

Abstract
Glioblastoma, the most aggressive primary brain tumor, thrives in a microenvironment of relative immunosuppression within the relatively immune-privileged central nervous system. Despite treatments with surgery, radiation therapy, and chemotherapy, prognosis remains poor. The recent success of immunotherapy in the treatment of other cancers has renewed interest in vaccine therapy for the treatment of gliomas. In this article, we outline various immunotherapeutic strategies, review recent clinical trials data, and discuss the future of vaccine therapy for glioblastoma.
Keywords: Glioblastoma, vaccine, immunotherapy, high-grade glioma
Introduction
Glioblastoma, the most frequent and malignant primary brain tumor, stands apart from other neoplasms by its biology and location within the central nervous system (CNS). In spite of aggressive multimodal treatment including surgical resection, radiation therapy, and cytotoxic chemotherapy, the disease remains incurable with a median survival under fifteen months, and a 2 year survival of 26.5% 1. The failure of conventional oncologic treatment to selectively eradicate glioblastoma cells has prompted investigators to look for new and more targeted therapeutic options, as well as for improved prognostic biomarkers that will help us better understand the variation of outcomes.
Immunotherapy offers a different mechanistic approach from chemotherapy, targeted therapy, radiation and surgery, and its recent success in the treatment of other cancers has fueled a resurgence of interest in this approach. Currently there are 17 FDA approved immunologic products used in treatment of human malignancies 2. Most of the available immunologic treatments are antibody based therapies; however, in 2011 the first cell-based therapy was approved for the treatment of prostate cancer, opening the door for other immunologic approaches. In the case of gliomas, there are several tumor vaccine strategies that have been explored in clinical trials. Although the initial results are encouraging, the small non-controlled studies preclude definitive proof of improvement in survival.
In this article, we will give a brief overview of the immune system and its relation to the nervous system and cancer, as it provides the rationale for the use of immunotherapy in brain tumors. We will discuss promising immune based therapies, focusing on the outcomes and limitations of ongoing clinical trials that employ vaccines to treat patients with glioblastoma. Finally, we focus on strategies that could refine these vaccine approaches to enhance the potential benefits and become part of the conventional armamentarium to fight glioblastoma.
Overview of the immune system
The primary role of the immune system is to discriminate between self- and non-self in order to recognize foreign invaders and defend against them. The immune system can be divided into two main branches: the innate and the adaptive immune systems 3–4. The innate immune system, the first line of defense, recognizes pathogen-associated molecular patterns, or PAMPs, via engagement of Toll-like receptors (TLRs) and other pattern recognition receptors (PRRs) which are present before an infection takes hold. The innate immune system consists of macrophages, monocytes, neutrophils, natural killer (NK) cells, basophils, eosinophils, and complement.
The adaptive immune system, by contrast, must be activated by antigens. The adaptive immune system consists of T and B lymphocytes, and antigen presenting cells. T-cells, so-named because they mature in the thymus, fall into 2 large subcategories of cytotoxic T cells and helper T cells. The cytotoxic T cells express CD8 receptor that binds to antigens presented in the context of human leukocyte antigen (HLA) major histocompatibility complex (MHC) class I molecules, and along with a second signal mediated by CD28 binding to its ligand B7, leads to cell mediated killing. The CD4+ helper T cells bind to antigens presented on HLA MHC class II molecules. The CD4+/MHC class II interaction leads to cytokine release and recruitment of other immune cells. By contrast, B cells mature in the bone marrow and are involved in production of antibody and antibody-dependent cell mediated cytotoxicity. Once they see antigen, B cells mature into plasmatic cells that secrete antibodies that then bind to antigens. The antigen-antibody complex signals immune detection and triggers killing by a variety of cells including NK cells.
The most active antigen-presenting cells (APCs) are dendritic cells (DCs) that reside as immature cells in almost every organ and tissue 5, sit at the interface of pathogen entry sites and continuously sample antigens. Antigen sampling results in effective antigen presentation when the DCs are triggered by other “danger signals”. “Danger signals” are signs of tissue damage or inflammation 5. Danger-triggered DCs start to mature and become activated. Activated DCs up-regulate their chemokine-receptors, allowing trafficking to lymph nodes where they can induce T cell responses (Figure 1).
Figure 1.
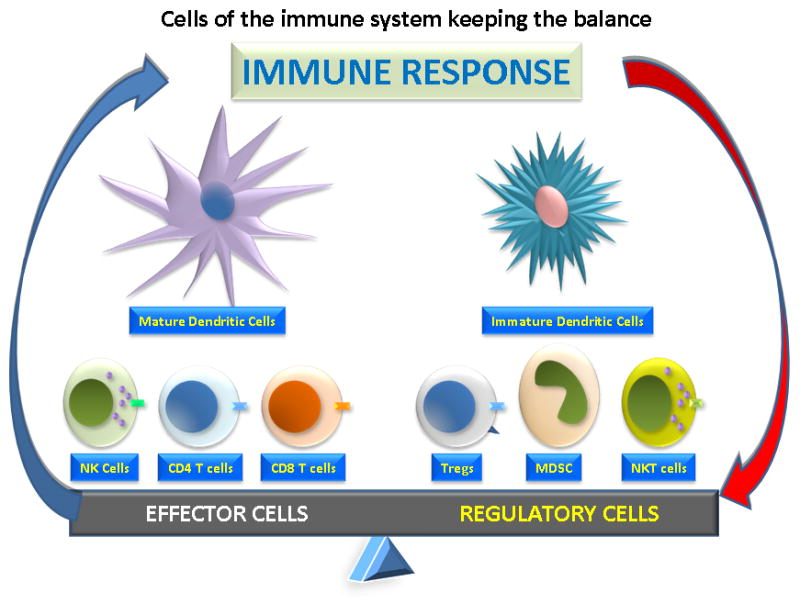
Cells of the immune system include effector cells and regulatory cells. Among the effector cells are CD4+ helper T cells, CD8+ cytotoxic T lymphocytes (CTL), natural killer cells (NK) and dendritic cells (DC), which are the antigen presenting cells. The regulatory cells include regulatory T cells (Treg), which express CD4, CD25 and CTLA4, and myeloid-derived suppressor cells (MDSC).
Regulation of the immune system
Though the immune system is designed to recognize between foreign invaders and self, with the constant antigen sampling, some foreign antigens, just by chance, are bound to resemble some antigens inherent in the body. The immune system has ways of regulating itself to put on a brake and prevent autoimmunity. When T cells are activated, they will upregulate membrane cytotoxic T-lymphocyte associated antigen 4 (CTLA4) and program cell death 1 (PD-1) proteins. CTLA4 competes with CD28 to bind B7 and PD-1 will bind to its ligand PD-L1, both signals will inhibit ongoing T cell activation. Another mechanism to brake immune activation is Regulatory T cells (Tregs) which were first proposed in 1972 6 and discovered in 1995 7. They are now recognized as a key regulatory pathway in tumor tolerance and have a unique cell surface signature, with expression of both CD4 and CD25 (the alpha chain of the interleukin-2 (IL-2) receptor) 7. These cells express other cell surface markers, including CTLA4 8 and glucocorticoid-induced tumor necrosis factor (GITR) receptor 9 and are regulated by a transcription factor called forkhead box protein 3 (FOXP3)cells 10. T-regs actively inhibit conventional CD4+ T cells, CD8+ T cells, DCs, NK cells 11, thus dampening immune responses. Other immune regulatory pathways such as immune suppressive cytokines, myeloid-derived suppressor cells, regulatory B cells and natural killer cells also play important roles in generating and maintaining tumor tolerance and, along with T-regs, are considered targets of immune therapy 12–13 (Figure 1).
