and its presence in the extracellular matrix is sufficient to induce a contractile myofibroblast phenotype. Previous reports support a role for periostin in gel contraction. Incorporation of purified periostin increased the ability of atrioventricular cushion mesenchymal cells to contract a collagen gel (Butcher et al., 2007). Similarly, addition of rhPN to collagen gels increased the contractility of fibroblasts isolated from patients suffering from Dupuytren's disease – a disease of connective tissue contracture (Vi et al., 2009). In both of these studies, increased contraction was associated with increased α-SMA protein.
Culture of isolated dermal fibroblasts on collagen-1-coated tissue culture plates resulted in abundant α-SMA production, irrespective of periostin expression, showing that Postn−/− fibroblasts are capable of α-SMA expression. The pattern of α-SMA expression within day 7 Postn−/− wounds must, therefore, be due to the environment provided by the granulation tissue itself. Mechanical tension (Cevallos et al., 2006; Hinz et al., 2001) and TGFβ (Desmouliere et al., 1993) induce α-SMA expression and promote myofibroblasts differentiation (Tomasek et al., 2002). Induction of α-SMA by TGFβ is primarily through activation of canonical TGFβ signaling, specifically Smad3 (Gu et al., 2007). Interestingly, nuclear Smad2/3–P was detected at equal levels within the granulation tissue of Postn+/+ and Postn−/− wounds. Although we cannot rule out Smad-independent TGFβ pathways (Derynck and Zhang, 2003), TGFβ signaling appears to be active within the granulation tissue of Postn−/− wounds. Therefore, we suspected that the mechanical environment might be the determinant factor for periostin-induced myofibroblast differentiation.
Mathematical models of fibroblast-driven dermal wound repair predict that the wound border is a region of peak matrix stiffness (Murray, 2003). Of particular importance, the tangential stiffness modulus of this region is greatly elevated over that of the central granulation tissue (Murray, 2003). The potential correlation between predicted tissue stiffness and α-SMA immunoreactivity in Postn−/− wounds prompted us to test whether periostin influences fibroblast behaviour in a mechanically dependent manner. On collagen-coated tissue culture plates, no difference in cell morphology or α-SMA expression was apparent. In contrast to 2D culture, when isolated dermal fibroblasts were cultured on the more compliant collagen gels a greater percentage of Postn−/− cells adopted a dendritic phenotype and α-SMA protein was reduced. Incorporation of rhPN into the collagen gels reversed both of these effects. Tissue culture plates provide an enormously stiff environment for cell growth with a reported elastic modulus of 2.78 GPa (Callister and Rethwisch, 2000), whereas 2 mg/ml collagen gels have an elastic modulus of approximately 300–400 Pa (Marenzana et al., 2006; Paszek et al., 2005). The elastic modulus of anchored collagen gels has not been reported. However, they are believed to more closely represent the mechanical environment of the granulation tissue (Grinnell, 1994), which has been reported to have an elastic modulus of 18.5 kPa in day 7 rat wounds (Goffin et al., 2006).
We propose that periostin facilitates myofibroblast differentiation and matrix contraction in a compliant 3D environment, such as in the granulation tissue. In a rigid environment, such as in 2D culture or at the wound border, however, the effects of periostin are overshadowed by the influence of mechanical tension. Multiple studies looking at the effect of matrix stiffness on fibroblasts morphology agree that above 3–6 kPa, fibroblasts assemble stress fibers, and above 15 kPa, incorporation of α-SMA occurs (Solon et al., 2007; Wells, 2005; Yeung et al., 2005). To refine our assessment of myofibroblast differentiation in the context of matrix stiffness, we adopted the widely used matrix-coated flexible polyacrylamide substrate (Aplin and Hughes, 1981; Pelham and Wang, 1997). Although the stiffness of collagen gels can be modulated by increasing or decreasing the collagen concentration, this method introduces ligand density as a confounding variable (Paszek et al., 2005). In this study, all substrates were coated with the same concentration of collagen, thereby ensuring that differences in cell differentiation were due to substrate stiffness and not ligand density. Using collagen-coated polyacrylamide gels of varying stiffness, we observed a significant reduction in the proportion of Postn−/− fibroblasts positive for α-SMA, compared with levels in wild-type controls. This reduction was present on the more compliant 4800 Pa and 19,200 Pa substrates, but not on the stiff 50,000 Pa substrates, thus clearly showing that the lack of periostin can be compensated for by matrix stiffness. Furthermore, this supports the hypothesis that increased matrix stiffness is responsible for α-SMA expression at the borders of day 7 Postn−/− wounds.
