

Abstract
Surface-grafted water soluble polymer brushes are being intensely investigated for preventing protein adsorption to improve biomedical device function, prevent marine fouling, and enable applications in biosensing and tissue engineering. In this contribution, we present an experimental-theoretical analysis of a peptidomimetic polymer brush system with regard to the critical brush density required for preventing protein adsorption at varying chain lengths. A mussel adhesive-inspired DOPA-Lys pentapeptide surface grafting motif enabled aqueous deposition of our peptidomimetic polypeptoid brushes over a wide range of chain densities. Critical densities of 0.88 nm−2 for a relatively short polypeptoid 10-mer to 0.42 nm−2 for a 50-mer were identified from measurements of protein adsorption. The experiments were also compared with the protein adsorption isotherms predicted by a molecular theory. Excellent agreements in terms of both the polymer brush structure and the critical chain density were obtained. Furthermore, atomic force microscopy (AFM) imaging is shown to be useful in verifying the critical brush density for preventing protein adsorption. The present co-analysis of experimental and theoretical results demonstrates the significance of characterizing the critical brush density in evaluating the performance of an anti-fouling polymer brush system. The high fidelity of the agreement between the experiments and molecular theory also indicate that the theoretical approach presented can aid in the practical design of antifouling polymer brush systems.
1. Introduction
Non-specific protein adsorption on biomedical surfaces initiates a range of adverse responses that can compromise function and, in some cases, lead to biomedical device failure. For example, thrombus formation on cardiovascular devices can be initiated by plasma protein adsorption and activation of the coagulation system,1–3 and can trigger immunological cascades that lead to chronic inflammation and fibrous encapsulation of orthopedic and other biomedical devices.4, 5 Protein adsorption also dominates the initial stages of bacteria adhesion and biofilm formation.6, 7 Further, blood-contacting devices, biosensors and tissue engineering constructs that deploy biorecognition surface groups often require an “inert” background surface free from non-specific adsorption for proper function.8, 9
Surface-grafted water soluble polymer brushes are being intensely investigated for preventing protein adsorption. In order for proteins to adsorb onto a brush coated surface, a large energetic penalty is exacted to distort the balance between volume exclusion, conformational entropy, and segment interactions of the polymer brush.10–13 Although polyethylene glycol (PEG) brushes have met with much success,14–18 PEG degradation can occur19–21 and specific protein interactions with PEG are also possible.22 Considerable attention has been focused on anti-fouling surface initiated polymerizable systems to attain high brush densities and brush lengths.23–26 Also, zwitterionic27–31 and peptidomimetic brushes32–36 have gained considerable interest. In our research, poly-N-substituted glycine “polypeptoid” brushes are being investigated for their ability to resist protein and cell fouling, resist enzymatic degradation and for their flexibility in incorporating novel chemical moieties such as antibacterial peptoid sequences.32–34
The amount of protein adsorption has conventionally been used to evaluate the performance of antifouling surface-grafted brush surfaces. The wide adoption of surface sensitive tools such as surface plasmon resonance, ellipsometry, quartz-crystal microbalance, etc., has enabled convenient and sensitive measurements of the protein adsorption amount over a (relatively short) duration of minutes to hours.37, 38 Frequently, success of a particular brush design is highlighted if the amount of proteins adsorbed falls near or below the sensitivity limits of the surface measurement.32, 39, 40 The amount of proteins adsorbed is also used as a metric to compare the performance of brushes with different polymer chemistries and physical brush parameters.24, 39–42
From a theoretical perspective, higher tethered surface chain densities and longer polymer chain lengths are recognized to be the main determinants in enhancing the performance of polymer brushes in resisting protein adsorption.10, 12, 13 At low brush densities, the time interval before protein adsorption is initiated is predicted to increase exponentially for increasing chain lengths, and at a high enough brush density and length, the adsorption kinetics can be sufficiently slow to completely prevent protein adsorption for any realistic time scale.43 Therefore quantitative analysis of protein adsorption on brush systems depends critically on information such as the density and length of the grafted chains.
Given the plethora of novel antifouling brush systems under investigation, the protein resistances of relatively few systems have been characterized with respect to the polymer chain length and grafting density.11, 14, 15, 35 Although the chain length can be controlled during polymerization of the brush polymers, the control over the brush surface density is non-trivial. Techniques for surface functionalization are frequently substrate dependent.44 In the case of “grafted-from” brush systems, the chain density is controlled through the initiator site density. Site density control via mixed self-assembled monolayers with varying ratios of an initiator-functionalized moiety and a diluent is an often-used strategy.45–47 Careful control of the initiator density by vapor phase deposition48 or other means is also possible.49–51 For “grafted-to” systems, end-functionalized PEG of pre-defined chain lengths can be coupled to specific activated substrates at varying densities depending on the concentration of the precursor PEG.14, 52 Copolymers of PEG with hydrophobic blocks can be grafted by physical adsorption at varying densities onto hydrophobic substrates,11, 16 but the hydrophobic blocks can limit the brush density and homogeneity.11 PEG or Poly-2-methyl-2-oxazoline (PMOXA) grafted to a positively charged poly-L-Lysine (PLL) backbone have also been surface immobilized onto negatively charged oxides at varying densities.15, 35, 41 However, this approach involves a trade-off between grafting stability and the brush grafting density.
In this contribution, we present an experimental-theoretical analysis of a polymer brush system with regard to the critical brush density required for preventing protein adsorption. We previously introduced a mussel adhesive-inspired pentapeptide element for surface grafting of antifouling peptidomimetic polypeptoid brushes,32 and we now demonstrate the versatility of this single element for controlling polypeptoid grafting over a range of chain densities and chain lengths. Dynamic water contact angle and spectroscopic ellipsometry were used to characterize the grafted polymer layers, and we directly measure the critical brush densities of our peptidomimetic system for preventing protein adsorption. Experimental results were also used to verify the brush structure predictions of a molecular theory.11, 13, 34 In turn, protein adsorption isotherms on the polypeptoid-modified surfaces were generated by the molecular theory and used to analyze the brush density and brush length dependences of protein adsorption. The polypeptoids were prepared by solid phase synthesis53 to obtain monodisperse chain lengths and provide strict control over composition to allow one-to-one comparison with theory. Finally, we show that atomic force microscopy (AFM) imaging of model protein adsorption54, 55 can be extremely sensitive at the levels relevant to the determination of the critical brush density. The present co-analysis of experimental and theoretical results demonstrates the significance of characterizing the critical brush density in evaluating the performance of an anti-fouling polymer brush system. The high fidelity of the match between the experimental results and theoretical predictions, together with previous quantitative predictions of protein adsorption isotherms from the theory,11, 13 also indicate that the molecular theory presented can aid in the practical design of antifouling polymer brush systems.
2. Experimental
2.1 Materials
Acetic anhydride, bromoacetic acid, diisopropyl-carbodiimide, Methoxyethylamine, trifluoroacetic acid (TFA), tri-isopropylsilane (TIPS), N-morpholinopropanesufonic acid (MOPS) buffer salt, lyophilized human fibrinogen, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffer salt, were purchased from Aldrich (Milwaukee, WI) and used without further purification. N-methylpyrrolidone, dimethyl-formamide, acetonitrile, diethyl ether, 2-propanol, and acetone were purchased from VWR (Radnor, PA). Rink amide-MBHA resin LL, Fmoc-Lys(Boc)-OH, Fmoc-DOPA(acetonide)-OH were purchased from Novabiochem/EMD Chemicals (San Diego, CA). Silicon wafers were purchased from Silicon Quest (Reno, NV). Ultrapure water (UP H2O) (resistivity = 18.3 MΩ cm; total organic content of 5 ppb) was obtained from a NANOpure Infinity System from Barnstead (Dubuque, IA).
