

Abstract
Peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), a key regulator of energy metabolism and lipid homeostasis in multiple highly oxidative tissues, has been implicated in the metabolic derangements of diabetes and obesity. However, relatively less is known regarding its role in neurological functions. Using shotgun lipidomics, we investigated the lipidome of mouse cerebral cortex with generalized deficiency of PGC-1α (PGC-1α−/−) versus wild-type (WT) mice under standard diet and chronically calorically restricted conditions. Specific deficiency in sulfatide, a myelin-specific lipid class critically involved in maintaining neurological function, was uncovered in the cortex of PGC-1α−/− mice compared with WT mice at all ages examined. Chronic caloric restriction (CR) for 22 months essentially restored the sulfatide reduction in PGC-1α−/− mice compared with WT, but sulfatide reduction was not restored in PGC-1α−/− with CR for a short term (i.e., 3 months). Mechanistic studies uncovered and differentiated the biochemical mechanisms underpinning the two conditions of altered sulfatide homeostasis. The former is modulated through PGC-1α-MAL pathway, whereas the latter is under the control of LXR/RXR-apoE metabolism pathway. These results suggest a novel mechanistic role of PGC-1α in sulfatide homeostasis, provide new insights into the importance of PGC-1α in neurological functions, and indicate a potential therapeutic approach for treatment of deficient PGC-1α-induced alterations in sulfatide homeostasis.
Keywords: Alzheimer's disease, apolipoprotein E, shotgun lipidomics, sphingolipidomics
Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) plays a vital role in mitochondrial biogenesis and bioenergetics (1–3). PGC-1α is responsive to numerous metabolic signals and can mediate the effects of a myriad of extracellular and physiological cues on broad and powerful genetic programs (see Ref. 4 for a recent review). Overexpression of PGC-1α in cardiac cells induces expression of hundreds of genes encoding key enzymes for all major metabolic pathways that regulate high-efficiency ATP production in the mitochondrion (4, 5). These target genes include numerous subunits of complexes I to IV, ATPase complexes, all eight enzymes of the citric acid cycle, enzymes in fatty acid β-oxidation as well as fatty acid transport proteins and lipases responsible for turnover of the intracellular triacylglycerol pool, and those genes mediating metabolism of lactate and ketone bodies. In addition, constitutive overexpression of PGC-1α in mouse myocardium triggers profound mitochondrial biogenesis (6). On the other hand, PGC-1α knockout (PGC-1α−/−, KO) mice exhibit impaired bioenergetics, moderate defects in cardiac function, and reduced cardiolipin levels, which are markedly worsened by hemodynamic and metabolic challenges (7–10). The PGC-1α−/− mice also exhibit reduced skeletal muscle mitochondrial number and respiratory capacity (e.g., state 3 respiration rates), leading to compromised muscle performance and exercise capacity at around 6 months of age.
PGC-1α has emerged as a molecular link between transcriptional dysregulation and neurodegeneration (11, 12). For example, increased PGC-1α level suppresses reactive oxygen species (ROS) and neurodegeneration through induction of many ROS-detoxifying enzymes (e.g., GPx1 and SOD2) (13). PGC-1α−/− mice display many phenotypic similarities to transgenic mouse models of Huntington's disease and the gene expression analysis of tissues from Huntington's disease patients revealed a disruption of the PGC-1α regulatory pathway (7, 8). This phenotype has recently been demonstrated in neuronal PGC-1α−/− mice, which show neurodegenerative lesions in their striatum (14). Furthermore, recent studies with human postmortem brain samples and Tg2576 Alzheimer's disease (AD) mouse neurons have revealed that brain PGC-1α expression decreases in individuals with AD as a function of dementia (15). However, the causal factor as well as sequela of this correlation remains unknown.
Our previous studies have showed that i) the content of sulfatide (a class of myelin-specific sphingolipid) is dramatically depleted at the earliest clinically recognizable stages of AD (mild cognitive impairment) in both postmortem brain (16, 17) and freshly collected cerebrospinal fluid of AD patients compared with age-matched cognitively normal controls (18); ii) the sulfatide content in the central nervous system is modulated by apolipoprotein E (apoE) in an isoform-dependent manner through the same metabolic pathways that regulate apoE-associated particles (19); and iii) apoE mediates sulfatide depletion in mouse AD models (20, 21).
Following these lines of evidence that PGC-1α levels and apoE-mediated sulfatide levels are both associated with AD dementia, we reasoned that PGC-1α might influence the brain sulfatide levels and that altered energy expenditure might have an impact on sulfatide homeostasis, which might involve apoE metabolism. Accordingly, we conducted lipidomic analysis of brain cortices of PGC-1α−/− mice with and without caloric restriction (CR) by using our multidimensional mass spectrometry-based shotgun lipidomics (MDMS-SL) platform (22–24) to interrogate the linkages among apoE, PGC-1α, and sulfatide. We found that i) sulfatide is remarkably and specifically reduced in PGC-1α−/− mouse cortex, which appears modulated through a PGC-1α-MAL pathway; ii) apoE-mediated sulfatide metabolism is independent of PGC-1α; and iii) chronic CR restores normal cortical sulfatide levels in PGC-1α−/− mice, which appear mediated through a liver X receptor (LXR)/retinoid X receptor (RXR)-apoE pathway. Collectively, the current study reveals a new mechanism responsible for sulfatide homeostasis, which might contribute to the sulfatide depletion in AD. Our results indicate that chronic CR might be a useful therapeutic strategy for reduction of dementia prevalence.