Interestingly, Sidhu and colleagues recently reported that incorporation of periostin, in the absence of any cell type, was sufficient to increase the elastic modulus of collagen type 1 gels (Sidhu et al., 2010). Based on this evidence, it is possible that periostin itself might contribute to the stiffness of the granulation tissue by collagen cross-linking, thereby indirectly promoting myofibroblast differentiation. Such an effect would surely be a confounding variable in the collagen gel contraction assays used here, although the concentrations of periostin used by Sidhu and colleagues were substantially higher (20 and 200 μg/ml) than the concentration used in our assays (5 μg/ml) (Sidhu et al., 2010). If the mechanism by which periostin facilitates myofibroblast differentiation is by simply increasing matrix stiffness, we would expect Postn+/+ and Postn−/− fibroblasts to behave similarly on 2D-matrix-coated polyacrylamide substrates, where stiffness is controlled independent of collagen cross-linking. Our results, however, do not support such a mechanism, because Postn−/− fibroblast differentiation was significantly lower on soft substrates. Our results, instead, favour a mechanism where periostin facilitates myofibroblast differentiation in compliant substrates and its influence is overshadowed by increased matrix stiffness.
Although we had established a ‘niche’ in which periostin promotes myofibroblast differentiation in compliant matrices, the mechanism by which it does this remained unclear. In gel contraction assays, we found that inhibition of β1-integrin ligation and inhibition of Src–FAK signaling reversed the periostin-induced increase in gel contraction. Moreover, coating polyacrylamide substrates with a combination of collagen and rhPN increased the level of α-SMA protein and phosphorylation of FAK(Y397). The rhPN-induced increase in α-SMA protein was attenuated by PP2. In addition to providing attachment to the ECM, integrin ligation is known to activate numerous signaling pathways (Giancotti and Ruoslahti, 1999). We cannot rule out the possibility that β1-integrin function is required for gel contraction and myofibroblast differentiation through a mechanism parallel to, but independent of, periostin. However, periostin has been shown to bind β1-integrins – an event that is required for cushion mesenchyme cell invasion into collagen-1 gels (Butcher et al., 2007).
Periostin was initially classified as an adhesion molecule (Horiuchi et al., 1999; Takeshita et al., 1993). However, more recent work (Katsuragi et al., 2004), as well as adhesion data presented in this report (supplementary material Fig. S4), do not support the notion that periostin is an adhesion molecule per se. We suggest, instead, that the binding of periostin to surface receptors in vivo serves to modulate intracellular signaling, and that the role of periostin is not strictly to increase attachment. Ligation of most integrin pairs results in the activation of FAK (Giancotti and Ruoslahti, 1999). Indeed, periostin has been shown to influence intracellular FAK activation in an integrin-dependant manner (Li et al., 2010; Shimazaki et al., 2008). Our data favours a mechanism by which periostin influences intracellular signaling of myofibroblast differentiation in a β1-intergrin- and FAK-dependent manner.
Myofibroblasts are at the core of many fibrotic diseases, including systemic sclerosis (Leask, 2010), hypertrophic scars (Baur et al., 1975) and subepithelial fibrosis in bronchial asthma (Brewster et al., 1990). To date, periostin has been implicated in subepithelial fibrosis (Takayama et al., 2006) and hypertrophic scars (Wang et al., 2007a; Zhou et al., 2010). The role of periostin in these diseases is not fully understood; however, our data supports the hypothesis that periostin facilitates myofibroblast differentiation, thereby contributing to disease progression. The ability of periostin-containing electrospun collagen scaffolds to increase α-SMA immunoreactivity in Postn−/− wounds might have important implications for treatment of non-healing skin lesions, or chronic wounds. By definition, chronic wounds are unable to close, and therefore represent a massive burden on healthcare systems (Elliott and Hamilton, 2011). There is an immediate need for therapeutics capable of expediting wound closure. Using recombinant material and collagen scaffolds, we have demonstrated the feasibility of using periostin to influence wound repair. Future work will focus on the use of periostin-containing electrospun collagen scaffolds to determine whether the increased α-SMA immunoreactivity translates into increases in wound-closure rates.
Materials and Methods
Animals
All animal procedures were in accordance with protocols approved by the University Council on Animal Care at The University of Western Ontario. Periostin-knockout mice (Postn−/−) were generated as described previously (Rios et al., 2005). Heterozygous mice were crossed with C57BL/6J (JAX Mice and Services, Bar Harbor, Maine) for a minimum of six generations to ensure an incipient congenic strain. Backcrossed heterozygous mice were used for breeding and offspring were genotyped as described previously (Rios et al., 2005). Postn−/− mice and sex-matched littermate Postn+/+ control mice were weaned at 3 weeks and provided powder food to reduce the effects of tooth defects on growth rate (Rios et al., 2005). All animals were subjected to a 12 hour light/dark cycle and temperature in accordance with the guidelines of the Canadian Council on Animal Care.
Punch wounds
For experiments, Postn−/− and sex-matched littermate Postn+/+ mice (12 weeks of age weighing approximately 25 g) were anesthetized with an intraperitoneal injection of buprenorphine (50 μg/kg), followed by an injection of ketamine (100 mg/kg) and xylazine (5 mg/kg). Backs were shaved, depilated and sterilized with iodine. Two full-thickness excisional wounds were made on each side of the dorsal midline with a 6 mm punch biopsy. Removed tissue was considered day 0 and was retained for further analysis. Wounds were photographed immediately after wounding and again at 3, 5, 7, 9 and 11 days post wounding. Wound area was assessed from photographs using Northern Eclipse v7.0 software (Empix Imaging, Mississauga, Ontario) and expressed as a fraction of initial area. Mice were caged individually following wounding and were sacrificed at day 11 for histology and gene expression assays.