2.2 Synthesis of peptidomimetic polymers
PMP1 polymers were synthesized as described previously32, 56 using a C S Bio 036 automated peptide synthesizer (C S Bio, Menlo Park, CA). Briefly, the C-terminal DOPA-Lys-DOPA-Lys-DOPA peptide anchor was first synthesized on a low loading rink amide resin using conventional Fmoc strategy of solid-phase peptide synthesis; the polypeptoid portion was then synthesized using a submonomer protocol.53 Acetic anhydride was used to acetylate the N-terminus of the polypeptoid chain upon removal from the reaction vessel. A portion of the resin already functionalized with the DOPA-Lys pentapeptide was taken out of the reaction vessel before the synthesis of the polypeptoid sequence to obtain the standalone control molecule. This pentapeptide was also N-acetylated with acetic anhydride. Cleavage of all the polymers from the resin and deprotection of the amino acid sidechains was accomplished by treating the resin with 95% TFA (v/v), 2.5% UP H2O, and 2.5% TIPS for 20 min. The cleaved polymer was then removed by filtering and rinsing with TFA, and the solvent was removed using a rotary evaporator; the oily product was precipitated in diethyl ether for initial purification, then dissolved in water, frozen, and lyophilized. The lyophilized products were purified by preparative reversed-phase high performance liquid chromatography (RP-HPLC) (Waters, Milford, MA) using a Vydac C18 column, and the purified fractions were frozen and lyophilized. The purity of each final product was confirmed by RP-HPLC and matrix-assisted laser desorption ionization mass spectrometry (Autoflex MALDI-TOF, Bruker Daltonics Inc., Billerica, MA).
2.3 Preparation of Brush Surfaces
3.5 nm thick TiO2 native oxide films were prepared on Si wafers from the corresponding thickness of Ti films deposited by electron beam evaporation (Edwards Auto500; 6×10−6 Torr, 0.1 nm/s). The wafers were cleaved into 11 × 12 mm2 pieces for individual measurements. Chemical shifts of the Ti2p and O1s signals measured by X-ray photoelectron spectroscopy confirmed that the films composed of pure TiO2. The substrates were cleaned in successive water, acetone and 2-propanol ultrasonic baths for 3 min each and dried under N2. Surfaces were then exposed to a reactive O2 plasma (Harrick Scientific, Ossining, NY) at 120 mTorr and 100 W for 3 min. To prepare the PMP1 coatings, the TiO2 wafer samples were individually marked and characterized by ellipsometry in air before immersion into PMP1 buffer solutions (3M NaCl buffered with 0.1M MOPS, pH = 6) maintained at 50°C. All wafers within a batch of preparation were placed face up in sealed cell culture flasks and the coating solution was constantly stirred within a heated orbital shaker. After coating, the substrates were extensively rinsed with UP H2O and dried in a stream of filtered N2. Identical wafer preparation and buffer conditions were used for the preparation of the control DOPA-Lys surfaces. The coating duration was fixed at 20 h and 0.3 mM coating solutions were used. The resulting DOPA-Lys pentapeptide films had a thickness of 7.0 nm.
2.4 Water Contact Angle
Advancing and receding water contact angles were measured using the dynamic sessile drop method (Model 190 CA, Ramé-Hart, Succasunna, NJ). The contact angle was determined by automated image analysis of live video images of the water drop on the surface in profile (DropImage, Ramé-Hart, Succasunna, NJ). An initial 1 μl drop of UP H2O was brought into contact with the sample surface. The drop volume was adjusted at 0.08 μl/step using an automated pipette. The advancing angle was taken as the largest angle measured without increasing the drop solid/liquid interfacial area. After the drop volume was increased to 4.2 μl, water was removed to measure the receding angle—the smallest possible angle without changing the drop size.
2.5 PMP1 Characterization
Film thickness measurements were performed using an M-2000 spectroscopic ellipsometer (J.A. Woollam, Lincoln, NE). Measurements were made at 68° and 76° using wavelengths from 377 to 1000 nm. The spectra were fitted with multilayer slab models in the CompleteEASE software (J.A. Woollam). Each individual TiO2 sample was measured and characterized in air by the fitted thickness and optical response (nTiO2 was generally measured to be ~ 2.3 + 0.04/λ2, with slight deviations between deposition batches and across the original 4″ Si substrates).
The bulk PMP1 optical response was measured by ellipsometry from 100 nm thick model PMP1 films spin-coated on bare 1 × 1 in2 Si wafers (15 mg/ml, using 1:1:1 H2O:methanol:isopropanol as the solvent; 1000 rpm, 20s). The sub-micron thickness of the model PMP1 layer enabled independent determination of both the film thickness and refractive index wavelength response. The spin-coated film thickness was also verified by stylus profilometer measurements. Due to the cost of peptide synthesis precursors, the model coatings employed PMP1-20 molecules with only the polypeptoid sequence and no DOPA or lys residues. The measured polypeptoid refractive index was nPMP1(λ) = 1.489 + 0.0055/λ2, which is similar to the refractive indices measured for proteins.37
The volumetric mass density of PMP1 coatings was determined by calibrated UV-vis absorption measurements of the spin coated films. The volume of a spin coated film was calculated from the measured ellipsometry thickness and the surface area measured from optical images. The polymer composing the film was dissolved in a calibrated volume of UP H2O and the optical absorption at 214 nm was measured (600E Autosampler and UV-vis spectrometer, Waters, Milford, MA) and compared with a range of PMP1 calibration solutions of known concentrations. The mass density was measured to be 150.0 ± 9 ng/cm2/nm. Since the molecular weight of the polymer and the mass of the spin-coated films were known, the average monomer volume could also be calculated to be 0.136 nm3, which is consistent with the value used in molecular dynamics (MD) simulations (0.139 nm3).57
The surface chain density of the PMP1 layers (σ) was calculated from the measured ellipsometer dry thickness and volumetric mass density:
| (1) |
where NA is Avogadro’s number, mv is the measured volumetric mass density, d is the film thickness and Mw is the molecular weight, which is precisely known for the monodisperse polymers prepared from solid phase synthesis. This approach of calculating grafting density from dry thickness was also previously demonstrated.14
2.6 Atomic Force Microscopy
AFM imaging was performed in tapping mode on a MFP3D instrument (Asylum, Santa Barbara, CA). Silicon cantilevers (60 kHz, 2 N/m; Mikromasch, Santa Barbara, CA) were used for tapping-mode measurements in air.
2.7 Protein Adsorption Experiments
Lyophilized human fibrinogen was dissolved at 3 mg/ml concentrations in 10 mM HEPES with 150 mM NaCl buffered at pH = 7.4. PMP1 coated TiO2 and bare TiO2 control samples were placed individually and vertically in separate wells in sterile polystyrene multi-well plates. Protein solution and control buffer solutions were then added to the corresponding wells and incubated at 37°C for the duration of the experiment (20 min or 18 h). No difference in results was observed for PMP1 samples temporarily pre-wetted with unloaded buffer before the addition of protein solutions. The supplied lyophilized fibrinogen also contained a significant concentration of citrate salt (~1.3 mM for 3 mg/ml proteins). This amount of citrate ions was added to the buffer in the control experiments. At the end of the adsorption experiment, the solutions in the wells were rinsed with unloaded buffer and then UP H2O or with UP H2O alone. The final adsorbed dry thickness was measured by ellipsometry. No significant difference in results was observed between the two rinsing protocols. The previously measured PMP1 layer thickness for each individual sample was subtracted from the final dry thickness to obtain the amount of protein adsorption. We have previously measured a mass density of 521 ng/cm2 on bare TiO2.34 This value was used to scale the current thickness measurements in terms of adsorbed mass densities and corresponds to a thickness of 3.7 nm (assuming nprotein = 1.45 + 0.01/λ2).