MATERIALS AND METHODS
Animals studies
The protocols for animal experiments were conducted in accordance with the National Institutes of Health guidelines for humane treatment of animals and were reviewed and approved by the Animal Studies Committee of Washington University. The generation and initial characterization of PGC-1α−/− mice has been described previously (8). PGC-1α−/− and wild-type (WT) mice were fed a standard chow and water ad libitum, unless otherwise noted. Mice that were calorically restricted 33% from their standard food intake were fed a restricted diet at 2 months of age. Mice were euthanized at 5 and 24 months of age under both ad libitum and calorically restricted conditions, and the cortex was dissected and immediately frozen in liquid nitrogen.
Lipid analysis with MDMS-SL
Lipids were extracted from dissected cortical gray matter by a modified Bligh and Dyer method as previously described (25). Internal standards for quantification of individual molecular species of lipid classes were added prior to lipid extraction (25). MDMS-SL analyses were performed with a QqQ mass spectrometer (Thermo Fisher TSQ Quantum Ultra) equipped with an automated Nanomate device (Advion Biosciences) and operated under Xcalibur software as previously described (26, 27). Identification and quantification of lipid molecular species were performed using automated software as previously described (24). To accurately analyze low abundance sphingolipid species, an aliquot of each lipid extract based on 0.5 mg protein of original brain sample was taken, and alkaline methanolysis was conducted to enrich sphingolipids and remove phospholipids from the extract as previously described (28).
Western blot analysis of WT and PGC-1α−/− cortices
Protein concentration of homogenized cortices was determined by the BCA protein assay using BSA standards (Pierce, Thermo Fisher Scientific). Total protein (10 μg) was loaded on 4–15% Tris-HCL Gel system (Bio-Rad Laboratories) and analyzed by electrophoresis. Proteins were transferred to an immobilon™-P membrane (Millipore) for 2 h at 80 V at 4°C. The immobilon™-P membrane was then washed with Tris-buffered saline (TBS) and blocked with 5% nonfat powdered milk in TBS with Tween 20, pH 7.6 for 1 h. Primary antibodies used were diluted as follows: myelin basic protein (MBP, 1:500, Millipore, MAB386); proteolipid protein (PLP, 1:4000, Novus Biological, NB100-74503); myelin-associated oligodendrocyte basic protein (MOBP, 1:1000, Santa Cruz, P18); glyceraldehydes 3-phosphate dehydrogenase (GAPDH, 1:2000, Santa Cruz, FL-335); cerebroside sulfotransferase (CST, 1:500, Sigma, HPA001220); arylsulsulfatase A (ASA, 1:750, Everest Biotech, EB07457); saposin B (Sap B, 1:1000, gift from Dr. Ying Sun, Cincinnati Children's Hospital); apoE (1:1000, Calbiochem, 178479); myelin and lymphocyte protein (MAL, 1:200, Santa Cruz, H-93). The blots were incubated with corresponding secondary antibodies based on manufacturers’ recommendations. The bands of Western blots were visualized with the Pierce chemiluminescence detection system (Pierce) and Kodak Imaging Station 440 CF. Band densities were quantified with Kodak 1D v3.5 software.
RESULTS
Global shotgun lipidomic analysis reveals a depletion of cortical sulfatide content in PGC-1α−/− mice compared with wild-type mice
MDMS-SL analysis of the cortical lipidome of PGC-1α−/− mice at 24 months of age revealed a selective loss of the mass levels of sulfatide (a class of myelin-specific sphingolipid) compared with their WT littermates. No other changes in phospholipid content or other enriched myelin lipids, such as cerebrosides and sphingomyelin, were detected. For example, the total content of 27 sphingomyelin species determined was 22.9 ± 2.5 and 23.6 ± 3.9 nmol/mg protein present in WT and PGC-1α KO cortex, respectively. An MDMS-SL sphingolipidomics approach utilizing lithium methoxide treatment was employed to enrich, enhance, and confirm the decrease of sulfatide (Fig. 1A, B). This approach removed all ester-linked lipids from each individual lipid extract by alkaline methanolysis followed by back extraction. Overall, there was 35% reduction in sulfatide mass content in all sulfatide molecular species in the cortices of PGC-1α−/− mice compared with that in WT mice. Analysis of 5-month-old mice also revealed a 30% decrease in sulfatide content in the PGC-1α−/− mice compared with WT mice (Fig. 1C), suggesting that PGC-1α ablation, which results in an altered metabolic environment in the brain, alters sulfatide homeostasis.
Fig. 1.

Global shotgun lipidomic analysis reveals a depletion of sulfatide content in the cortex of PGC-1α knockout mice. Mass spectrometric analysis reveals a significant decrease of mass levels of sulfatide species in cortices of PGC-1α KO mice (B) compared with WT mice (A) at 24 months of age. All assigned sulfatide species were identified by tandem mass spectrometric analysis. N16:0 sulfatide at m/z 778.5 was used as an internal standard (5 nmol/mg protein). A significant reduction of sulfatide content was also determined in cortices of PGC-1α KO mice compared with WT mice (C). Values represent the mean sulfatide content (nmol/mg protein) ± SE (n = 3–4 cortices per group). Statistical analysis was performed with a two-tailed Student t-test; *P < 0.05.