Tissue preparation and immunohistochemistry
Additional animals were wounded as described above and sacrificed at days 5 and 7. At various time-points, wounds were excised and either snap frozen in liquid nitrogen, or fixed in 10% neutral buffered formalin (Sigma Aldrich). Tissues were stained as previously described (Jackson-Boeters et al., 2009). Sections were blocked with 10% horse serum and incubated with goat anti-periostin (sc49480, Santa Cruz Biotechnology, Santa Cruz, CA) primary antibody overnight at 4°C. For negative controls, periostin primary antibody was pre-absorbed for 1 hour with the immunizing peptide. Detection was by ImmPRESS Ig peroxidase kits (Vector Laboratories, Burlingame, California) and visualized with 3,3-diaminobenzidine (Vector Laboratories). Sections were counterstained with haematoxylin. Staining of α-SMA was carried out with the rabbit anti-α-SMA primary antibody (ab5694, Abcam plc, Cambridge, United Kingdom), fibroblast-specific protein-1 was with anti-FSP1/S100A4 (Millipore, Billerica, MA), CD68 was with MCA1957 (AbD Serotec, Oxford, UK), neutrophil elastase was with ab68672 (Abcam). Negative controls excluded the primary antibody. For immunofluorescence, nuclei were labeled with DAPI (Vector Laboratories). Detection of phosphorylated Smad2/3 (sc11769-R, Santa Cruz Biotechnology)-positive nuclei on paraffin sections was carried out as above excluding haematoxylin counterstaining. Trichrome staining was carried out as previously described (Liu et al., 2008). For assessment of re-epithelialization, sections from the centre of the wounds were stained with haematoxylin and photographed. Wound size and epithelial migration distance were measured using Northern Eclipse v7.0 software. Epithelial migration distance was defined as the unilateral distance between the wound border and the migrating front of keratinocytes. Percentage epithelialization was determined from bilateral epithelial migration distance, normalized to wound size.
In situ hybridization
In situ hybridization for periostin message was performed on 5 μm paraffin serial skin sections using [35S]-labeled riboprobe transcribed from a 574 bp fragment of mouse periostin cDNA (Kruzynska-Frejtag et al., 2001). Hybridization with sense probe was performed in parallel as negative control.
RT-qPCR
Snap frozen tissue samples were homogenized in 1 ml of TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA was extracted as per the manufacturer's recommendations. Real-time quantitative PCR was carried out on 50 ng of total RNA using TaqMan One-Step RT-PCR Master Mix and gene-specific TaqMan probes (Applied Biosystems, Carlsbad, CA). Postn and Acta2 gene expression was normalized to the endogenous control gene, 18S. PCR efficiency was verified to fall between 90 and 110% by dilution series, and relative expression was calculated using the ΔΔCT method (Livak and Schmittgen, 2001).
Hydroxyproline
Hydroxyproline content of excised dermal wounds was determined as an indicator of collagen content essentially as previously described (Samuel, 2008). Hydroxyproline content was determined using a standard curve and normalized to tissue dry weight. Values were expressed as g of hydroxyproline per 100 g tissue.
Isolation of primary dermal fibroblasts
Excised tissue from punch wounds were immediately transferred to Dulbecco's modified Eagle's medium (High Glucose) supplemented with 10% fetal bovine serum and 2% AA (200 U penicillin, 200 μg streptomycin, 0.5 μg/ml amphotericin B) (Gibco, Carlsbad, CA). Skin was washed with five changes of medium then incubated at 37°C, 5% CO2 to allow fibroblasts to migrate onto the culture surface. Skin was removed and cells were cultured for two to three passages before use.
Gel contraction
Gel contraction assays were conducted essentially as previously described (Shi-Wen et al., 2004). Collagen was prepared as follows: 10% 0.2 M HEPES (pH 8), 40% bovine collagen (Advanced BioMatrix, San Diego, CA) and 50% 2× Dulbecco's modified Eagle's medium (high glucose). Dermal fibroblasts were suspended in 0.5% FBS DMEM and mixed 1:1 with the collagen preparation to a final density of 100,000 cells/ml. Either 5 μg/ml rhPN (R&D Systems, Minneapolis, MN) or an equivalent volume of PBS was incorporated into the collagen and cell mix. 24-well tissue culture plates were precoated with BSA overnight then washed with PBS. 0.5 ml of collagen and cell mix was added to each well and allowed to set at 37°C. Following polymerization, wells were flooded with 1 ml 0.5% FBS in DMEM. After 24 hours, gels were separated from the surface of the plate and incubated for an additional 24 hours. To ensure that contraction of gels horizontally and vertically was accounted for; quantification of gel contraction was assessed by loss of gel weight, whereby contraction of the collagen matrix excluded growth medium, thus reducing the weight of the gel (Tingstrom et al., 1992).
The Src and FAK inhibitor, PP2 (Calbiochem, Darmstadt, Germany), was added at 10 μM to the collagen and cell mix during preparation and to the medium following polymerization. An equal volume of dimethyl sulfoxide (DMSO) (Sigma) was added to untreated control gels. Additionally, integrin β1 signaling was inhibited by addition of 10 μg/ml blocking antibody (MA2253, Millipore), or mouse IgG (PP100, Millipore) in controls, to the collagen and cell mix.