3. Theoretical Approach
The theoretical approach applied10 is a molecular theory that has been shown to provide accurate information as confirmed by experimental observations for the structure and thermodynamics of tethered polymer layers, as well as in the determination of the amount of protein adsorption on surfaces with grafted PEG.11 The agreement with experimental observations is for oligomeric chains13 as well as long polymers11 at a range of polymer-grafting densities. The basic idea is to consider conformations of the chain molecules and proteins. By minimization of the system’s free energy, the probability of each of those conformations is determined depending on the solution conditions. In this way, the theory enables the study of both structural and thermodynamic properties of the equilibrium state for the explicit molecular system studied for a range of chain densities within the mushroom and brush regimes, including all the relevant range between these two regimes (see Figure S2). We present below the main points of the theory and refer the reader to Refs. 10, 58 for discussion and technical details.
First, we consider the calculation of polymer adsorption on the surface with a binding energy. PMP1 was considered to have one large headgroup representing the DOPA-Lys pentapeptide anchor and smaller peptoid normal segments. The head group has a strong binding energy with the surface. Here the volume of the peptoid monomer, vp, within the chain is 0.139 nm3, as obtained from experiments and MD calculations. The volume of the headgroup, vh, is chosen as 0.3 nm3. The free energy per unit area of polymer layer is:
| (2) |
The first term is the conformational entropy of the peptoids, where P(α) is the probability of finding the peptoid in conformation α. The second term is the two-dimensional (surface) translational entropy of the peptoids, with representing the peptoid’s de Broglie wavelength, where h is the Planck constant and m is the peptoid’s mass. The third term is the translational (mixing) entropy of the water, with ρw(z) being the position dependent water density and vw the molecular volume of a water molecule. The fourth term is the van der Waals attractions among peptoids, with the strength of the monomer-monomer attraction being χ. The fifth term represents the repulsive intermolecular interactions due to the excluded volume interactions among different species, where π(z) is the repulsive field and φi(z) are the position dependent volume fractions of species i = p, h and w for peptoid tail, head, and water, respectively. The last term reflects the binding between the surface and the peptoid head. The specific expressions of P(α) and φi(z) are obtained by minimizing the free energy. The only unknowns are the position dependent repulsive interactions π(z), which are determined by packing constraint that ensure that at all distances from the surface, polymer or solvent must occupy the available volume. For the details on how the chain conformations are generated and the equations solved, please see Refs. 10 and 58. To determine the amount of peptoid that end-tethers to the surface, we need to determine the surface coverage that corresponds to the same chemical potential as the peptoid in the solution. The parameters χ and Fbinding are determined by comparing the amount of peptoid adsorbed with the experimental observations for PMP1-20 and then used for all the other systems and conditions. We obtained χ = −140/T and Fbinding = − 84 kBT. To quantify the thickness of the polymer layer we use the first moment of the polymer volume fraction profile by:
| (3) |
When the surface coverage of the polymer layer is varied from low to high, there is a stretching of the polymer chains due to the lateral repulsions associated with the packing. For long polymer chains this is usually described in terms of the mushroom-brush transition. However, for the typical chain lengths treated here it has been demonstrated that the transition is very smooth and therefore it is not precisely defined.59 Rather, the stretched chain conformations take, gradually, a larger fraction of the probability leading to an increase in the height with surface coverage. The theory explicitly accounts for the different conformations of the polymers and their probability, P(α), that minimizes the free energy changes in order to minimize the repulsions and lading to more stretched configuration with increasing surface coverage. These changes in the structure of the peptoid layer are shown in the Supplementary Information (Figure S2).
The next step is to determine the amount of protein adsorbed, in particular fibrinogen adsorption on the PMP1 layers that are the experimental systems under study. The protein is modeled as a molecule composed of three spheres, which represents the 3-lobed elongated structure of the actual protein (Figure 7A). Two of the spheres have the same size with the diameter d = 6.5 nm, corresponding to the terminal lobes comprising of D domains. The sphere representing the central domain in the middle has the diameter d = 5.0 nm. There are two conformations adopted by proteins, perpendicular to the surface and parallel to the surface. This model is based on previous success in predicting experimental observations.11, 13 The former has the attraction between protein and the surface: εads(1) = −275 kBT, and the latter has εads(2) = 2.5εads(1).
Figure 7.
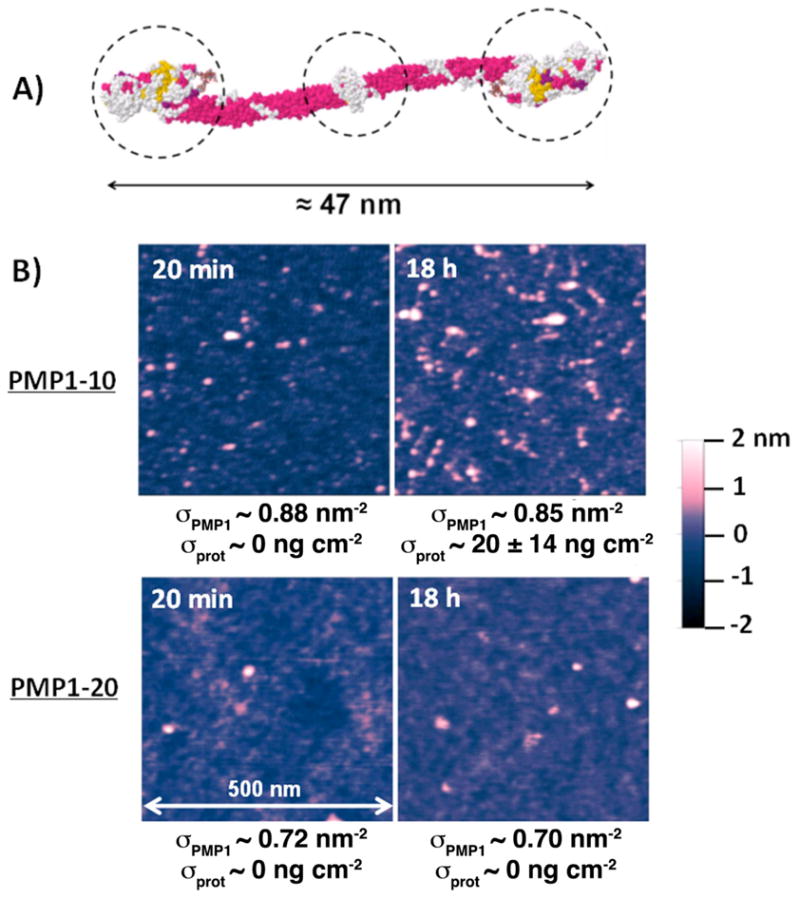
A) Space-filling molecular model of fibrinogen. The dashed lines indicate the overall 3-lobed profile of the protein. B) AFM tapping mode images of PMP1-10 and PMP1-20 brush coated TiO2 surfaces after immersion in fibrinogen solutions for the indicated durations. Individual adsorbed fibrinogen proteins were identified by their 3-lobed profiles (triple protrusions in close proximity to each other). All images show scan areas of 500×500 nm2. The brush densities of each sample and the measured fibrinogen adsorption are indicated under each panel. The samples were rinsed in ultra-pure water and then imaged in air.
The free energy of fibrinogen solution in contact with the grafted peptoid layer is given by:
| (4) |
Comparing with Eq. (2), Eq. (4) includes the additions of terms related to the protein. The third term in Eq. (4) corresponds to the conformational entropy and the adsorption energy of fibrinogen in conformation γ. The fourth term represents the translational entropy of the adsorbed fibrinogen together with the constant chemical potential requirement that ensures that the adsorbed proteins are in equilibrium with the proteins in the bulk solution. The seventh term is the effective interactions between the fibrinogen and the peptoid layer. Here χ′ is fixed as −260/T. Again, the different probabilities and density profiles are obtained through a free energy minimization. The details of the calculations can be found in references 10, 34, 58.