Depletion of sulfatide content in PGC-1α−/− cortex was not associated with cortical myelin proteins
To identify the causal factors responsible for deficiency in sulfatide levels in cortex induced by PGC-1α ablation, we first examined the expression levels of various myelin proteins, as numerous previous studies have shown that various cortical myelin proteins are associated with the regulation of sulfatide content in the cortex (29–31). Western blot analysis revealed no changes in the content of the isoforms of myelin basic protein (MBP), proteolipid protein (PLP), or myelin-associated oligodendrocyte basic protein (MOBP) (Fig. 2) by densitometry followed by statistical analysis. These results indicate that the loss of sulfatide content was unlikely due to the loss of myelin or the effects of dysregulation of the major myelin proteins induced by PGC-1α ablation.
Fig. 2.
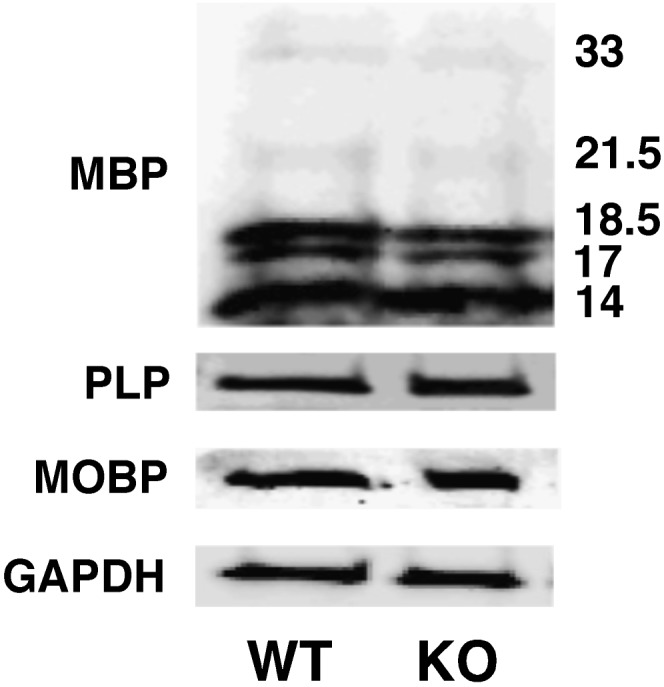
Western blot analysis of the content of myelin-specific proteins. The expression levels of MBP (splice variants 14, 17, 18.5, 21.5, and 33 kDa), PLP, and MOBP in cortices of PGC-1α KO mice compared with WT mice were analyzed with Western blot (see Materials and Methods) and normalized with GAPDH. Selected Western images represent (N = 4) the analysis of cortices of WT and PGC-1α KO mice at 24 months of age. The analysis data indicate that there is no difference between the KO and WT groups.
Expression level of enzymes involved in sulfatide metabolism were unchanged in PGC-1α−/− mice compared with wild-type mice
Next, we determined the mass content of the key enzymes that are involved in sulfatide biosynthesis and degradation. Sulfatide is synthesized by transferring a sulfate group from 3′-phosphoadenosine-5′-phosphosulfate to galactosylceramide (i.e., cerebroside) by the enzyme cerebroside sulfotransferase (CST) following the transport of cerebroside into the Golgi. Degradation of sulfatide involves two proteins saposin B (Sap B) and arylsulfatase A (ASA). Sap B (a coenzyme for ASA hydrolysis of sulfatide) (32) acts to transport sulfatide from lipoprotein particles, carrying sulfatide to ASA, which hydrolyzes the sulfate group in lysosome. Western blot analysis of CST, ASA, prosaposin (the precursor protein for Sap B), and Sap B revealed no changes in the overall content of these proteins by densitometry followed by statistical analysis, suggesting that the decrease in sulfatide mass levels was not due to a dysregulation of the proteins involved in sulfatide biosynthesis and degradation (Fig. 3). Furthermore, the unchanged mass levels of both ceramide and cerebroside in PGC-1α−/− mouse cortex as determined by MDMS-SL (see above) indicate that other biosynthesis and degradation pathways that may be associated with sulfatide mass levels are not perturbed due to PGC-1α ablation.
Fig. 3.

Western blot analysis of expression of enzymes involved in the biosynthesis and catabolism of sulfatide. Expressions of CST, ASA, prosaposin, and Sap B in the cortices of PGC-1α KO and WT mice at 24 months of age were analyzed by Western blot analysis using GAPDH as a protein loading control. Four different cortices of WT and KO mice at 24 months of age were examined. The analysis revealed no change in expression levels of these proteins in the cortices of PGC-1α KO mice compared with those in WT mice.
Cortical MAL expression was decreased in the PGC-1α−/− mice
It is well known that myelin and lymphocyte protein (MAL) is associated with formation of microdomains in myelin membranes and sulfatide trafficking (33–36). Thus, Western blot analysis of the protein expression of MAL was next performed to determine the altered expression levels of MAL in PGC-1α−/− mouse cortex, and it revealed a significant decrease in the null mouse cortex compared with that in WT mice at both 5 months (Fig. 4A, B) and 24 months (Fig. 4C, 4D) of age. These results indicate that ablation of PGC-1α results in a significantly decreased expression level of MAL protein, which could lead to the selected deficiency in sulfatide content based on the function of MAL protein (33–36).