Fibroblast-populated collagen lattice (FPCL) contraction assay
Experiments were performed essentially as previously described (Shi-Wen et al., 2004). Briefly, cells (1×106 cells/ml) were seeded into a collagen gel (First Link Ltd., Birmingham, UK) floated in DMEM with 0.5% FBS. Gels were anchored at one end and attached to a force transducer at the other end. Forces generated across the collagen lattice were measured over a 24 hour period. Graphical readings are produced every 15 seconds providing continuous measurements of generated forces (Dynes, 1×10−5 N) which are logged into a personal computer.
Adhesion
Tissue culture treated 96-well plates were coated overnight at 4°C with 10 μg/ml human fibronectin (Sigma), 10 μg/ml bovine collagen type 1 (Advanced BioMatrix), 10 μg/ml rhPN (R&D Systems) or a combination of periostin and collagen or fibronectin. Plates were subsequently blocked with 3% BSA at 37°C for 2 hours. Dermal fibroblasts were suspended in serum-free medium, seeded and allowed to attach for 2 hours. Adherent cells were fixed with 10% neutral buffered formalin (Sigma) and stained with Methylene Blue. Adhesion was quantified by dye extraction and measurement of absorbance at 650 nm (Oliver et al., 1989). Cell number was determined from a standard curve.
Migration
Migration was assessed by scratch wound assays essentially as previously described (Liang et al., 2007). Dermal fibroblasts were seeded on glass bottom culture dishes and allowed to reach confluence. Scratches were created with a P200 tip and disrupted cells were washed away with PBS. Growth medium was replaced with 0.5% FBS in DMEM to reduce proliferation. Closure of scratches was documented by time-lapse video microscopy.
Immunocytochemistry
Dermal fibroblasts were suspended in 0.5% FBS DMEM and seeded at 30,000 cells/well on pre-coated collagen-1 six-well plates (BD Biosciences, Franklin Lakes, NJ). Cells were left to attach overnight before treatment with 5 ng/ml recombinant human TGFβ1 (R&D Systems) for 24 hours. Alternatively, cells were seeded on top of polymerized collagen gels (prepared as described above) in 24-well plates and incubated for 48 hours. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 3% BSA. Filamentous actin was visualized using Rhodamine-conjugated Phalloidin (Molecular Probes, Carlsbad, CA). α-SMA was labeled with a mouse monoclonal primary antibody (A5228, Sigma) and detected with a goat anti-mouse IgG conjugated to Alexa Fluor 488 secondary antibody (Molecular Probes). Fibroblast morphology was assessed as dendritic or planar by a blinded observer from random fields of view. Fibroblasts were considered as displaying the dendritic morphology based on the lack of stress fibers and extension of thin branching cytoplasmic extensions. Cells displaying planar morphology were well spread and showed prominent stress fibers.
Western blotting
Cell lysates were harvested at 48 hours with RIPA buffer (Sigma) containing protease and phosphatase inhibitor cocktails (Sigma). Protein concentration was determined by BCA assay (Pierce, Waltham, MA). For 3D collagen gels, gels were homogenized and cells were lysed by sonication. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were washed with Tris-buffered saline containing 0.05% Tween-20 (TBST). Membranes were blocked with 5% milk in TBST. Primary antibodies were anti-α-SMA (ab5694, Abcam), anti-FAK(Y397) (BD611722, BD Biosciences), anti-FAK (sc-558, Santa Cruz Biotechnology) and anti-GAPDH (MAB374, Millipore). Detection was with appropriate peroxidase-conjugated secondary antibodies (Jackson ImmunoReaserch, West Grove, PA) and enhanced chemiluminescence (Pierce). Bands were quantified using ImageJ software, using GAPDH to correct for loading.
Polyacrylamide substrates
Matrix-coated flexible polyacrylamide substrates were created on glass coverslips using methods described previously (Bhana et al., 2010; Pelham and Wang, 1997; Tilghman et al., 2010). Polyacrylamide gels of varying stiffness were prepared with 7% (4800 and 19,200 Pa) or 15% (50,000 Pa) acrylamide (Sigma) and 0.05% (4800 Pa), 0.24% (19,200 Pa) or 0.3% (50,000 Pa) N,N′-methylenebis(acrylamide) (Sigma) (Bhana et al., 2010; Tilghman et al., 2010). Gels were coated with either 100 μg/ml collagen 1 (Advanced BioMatrix) or 100 μg/ml collagen 1 and 5 μg/ml rhPN (R&D Systems) using the heterobifunctional crosslinker Sulfo-SANPAH (Pierce). Gels were washed with PBS and equilibrated with growth medium before seeding cells.