4. Results and Discussion
4.1 Characterization of PMP1 Brushes
Figure 1 shows the peptidomimetic polymers (PMP1-n) used to prepare the anti-fouling brush system under investigation. The polymers consist of a mussel adhesive-inspired DOPA-Lys pentapeptide surface grafting anchor coupled to a poly-N-substituted glycine “polypeptoid” sequence with methoxyethyl sidechains. Chain lengths of n = 5 to 50 peptoid units were studied. In peptoids, side chains are joined to the amide nitrogens, unlike peptides that have side chains attached to the alpha carbon. This shift is believed to be responsible for conferring resistance to enzymatic cleavage of the peptoid backbone32, 60 and is advantageous for long-term stability in biomedical applications. As in previous studies,32, 33, 56, 61 we employ the mussel-inspired DOPA-Lys pentapeptide element to graft PMP1 chains onto TiO2 model substrates for the current experiments. DOPA (3,4-dihydroxy-phenylalanine) and lysine (lys) amino acids are found enriched in the adhesive plaques that marine mussels use to attach strongly on a variety of wet surfaces. TiO2 was chosen as a model material of biomedical implants. Solid phase synthesis and HPLC were used to obtain polymers possessing monodisperse chain sequences and exact lengths for comparison with theory calculations.
Figure 1.

Chemical structures of the peptidomimetic polypeptoid (PMP1-n), where n = 5, 10, 20, 50, and the acetyl-capped dopa-Lys surface anchor segment used in control experiments.
PMP1-n were dissolved in high salt buffers for solution deposition and grafting onto TiO2 surfaces near cloud point conditions. Deposition was carried out at 50°C, <5~10°C below the cloud point measured for PMP156 to prevent indiscriminate precipitation of the polymer but sufficiently close to the solubility limit to minimize the polymer hydrodynamic volume and steric hindrance during surface grafting. To promote monolayer brush grafting, the depositions were performed at pH 6 to prevent the catechol-derived crosslinking and polymerization that is observed at higher pH and exploited in other mussel adhesive-inspired surface modification strategies.44, 62
Figure 2 shows an example of the range of brush densities that is achieved by the DOPA-Lys pentapeptide anchor, for grafting of PMP1-20 on TiO2. The maximum brush density of 0.86 nm−2 was obtained from deposition in 3 mM solution for 20 h and the minimum density of 0.22 nm−2 was obtained from deposition in 3 μM solution for 1 min. The dependence of the final (pseudo-equilibrium) density after long deposition times on the solution concentration is log-linear. The density of 0.86 nm−2 corresponds to a dry thickness of 3.0 nm. The corresponding surface separation between chains is 1.2 nm, assuming that the DOPA-Lys anchors of the grafted PMP1 chains are arranged in a hexagonal lattice. This separation is similar to the estimated footprint diameter of the DOPA-Lys pentapeptide segment.63 In contrast, a dry thickness = 7 nm was measured for a film prepared from a stand-alone DOPA-Lys pentapeptide control (Figure 1) deposited from 0.3 mM solution under analogous conditions. Given the expected antifouling properties of the PMP1 polypeptoid brush sequence, after an initial layer of PMP1 is grafted, the probability of further DOPA-Lys anchors binding to the TiO2 substrate is decreased at lower PMP1 solution concentrations and for shorter deposition durations, and lower brush densities are obtained. The maximum chain density achieved for the shortest polymer studied, PMP1-5, was 0.96 nm−2 (Figure 5). No significant increase in the brush density was achieved at even longer deposition durations for all chain lengths, which further indicates monolayer grafting.
Figure 2.

Deposited PMP1-20 chain density (left axis) and dry layer thickness (right axis) as functions of coating solution concentration and deposition duration. All results correspond to depositions from cloud point buffers at 50°C and pH 6. The trend-line is a log fit to the 20 h deposition data. The error bars indicated ±1 SD.
Figure 5.

Comparison between the deposited chain density of PMP1-n brush films (σ) using a coating solution of 0.3 mM and 20 h deposition duration, and the corresponding theoretical prediction at equilibrium. The theoretical values are the same as those shown in the legend of Figure 4. The error bars indicated ±1 SD.
The water wetting behavior of the grafted PMP1 was also characterized to provide insight into the structure of the brush layers. Figure 3 shows that both the advancing and receding water contact angles on PMP1-20 and PMP1-50 surfaces tended towards a “terminal” contact angle of 39° at higher brush densities (> 0.3 nm−2 for PMP1-20 and > 0.1 nm−2 for PMP1-50). This lack of contact angle hysteresis is consistent with the presence of highly homogeneous brush surfaces. Although the “terminal” contact angle coincided with the advancing angle of a control film surface composed of only the DOPA-Lys anchor element, the wetting behavior of the DOPA-Lys surface was vastly different, exhibiting a very low receding angle. The uniqueness of the “terminal” contact angle (39°) for both chain lengths also indicates surfaces composed of a single chemical component. In addition, the difference in wetting behavior even for brushes at lower densities was small, with no change in the receding angles and only slightly elevated advancing angles, which could indicate molecular scale inhomogeneities. The wetting behavior of PMP1-10 was also observed to be similar, with a “terminal” contact angle of ~41° (see Figure S1). However, the receding angle at low densities decreased and indicated the influence of the underlying DOPA-Lys chemistry. In summary, our observations are consistent with the successful preparation of a range of well-oriented brush layers with wetting behavior dominated by the chemical nature of peptoid monomers with methoxyethyl sidechains, with the polypeptoid sequences extending away from the substrate.
Figure 3.

The dynamic water contact angles measured on TiO2 functionalized with (A) PMP1-20 and (B) PMP1-50 brushes of varying chain densities. The contact angles of TiO2 coated with a thin film of only the DOPA-Lys mussel-mimetic anchor segment are overlaid for comparison. The DOPA-Lys surface exhibited a very small receding angle (~5°, too small to be accurately measured).
Molecular theory calculations of the equilibrium PMP1 brush structures at different chain lengths are presented in Figure 4. The profiles show high volume fraction regions adjacent to the substrate (z = 0 nm) that correspond to the DOPA-Lys surface anchor segments. The profiles then extend away from the substrate at lower densities. Interestingly, the longer polypeptoid chains show relatively extended volume density regions through much of the thickness of the brush layers. This is due to the bulky nature of the chains that induce lateral repulsions that lead to density profiles whose shape are not exactly parabolic-like as in classical brushes. For example, the polypeptoid volume density for PMP1-50 is maintained above 0.35 for brush height positions z = 1.5 ~ 7 nm, and that for PMP1-20 is maintained above ~0.4 for z = 2 ~ 4.5 nm. Moreover, the polypeptoid profile extensions for all chain lengths longer than PMP1-5 exceed more than twice the expected radii of gyration (Table 1). These conditions indicate extended chains at the indicated brush densities for PMP1-10, 20 and 50. Therefore experimental protein adsorption experiments were focused on these longer chain lengths (see section 4.2), where maximal protein repulsion is expected. At lower chain densities, a plateau region is not predicted. For example, the extended plateau configurations occurs between 0.1~0.3 nm−2 for PMP1-50 (see Figure S2), which corroborates the contact angle measurements.
Figure 4.