Neuroimmunology
It was once thought that the nervous system was an immune privileged organ, devoid of normal immunologic function 3, 14–15. The CNS features in support of this theory included the blood brain barrier that allows for selective entry of immune cells from the peripheral blood into the brain parenchyma, the lack of lymphatic vessels and lymph nodes within the CNS, and the low numbers of circulating T cells in the CNS. Furthermore, there is less HLA presentation and absence of traditional APCs in the CNS when compared to other tissues. Nevertheless, under physiologic conditions, the brain hosts several populations of immune cells 15. Microglia arise from hematopoietic cells and colonize CNS during embryonic development. The microglia constitutes an early line of defense for the brain. Microglial cells migrate to inflammatory zones in the CNS and become activated. Once activated they have phagocytic and antigen presenting cell properties, as well as the ability to recruit other immune cells by secreting cytokines and chemokines 16. Macrophages and DCs both arise from monocytes. They are found in perivascular zones, choroid plexus, and meninges. Because of their role as professional APCs, DC based vaccine therapy has been the most studied approach for high grade gliomas. T cells are found in the CNS only in the activated form. Naïve T cells are not present in the CNS 17–18. T cells are activated in cervical lymph nodes and then move into the CNS. It is unclear how antigens are transported from the brain to the cervical nodes to activate the T cells.
Cancer immunotherapy has unique challenges in the CNS due to the relative immune-privilege of the brain and the immune-suppression caused by high grade gliomas. The blood brain barrier, low numbers of T lymphocytes, and lack of a lymphatic systems make it challenging for immune cells to enter the CNS. In patients with glioblastoma, the blood brain barrier is disorganized 15, 19. Tissue injury leads to breakdown of the tight junctions between endothelial cells that facilitate migration of leukocytes into the CNS. Though the CNS is devoid of traditional lymphatic vessels, CSF drains via Virchow-Robin spaces to the deep cervical lymphatics 17, 20. T cells located in the cervical lymph nodes are activated and can patrol the CNS 17–18. Activated T cells that encounter their antigen are retained in the CNS. HLA presentation occurs on astrocytes, microglia, and endothelial cells 3, 14. The net balance is that CNS immune surveillance still occurs 3.
Patients with glioblastoma exhibit a relative systemic immune suppression compared to the general population. Adaptive immune responses are deficient 17. The tumor microenvironment is rich with immunosuppressive factors secreted by the tumor 17, like transforming growth factor beta (TGF-β) and vascular endothelial growth factor (VEGF) that suppresses T cell proliferation and cytotoxic function 21–22. VEGF inhibits the maturation of DCs. In glioma patients, there are diminished absolute counts of CD4+ T cells with increased fraction of T-regs 3, 23. This immunosuppression likely plays an important role in tumor progression in patients with glioblastoma. In addition to being immunosuppressed from having a malignancy, older age, cytotoxic chemotherapy and exogenous administration of corticosteroids are other factors that contribute to systemic immune suppression in this population (Figure 2). It stands to reason that if the immune suppression could be reversed allowing effective immune targeting of glioma, then patients with glioma might have less tumor progression and improved outcomes.
Figure 2.
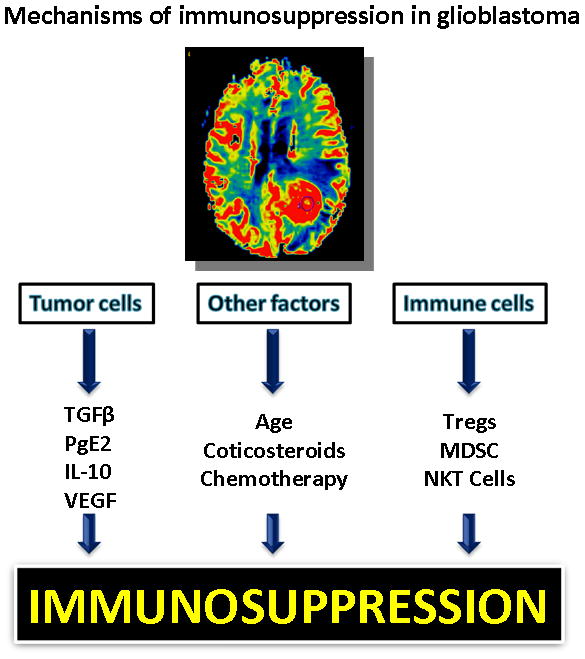
Factors contributing to immunosuppression in glioblastoma include tumor factors, exogenous factors, and immune factors. The glioma cells secrete immunosuppressive cytokines, such as TGFβ, prostaglandin E2, IL-10, and VEGF. Age, exogenous steroids, and chemotherapy all contribute to exogenous immune suppression; while regulatory immune cells such as regulatory T cells (Treg) and myeloid derived suppressor cells (MDSC), also dampen the immune response.
Rationale for immunotherapy
It has been long observed that changes in the immune system relate with cancer survival. Neurologists and neurosurgeons provide anecdotal reports that glioma patients who suffer postoperative infections near the tumor bed seem to do better than the average patient similar to the observations made over a century ago by Coley 24. Recently de Bonis and colleagues investigated the idea that postoperative infection may confer a survival advantage to patients with malignant glioma 25. They reviewed 197 cases of newly diagnosed glioblastoma, 10 of whom had peri-operative infections. The infection group had a significant advantage in the median survival; 30 months compared to 15 months in the non-infected tumor patients. We now understand that infection can contribute to activation of immune pathways via PAMPs and activation of TLR on the innate immune system and subsequently initiate anti-tumor immunity. Indeed, a direct correlation between survival of patients with primary glioblastoma and tumor infiltration of cytotoxic and helper T cells has been observed 26. As noted previously, glioblastoma patients are relatively immunosuppressed compared to the general population. The degree of immunosuppression also correlates with survival. Grossman and colleagues recently followed a group of 96 patients with newly diagnosed high grade gliomas through surgery, radiation therapy, and chemotherapy with temozolomide 27. The patients had a normal CD4+ count at diagnosis, but hit a nadir 2 months post-treatment. Forty percent of the study population had CD4+ counts of <200, and those patients had a significantly shorter median survival at 13.1 months compared to 19.7 months in the patients with higher counts. All of these observations taken together suggest that if the immune effectors were better activated in glioblastoma, patients might have better outcomes.
Strategies for using immunotherapy in the treatment of gliomas
As with immunization for infections, immunization against tumors can theoretically occur in the form of passive or active immunotherapy 15. In passive immunotherapy, a patient is given immune cells or antibodies capable of targeting the tumor cell. Passive immunotherapy does not require activation of the patient’s own immune system, but instead immune cells are active in vitro and injected into the patient. By contrast, active immunotherapy provides a boost to the patient’s native immune system (Figure 3).
Figure 3.