Scaffolds
Type I collagen (Sigma) was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol to make a 15% (w/v) solution. Periostin (R&D Systems) was dissolved in PBS to make a 1 mg/ml solution. 20 μl of the periostin (concentration of 100 μg/ml) was mixed with 3 ml of collagen solution, and the mixture injected at a speed of 1 ml/hour by a syringe pump into a capillary charged with a voltage of +15 kV. The generated nanofibers were collected on a negatively charged (−10 kV) rotation mandrel. Control scaffolds contained 20 μl of PBS. To crosslink the scaffolds, they were immersed in 5% glutaraldehyde in ethanol solution for 30 minutes. Scaffolds were spun onto aluminium foil, and a 6 mm biopsy punch was used to cut the scaffolds to ensure they were the same size as the wound. Each scaffold was sterilized in 100% ethanol and rinsed three times with PBS. Two 6-mm-diameter scaffolds were inserted into each wound immediately following wounding.
Statistical methods
Statistical analysis was by one-way or two-way ANOVA, as appropriate, followed by a Bonferroni correction, using Graphpad Software v4 (Graphpad Software, La Jolla, CA) (P≤0.05 was considered significant). Student's t-tests were used for phosphorylated Smad2/3 and hydroxyproline data. A Dunnett's multiple comparison test was used for gel contraction assays including inhibitors to compare all treatments with the reference group. In total, 16 Postn−/− and 16 Postn+/+ wounds were tracked for wound area over the 11-day time course. Data are expressed as a fraction of the original wound area (mean ± s.d.). In vivo gene expression data represents the mean ± s.e.m. of at least five Postn−/− and five Postn+/+ wounds for each time point. In vitro data is expressed as the mean ± s.d. of three individual experiments with three independent sex-matched littermate primary cultures. Individual experiments included at least three replicates.
Supplementary Material
Acknowledgements
We thank Dr Weiyan Wen, Dr Shangxi Liu and Linda Jackson-Boeters for technical support.
Footnotes
Funding
This work was funded by the Canadian Foundation for Innovation Leaders Opportunity Fund [grant number 18742], the Canadian Institutes of Health Research Institute of Musculoskeletal Health and Arthritis operating grants [grant number IMH-94010] to D.W.H. and the National Sciences and Engineering Research Council of Canada Canadian Graduate Scholarship program to C.G.E. We would also like to acknowledge Arthritis Research UK for funding. These studies were also supported, in part, by Riley Children's Foundation, Indiana University Department of Pediatrics (Neonatal-Perinatal Medicine] and National Institute of Health [grant number HL092508] to S.J.C.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.087841/-/DC1
References
- Aplin J. D., Hughes R. C. (1981). Protein-derivatised glass coverslips for the study of cell-to substratum adhesion. Anal. Biochem. 113, 144-148 [DOI] [PubMed] [Google Scholar]
- Arora P. D., Narani N., McCulloch C. A. (1999). The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am. J. Pathol. 154, 871-882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M., Diegelmann R., Oliver N. (1992). Keloid fibroblasts exhibit an altered response to TGF-beta. J. Invest. Dermatol. 99, 650-655 [DOI] [PubMed] [Google Scholar]
- Bao S., Ouyang G., Bai X., Huang Z., Ma C., Liu M., Shao R., Anderson R. M., Rich J. N., Wang X. F. (2004). Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 5, 329-339 [DOI] [PubMed] [Google Scholar]
- Baril P., Gangeswaran R., Mahon P. C., Caulee K., Kocher H. M., Harada T., Zhu M., Kalthoff H., Crnogorac-Jurcevic T., Lemoine N. R. (2007). Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene 26, 2082-2094 [DOI] [PubMed] [Google Scholar]
- Baur P. S., Larson D. L., Stacey T. R. (1975). The observation of myofibroblasts in hypertrophic scars. Surg. Gynecol. Obstet. 141, 22-26 [PubMed] [Google Scholar]
- Bhana B., Iyer R. K., Chen W. L., Zhao R., Sider K. L., Likhitpanichkul M., Simmons C. A., Radisic M. (2010). Influence of substrate stiffness on the phenotype of heart cells. Biotechnol. Bioeng. 105, 1148-1160 [DOI] [PubMed] [Google Scholar]
- Blumbach K., Zweers M. C., Brunner G., Peters A. S., Schmitz M., Schulz J. N., Schild A., Denton C. P., Sakai T., Fassler R., et al. (2010). Defective granulation tissue formation in mice with specific ablation of integrin-linked kinase in fibroblasts – role of TGFbeta1 levels and RhoA activity. J. Cell Sci. 123, 3872-3883 [DOI] [PubMed] [Google Scholar]
- Bornstein P. (1995). Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J. Cell Biol. 130, 503-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster C. E., Howarth P. H., Djukanovic R., Wilson J., Holgate S. T., Roche W. R. (1990). Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 3, 507-511 [DOI] [PubMed] [Google Scholar]
- Burridge K., Chrzanowska-Wodnicka M. (1996). Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 12, 463-518 [DOI] [PubMed] [Google Scholar]