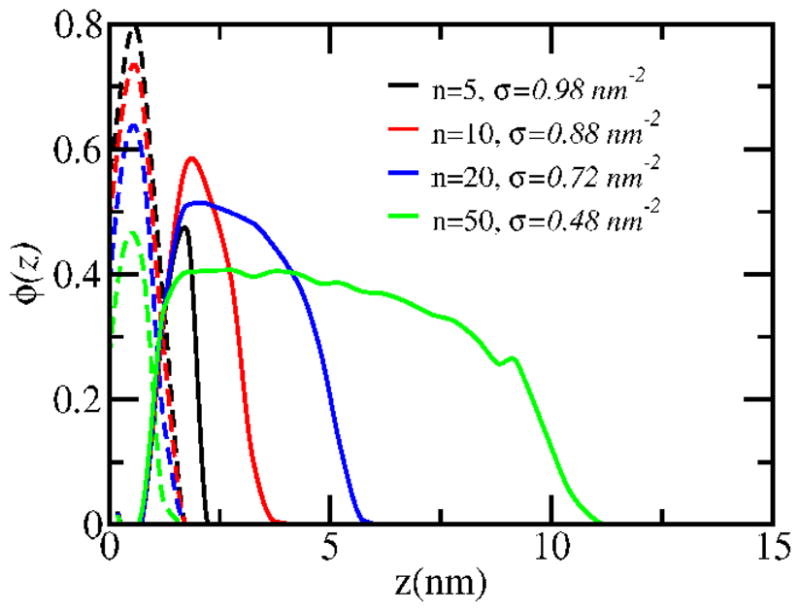
The volume fraction profiles of PMP1 at varying chain lengths. The substrate surface is indicated by z = 0 nm. The corresponding equilibrium chain densities calculated from molecular theory is indicated in the legend. The solid lines refer to the polypeptoid sequences, and the dashed lines refer to the DOPA-Lys anchor segments.
Table 1.
The molecular weight of the monodisperse PMP1 polymers at different chain lengths, and their radii of gyration (MD calculated values obtained from ref. 57).
| Polymer | Molecular weight | Radius of gyration of polypeptoid sequence |
|---|---|---|
| PMP1-5 | 900 | 0.544 nm |
| PMP1-10 | 2003 | 0.655 nm |
| PMP1-20 | 3156 | 0.869 nm |
| PMP1-50 | 6610 | 1.363 nm |
Another interesting feature of the predicted volume fraction profiles, for polypeptoid chain lengths > 5, relate to the lower maximal densities and thicker brush layers as the chain length increases. For the case of short chain length, the loss of conformational entropy during the formation of the polymer layer is smaller than that of the long chain case. Therefore, a larger number of short polymers can be adsorbed under the same strong DOPA-Lys surface binding, which leads to the higher maximum volume fraction. On the other hand, the thickness of polymer brush usually is expected to be proportional to the chain length, causing the thicker brush layers in the case of longer chains.
The accuracy of the molecular theory for PMP1 brushes was verified by comparing the experimental measurements of the grafted brush density with the predicted equilibrium densities. The calculated densities correspond to the volume fraction profiles shown in Figure 4 and are in excellent agreement with the grafted densities measured for 20 h depositions from 0.3 mM PMP1 solutions (Figure 5). However, it is noted that brush densities slightly higher than those predicted for equilibrium conditions have also been obtained in experiments (e.g. PMP1-20 brush density at 0.86 nm−2; Figure 2). This could indicate the possibility of a different regime of DOPA-Lys anchor binding on the TiO2 surface, under which partial overlap of the DOPA-Lys pentapeptide elements promoted by high solution concentrations could accommodate chain packing higher than the ideal equilibrium density.
4.2 Protein Adsorption Studies
The experimentally measured mass densities of fibrinogen adsorbed on the PMP1 modified surfaces at varying grafting densities and chain lengths are shown in Figure 6, and are compared with the theoretically predicted isotherms. Fibrinogen was used as a model protein because of the significant biological roles it plays in platelet and monocyte adhesion, in the coagulation pathway and in inflammation.64, 65 In the experimental data, one can clearly observe that the amount of protein adsorption decreased monotonically with increasing peptoid chain density (Figure 6A). Furthermore, parallel trends can be delineated for different chain lengths, with the prevention of protein adsorption occurring at higher chain densities for shorter chain lengths. Importantly, it was also observed that above a certain “critical” chain density particular for each chain length, the amount of protein adsorption falls to a very low level, close to the uncertainties of the current measurements. An adsorbed fibrinogen level of 10 ng/cm2 has been identified as sufficient for initiating platelet adhesion.64, 66 Based on this threshold level of fibrinogen adsorption, the critical chain densities are identified in Figure 6B. Results for PMP1-20 most clearly demonstrates the existence of this chain length specific value. It is seen that above ~0.54 nm−2, the amount of adsorbed fibrinogen was, within error, consistently below the threshold adsorption. Incidentally, the experimental data in Figure 6A consist of results from both 20 min and 18 h adsorption experiments. After extended immersion of the brush coated substrates in protein solution, we have measured from control samples consistent, albeit slight, decreases in the PMP1 chain densities. The chain densities plotted in Figure 6 correspond to these final densities. However, no obvious difference in the trends of protein adsorption vs. chain density was observed between the 20 min and 18 h data sets (see Figure S3 for results plotted separately).
Figure 6.

A) Comparison between the adsorbed fibrinogen mass densities obtained experimentally by ellipsometry (symbols) and from molecular theory (line traces). The error bars indicated ±1 SD. B) Comparison between the theoretical and experimental critical chain densities identified from (A). The error bars indicate a ±10% uncertainty in the experimental results. The corresponding adsorption isotherm for PMP1-30 is shown in Figure S4.
Figure 6 also illustrates the excellent overall agreement between the theoretical predictions and the experimental data. In the model, resistance to protein adsorption results from excluded volume repulsions and the reduction in the number of available chain conformations as proteins insert into the tethered peptoid layer. Therefore it is expected that, at each PMP1 chain length, the amount of protein adsorption decreases with increasing chain density. Furthermore, in agreement with experiments, the theory predicts that at each particular chain length, protein adsorption abruptly becomes very small above certain critical chain densities (Figure 6B). Fortuitously, these chain densities are below the maximum densities that can be theoretically achieved (and were obtained in practice; Figure 5). Significantly, the theory retraces the experimentally measured trends for the measured protein adsorption, and predicted similar critical chain densities. Actually, the whole adsorption isotherms are very well predicted by the theory.
The chain length dependence of the critical chain density is an important experimental result and a important theoretical prediction. As supported by the experiments, the critical chain density decreases sharply at shorter chain lengths but this chain length dependence grows weaker for longer chain lengths (Figure 6). In fact, the predicted adsorption isotherm for PMP1-30 approaches that for PMP1-50 (Figure 6B and Figure S4). This behavior is also in agreement with previous investigations of PEG brushes, which predicted that the position of the adsorption isotherm becomes independent of the polymer chain length once the brush thickness is larger than the size of the protein10, 58 (although kinetic effects may still be strongly dependent on polymer chain length)43. The largest domain of fibrinogen is approximately 6.5 nm in width (Figure 7A). As surmised from Figure 4, the thickness of the PMP1-50 brush at the critical chain density of 0.42 nm−2 is significantly larger than this domain size. Therefore we expect that the adsorption isotherm is only weakly dependent on chain length for polypeptoid brushes with more than ~50 monomer length, as suggested by the trend shown in Figure 6B.
The values of certain molecular parameters of the polymer molecules can significantly influence the theoretical predictions of the chain density dependence of protein adsorption. The current theoretical results use a more refined model for the peptoids than previous calculations34 by using a value for the PMP1 monomer volume obtained from detailed fully atomistic MD simulations57 (see Theoretical Approach). In addition, the predicted critical chain densities depend strongly on the size of the protein and on the effective interaction between protein and polymer. For larger proteins, steric repulsions are strengthened and the critical brush density is decreased for any given chain length. As for the effective protein-polymer attraction, the reduction in protein adsorption with increasing chain densities would be weakened and the critical brush density increased if this interaction were strengthened. Protein-polymer interactions are complex and the strength of this interaction was estimated from the match between measured data and the theory. The molecular parameters used in the present study on peptidomimetic polypeptoids are different from those on PEG11, 13 due to the different characteristics of the two molecular systems. The difference in the measured and predicted adsorption isotherms in these two different polymer layers is a manifestation of the dependence of protein adsorption on the molecular characteristics of the polymer layer. Another unknown parameter is the protein-substrate interaction strength. We have obtained it by fitting the theoretical predictions with the amount of fibrinogen adsorption measured on the uncoated TiO2. Our experimentally measured value of 521 ± 60 ng/cm2 is consistent with values measured by other groups15 and also with that predicted by a random sequential model of monolayer fibrinogen adsorption.67 In case of higher substrate interactions, to prevent protein adsorption, higher brush densities would be required to induce the requisite repulsive steric interactions. In such a case, the predicted trends for different chain lengths may be spaced farther apart and generally shifted towards higher values on the chain density axis.