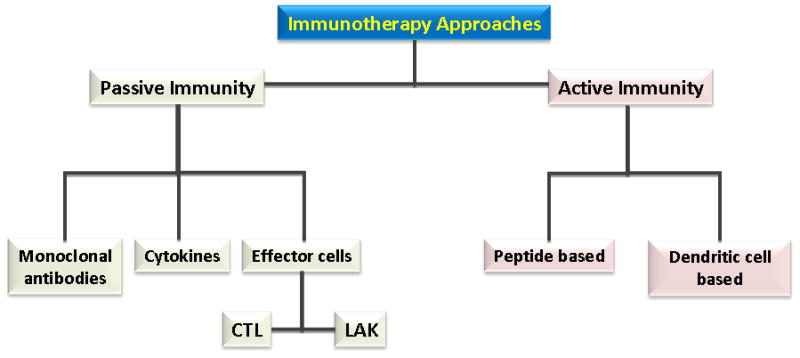
Approaches to immunotherapy can be divided into passive immunity and active immunity. Passive immunotherapy approaches include direct administration of monoclonal antibodies or cytokines or adoptive immunity with cytotoxic T lymphocytes (CTL) or lymphocyte activated killer (LAK) cells. Active immunity includes peptide based immunotherapy and dendritic cell therapy.
In broad terms, passive immunotherapy can be further divided into 3 approaches 5 (Figure 3). The first is the direct injection of monoclonal antibodies 2–3. In this approach antibodies that are known to interact with an antigen specific to or associated with a tumor are administered to the patient. In glioblastoma, only one monoclonal antibody has been approved for treatment. Bevacizumab is a humanized IgG1 monoclonal antibody that binds to and neutralizes the vascular endothelial growth factor (VEGF) ligand 2, 28–29. VEGF is a tumor-associated protein, which is found on a variety of malignancies, including glioblastoma. VEGF is the central mediator of tumor angiogenesis. Although bevacizumab retains its ability to bind complement and Fc receptor, its action is through blocking the ligand that activates the VEGF receptor on tumor blood vessels thereby inhibiting angiogenesis and tumor growth. Bevacizumab may also be associated with afferent vascular dilatation and efferent vascular constriction of tumor vessels. This may give the additional benefit of concentrating chemotherapy at tumor site.
The second approach to cancer passive immunotherapy is to stimulate the immune system with cytokines. Cytokine stimulation with IL-2 have been studied in a variety of cancers, and while it has been a successful approach in melanoma and renal cell cancer, it has not shown benefit in glioblastoma 2.
A third strategy to passive immunotherapy is treatment with stimulated immune effector cells. This approach has also been called adoptive immunity, or cell based therapy immunotherapy 15. In adoptive immunity, immune cells activated ex-vivo are administrated to patient, either by systemic injection or directly into the tumor or tumor resection cavity. Both lymphocyte-activated killer cells (LAK) and cytotoxic T lymphocytes (CTL) have been used. LAK cells are generally obtained by cultivating autologous peripheral lymphocytes in the presence of IL-2. The culture yields both T and NK cells. The immune reaction provided by LAK cells is non-specific cytotoxicity, which is not necessarily tumor-directed. By contrast, CTLs are prepared by collecting peripheral blood mononuclear cells or tumor infiltrating lymphocytes and then stimulating them ex vivo with antigens. For cancer immunotherapy, autologous tumor cells are also used for the antigen stimulation, thus yielding CTLs that have been activated with specificity to the tumor.
Active immunotherapy boosts the patient’s immune system by priming it with antigen exposure. There are two approaches to active immunization: peptide based therapy and cell based therapy 30 (Figure 3). In peptide based therapy, peptides are injected as a vaccine to induce immune activation. Tumors express various antigens, some of which are tumor-specific, and others which are tumor associated. Tumor antigens can be also categorized as cancer-testes antigens, tumor differentiation antigens, viral related antigens, or mutated oncogenic proteins. The peptides selected for cancer vaccines are typically small, around nine amino acids in length and are capable of binding to MHC class I molecules, which leads to activation of CTL.
Cell based active immunotherapy uses antigen presenting cells activated by tumor antigens to prime the immune system rather than the antigen itself. Since DCs are professional APCs, they are an obvious choice for active immunotherapy and have to date been the most studied cell based vaccine 5, 15, 30. In most cases, DCs are prepared from autologous peripheral blood mononuclear cells and are cultivated in the presence of growth factors, such as granulocyte macrophage colony-stimulating factor (GM-CSF) and interleukin-4. The DCs are then matured and activated by antigens, which can be prepared from various sources, including tumor lysates, peptides eluded from activated tumor cells, defined peptides such as those used in peptide-based immunotherapy, viral antigens, mRNA derived from activated tumor cells, and whole tumor cells. The activated DCs are then given back to the patient, either injected intradermally, in the lymph nodes or locally at the tumor site.
Clinical reports of immunotherapy
Interpretation and comparison of the results of clinical trials using immune therapy against glioblastoma is extremely difficult because of heterogeneity in study design, therapeutic approach used, immune endpoints measured, and patient eligibility criteria. The classic design of cancer clinical trials does not fit the immune therapy model. For example although most vaccine trials are phase I, the “dose escalation” design does not apply since treatments usually have minimal toxicity; the dose limitation is the availability of cells and not the appearance of adverse events 17. The few phase II clinical trials published are not randomized and use historic controls to compare outcomes. Most clinical trials included patients with recurrent glioblastoma, who may have a poor functional status, large tumor burden and been heavily pre-treated making less likely to benefit of immune therapy. In the clinical trials there are exclusion criteria that render a highly selected population of evaluable patient 31. Furthermore, some trials include patients with newly diagnosed and recurrent disease, and on occasions, include patients with anaplastic gliomas. Surrogate endpoints, like immunologic assays and brain imaging studies, have not been harmonized and validated in most cases.
Passive immunotherapy
The earliest attempts at immune therapy in the late 1980s into the 1990s focused on passive immunization by infusion of LAK cells directly into the tumor bed in the peri-operative and post-operative period 32–38. Side effects from this strategy included cerebral edema, increased intracranial pressure, headaches, fever, and confusion 32, 36, but in general the treatment was well tolerated. In most instances there was no impact on survival 32, 34, 36–37; though Hayes and colleagues and others suggested an improved median survival compared to contemporary controls in patients with recurrent glioblastoma treated with LAK cells and IL-2 38–41.
In addition to LAK cells, CTL are the other group of cells, studied in adoptive immune responses for glioblastoma 42–50. The first study of CTLs was by Kitahara and colleagues in 1987 48. Two of five patients had a reduction in tumor size by imaging following intratumoral CTL treatment. Others have also reported responses to intratumoral and systemic administration of adoptive T cell therapy 42–44, 46, 49–51.
Active Immunotherapy
In the realm of active immunity, both cell based therapies and peptide based therapies have been studied. Patients have be given individual peptides 52–54, whole tumor lysate 55, or some combination of tumor antigens and cytokines 56.
Epidermal Growth Factor Receptor variant III (EGFRvIII) is a tumor-specific antigen commonly expressed by glioblastoma cells but not on normal tissue 57. PEP-3 is a 14 amino acid peptide from EGFRvIII, which, when coupled with a foreign “helper molecule” keyhole limpet hemocyanin (KLH) (PEP-3-KLH) has been used as a vaccine to generate EGFRvIII specific antibodies 57–58. Sampson and colleagues have studied the EGFRvIII peptide, PEPvIII-KLH in 18 glioma patients expressing EGFRvIII on their tumors 53, 59. The vaccinated patients had improved 6 month progression free survival (the primary endpoint) and improved overall survival compared to contemporary controls 52. Of interest, sampling of the tumor in some cases at recurrence revealed loss of EGFRvIII expression.