- Butcher J. T., Norris R. A., Hoffman S., Mjaatvedt C. H., Markwald R. R. (2007). Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev. Biol. 302, 256-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callister W. D., Rethwisch D. G. (2000). Fundamentals of Materials Science and Engineering: an Interactive E-Text, Fifth Edition, pp. 153-160 New Jersey: John Wiley and Sons, Inc. [Google Scholar]
- Cevallos M., Riha G. M., Wang X., Yang H., Yan S., Li M., Chai H., Yao Q., Chen C. (2006). Cyclic strain induces expression of specific smooth muscle cell markers in human endothelial cells. Differentiation 74, 552-561 [DOI] [PubMed] [Google Scholar]
- Conway S. J., Molkentin J. D. (2008). Periostin as a heterofunctional regulator of cardiac development and disease. Curr. Genomics 9, 548-555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Zhang Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577-584 [DOI] [PubMed] [Google Scholar]
- Desmouliere A., Geinoz A., Gabbiani F., Gabbiani G. (1993). Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott C. G., Hamilton D. W. (2011). Deconstructing fibrosis research: do pro-fibrotic signals point the way for chronic dermal wound regeneration? J. Cell Commun. Signal. [In press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan M., Kleeff J., Gorbachevski A., Reiser C., Mitkus T., Esposito I., Giese T., Buchler M. W., Giese N. A., Friess H. (2007). Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 132, 1447-1464 [DOI] [PubMed] [Google Scholar]
- Gabbiani G., Ryan G. B., Majne G. (1971). Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549-550 [DOI] [PubMed] [Google Scholar]
- Gabbiani G., Hirschel B. J., Ryan G. B., Statkov P. R., Majno G. (1972). Granulation tissue as a contractile organ. A study of structure and function. J. Exp. Med. 135, 719-734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti F. G., Ruoslahti E. (1999). Integrin signaling. Science 285, 1028-1032 [DOI] [PubMed] [Google Scholar]
- Gillan L., Matei D., Fishman D. A., Gerbin C. S., Karlan B. Y., Chang D. D. (2002). Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 62, 5358-5364 [PubMed] [Google Scholar]
- Goetsch S. C., Hawke T. J., Gallardo T. D., Richardson J. A., Garry D. J. (2003). Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol. Genomics 14, 261-271 [DOI] [PubMed] [Google Scholar]
- Goffin J. M., Pittet P., Csucs G., Lussi J. W., Meister J. J., Hinz B. (2006). Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J. Cell Biol. 172, 259-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorin D. R., Cordts P. R., LaMorte W. W., Manzoian J. O. (1996). The influence of wound geometry on the measurement of wound healing rates in clinical trials. J. Vasc. Surg. 23, 524-528 [DOI] [PubMed] [Google Scholar]
- Grinnell F. (1994). Fibroblasts, myofibroblasts, and wound contraction. J. Cell Biol. 124, 401-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell F. (2003). Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 13, 264-269 [DOI] [PubMed] [Google Scholar]
- Gu L., Zhu Y. J., Yang X., Guo Z. J., Xu W. B., Tian X. L. (2007). Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta. Pharmacol. Sin. 28, 382-391 [DOI] [PubMed] [Google Scholar]
- Hinz B., Mastrangelo D., Iselin C. E., Chaponnier C., Gabbiani G. (2001). Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 159, 1009-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K., Amizuka N., Takeshita S., Takamatsu H., Katsuura M., Ozawa H., Toyama Y., Bonewald L. F., Kudo A. (1999). Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J. Bone Miner. Res. 14, 1239-1249 [DOI] [PubMed] [Google Scholar]
- Iekushi K., Taniyama Y., Azuma J., Katsuragi N., Dosaka N., Sanada F., Koibuchi N., Nagao K., Ogihara T., Morishita R. (2007). Novel mechanisms of valsartan on the treatment of acute myocardial infarction through inhibition of the antiadhesion molecule periostin. Hypertension 49, 1409-1414 [DOI] [PubMed] [Google Scholar]
- Ignotz R. A., Massague J. (1986). Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 261, 4337-4345 [PubMed] [Google Scholar]
- Jackson-Boeters L., Wen W., Hamilton D. W. (2009). Periostin localizes to cells in normal skin, but is associated with the extracellular matrix during wound repair. J. Cell Commun. Signal. 3, 125-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J. I., Lau L. F. (2010). Cellular senescence controls fibrosis in wound healing Aging 2, 627-631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima T. G., Nishiyama T., Shimazu K., Shimazaki M., Kii I., Grigoriadis A. E., Fukayama M., Kudo A. (2009). Periostin, a novel marker of intramembranous ossification, is expressed in fibrous dysplasia and in c-Fos-overexpressing bone lesions. Hum. Pathol. 40, 226-237 [DOI] [PubMed] [Google Scholar]
- Katsuragi N., Morishita R., Nakamura N., Ochiai T., Taniyama Y., Hasegawa Y., Kawashima K., Kaneda Y., Ogihara T., Sugimura K. (2004). Periostin as a novel factor responsible for ventricular dilation. Circulation 110, 1806-1813 [DOI] [PubMed] [Google Scholar]
- Kruzynska-Frejtag A., Machnicki M., Rogers R., Markwald R. R., Conway S. J. (2001). Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech. Dev. 103, 183-188 [DOI] [PubMed] [Google Scholar]