Fig. 4.

Cortical expression of myelin and lymphocyte protein was decreased in PGC-1α KO mice. Expression levels of MAL in PGC-1α KO compared with WT mice under AL and CR at 5 months (A, B) and 24 months (C, D) of age were analyzed by Western blot analysis (see Materials and Methods). Data represent mean ± SE of four animals per group after normalization to loading control GAPDH. Statistical significance was determined by a two-tailed Student t-test; **P < 0.01.
In addition, we further assessed the mass levels of MAL in cortices of WT and PGC-1α−/− mice that were calorically restricted for 3 and 22 months (see below). Western blot analysis revealed that CR did not induce any changes of MAL content in either WT or PGC-1α−/− mouse cortex (Fig. 4). This result indicates that MAL is not involved in the altered sulfatide homeostasis induced with chronic CR (see below).
Chronic caloric restriction attenuates the loss of sulfatide cortical content in PGC-1α−/− mice
To investigate the relationship of energy intake with PGC-1α expression, we calorically restricted WT and PGC-1α−/− mice 33% from their standard food intake at 2 months of age for a short term (3 months) and a long term (22 months), and we examined the effects of CR on sulfatide homeostasis in the mouse cortices. MDMS-SL analysis revealed a significant attenuation of the loss of sulfatide content in PGC-1α−/− mice compared with that in WT mice after 22 months of CR (Fig. 5A, B). CR for 22 months also caused a slight increase in sulfatide content in WT mouse cortex, which is not significant compared with that in WT mice fed a standard diet ad libitum (Fig. 5C). Furthermore, short-term CR did not alter the decrease in sulfatide content in PGC-1α−/− mice compared with WT mice (Fig. 5C). Thus, it appears that long-term CR attenuates the decreased sulfatide to levels near WT mice fed a standard diet ad libitum for 24 months (Fig. 5C). It is worth noting that the mass levels of all other lipid classes and molecular species determined with MDMS-SL were not affected significantly with CR for 22 months.
Fig. 5.

The effects of caloric restriction on cortical sulfatide homeostasis. Mass spectrometric analysis was performed (see Materials and Methods) and revealed an alleviation of sulfatide depletion in the cortices of 24-month old PGC-1α KO (B) mice compared with WT (A). However, no alleviation of sulfatide depletion was observed in short-term caloric restriction (5-month-old mice) (C, compared with Fig. 1C). Chronic caloric restriction alleviated the depletion of sulfatide in PGC-1α KO mice, closer to the cortical sulfatide content in WT mice fed a standard diet (AL). Data represent mean ± SE (nmol/mg protein) averaged from those of at least three separate animals. Statistical significance was determined by a two-tailed Student t-test; *P < 0.05.
ApoE expression was decreased in the cortex during chronic caloric restriction
To underpin the biochemical mechanism(s) responsible for the CR-induced sulfatide regulation, we determined mass levels of myelin proteins, enzymes involving sulfatide biosynthesis and degradation, as well as MAL by Western blot analyses, and we did not observe any significant mass level changes of these proteins (e.g., Fig. 4). Our previous studies showed that the expression as well as haplotype of apoE has a close association with sulfatide homeostasis (20, 37–39). Additionally, it has been found that long-term CR can lead to alterations in the expression of apoE in brain (40). Thus, we examined by Western blot analysis the expression of apoE in the cortex of 5- and 24-month-old WT and PGC-1α−/− mice that were calorically restricted from 2 months of age compared with mice that were fed ad libitum standard diet. We found that apoE expression was not changed in mice calorically restricted for 3 months (Fig. 6A). However, the expression of apoE was approximately 40% lower in both WT and PGC-1α−/− mice that were calorically restricted for 22 months than that it was in age-matched, ad libitum-fed mice (Fig. 6B, C). These results indicate that long-term CR, but not short-term, can lead to a reduction of apoE expression in mouse cortex, which is sufficient to account for the increased mass levels of sulfatide, particularly in PGC-1α−/− mice.
Fig. 6.

Chronic long-term caloric restriction, not short-term caloric restriction, decreases apoE expression in mouse cortices. Expression levels of apoE in the cortices of WT and PGC-1α KO mice under AL or CR conditions at 5 months (A) and 24 months (B) of age were determined by Western blot analysis (see Materials and Methods). Expression of apoE was decreased by 40% under CR in both WT and KO mice at 24 months of age, but it was not changed at short-term CR, compared with WT and KO mice fed AL (C). Values represent the mean ratio of apoE expression levels in mouse cortices after normalization to the loading control GAPDH from four separate animals. Statistical significance was determined by a two-tailed Student t-test compared with that of WT mice under AL conditions; **P < 0.01.