Though Epidermal Growth Factor Receptor related peptides have received the most attention, there are other peptides that are tumor associated which may be useful in inducing immune responses. Yajima at colleagues created “personalized peptide vaccines” 54. They treated 21 HLA-A2 or HLA-A24 patients with GBM with a combination of 23-25 tumor associated peptides known to bind to either HLA-A2 or HLA-A24. Among them, 5 patients had a partial response, 8 patients had stable disease, and 8 patients had progressive disease. Peptide specific IgG antibody responses were detected in the tumor bed or spinal fluid of all patients tested.
Terasaki and colleagues also explored this idea of “antigen soup” as a peptide-immunotherapy approach 60. They enrolled 12 HLA-A24 positive patients with glioblastoma and HLA-A24. The patients were vaccinated with HLA-A24 restricted (ITK-1) peptides. ITK-1 consisted of 14 peptides known to be expressed at high levels in cancer cells and low levels in normal cells. The 14 chosen peptides were capable of inducing peptide-specific cellular immunity and humoral immunity in HLA-A24 patients with glioblastoma. Of the 12 pts enrolled, 2 patients had a partial clinical response, 5 had stable disease, and 5 had progressive disease. All patients tolerated the vaccination without any serious adverse reactions 60.
Another approach being explored is to vaccinate with a heat shock protein in complex with autologous tumor derived peptides. Clinical trials in patients with recurrent glioblastoma have revealed both an adaptive and innate immune response with the treatment being well tolerated and suggesting an improvement in survival when compared to historic controls 61.
It is unclear if individual peptides or whole tumor lysate induce a better immune response, as they have never been studied head to head. Autologous tumor prepared vaccines alone or in combination with cytokines have also demonstrated isolated clinical response 55–56.
As DCs are critical in initiating antigen-specific immunity, we and others have used autologous DC vaccination as an approach to treat patients with glioblastoma. The basic strategy for DC vaccination is to give autologous DCs which have been manipulated ex vivo to present autologous tumor antigens 54, 62–66. DC administration has varied by route (intradermal, subcutaneous, intranodal, intratumoral), schedule and combination with other treatment modalities. 67. Most trials are phase I studies that included patients with recurrent high grade gliomas (variable number of glioblastomas) and in general, vaccination with DCs has been well tolerated 63–65, 68–74. Some studies have been able to show clinical responses, either in tumor regression 62, 66, 73–74 or improved survival compared to historical or contemporary controls 70, 74–75. Immune responses have also been demonstrated with use of surrogate endpoints. In patients who underwent reoperation after vaccination with DCs, some have infiltration of CTL within the tumor 63, 68–69.
Some studies enrolled patients with both recurrent and newly diagnosed glioblastoma, but recent series have included patients with newly diagnosed glioblastoma and in combination with other therapeutic modalities (Table 1) 31, 67–69, 75–78. As can be seen in Table 1, of the 73 patients with newly diagnosed glioblastoma treated with DCs immune therapy as part of the first-line therapy only 2 developed severe toxicity including one report of a patient who developed a cutaneous glioblastoma at a lymph node injection site 75. One additional high grade toxicity was seen in the 156 patients with recurrent glioblastoma treated with DC vaccines (Table 2).
Table 1.
Published clinical trials using DC vaccine for newly diagnosed GBM; studies that included more than 3 patients. In total 73 patients have been given DC vaccines and 2 had grade III+ toxicities.
| Author | Year | Total | Pre-vaccine therapy | Vaccine | Route | Age (Median) | Overall survival (months) | Toxicity Grade III+ |
|---|---|---|---|---|---|---|---|---|
| Yu68 | 2001 | 7 | RT | DC/Eluted peptides | ID Deltoid | 50 | 14 | No |
| Liau69 | 2005 | 7 | RT | DC/Eluted peptides | ID Deltoid | 33 | 23 | No |
| Walker78 | 2008 | 7 | No RT 1 RT/TMZ |
DC/Lysate | ID Abdomen | 53 | 11 | No |
| Wheeler75 | 2008 | 11 | ? | DC/Lysate | ID? | ? | ? | 1(*) |
| Ardon72 | 2010 | 8 | RT/TMZ | DC/Lysate | ID Deltoid | 50 | 24 | 1(edema) |
| Chang70 | 2011 | 8 | RT | DC/Lysate | SC Axilla | 60 | 24 | No |
| Prins76 | 2011 | 15 | RT/TMZ | DC/Lysate | ID Axilla | 48 | 36 | No |
| Fadul31 | 2011 | 10 | RT/TMZ | DC/Lysate | Cervical Lymph Node | 60 | 28 | No |
Cutaneous glioblastoma at the site of irradiated tumor cell inoculation for DTH testing. Same as in Table 2. RT indicates radiation therapy; TMZ, Temozolomide; DC, Dendritic cells; ID, Intradermal; SC, Subcutaneous
Table 2.
Published clinical trials using DC vaccines for recurrent glioblastoma. 159 patients have been treated with only 2 grade III/IV toxicities.
| Author | Year | Total | Pre-vaccine therapy | Vaccine | Route | Age (Median) | Overall survival (months) | Toxicity Grade III+ |
|---|---|---|---|---|---|---|---|---|
| Kikuchi62 | 2001 | 5 | RT/Chemo | DC/Glioma fusion | ID | 42 | ? | No |
| Yamanaka73 | 2003 | 8 | RT | DC/Lysate | ID/Tumor | 46 | ? | No |
| Yu63 | 2004 | 9 | Chemo | DC/Lysate | Deltoid | 45 | 33 | No |
| Kikuchi66 | 2004 | 6 | RT/Chemo | DC/Glioma fusion | ID | 45 | 10 | No |
| Liau69 | 2005 | 5 | RT/TMZ | DC/Eluted Peptides | ID Deltoid | 40 | 23 | No |
| Yamanaka74 | 2005 | 24 | RT/Chemo | DC/Lysate | ID/Tumor | 49 | 16 | No |
| Rutkowski96 | 2005 | 7 | RT/Chemo | DC/Lysate | ID | 55 | ? | 1 (edema) |
| Walker78 | 2008 | 2 | RT/TMZ | DC/Lysate | ID Abdomen | ? | 5 | No |
| Wheeler75 | 2008 | 23 | RT/TMZ | DC/Lysate | ID? | ? | ? | 1(*) |
| Vleeschouwer64 | 2008 | 56 | RT/TMZ | DC/Lysate | ID | 45 | 9 | No |
| Chang70 | 2011 | 6 | RT | DC/Lysate | SC | 37 | 39 | No |
| Prins76 | 2011 | 8 | RT/TMZ | DC/Lysate | ID Axilla | 48 | 21 | No |
Cutaneous glioblastoma at the site of irradiated tumor cell inoculation for DTH testing. Same as in Table 1. RT indicates radiation therapy; TMZ, Temozolomide; DC, Dendritic cells; ID, Intradermal; SC, Subcutaneous; Chemo, Chemotherapy
We evaluated the immunologic response to cervical intranodal vaccination with autologous tumor lysate-loaded DCs in 10 patients with newly diagnosed glioblastoma after concomitant radiation therapy and chemotherapy and before starting adjuvant temozolomide 31 (Figure 4). We explored immunologic endpoints in a novel approach using hierarchical clustering analysis of the results of 5 immune assays measured before and after vaccination (Figure 5). Immune activation as determined by this methodology was associated with improved survival.
Figure 4.