In the current protein adsorption experiments, the aforementioned threshold protein adsorption was 10 ng/cm2, which was also the level of uncertainty in the ex situ ellipsometry measurements. Although this is much lower than the monolayer fibrinogen coverage on TiO2 ~521 ng/cm2, it corresponds to a relatively high number density of ~200 fibrinogen/μm2 (the molecular mass of fibrinogen is 340 kDa). The 3-lobed structure of the relatively large fibrinogen molecule (Figure 7A) can be used in AFM imaging to positively identify the adsorbed fibrinogen,55, 68 and the technique was used to corroborate the association between the measured critical chain density and protein adsorption.
Figure 7B shows example AFM images of PMP1-10 and PMP1-20 after protein adsorption experiments. It is seen that a small decrease in the chain density of the PMP1-10 surface from 0.88 nm−2 to 0.85 nm−2, resulting from 18 h as opposed to 20 min immersion in the protein buffer, led to a substantial increase in the number of fibrinogen adsorbed. Although the increase in fibrinogen adsorption could be interpreted as a kinetic effect, in light of the behavior of higher protein adsorption at lower chain densities identified in Figure 6, it is plausible that the dramatic difference in the number of proteins adsorbed could be attributed to a cross-over at the critical chain density (Figure 6B). The measured protein surface coverage (θ ) after 18 h adsorption was 0.039 ± 0.026, corresponding to a number density of 360 ± 240 fibrinogen/μm2 (approximately 40 fibrinogen molecules are visible in the 0.5 × 0.5 μm2 panel). This level of adsorption is plausibly more than sufficient to elicit cellular recognition. In contrast, little observable fibrinogen was visible on the PMP1-20 surfaces with chain densities from 0.70 (18 h) to 0.72 nm−2 (20 min), high above the theoretical threshold of 0.57 nm−2. The high resolution of AFM imaging can conceivably allow detection levels of ~1 fibrinogen/μm2, corresponding to ~0.06 ng/cm2. Although AFM measurements are more time-consuming than the optical techniques generally used for measuring protein adsorption, the high sensitivity and specificity to fibrinogen detection may lend the technique to the verification of the critical chain density of protein resistant polymer brushes.
5. Conclusions
Peptidomimetic polypeptoid polymer brushes were demonstrated to form an excellent experimental platform to quantitatively study the dependence of protein adsorption on the polymer surface chain density. The critical chain densities required to prevent protein adsorption for brushes with different chain lengths were measured. They ranged from 0.88 nm−2 for the relatively short PMP1-10 to 0.42 nm−2 for PMP1-50. Importantly, a single mussel adhesive-inspired DOPA-Lys pentapeptide surface grafting motif enabled the convenient aqueous deposition of the PMP1 brushes over a wide range of chain densities.
A predictive molecular theory can be an invaluable component in the design of non-fouling polymer systems. We have shown that our theory correctly calculates the surface chain densities of the polypeptoid polymer brushes obtained experimentally for a range of chain lengths, and the predicted brush structures are consistent with the experimental water contact angle and ellipsometer results. The theory further predicted the experimentally measured adsorption isotherms and in particular the critical brush densities required to prevent protein adsorption. Our experimental-theoretical analysis demonstrates how the resistance to protein adsorption of a polymer brush critically depends on the grafted chain density. Furthermore, AFM measurements show that even “low” mass densities of adsorbed proteins near the detection limit of conventional optical measurements of protein adsorption may correspond to relatively high protein number densities. In this respect, measurements of the critical brush density, which can be identified by the substantial increase in protein adsorption towards sub-critical brush densities, does not depend on single protein adsorption measurements that challenge the sensitivity limits of instrumentation. Practical protein resistant polymer brushes can then be designed with a brush density above the critical value to an extent commensurate with the importance of the application. Finally, the ability of the theoretical approach to a priori identify the chain length dependent critical chain densities of a polymer layer, for different polymer systems, could significantly guide the design choices concerning grafting density, molecular weight and monomer chemistry for achieving the desired anti-fouling performance.
Supplementary Material
Acknowledgments
This research was supported by grant number EB005772 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) at the National Institutes of Health (NIH). K.H.A.L. acknowledges support by fellowship number HL104966 from the National Heart, Blood and Lung Institute (NHBLI) at the NIH. C.R. acknowledges support by the National Natural Science Foundation of China under Grant 10804045. The authors thank Erica Pollock and Kevin Anderson for technical support. XPS and stylus profilometer measurements were performed at KeckII/NUANCE at Northwestern University. MALDI-MS was performed at IMSERC at Northwestern University.
Footnotes
Supporting Information Available. Detailed water contact angle data, theoretical protein adsorption isotherms and brush profiles at different chain densities and protein adsorption results after different experimental durations are available as Supporting Informaiton, available free of charge at http://pubs.acs.org/langmuir.
Contributor Information
K.H. Aaron Lau, Email: aaronlau@northwestern.edu.
Chunlai Ren, Email: chunlair@nju.edu.cn.
Sung Hyun Park, Email: parksh@northwestern.edu.
Igal Szleifer, Email: igalsz@northwestern.edu.
Phillip B. Messersmith, Email: philm@northwestern.edu.
References
- 1.Siddiqui RF, Abraham JR, Butany J. Bioprosthetic heart valves: modes of failure. Histopathology. 2009;55(2):135–144. doi: 10.1111/j.1365-2559.2008.03190.x. [DOI] [PubMed] [Google Scholar]
- 2.Zilla P, Bezuidenhout D, Human P. Prosthetic vascular grafts: Wrong models, wrong questions and no healing. Biomaterials. 2007;28(34):5009–5027. doi: 10.1016/j.biomaterials.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 3.Venkatraman S, Boey F, Lao LL. Implanted cardiovascular polymers: Natural, synthetic and bio-inspired. Progress in Polymer Science. 2008;33(9):853–874. [Google Scholar]
- 4.Ratner B. A Perspective on Titanium Biocompatibility. In: Brunette DM, Tengvall P, Thomsen P, Textor M, editors. Titanium in Medicine: Material Science, Surface Science, Engineering, Biological Responses and Medical Applications. Springer; Berlin amd Heidelberg: 2000. pp. 1–12. [Google Scholar]
- 5.Anderson JM. Biological Responses to Materials. Annual Review of Materials Research. 2001;31(1):81–110. [Google Scholar]