DISCUSSION
In addition to the vital role of PGC-1α in mitochondrial biogenesis and bioenergetics (1–3), PGC-1α is also associated with energy balance, lipid homeostasis, and numerous neurological functions (4, 9, 14). In the current study, we aimed to identify the altered cortical lipids induced with PGC-1α ablation. By using a global lipidomic analysis approach (i.e., MDMS-SL) (23, 24), we uncovered the significant reduction of cortical sulfatide mass in PGC-1α−/− mice relative to their WT littermates. We examined all the pathways that are involved in sulfatide metabolism, trafficking, and homeostasis, including proteins linked to the integration of sulfatide-containing membrane (i.e., numerous myelin proteins), enzymes connected with sulfatide biosynthesis and degradation (i.e., CST, ASA, prosaposin, and Sap B), and proteins associated with sulfatide transport (i.e., MAL and apoE). We found that the PGC-1α-mediated MAL pathway is the key factor for the reduced sulfatide mass in PGC-1α−/− mice. Intriguingly, previously published microarray analysis of the PGC-1α−/− mouse cortex revealed an increased expression of MAL gene level (Gene Expression Omnibus GDS2391) compared with WT cortex (41). This increased gene expression in combination with the lower protein expression of MAL found in the PGC-1α−/− mice strongly suggests an increased turnover rate of MAL, which sufficiently leads to the deficiency in sulfatide content. Collectively, one of the findings in the current study is the uncovering of PGC-1α-linked, MAL-mediated sulfatide homeostasis.
Sulfatide is a class of sulfated galactocerebroside (i.e., cerebroside) (Fig. 3) that mediates diverse biological processes (42–45). Sulfatides are almost exclusively synthesized by oligodendrocytes in the CNS and are present predominantly in the myelin sheath surrounding axons (42). Accumulation of sulfatides, due to a deficiency in ASA or its coenzyme (i.e., Sap B) in lysosomes, is responsible for metachromatic leukodystrophy (46, 47). Previously, MDMS-SL analysis also revealed sulfatide accumulation in the brain of subjects with Parkinson's disease (17). Mice deficient in cerebroside and sulfatide resulting from genetic ablation of ceramide galactosyltransferase generally die by 3 months of age and demonstrate multiple neurological abnormalities [e.g., abnormal axonal function, dysmyelinosis, and loss of axonal conduction velocity (48–51)]. Severe myelin developmental abnormalities, myelin sheath degeneration, and significant increases in the deterioration of nodal/paranodal structures are manifest in sulfatide-deficient mice generated by nulling CST (52). Importantly, we and others have found a substantial loss of sulfatide content in postmortem brain tissues of patients with Alzheimer's disease relative to cognitively normal individuals (16, 17, 53). Accordingly, the discovery of the link between PGC-1α and sulfatide homeostasis is of importance in two aspects. First, PGC-1α can play a major role in neuronal functions through modulation of sulfatide mass levels in addition to its role related to mitochondrial functions, indicating that PGC-1α is a connection node of metabolism in regulation of neurological functions. Second, PGC-1α could be one of the causal factors for sulfatide depletion in AD as evidenced by the fact that brain PGC-1α expression decreases in individuals with AD as a function of dementia (15).
Because of the role of PGC-1α in energy homeostasis, we also investigated the influence of changed energy expenditure (i.e., CR) on PGC-1α-induced brain lipid homeostasis. MDMS-SL analysis demonstrated the restoration of the reduced sulfatide mass in PGC-1α mouse cortex after chronic CR without alterations in content and/or composition of other lipid classes. Mechanistic studies revealed that only the decreased apoE mass levels induced by chronic CR could account for the increased mass levels of sulfatide in both PGC-1α−/− and WT mouse cortices, as our previous studies showed that apoE can modulate brain sulfatide content (19).
Intriguingly, our studies also revealed that chronic CR-induced apoE mass level changes are independent of PGC-1α, because the decreased mass levels of apoE induced by chronic CR are essentially identical in both PGC-1α−/− and WT mouse cortices (Fig. 6). We recognized an outcome that the increase in mass levels of sulfatide in PGC-1α−/− mice induced by chronic CR is much larger than that in WT mice. This is more likely due to the fact that sulfatide mass content is strictly controlled, and both higher and lower mass levels of sulfatide are linked with pathological conditions [i.e., metachromatic leukodystrophy (46, 47) and AD (16), respectively].
Our finding that chronic CR modulates cortical sulfatide mass content through reduction of the apoE metabolism pathway is of importance in two respects. First, our results suggest that sulfatide homeostasis can be maintained through PGC-1α and/or LXR/RXR nuclear receptors, indicating a connection of energy metabolism and lipid homeostasis. Second, dietary control may be a therapeutic approach for a secondary treatment of sulfatide-associated neuropathological conditions.
It has been identified that apoE expression is mediated by LXR/RXR (54), likely through ATP-binding cassette transporter ABCA1 (55, 56) and interaction with peroxisome proliferator-activated receptors (PPAR) (57–60). Chronic CR leads to downregulation and/or fine modulation of this process (61). These results are consistent with our current findings and can be readily integrated into our previously uncovered sulfatide metabolism pathway (Fig. 7).
Fig. 7.