Four weeks after complete combined RT-TMZ, patients had a prevaccination (V) aphesis, DTH panel placement, and MRI. One week later, the first vaccination (V1) was administered, and 2 additional vaccinations were given 2 weeks apart. Two weeks after the third vaccine patients had a post-V apheresis, DTH panel placement, and MRI, followed by 12 cycles of adjuvant TMZ. DDPA indicates dye dilution proliferation assay; DTH, delayed-type hypersensitivity reaction; ELISPOT, enzyme-liked immunosorbent spot assay; PBMNC, peripheral blood mononuclear cells; POST-V, postvaccination; PRE-V, prevaccination; RT, radiation therapy; TMZ, temozolomide; v, vaccination. Printed with permission from Fadul et al, 2011 31.
Figure 5.
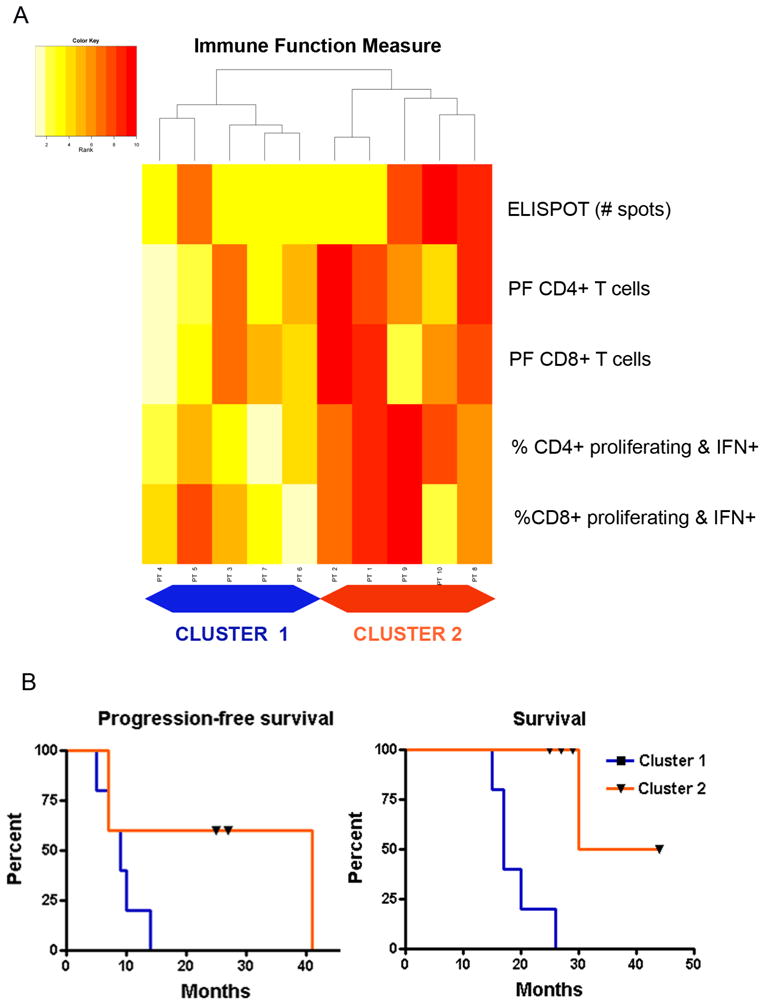
(5A) Heatmap of hierarchical clustering analysis of the postvaccination immune responses. Five patients with generally low ranks in immune function (pale yellow colors) formed cluster 1 on the left. Five other patients with higher ranks in immune function measures (dark red colors) formed cluster 2 on the right. (5B) Kaplan-Meier curve of overall survival for the 2 clusters. The overall survival was significantly different between cluster 1 (median = 17 mo) and cluster 2 (median = not achieved) (P = 0.002). ELISPOT indicates enzyme-linked immunosorbent spot assay; IFN, interferon; PF, precursor frequency. Printed with permission from Fadul et al, 2011 31.
Other approaches to vaccine therapy deserve mention. In 2001, Schneider and colleges were able to deliver autologous tumor cells via a viral vaccine vector using Newcastle Disease Virus (NDV) 79. The advantage of NDV is that it is a single stranded RNA virus that poses little health hazard to humans and has the ability to selectively kill human tumor cells. Other approaches include use of autologous tumor transfected with cytokine genes to express cytokine or DC-tumor cell fusions 80–81.
To date, all vaccine and immune therapy studies in patients with glioblastoma including ours, suffer from small sample size and thus bias induced by patient selection. There are many variables in the design of the studies without consensus in the optimal method to prepare and condition the DCs, antigen to use, site of administration, and immune assays to monitor 82. Careful clinical trial design that would provide information on the most favorable dose, frequency of administration, and timing to introduce DC based vaccine in the multimodality treatment scheme of patients with glioblastoma is needed. Nevertheless, the observations of induction of tumor specific immune responses, clinical response, prolonged survival in a few patients, and low toxicity is encouraging and supports the continued investigation of immune therapies in patients with glioblastoma.
Endpoints and outcomes
While primary endpoints of overall survival and progression free survival are most important in the development of a new therapy, surrogate endpoints can be very helpful in predicting clinical outcomes and fueling further research. Surrogate endpoints for vaccine therapy include a variety of immune responses 5. Often reported are delayed-type hypersensitivity responses, interferon-gamma (IFNγ) release from peripheral blood mononuclear cells as measured by flow cytometry or IFNγ enzyme-linked immunosorbent spot (ELISPOT) assays. Delayed-type hypersensitivity reactions are used as a measure of antigen recall and correlate with peripheral blood antigen-specific T cell responses 83–84. Nevertheless, patients with glioblastoma are frequently anergic and it is of limited use for monitoring these patients 31, 75. IFNγ is released by Type I CD4+ T cells 83. The IFNγ ELISPOT assay is one of the common methods of assessing adaptive immune responses. Recent studies have reported a positive correlation between immune response, measured by ELISPOT assays, and clinical outcomes 83.
We have used the Dye Dilution Proliferation Assay (DDPA) in immunotherapy clinical trials to evaluate immune response by monitoring tumor-specific CD8+ and CD4+ precursor frequency 85. This method also allows measurement of the proportion of CD4+ and CD8+ IFNγ producing cells for immune monitoring. The median values for the results of 5 immune response measurements (DDPA and ELISPOT) before and after DC based vaccination in 10 patients with glioblastoma was analyzed using hierarchical clustering analysis 31. A measurable immune response by this composite method was associated with improved survival. There is need to standardize and prospectively validate immune monitoring assays if immunologic surrogate endpoints are to be used in larger clinical trials involving multiple sites.
Radiologic assessment by MRI has been the mainstay in the evaluation of the response to therapy in glioblastoma and used as the primary endpoint in many phase II trials. The appearance of new and more varied oncologic treatment modalities has underscored the limitations of the criteria used to assess response in clinical trials 86. Specifically, in immune therapy trials for high grade gliomas, increased size of gadolinium-enhanced lesions on MRI studies, suggestive of recurrent tumor, have revealed inflammatory infiltration without active tumor 87. Furthermore, the modest response on MRI does not correlate with clinical endpoints 88. Therefore, radiologic criteria to evaluate brain tumor immunotherapy have to be refined and the use of more advanced techniques to image inflammation and immune response are needed 89.