- Leask A. (2010). Towards an anti-fibrotic therapy for scleroderma: targeting myofibroblast differentiation and recruitment. Fibrogenesis Tissue Repair 3, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenga Y., Koh A., Perera A. S., McCulloch C. A., Sodek J., Zohar R. (2008). Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 102, 319-327 [DOI] [PubMed] [Google Scholar]
- Li G., Oparil S., Sanders J. M., Zhang L., Dai M., Chen L. B., Conway S. J., McNamara C. A., Sarembock I. J. (2006). Phosphatidylinositol-3-kinase signaling mediates vascular smooth muscle cell expression of periostin in vivo and in vitro. Atherosclerosis 188, 292-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Jin R., Norris R. A., Zhang L., Yu S., Wu F., Markwald R. R., Nanda A., Conway S. J., Smyth S. S., et al. (2010). Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 208, 358-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C. C., Park A. Y., Guan J. L. (2007). In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329-333 [DOI] [PubMed] [Google Scholar]
- Lindner V., Wang Q., Conley B. A., Friesel R. E., Vary C. P. (2005). Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler. Thromb. Vasc. Biol. 25, 77-83 [DOI] [PubMed] [Google Scholar]
- Liu S., Kapoor M., Shi-wen X., Kennedy L., Denton C. P., Glogauer M., Abraham D. J., Leask A. (2008). Role of Rac1 in a bleomycin-induced scleroderma model using fibroblast-specific Rac1-knockout mice. Arthritis Rheum. 58, 2189-2195 [DOI] [PubMed] [Google Scholar]
- Liu S., Shi-Wen X., Blumbach K., Eastwood M., Denton C. P., Eckes B., Krieg T., Abraham D. J., Leask A. (2010). Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J. Cell Sci. 123, 3674-3682 [DOI] [PubMed] [Google Scholar]
- Liu S., Shi-Wen X., Abraham D. J., Leask A. (2011). CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum. 63, 239-246 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402-408 [DOI] [PubMed] [Google Scholar]
- Majno G., Gabbiani G., Hirschel B. J., Ryan G. B., Statkov P. R. (1971). Contraction of granulation tissue in vitro: similarity to smooth muscle. Science 173, 548-550 [DOI] [PubMed] [Google Scholar]
- Marenzana M., Wilson-Jones N., Mudera V., Brown R. A. (2006). The origins and regulation of tissue tension: identification of collagen tension-fixation process in vitro. Exp. Cell Res. 312, 423-433 [DOI] [PubMed] [Google Scholar]
- Midwood K. S., Williams L. V., Schwarzbauer J. E. (2004). Tissue repair and the dynamics of the extracellular matrix. Int. J. Biochem. Cell Biol. 36, 1031-1037 [DOI] [PubMed] [Google Scholar]
- Murray J. D. (2003). Model for fibroblast-driven wound healing: residual strain and tissue remodelling. In Mathematical Biology II: Spatial Models and Biomedical Applications, Vol. 2 (ed. Antman S. S., Marsden J. E., Sirovich L., Wiggins S.), pp. 503-510 New York: Springer-Verlag Inc. [Google Scholar]
- Naitoh M., Kubota H., Ikeda M., Tanaka T., Shirane H., Suzuki S., Nagata K. (2005). Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells 10, 1081-1091 [DOI] [PubMed] [Google Scholar]
- Nakazawa T., Nakajima A., Seki N., Okawa A., Kato M., Moriya H., Amizuka N., Einhorn T. A., Yamazaki M. (2004). Gene expression of periostin in the early stage of fracture healing detected by cDNA microarray analysis. J. Orthop. Res. 22, 520-525 [DOI] [PubMed] [Google Scholar]
- Nishiyama T., Kii I., Kashima T. G., Kikuchi Y., Ohazama A., Shimazaki M., Fukayama M., Kudo A. (2011). Delayed re-epithelialization in periostin-deficient mice during cutaneous wound healing. PLoS ONE 6, e18410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T., Xu J., Kaiser R. A., Melendez J., Hambleton M., Sargent M. A., Lorts A., Brunskill E. W., Dorn G. W., 2nd, Conway S. J., et al. (2007). Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 101, 313-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku E., Kanaji T., Takata Y., Oshima K., Seki R., Morishige S., Imamura R., Ohtsubo K., Hashiguchi M., Osaki K., et al. (2008). Periostin and bone marrow fibrosis. Int. J. Hematol. 88, 57-63 [DOI] [PubMed] [Google Scholar]
- Oliver M. H., Harrison N. K., Bishop J. E., Cole P. J., Laurent G. J. (1989). A rapid and convenient assay for counting cells cultured in microwell plates: application for assessment of growth factors. J. Cell Sci. 92, 513-518 [DOI] [PubMed] [Google Scholar]
- Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D., et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241-254 [DOI] [PubMed] [Google Scholar]
- Pelham R. J., Jr, Wang Y. (1997). Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 94, 13661-13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios H., Koushik S. V., Wang H., Wang J., Zhou H. M., Lindsley A., Rogers R., Chen Z., Maeda M., Kruzynska-Frejtag A., et al. (2005). periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol. Cell Biol. 25, 11131-11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. B., Sporn M. B., Assoian R. K., Smith J. M., Roche N. S., Wakefield L. M., Heine U. I., Liotta L. A., Falanga V., Kehrl J. H., et al. (1986). Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 83, 4167-4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. (1968). The fibroblast and wound repair. Biol. Rev. Camb. Philos. Soc. 43, 51-96 [DOI] [PubMed] [Google Scholar]