A schematic diagram of the metabolism pathway of apoE-associated lipoproteins that mediate sulfatide transport and homeostasis. In this model, apoE expression and secretion are mediated by LXR/RXR heterodimers; apoE-associated lipoprotein particles are lipidated through ABCA1-mediated lipid translocation and released from astrocytes, whereas cross-talk between PPARs and LXR/RXR heterodimers mediates the homeostasis of those lipids as well as the expression of apoE. The released apoE-associated lipoprotein particles likely acquire sulfatides from myelin sheath through MAL, the levels of which are mediated by PGC-1α. The sulfatide-containing, apoE-associated lipoproteins can then be either metabolized through the endocytotic pathway via the LDL receptor superfamily [e.g., LDL receptor-related protein (LRP)] or transported to cerebrospinal fluid. This diagram has been modified from previously published ones (20, 38) based on the observations uncovered in the current study.
In the current study, we exploited our MDMS-SL technology platform to perform a global analysis of the lipidome in brain cortices of PGC-1α−/− mice either fed a standard diet ad libitum or subjected to chronic CR. Shotgun lipidomics revealed that significant and specific sulfatide depletion exists in PGC-1α−/− mouse cortex compared with that of its WT littermates and that chronic CR leads to restoration of the reduced sulfatide mass content in PGC-1α−/− mice. Further studies uncovered and differentiated the biochemical mechanisms underpinning the two conditions of altered sulfatide homeostasis. The former is modulated through a PGC-1α-MAL pathway, whereas the latter is under the control of LXR/RXR-apoE metabolism pathway (Fig. 7). We believe that uncovering these mechanisms are important in understanding the role of PGC-1α in neurological functions and in therapeutic treatment of deficient PGC-1α-induced alterations in sulfatide homeostasis.
Footnotes
Abbreviations:
- AD
- Alzheimer's disease
- AL
- ad libitum
- apoE
- apolipoprotein E
- ASA
- arylsulsulfatase A
- CR
- caloric restriction
- CST
- cerebroside sulfotransferase
- KO
- knockout
- LXR
- liver X receptor
- MAL
- myelin and lymphocyte protein
- MBP
- myelin basic protein
- MDMS-SL
- multidimensional mass spectrometry-based shotgun lipidomics
- MOBP
- myelin-associated oligodendrocyte basic protein
- PGC-1α
- peroxisome proliferator-activated receptor gamma coactivator-1α
- PLP
- proteolipid protein
- RXR
- retinoid X receptor
- Sap B
- saposin B
- WT
- wild-type
This work was supported by National Institute on Aging/National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 AG-31675. X.H. has a financial relationship with LipoSpectrum, LLC. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Puigserver P., Spiegelman B. M. 2003. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24: 78–90. [DOI] [PubMed] [Google Scholar]
- 2.Lai L., Leone T. C., Zechner C., Schaeffer P. J., Kelly S. M., Flanagan D. P., Medeiros D. M., Kovacs A., Kelly D. P. 2008. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 22: 1948–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finck B. N., Kelly D. P. 2006. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116: 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowe G. C., Jiang A., Arany Z. 2010. PGC-1 coactivators in cardiac development and disease. Circ. Res. 107: 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banke N. H., Wende A. R., Leone T. C., O'Donnell J. M., Abel E. D., Kelly D. P., Lewandowski E. D. 2010. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 107: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehman J. J., Barger P. M., Kovacs A., Saffitz J. E., Medeiros D. M., Kelly D. P. 2000. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jager S., Vianna C. R., Reznick R. M., et al. 2004. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 119: 121–135. [DOI] [PubMed] [Google Scholar]
- 8.Leone T. C., Lehman J. J., Finck B. N., Schaeffer P. J., Wende A. R., Boudina S., Courtois M., Wozniak D. F., Sambandam N., Bernal-Mizrachi C., et al. 2005. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3: e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehman J. J., Boudina S., Banke N. H., Sambandam N., Han X., Young D. M., Leone T. C., Gross R. W., Lewandowski E. D., Abel E. D., et al. 2008. The transcriptional coactivator PGC-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am. J. Physiol. Heart Circ. Physiol. 295: H185–H196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P. H., et al. 2005. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 1: 259–271. [DOI] [PubMed] [Google Scholar]
- 11.Handschin C., Spiegelman B. M. 2006. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 27: 728–735. [DOI] [PubMed] [Google Scholar]
- 12.Róna-Vörös K., Weydt P. 2010. The role of PGC-1alpha in the pathogenesis of neurodegenerative disorders. Curr. Drug Targets. 11: 1262–1269. [DOI] [PubMed] [Google Scholar]
- 13.St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., et al. 2006. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 127: 397–408. [DOI] [PubMed] [Google Scholar]