Future Directions
The challenges of the future of immune therapies relate to enhancing antigen presentation capabilities, effectively breaking tumor-induced immune tolerance, improving activation of tumor-specific cytolytic effector cells, and the standardization and upscale production of cell based therapy. At the same time there is concern that further boosting of the immune response, albeit more effective, may result in serious adverse events secondary to brain edema and auto-immunity.
A strong and long lasting anti-tumor T cell response that confers clinical benefit is the goal on DC based immunotherapy. Enhancing the antigen presenting cell cap abilities by polarizing the cell towards a more effective (α-type) 1 DC phenotype has been reported, but there is still controversy on the culture conditions to obtain the most activated and potent DCs. A clinical trial examined using the fusion of dendritic and glioma cells combined with recombinant human interleukin 12 (rhIL-12) for the treatment of malignant glioma 66. No serious side effects and a few responses on MRI were observed. Combination of DC vaccination with an immunoadjuvant polyinosinic-polycytidylic acid [poly (I: C)] stabilized by lysine and carboxymethylcellulose (poly-ICLC), a PAMP that activates DCs, was safe 87. Finally the optimal route of administration of DCs for brain tumors has not been established, although animal 90 and clinical studies 91 suggest that intranodal injection is the most effective.
One of the challenges with vaccine therapy is the self-imposed “brake” on the immune system in glioma patients, which may limit the immune system’s response to a vaccine. There are a few potential targets that could remove the brake of the immune system. One strategy might be to deplete the regulatory T cells. To that end, an antibody against CD-4 or CD25 could be used to target Tregs, or more general immunotoxins could be used. A study suggests that administration of daclizumab, an antibody against IL-2Rα, when patients are lymphopenic after administration of temozolomide enhances the antitumor immunity of vaccination by depleting Treg. Once T cells are activated, they up regulate molecules such as CTLA4 and PD-1 to limit their activity (Figure 6). Use of blocking humanized monoclonal antibodies such as ipilimumab (anti-CTLA4) to these check point molecules appear very promising and have already made it to the clinic in treating patients with metastatic melanoma 92. A phase I clinical trial using a vaccine comprising of autologous tumor cells genetically modified by a transforming growth factor–β2 (TGF-β2) antisense vector in 6 patients with recurrent glioblastoma was well tolerated with indication of anti-tumor induced immunity 93. Use of additional inflammatory cytokines such as IL-12, IL-7, and IL-15, activating antibodies to costimulatory molecules such as CD40, or blocking antibodies to immune inhibitory cytokines such as IL-10 or TGFβ in combination with DC vaccination can potentially enhance clinical activity 94. Which of these strategies in combination with vaccination will yield the best therapeutic ratio (most effective and less toxic) is to be determined.
Figure 6.
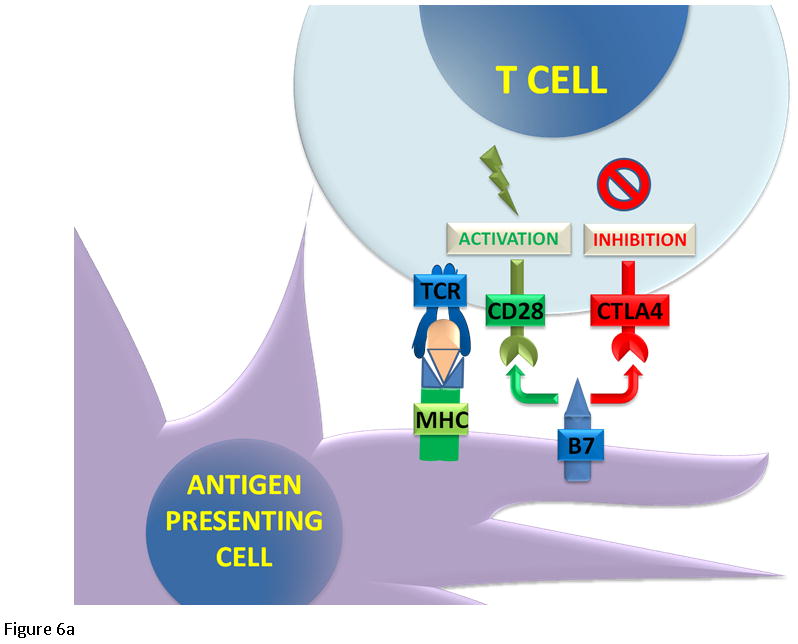
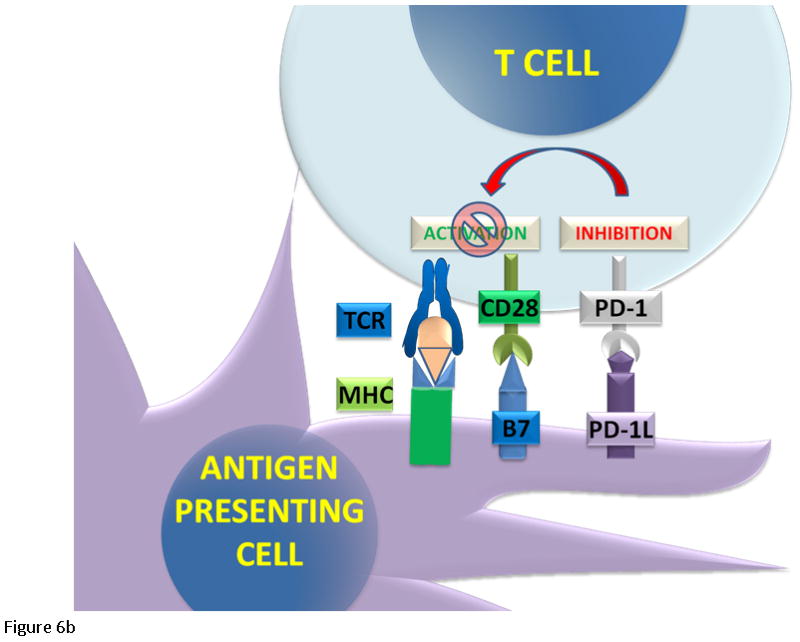
T cell regulation. (6A) The cytotoxic T cells express CD8 receptor that binds to the MHC receptor on the APC, along with a second signal mediated by CD28 binding to its ligand B7. The binding of the T cell receptor and CD28 lead to activation and cell mediated killing. Activated T cells upregulate CTLA4 and PD-1 proteins. CTLA4 competes with CD28 to bind B7. (6B) When PD-1 binds to its ligand PD-L1, it inhibits T cell activation.
Treatment for high grade glioma involves a multi-disciplinary approach using surgery, radiation therapy, and chemotherapy. Clarifying when and how immune therapy should be given with these other modalities and the role of steroid use in this population of patients will require well designed and appropriately powered clinical trial. In small studies, we and others have demonstrated that DC vaccines can be given safely to patient with glioblastoma undergoing temozolomide chemotherapy alone or in combination with radiotherapy 31. The studies confirm that tumor-specific immune responses in these patients can be induced 31,58, 69,75. The rational to give chemotherapy with immunotherapy may relate to the chemotherapy effects on tumor release of relevant antigens, on inhibiting the regulatory compartment, and on the ability to change the tumor vasculature providing better access for effector cells 69, 95–97. Another possibility is that vaccination sensitizes the tumor to chemotherapy 75, 95, 97.