- Samuel C. S. (2008). Determination of collagen content, concentration, and sub-types. In Methods in Molecular Biology, Vol. 466: Kidney Research: Experimental Protocols (ed. Becker G. J., Hewitson T. D.), pp. 223-235 New Jersey: Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- Serini G., Bochaton-Piallat M. L., Ropraz P., Geinoz A., Borsi L., Zardi L., Gabbiani G. (1998). The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 142, 873-881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R., Bao S., Bai X., Blanchette C., Anderson R. M., Dang T., Gishizky M. L., Marks J. R., Wang X. F. (2004). Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol. Cell. Biol. 24, 3992-4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X., Chen Y., Denton C. P., Eastwood M., Renzoni E. A., Bou-Gharios G., Pearson J. D., Dashwood M., du Bois R. M., Black C. M., et al. (2004). Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol. Biol. Cell 15, 2707-2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-wen X., Stanton L. A., Kennedy L., Pala D., Chen Y., Howat S. L., Renzoni E. A., Carter D. E., Bou-Gharios G., Stratton R. J., et al. (2006). CCN2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J. Biol. Chem. 281, 10715-10726 [DOI] [PubMed] [Google Scholar]
- Shimazaki M., Nakamura K., Kii I., Kashima T., Amizuka N., Li M., Saito M., Fukuda K., Nishiyama T., Kitajima S., et al. (2008). Periostin is essential for cardiac healing after acute myocardial infarction. J. Exp. Med. 205, 295-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu S. S., Yuan S., Innes A. L., Kerr S., Woodruff P. G., Hou L., Muller S. J., Fahy J. V. (2010). Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc. Natl. Acad. Sci. USA 107, 14170-14175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P., Standley K. N., Wang J., Azhar M., Doetschman T., Conway S. J. (2009). Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 105, 934-947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodek J., Ganss B., McKee M. D. (2000). Osteopontin. Crit. Rev. Oral Biol. Med. 11, 279-303 [DOI] [PubMed] [Google Scholar]
- Solon J., Levental I., Sengupta K., Georges P. C., Janmey P. A. (2007). Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 93, 4453-4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama G., Arima K., Kanaji T., Toda S., Tanaka H., Shoji S., McKenzie A. N., Nagai H., Hotokebuchi T., Izuhara K. (2006). Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 118, 98-104 [DOI] [PubMed] [Google Scholar]
- Takeshita S., Kikuno R., Tezuka K., Amann E. (1993). Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem. J. 294, 271-278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaoki M., Imanaka-Yoshida K., Yokoyama K., Nishioka T., Inada H., Hiroe M., Sakakura T., Yoshida T. (2005). Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am. J. Pathol. 167, 71-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman R. W., Cowan C. R., Mih J. D., Koryakina Y., Gioeli D., Slack-Davis J. K., Blackman B. R., Tschumperlin D. J., Parsons J. T. (2010). Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS ONE 5, e12905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingstrom A., Heldin C. H., Rubin K. (1992). Regulation of fibroblast-mediated collagen gel contraction by platelet-derived growth factor, interleukin-1 alpha and transforming growth factor-beta 1. J. Cell Sci. 102, 315-322 [DOI] [PubMed] [Google Scholar]
- Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., Brown R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349-363 [DOI] [PubMed] [Google Scholar]
- Vi L., Feng L., Zhu R. D., Wu Y., Satish L., Gan B. S., O'Gorman D. B. (2009). Periostin differentially induces proliferation, contraction and apoptosis of primary Dupuytren's disease and adjacent palmar fascia cells. Exp. Cell Res. 315, 3574-3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Nie F. F., Zhao X., Qin Z. L. (2007a). The expression of periostin in hyperplasic scars and the relations to TGF-beta1 and its receptors. Zhonghua Zhengxing Waike Zazhi 23, 229-232 [PubMed] [Google Scholar]
- Wang Z., Gao Z., Shi Y., Sun Y., Lin Z., Jiang H., Hou T., Wang Q., Yuan X., Zhu X., et al. (2007b). Inhibition of Smad3 expression decreases collagen synthesis in keloid disease fibroblasts. J. Plast. Reconstr. Aesthet. Surg. 60, 1193-1199 [DOI] [PubMed] [Google Scholar]
- Wells R. G. (2005). The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J. Clin. Gastroenterol. 39, S158-S161 [DOI] [PubMed] [Google Scholar]
- Yeung T., Georges P. C., Flanagan L. A., Marg B., Ortiz M., Funaki M., Zahir N., Ming W., Weaver V., Janmey P. A. (2005). Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton 60, 24-34 [DOI] [PubMed] [Google Scholar]
- Zhou H. M., Wang J., Elliott C., Wen W., Hamilton D. W., Conway S. J. (2010). Spatiotemporal expression of periostin during skin development and incisional wound healing: lessons for human fibrotic scar formation. J. Cell Commun. Signal. 4, 99-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}