- 6.An YH, Friedman RJ. Concise review of mechanisms of bacterial adhesion to biomaterial surfaces. Journal of Biomedical Materials Research. 1998;43(3):338–348. doi: 10.1002/(sici)1097-4636(199823)43:3<338::aid-jbm16>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 7.Kuijer R, Jansen EJP, Emans PJ, Bulstra SK, Riesle J, Pieper J, Grainger DW, Busscher HJ. Assessing infection risk in implanted tissue-engineered devices. Biomaterials. 2007;28(34):5148–5154. doi: 10.1016/j.biomaterials.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Rastogi A, Nad S, Tanaka M, Mota ND, Tague M, Baird BA, Abruña HcD, Ober CK. Preventing Nonspecific Adsorption on Polymer Brush Covered Gold Electrodes Using a Modified ATRP Initiator. Biomacromolecules. 2009;10(10):2750–2758. doi: 10.1021/bm900564t. [DOI] [PubMed] [Google Scholar]
- 9.Ostuni E, Chapman RG, Holmlin RE, Takayama S, Whitesides GM. A Survey of Structure; Property Relationships of Surfaces that Resist the Adsorption of Protein. Langmuir. 2001;17(18):5605–5620. doi: 10.1021/la0015258. [DOI] [PubMed] [Google Scholar]
- 10.Szleifer I. Protein Adsorption on Surfaces with Grafted Polymers: A Theoretical Approach. Biophys J. 1997;72(2_Pt_1):595–612. doi: 10.1016/s0006-3495(97)78698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McPherson T, Kidane A, Szleifer I, Park K. Prevention of Protein Adsorption by Tethered Poly(ethylene oxide) Layers: Experiments and Single-Chain Mean-Field Analysis. Langmuir. 1998;14(1):176–186. [Google Scholar]
- 12.Halperin A. Polymer Brushes that Resist Adsorption of Model Proteins: Design Parameters. Langmuir. 1999;15(7):2525–2533. [Google Scholar]
- 13.Satulovsky J, Carignano MA, Szleifer I. Kinetic and thermodynamic control of protein adsorption. Proceedings of the National Academy of Sciences. 2000;97(16):9037–9041. doi: 10.1073/pnas.150236197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sofia SJ, Premnath V, Merrill EW. Poly(ethylene oxide) Grafted to Silicon Surfaces: Grafting Density and Protein Adsorption. Macromolecules. 1998;31(15):5059–5070. doi: 10.1021/ma971016l. [DOI] [PubMed] [Google Scholar]
- 15.Kenausis GL, Voros J, Elbert DL, Huang N, Hofer R, Ruiz-Taylor L, Textor M, Hubbell JA, Spencer ND. Poly(l-lysine)-g-Poly(ethylene glycol) Layers on Metal Oxide Surfaces: Attachment Mechanism and Effects of Polymer Architecture on Resistance to Protein Adsorption. The Journal of Physical Chemistry B. 2000;104(14):3298–3309. [Google Scholar]
- 16.Bosker WTE, Iakovlev PA, Norde W, Cohen Stuart MA. BSA adsorption on bimodal PEO brushes. Journal of Colloid and Interface Science. 2005;286(2):496–503. doi: 10.1016/j.jcis.2005.01.091. [DOI] [PubMed] [Google Scholar]
- 17.Zou YQ, Rossi NAA, Kizhakkedathu JN, Brooks DE. Barrier Capacity of Hydrophilic Polymer Brushes To Prevent Hydrophobic Interactions: Effect of Graft Density and Hydrophilicity. Macromolecules. 2009;42(13):4817–4828. [Google Scholar]
- 18.Dalsin JL, Hu B-H, Lee BP, Messersmith PB. Mussel Adhesive Protein Mimetic Polymers for the Preparation of Nonfouling Surfaces. Journal of the American Chemical Society. 2003;125(14):4253–4258. doi: 10.1021/ja0284963. [DOI] [PubMed] [Google Scholar]
- 19.Han S, Kim C, Kwon D. Thermal/oxidative degradation and stabilization of polyethylene glycol. Polymer. 1997;38(2):317–323. [Google Scholar]
- 20.Kawai F. Microbial degradation of polyethers. Appl Microbiol Biotechnol. 2002;58(1):30–38. doi: 10.1007/s00253-001-0850-2. [DOI] [PubMed] [Google Scholar]
- 21.Li LY, Chen SF, Jiang SY. Protein interactions with oligo(ethylene glycol) (OEG) self-assembled monolayers: OEG stability, surface packing density and protein adsorption. J Biomater Sci-Polym Ed. 2007;18(11):1415–1427. doi: 10.1163/156856207782246795. [DOI] [PubMed] [Google Scholar]
- 22.Halperin A, Fragneto G, Schollier A, Sferrazza M. Primary versus Ternary Adsorption of Proteins onto PEG Brushes. Langmuir. 2007;23(21):10603–10617. doi: 10.1021/la701007j. [DOI] [PubMed] [Google Scholar]
- 23.Senaratne W, Andruzzi L, Ober CK. Self-Assembled Monolayers and Polymer Brushes in Biotechnology: Current Applications and Future Perspectives. Biomacromolecules. 2005;6(5):2427–2448. doi: 10.1021/bm050180a. [DOI] [PubMed] [Google Scholar]
- 24.Mei Y, Gerecht S, Taylor M, Urquhart AJ, Bogatyrev SR, Cho S-W, Davies MC, Alexander MR, Langer RS, Anderson DG. Mapping the Interactions among Biomaterials, Adsorbed Proteins, and Human Embryonic Stem Cells. Advanced Materials. 2009;21(27):2781–2786. doi: 10.1002/adma.200803184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fristrup CJ, Jankova K, Hvilsted S. Surface-initiated atom transfer radical polymerization-a technique to develop biofunctional coatings. Soft Matter. 2009;5(23):4623–4634. [Google Scholar]
- 26.Banerjee I, Pangule RC, Kane RS. Antifouling Coatings: Recent Developments in the Design of Surfaces That Prevent Fouling by Proteins, Bacteria, and Marine Organisms. Advanced Materials. 2011;23(6):690–718. doi: 10.1002/adma.201001215. [DOI] [PubMed] [Google Scholar]
- 27.Nakaya T, Li Y-J. Phospholipid polymers. Progress in Polymer Science. 1999;24(1):143–181. [Google Scholar]
- 28.Lewis AL, Hughes PD, Kirkwood LC, Leppard SW, Redman RP, Tolhurst LA, Stratford PW. Synthesis and characterisation of phosphorylcholine-based polymers useful for coating blood filtration devices. Biomaterials. 2000;21(18):1847–1859. doi: 10.1016/s0142-9612(00)00055-7. [DOI] [PubMed] [Google Scholar]
- 29.West SL, Salvage JP, Lobb EJ, Armes SP, Billingham NC, Lewis AL, Hanlon GW, Lloyd AW. The biocompatibility of crosslinkable copolymer coatings containing sulfobetaines and phosphobetaines. Biomaterials. 2004;25(7–8):1195–1204. doi: 10.1016/j.biomaterials.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Zhang M, Chen S, Horbett TA, Ratner BD, Jiang S. Blood compatibility of surfaces with superlow protein adsorption. Biomaterials. 2008;29(32):4285–4291. doi: 10.1016/j.biomaterials.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 31.Ladd J, Zhang Z, Chen S, Hower JC, Jiang S. Zwitterionic Polymers Exhibiting High Resistance to Nonspecific Protein Adsorption from Human Serum and Plasma. Biomacromolecules. 2008;9(5):1357–1361. doi: 10.1021/bm701301s. [DOI] [PubMed] [Google Scholar]
- 32.Statz AR, Meagher RJ, Barron AE, Messersmith PB. New Peptidomimetic Polymers for Antifouling Surfaces. J Am Chem Soc. 2005;127(22):7972–7973. doi: 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- 33.Statz AR, Park JP, Chongsiriwatana NP, Barron AE, Messersmith PB. Surface-immobilised antimicrobial peptoids. Biofouling. 2008;24(6):439–448. doi: 10.1080/08927010802331829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Statz AR. Experimental and theoretical investigation of chain length and surface coverage on fouling of surface grafted polypeptoids. Biointerphases. 2009;4(2):FA22. doi: 10.1116/1.3115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pidhatika B, Möller J, Benetti EM, Konradi R, Rakhmatullina E, Muehlebach A, Zimmermann R, Werner C, Vogel V, Textor M. The role of the interplay between polymer architecture and bacterial surface properties on the microbial adhesion to polyoxazoline-based ultrathin films. Biomaterials. 2010;31(36):9462–9472. doi: 10.1016/j.biomaterials.2010.08.033. [DOI] [PubMed] [Google Scholar]