Conclusion
Research over the last 10 years has demonstrated that immune therapy for glioblastoma triggers a measurable immune response in spite of poor tumor antigenicity and considerable immune suppression. If that antitumor effect is enough to translate in improvement in survival is still to be proven. The limited number of patients with glioblastoma, the lack of a cooperative group that can do large clinical trials for the study of brain tumor immunotherapy, and the variability in approaches and immune monitoring assays used are the major barriers to determine if immune therapy could be part of the standard of care. Furthermore, the challenge of immunotherapy is to understand the various regulatory and co-stimulatory factors in the patient and the tumor microenvironment and being able to manipulate these forces effectively to enhance anti-tumor immune response and clinical benefit. As immunotherapy evolves, prognostic and predictive biomarkers will be important to determine which patients will make the best candidates for vaccine therapies. There is need for harmonization and validation of immunologic endpoints as well as imaging techniques that allow adequate monitoring of patients with brain tumors receiving immune base therapies. With our expanded knowledge of immune pathways and the effects tumors have on immune function we will be better able to develop vaccine strategies for the future.
Acknowledgments
Supported in part by R01CA095648 and by P30CA023108
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Dillman RO. Cancer immunotherapy. Cancer Biother Radiopharm. 2011;26:1–64. doi: 10.1089/cbr.2010.0902. [DOI] [PubMed] [Google Scholar]
- 3.Kanaly CW, Ding D, Heimberger AB, Sampson JH. Clinical applications of a peptide-based vaccine for glioblastoma. Neurosurg Clin N Am. 2010;21:95–109. doi: 10.1016/j.nec.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbas . Diseases of Immunity. In: Fausto KA, editor. Robbins and Cotran Pathologic Basis of Disease. 7. Philadelphia: Elsevier Saunders; 2005. pp. 193–267. [Google Scholar]
- 5.Van Gool S, Maes W, Ardon H, Verschuere T, Van Cauter S, De Vleeschouwer S. Dendritic cell therapy of high-grade gliomas. Brain Pathol. 2009;19:694–712. doi: 10.1111/j.1750-3639.2009.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gershon RK, Cohen P, Hencin R, Liebhaber SA. Suppressor T cells. J Immunol. 1972;108:586–90. [PubMed] [Google Scholar]
- 7.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 8.Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McHugh RS, Whitters MJ, Piccirillo CA, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 10.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–84. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 11.Beyer M, Schultze JL. Immunoregulatory T cells: role and potential as a target in malignancy. Curr Oncol Rep. 2008;10:130–6. doi: 10.1007/s11912-008-0021-z. [DOI] [PubMed] [Google Scholar]
- 12.Begley J, Ribas A. Targeted therapies to improve tumor immunotherapy. Clin Cancer Res. 2008;14:4385–91. doi: 10.1158/1078-0432.CCR-07-4804. [DOI] [PubMed] [Google Scholar]
- 13.Ernstoff MS, Crocenzi TS, Seigne JD, et al. Developing a rational tumor vaccine therapy for renal cell carcinoma: immune yin and yang. Clin Cancer Res. 2007;13:733s–40s. doi: 10.1158/1078-0432.CCR-06-2064. [DOI] [PubMed] [Google Scholar]
- 14.Sehgal A, Berger MS. Basic concepts of immunology and neuroimmunology. Neurosurg Focus. 2000;9:e1. doi: 10.3171/foc.2000.9.6.2. [DOI] [PubMed] [Google Scholar]
- 15.Vauleon E, Avril T, Collet B, Mosser J, Quillien V. Overview of cellular immunotherapy for patients with glioblastoma. Clin Dev Immunol. 2010 doi: 10.1155/2010/689171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol. 2009;85:352–70. doi: 10.1189/jlb.0608385. [DOI] [PubMed] [Google Scholar]
- 17.Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol. 2011;13:3–13. doi: 10.1093/neuonc/noq169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–60. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 19.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–79. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 1992;2:269–76. doi: 10.1111/j.1750-3639.1992.tb00703.x. [DOI] [PubMed] [Google Scholar]
- 21.Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech. 2001;52:401–10. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 22.Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunol Today. 1991;12:370–4. doi: 10.1016/0167-5699(91)90068-5. [DOI] [PubMed] [Google Scholar]
- 23.Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 24.Nauts HC, McLaren JR. Coley toxins--the first century. Adv Exp Med Biol. 1990;267:483–500. doi: 10.1007/978-1-4684-5766-7_52. [DOI] [PubMed] [Google Scholar]
- 25.De Bonis P, MDA, MDG, et al. Post-operative infection may influence survival in patients with glioblastoma: simply a myth?: Glioblastoma, infection and survival. Neurosurgery. 2011 doi: 10.1227/NEU.0b013e318222adfa. [DOI] [PubMed] [Google Scholar]
- 26.Lohr J, Ratliff T, Huppertz A, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. 2011;17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 27.Grossman SA, Ye X, Lesser G, et al. Immunosuppression in Patients with High-Grade Gliomas Treated with Radiation and Temozolomide. Clin Cancer Res. 2011;17:5473–80. doi: 10.1158/1078-0432.CCR-11-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 29.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–5. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamanaka R. Cell- and peptide-based immunotherapeutic approaches for glioma. Trends Mol Med. 2008;14:228–35. doi: 10.1016/j.molmed.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Fadul CE, Fisher JL, Hampton TH, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34:382–9. doi: 10.1097/CJI.0b013e318215e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merchant RE, Grant AJ, Merchant LH, Young HF. Adoptive immunotherapy for recurrent glioblastoma multiforme using lymphokine activated killer cells and recombinant interleukin-2. Cancer. 1988;62:665–71. doi: 10.1002/1097-0142(19880815)62:4<665::aid-cncr2820620403>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 33.Jacobs SK, Wilson DJ, Kornblith PL, Grimm EA. Interleukin-2 or autologous lymphokine-activated killer cell treatment of malignant glioma: phase I trial. Cancer Res. 1986;46:2101–4. [PubMed] [Google Scholar]
- 34.Lillehei KO, Mitchell DH, Johnson SD, McCleary EL, Kruse CA. Long-term follow-up of patients with recurrent malignant gliomas treated with adjuvant adoptive immunotherapy. Neurosurgery. 1991;28:16–23. doi: 10.1097/00006123-199101000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Barba D, Saris SC, Holder C, Rosenberg SA, Oldfield EH. Intratumoral LAK cell and interleukin-2 therapy of human gliomas. J Neurosurg. 1989;70:175–82. doi: 10.3171/jns.1989.70.2.0175. [DOI] [PubMed] [Google Scholar]
- 36.Blancher A, Roubinet F, Grancher AS, et al. Local immunotherapy of recurrent glioblastoma multiforme by intracerebral perfusion of interleukin-2 and LAK cells. Eur Cytokine Netw. 1993;4:331–41. [PubMed] [Google Scholar]
- 37.Boiardi A, Silvani A, Ruffini PA, et al. Loco-regional immunotherapy with recombinant interleukin-2 and adherent lymphokine-activated killer cells (A-LAK) in recurrent glioblastoma patients. Cancer Immunol Immunother. 1994;39:193–7. doi: 10.1007/BF01533386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayes RL, Koslow M, Hiesiger EM, et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995;76:840–52. doi: 10.1002/1097-0142(19950901)76:5<840::aid-cncr2820760519>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 39.Hayes RL, Arbit E, Odaimi M, et al. Adoptive cellular immunotherapy for the treatment of malignant gliomas. Crit Rev Oncol Hematol. 2001;39:31–42. doi: 10.1016/s1040-8428(01)00122-6. [DOI] [PubMed] [Google Scholar]