- 14.Ma D., Li S., Lucas E. K., Cowell R. M., Lin J. D. 2010. Neuronal inactivation of peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) protects mice from diet-induced obesity and leads to degenerative lesions. J. Biol. Chem. 285: 39087–39095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin W., Haroutunian V., Katsel P., Cardozo C. P., Ho L., Buxbaum J. D., Pasinetti G. M. 2009. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 66: 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han X., Holtzman D. M., McKeel D. W., Jr, Kelley J., Morris J. C. 2002. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J. Neurochem. 82: 809–818. [DOI] [PubMed] [Google Scholar]
- 17.Cheng H., Xu J., McKeel D. W., Jr, Han X. 2003. Specificity and potential mechanism of sulfatide deficiency in Alzheimer's disease: an electrospray ionization mass spectrometric study. Cell. Mol. Biol. 49: 809–818. [PubMed] [Google Scholar]
- 18.Han X., Fagan A. M., Cheng H., Morris J. C., Xiong C., Holtzman D. M. 2003. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann. Neurol. 54: 115–119. [DOI] [PubMed] [Google Scholar]
- 19.Han X., Cheng H., Fryer J. D., Fagan A. M., Holtzman D. M. 2003. Novel role for apolipoprotein E in the central nervous system: modulation of sulfatide content. J. Biol. Chem. 278: 8043–8051. [DOI] [PubMed] [Google Scholar]
- 20.Cheng H., Zhou Y., Holtzman D. M., Han X. 2010. Apolipoprotein E mediates sulfatide depletion in amyloid precursor protein transgenic animal models of Alzheimer's disease. Neurobiol. Aging. 31: 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han X. 2010. The pathogenic implication of abnormal interaction between apolipoprotein E isoforms, amyloid-beta peptides, and sulfatides in Alzheimer's disease. Mol. Neurobiol. 41: 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han X., Gross R. W. 2005. Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of the cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 24: 367–412. [DOI] [PubMed] [Google Scholar]
- 23.Han X., Yang K., Gross R. W. 2011. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom. Rev. Epub ahead of print. July 13, 2011; doi:10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang K., Cheng H., Gross R. W., Han X. 2009. Automated lipid identification and quantification by multi-dimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 81: 4356–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christie W. W., Han X. 2010. Lipid Analysis: Isolation, Separation, Identification and Lipidomic Analysis. 4th edition. The Oily Press, Bridgwater, England. [Google Scholar]
- 26.Kiebish M. A., Han X., Cheng H., Lunceford A., Clarke C. F., Moon H., Chuang J. H., Seyfried T. N. 2008. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 106: 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han X., Yang K., Gross R. W. 2008. Microfluidics-based electrospray ionization enhances intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: development of an automated high throughput platform for shotgun lipidomics. Rapid Commun. Mass Spectrom. 22: 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang X., Cheng H., Yang K., Gross R. W., Han X. 2007. Alkaline methanolysis of lipid extracts extends shotgun lipidomics analyses to the low-abundance regime of cellular sphingolipids. Anal. Biochem. 371: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holz A., Schaeren-Wiemers N., Schaefer C., Pott U., Colello R. J., Schwab M. E. 1996. Molecular and developmental characterization of novel cDNAs of the myelin-associated/oligodendrocytic basic protein. J. Neurosci. 16: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishibashi T., Dupree J. L., Ikenaka K., Hirahara Y., Honke K., Peles E., Popko B., Suzuki K., Nishino H., Baba H. 2002. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J. Neurosci. 22: 6507–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith R. 1992. The basic protein of CNS myelin: its structure and ligand binding. J. Neurochem. 59: 1589–1608. [DOI] [PubMed] [Google Scholar]
- 32.Sun Y., Witte D. P., Ran H., Zamzow M., Barnes S., Cheng H., Han X., Williams M. T., Skelton M. R., Vorhees C. V., et al. 2008. Neurological deficits and glycosphingolipid accumulation in saposin B deficient mice. Hum. Mol. Genet. 17: 2345–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank M., van der Haar M. E., Schaeren-Wiemers N., Schwab M. E. 1998. rMAL is a glycosphingolipid-associated protein of myelin and apical membranes of epithelial cells in kidney and stomach. J. Neurosci. 18: 4901–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frank M. 2000. MAL, a proteolipid in glycosphingolipid enriched domains: functional implications in myelin and beyond. Prog. Neurobiol. 60: 531–544. [DOI] [PubMed] [Google Scholar]
- 35.Saravanan K., Schaeren-Wiemers N., Klein D., Sandhoff R., Schwarz A., Yaghootfam A., Gieselmann V., Franken S. 2004. Specific downregulation and mistargeting of the lipid raft-associated protein MAL in a glycolipid storage disorder. Neurobiol. Dis. 16: 396–406. [DOI] [PubMed] [Google Scholar]