- 36.Lin S, Zhang B, Skoumal MJ, Ramunno B, Li X, Wesdemiotis C, Liu L, Jia L. Antifouling Poly(beta-peptoid)s. Biomacromolecules. 2011;12(7):2573–2582. doi: 10.1021/bm200368p. [DOI] [PubMed] [Google Scholar]
- 37.Vörös J. The Density and Refractive Index of Adsorbing Protein Layers. Biophysical journal. 2004;87(1):553–561. doi: 10.1529/biophysj.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell CT, Kim G. SPR microscopy and its applications to high-throughput analyses of biomolecular binding events and their kinetics. Biomaterials. 2007;28(15):2380–2392. doi: 10.1016/j.biomaterials.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 39.Mi L, Bernards MT, Cheng G, Yu Q, Jiang S. pH responsive properties of non-fouling mixed-charge polymer brushes based on quaternary amine and carboxylic acid monomers. Biomaterials. 2010;31(10):2919–2925. doi: 10.1016/j.biomaterials.2009.12.038. [DOI] [PubMed] [Google Scholar]
- 40.Yang W, Xue H, Li W, Zhang J, Jiang S. Pursuing “Zero” Protein Adsorption of Poly(carboxybetaine) from Undiluted Blood Serum and Plasma. Langmuir. 2009;25(19):11911–11916. doi: 10.1021/la9015788. [DOI] [PubMed] [Google Scholar]
- 41.Pasche S, Voros J, Griesser HJ, Spencer ND, Textor M. Effects of Ionic Strength and Surface Charge on Protein Adsorption at PEGylated Surfaces. The Journal of Physical Chemistry B. 2005;109(37):17545–17552. doi: 10.1021/jp050431+. [DOI] [PubMed] [Google Scholar]
- 42.Chang Y, Shu S-H, Shih Y-J, Chu C-W, Ruaan R-C, Chen W-Y. Hemocompatible Mixed-Charge Copolymer Brushes of Pseudozwitterionic Surfaces Resistant to Nonspecific Plasma Protein Fouling. Langmuir. 2009;26(5):3522–3530. doi: 10.1021/la903172j. [DOI] [PubMed] [Google Scholar]
- 43.Fang F, Satulovsky J, Szleifer I. Kinetics of Protein Adsorption and Desorption on Surfaces with Grafted Polymers. Biophysical journal. 2005;89(3):1516–1533. doi: 10.1529/biophysj.104.055079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-Inspired Surface Chemistry for Multifunctional Coatings. Science. 2007;318(5849):426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma HW, Wells M, Beebe TP, Chilkoti A. Surface-initiated atom transfer radical polymerization of oligo(ethylene glycol) methyl methacrylate from a mixed self-assembled monolayer on gold. Adv Funct Mater. 2006;16(5):640–648. [Google Scholar]
- 46.Bao ZY, Bruening ML, Baker GL. Control of the density of polymer brushes prepared by surface-initiated atom transfer radical polymerization. Macromolecules. 2006;39(16):5251–5258. [Google Scholar]
- 47.Nagase K, Kobayashi J, Kikuchi AI, Akiyama Y, Kanazawa H, Okano T. Effects of graft densities and chain lengths on separation of bioactive compounds by nanolayered thermoresponsive polymer brush surfaces. Langmuir. 2008;24(2):511–517. doi: 10.1021/la701839s. [DOI] [PubMed] [Google Scholar]
- 48.Wu T, Efimenko K, Vlcek P, Subr V, Genzer J. Formation and properties of anchored polymers with a gradual variation of grafting densities on flat substrates. Macromolecules. 2003;36(7):2448–2453. [Google Scholar]
- 49.Liu Y, Klep V, Zdyrko B, Luzinov I. Polymer grafting via ATRP initiated from macroinitiator synthesized on surface. Langmuir. 2004;20(16):6710–6718. doi: 10.1021/la049465j. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto S, Ejaz M, Tsujii Y, Fukuda T. Surface interaction forces of well-defined, high-density polymer brushes studied by atomic force microscopy. 2. Effect of graft density. Macromolecules. 2000;33(15):5608–5612. [Google Scholar]
- 51.Kizhakkedathu JN, Janzen J, Le Y, Kainthan RK, Brooks DE. Poly(oligo(ethylene glycol)acrylamide) Brushes by Surface Initiated Polymerization: Effect of Macromonomer Chain Length on Brush Growth and Protein Adsorption from Blood Plasma. Langmuir. 2009;25(6):3794–3801. doi: 10.1021/la803690q. [DOI] [PubMed] [Google Scholar]
- 52.Kingshott P, Thissen H, Griesser HJ. Effects of cloud-point grafting, chain length, and density of PEG layers on competitive adsorption of ocular proteins. Biomaterials. 2002;23(9):2043–2056. doi: 10.1016/s0142-9612(01)00334-9. [DOI] [PubMed] [Google Scholar]
- 53.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. Journal of the American Chemical Society. 1992;114(26):10646–10647. [Google Scholar]
- 54.Soman P, Siedlecki CA. Effects of Protein Solution Composition on the Time-Dependent Functional Activity of Fibrinogen on Surfaces. Langmuir. 2011;27(17):10814–10819. doi: 10.1021/la201111r. [DOI] [PubMed] [Google Scholar]
- 55.Toscano A, Santore MM. Fibrinogen Adsorption on Three Silica-Based Surfaces: Conformation and Kinetics. Langmuir. 2006;22(6):2588–2597. doi: 10.1021/la051641g. [DOI] [PubMed] [Google Scholar]
- 56.Statz AR. Surface Modification with Antifouling Polymers Composed of Polypeptoids and Mussel Adhesive-Inspired Peptides. Northwestern University; Evanston: 2009. [Google Scholar]
- 57.Park SH, Szleifer I. Structural and Dynamical Characteristics of Peptoid Oligomers with Achiral Aliphatic Side Chains Studied by Molecular Dynamics Simulation. The Journal of Physical Chemistry B. 2011;115(37):10967–10975. doi: 10.1021/jp2025957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang, Szleifer I. Effect of Molecular Structure on the Adsorption of Protein on Surfaces with Grafted Polymers. Langmuir. 2002;18(14):5497–5510. [Google Scholar]
- 59.Szleifer I. Statistical thermodynamics of polymers near surfaces. Current Opinion in Colloid and Interface Science. 1996;1(3):416–423. [Google Scholar]
- 60.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev Res. 1995;35(1):20–32. [Google Scholar]
- 61.Statz AR, Barron AE, Messersmith PB. Protein, cell and bacterial fouling resistance of polypeptoid-modified surfaces: effect of side-chain chemistry. Soft Matter. 2008;4(1):131–139. doi: 10.1039/B711944E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ham HO, Liu Z, Lau KHA, Lee H, Messersmith PB. Facile DNA Immobilization on Surfaces through a Catecholamine Polymer. Angewandte Chemie International Edition. 2011;50(3):732–736. doi: 10.1002/anie.201005001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The effective diameter of the DOPA-lys pentapeptide segment was estimated from the aggregate geometric volumes of the 5 amino acids = 0.9 nm3.
- 64.Kwak D, Wu Y, Horbett TA. Fibrinogen and von Willebrand’s factor adsorption are both required for platelet adhesion from sheared suspensions to polyethylene preadsorbed with blood plasma. J Biomed Mater Res A. 2005;74A(1):69–83. doi: 10.1002/jbm.a.30365. [DOI] [PubMed] [Google Scholar]
- 65.Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25(26):5681–5703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 66.Shen M, Wagner MS, Castner DG, Ratner BD, Horbett TA. Multivariate Surface Analysis of Plasma-Deposited Tetraglyme for Reduction of Protein Adsorption and Monocyte Adhesion. Langmuir. 2003;19(5):1692–1699. [Google Scholar]
- 67.Adamczyk Z, Barbasz J, Ciesla M. Mechanisms of Fibrinogen Adsorption at Solid Substrates. Langmuir. 2011;27(11):6868–6878. doi: 10.1021/la200798d. [DOI] [PubMed] [Google Scholar]
- 68.Wasilewska M, Adamczyk Z. Fibrinogen Adsorption on Mica Studied by AFM and in Situ Streaming Potential Measurements. Langmuir. 2011;27(2):686–696. doi: 10.1021/la102931a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.