- 36.Schaeren-Wiemers N., Bonnet A., Erb M., Erne B., Bartsch U., Kern F., Mantei N., Sherman D., Suter U. 2004. The raft-associated protein MAL is required for maintenance of proper axon-glia interactions in the central nervous system. J. Cell Biol. 166: 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han X. 2004. The role of apolipoprotein E in lipid metabolism in the central nervous system. Cell. Mol. Life Sci. 61: 1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han X. 2007. Potential mechanisms contributing to sulfatide depletion at the earliest clinically recognizable stages of Alzheimer's disease: a tale of shotgun lipidomics. J. Neurochem. 103(Suppl. 1): 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng H., Jiang X., Han X. 2007. Alterations in lipid homeostasis of mouse dorsal root ganglia induced by apolipoprotein E deficiency: a shotgun lipidomics study. J. Neurochem. 101: 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruzdijic S., Perovic M., Mladenovic A., Milanovic D., Rakic L., Petanceska S., Kanazir S. 2005. The impact of aging, dietary restriction, and glucocorticoids on ApoE gene expression in rat brain. Ann. N. Y. Acad. Sci. 1053: 231–232. [DOI] [PubMed] [Google Scholar]
- 41.Cui L., Jeong H., Borovecki F., Parkhurst C. N., Tanese N., Krainc D. 2006. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 127: 59–69. [DOI] [PubMed] [Google Scholar]
- 42.Vos J. P., Lopes-Cardozo M., Gadella B. M. 1994. Metabolic and functional aspects of sulfogalactolipids. Biochim. Biophys. Acta. 1211: 125–149. [DOI] [PubMed] [Google Scholar]
- 43.Ishizuka I. 1997. Chemistry and functional distribution of sulfoglycolipids. Prog. Lipid Res. 36: 245–319. [DOI] [PubMed] [Google Scholar]
- 44.Ikami T., Ishida H., Kiso M. 2000. Synthesis and biological activity of glycolipids, with a focus on gangliosides and sulfatide analogs. Methods Enzymol. 311: 547–568. [DOI] [PubMed] [Google Scholar]
- 45.Marcus J., Popko B. 2002. Galactolipids are molecular determinants of myelin development and axo-glial organization. Biochim. Biophys. Acta. 1573: 406–413. [DOI] [PubMed] [Google Scholar]
- 46.von Figura K., Gieselmann V., Jaeken J. 2001. Metachromatic leukodystrophy: lysosomal disorders. The Metabolic and Molecular Bases of Inherited Diseases. Sachdev H. S., Beaudet A. L., Sly W. S., Valle D., McGraw-Hill, New York: 3695–3724. [Google Scholar]
- 47.Molander-Melin M., Pernber Z., Franken S., Gieselmann V., Mansson J. E., Fredman P. 2004. Accumulation of sulfatide in neuronal and glial cells of arylsulfatase A deficient mice. J. Neurocytol. 33: 417–427. [DOI] [PubMed] [Google Scholar]
- 48.Bosio A., Binczek E., Stoffel W. 1996. Functional breakdown of the lipid bilayer of the myelin membrane in central and peripheral nervous system by disrupted galactocerebroside synthesis. Proc. Natl. Acad. Sci. USA. 93: 13280–13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coetzee T., Fujita N., Dupree J., Shi R., Blight A., Suzuki K., Popko B. 1996. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 86: 209–219. [DOI] [PubMed] [Google Scholar]
- 50.Coetzee T., Dupree J. L., Popko B. 1998. Demyelination and altered expression of myelin-associated glycoprotein isoforms in the central nervous system of galactolipid-deficient mice. J. Neurosci. Res. 54: 613–622. [DOI] [PubMed] [Google Scholar]
- 51.Bosio A., Bussow H., Adam J., Stoffel W. 1998. Galactosphingolipids and axono-glial interaction in myelin of the central nervous system. Cell Tissue Res. 292: 199–210. [DOI] [PubMed] [Google Scholar]
- 52.Marcus J., Honigbaum S., Shroff S., Honke K., Rosenbluth J., Dupree J. L. 2006. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 53: 372–381. [DOI] [PubMed] [Google Scholar]
- 53.Colsch B., Afonso C., Fournier F., Portoukalian J., Tabet J-C., Baumann N. 2004. Characterization of the ceramide moieties of sphingoglycolipids containing sphingadienine from human brain with Alzheimer's disease by ESI-MS/MS. (Abstract in 52nd ASMS Conference, Nashville, TN, May 23–27, 2004). [DOI] [PubMed] [Google Scholar]
- 54.Liang Y., Lin S., Beyer T. P., Zhang Y., Wu X., Bales K. R., DeMattos R. B., May P. C., Li S. D., Jiang X. C., et al. 2004. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J. Neurochem. 88: 623–634. [DOI] [PubMed] [Google Scholar]
- 55.Hirsch-Reinshagen V., Maia L. F., Burgess B. L., Blain J. F., Naus K. E., McIsaac S. A., Parkinson P. F., Chan J. Y., Tansley G. H., Hayden M. R., et al. 2005. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280: 43243–43256. [DOI] [PubMed] [Google Scholar]
- 56.Koldamova R., Staufenbiel M., Lefterov I. 2005. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280: 43224–43235. [DOI] [PubMed] [Google Scholar]
- 57.Li A. C., Glass C. K. 2004. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 45: 2161–2173. [DOI] [PubMed] [Google Scholar]
- 58.Jiang Q., Heneka M., Landreth G. E. 2008. The role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in Alzheimer's disease: therapeutic implications. CNS Drugs. 22: 1–14. [DOI] [PubMed] [Google Scholar]
- 59.Chawla A., Boisvert W. A., Lee C. H., Laffitte B. A., Barak Y., Joseph S. B., Liao D., Nagy L., Edwards P. A., Curtiss L. K., et al. 2001. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 7: 161–171. [DOI] [PubMed] [Google Scholar]
- 60.Yoshikawa T., Ide T., Shimano H., Yahagi N., Amemiya-Kudo M., Matsuzaka T., Yatoh S., KitamineI T., Okazaki H., Tamura Y., et al. 2003. Cross-talk between peroxisome proliferator-activated receptor (PPAR) and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I. PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol. Endocrinol. 17: 1240–1254. [DOI] [PubMed] [Google Scholar]
- 61.Corton J. C., Apte U., Anderson S. P., Limaye P., Yoon L., Latendresse J., Dunn C., Everitt J. I., Voss K. A., Swanson C., et al. 2004. Mimetics of caloric restriction include agonists of lipid-activated nuclear receptors. J. Biol. Chem. 279: 46204–46212. [DOI] [PubMed] [Google Scholar]