- 40.Dillman RO, Duma CM, Schiltz PM, et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. 2004;27:398–404. doi: 10.1097/00002371-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Dillman RO, Duma CM, Ellis RA, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. 2009;32:914–9. doi: 10.1097/CJI.0b013e3181b2910f. [DOI] [PubMed] [Google Scholar]
- 42.Tsuboi K, Saijo K, Ishikawa E, et al. Effects of local injection of ex vivo expanded autologous tumor-specific T lymphocytes in cases with recurrent malignant gliomas. Clin Cancer Res. 2003;9:3294–302. [PubMed] [Google Scholar]
- 43.Quattrocchi KB, Miller CH, Cush S, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J Neurooncol. 1999;45:141–57. doi: 10.1023/a:1006293606710. [DOI] [PubMed] [Google Scholar]
- 44.Wood GW, Holladay FP, Turner T, Wang YY, Chiga M. A pilot study of autologous cancer cell vaccination and cellular immunotherapy using anti-CD3 stimulated lymphocytes in patients with recurrent grade III/IV astrocytoma. J Neurooncol. 2000;48:113–20. doi: 10.1023/a:1006456421177. [DOI] [PubMed] [Google Scholar]
- 45.Sloan AE, Dansey R, Zamorano L, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurg Focus. 2000;9:e9. doi: 10.3171/foc.2000.9.6.10. [DOI] [PubMed] [Google Scholar]
- 46.Tsurushima H, Liu SQ, Tuboi K, et al. Reduction of end-stage malignant glioma by injection with autologous cytotoxic T lymphocytes. Jpn J Cancer Res. 1999;90:536–45. doi: 10.1111/j.1349-7006.1999.tb00781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holladay FP, Heitz-Turner T, Bayer WL, Wood GW. Autologous tumor cell vaccination combined with adoptive cellular immunotherapy in patients with grade III/IV astrocytoma. J Neurooncol. 1996;27:179–89. doi: 10.1007/BF00177482. [DOI] [PubMed] [Google Scholar]
- 48.Kitahara T, Watanabe O, Yamaura A, et al. Establishment of interleukin 2 dependent cytotoxic T lymphocyte cell line specific for autologous brain tumor and its intracranial administration for therapy of the tumor. J Neurooncol. 1987;4:329–36. doi: 10.1007/BF00195603. [DOI] [PubMed] [Google Scholar]
- 49.Plautz GE, Barnett GH, Miller DW, et al. Systemic T cell adoptive immunotherapy of malignant gliomas. J Neurosurg. 1998;89:42–51. doi: 10.3171/jns.1998.89.1.0042. [DOI] [PubMed] [Google Scholar]
- 50.Kruse CA, Cepeda L, Owens B, Johnson SD, Stears J, Lillehei KO. Treatment of recurrent glioma with intracavitary alloreactive cytotoxic T lymphocytes and interleukin-2. Cancer Immunol Immunother. 1997;45:77–87. doi: 10.1007/s002620050405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plautz GE, Miller DW, Barnett GH, et al. T cell adoptive immunotherapy of newly diagnosed gliomas. Clin Cancer Res. 2000;6:2209–18. [PubMed] [Google Scholar]
- 52.Sampson JH, Heimberger AB, Archer GE, et al. Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor ReceptorVariant III Peptide Vaccination in Patients With NewlyDiagnosed Glioblastoma. J Clin Oncol. 2010 doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8:2773–9. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yajima N, Yamanaka R, Mine T, et al. Immunologic evaluation of personalized peptide vaccination for patients with advanced malignant glioma. Clin Cancer Res. 2005;11:5900–11. doi: 10.1158/1078-0432.CCR-05-0559. [DOI] [PubMed] [Google Scholar]
- 55.Ishikawa E, Tsuboi K, Yamamoto T, et al. Clinical trial of autologous formalin-fixed tumor vaccine for glioblastoma multiforme patients. Cancer Sci. 2007;98:1226–33. doi: 10.1111/j.1349-7006.2007.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clavreul A, Piard N, Tanguy JY, et al. Autologous tumor cell vaccination plus infusion of GM-CSF by a programmable pump in the treatment of recurrent malignant gliomas. J Clin Neurosci. 2010;17:842–8. doi: 10.1016/j.jocn.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 57.Choi BD, Archer GE, Mitchell DA, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19:713–23. doi: 10.1111/j.1750-3639.2009.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heimberger AB, Sampson JH. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther. 2009;9:1087–98. doi: 10.1517/14712590903124346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Terasaki M, Shibui S, Narita Y, et al. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen--A24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol. 2011;29:337–44. doi: 10.1200/JCO.2010.29.7499. [DOI] [PubMed] [Google Scholar]
- 61.See AP, Pradilla G, Yang I, Han S, Parsa AT, Lim M. Heat shock protein-peptide complex in the treatment of glioblastoma. Expert Rev Vaccines. 2011;10:721–31. doi: 10.1586/erv.11.49. [DOI] [PubMed] [Google Scholar]
- 62.Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50:337–44. doi: 10.1007/s002620100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–9. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 64.De Vleeschouwer S, Fieuws S, Rutkowski S, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14:3098–104. doi: 10.1158/1078-0432.CCR-07-4875. [DOI] [PubMed] [Google Scholar]
- 65.Caruso DA, Orme LM, Neale AM, et al. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro Oncol. 2004;6:236–46. doi: 10.1215/S1152851703000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kikuchi T, Akasaki Y, Abe T, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother. 2004;27:452–9. doi: 10.1097/00002371-200411000-00005. [DOI] [PubMed] [Google Scholar]
- 67.Ardon H, Van Gool S, Lopes IS, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neurooncol. 2010;99:261–72. doi: 10.1007/s11060-010-0131-y. [DOI] [PubMed] [Google Scholar]
- 68.Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–7. [PubMed] [Google Scholar]
- 69.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 70.Chang CN, Huang YC, Yang DM, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011 doi: 10.1016/j.jocn.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 71.Ardon H, De Vleeschouwer S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54:519–25. doi: 10.1002/pbc.22319. [DOI] [PubMed] [Google Scholar]
- 72.Ardon H, Van Gool S, Lopes IS, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neurooncol. 2010 doi: 10.1007/s11060-010-0131-y. [DOI] [PubMed] [Google Scholar]
- 73.Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89:1172–9. doi: 10.1038/sj.bjc.6601268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11:4160–7. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 75.Wheeler CJ, Black KL, Liu G, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68:5955–64. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 76.Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17:1603–15. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang CN, Huang YC, Yang DM, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18:1048–54. doi: 10.1016/j.jocn.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 78.Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008;15:114–21. doi: 10.1016/j.jocn.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 79.Schneider T, Gerhards R, Kirches E, Firsching R. Preliminary results of active specific immunization with modified tumor cell vaccine in glioblastoma multiforme. J Neurooncol. 2001;53:39–46. doi: 10.1023/a:1011856406683. [DOI] [PubMed] [Google Scholar]
- 80.Okada H, Lieberman FS, Edington HD, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of recurrent glioblastoma: preliminary observations in a patient with a favorable response to therapy. J Neurooncol. 2003;64:13–20. doi: 10.1007/BF02700016. [DOI] [PubMed] [Google Scholar]
- 81.Okada H, Lieberman FS, Walter KA, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med. 2007;5:67. doi: 10.1186/1479-5876-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 83.Disis ML. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunol Immunother. 2011;60:433–42. doi: 10.1007/s00262-010-0960-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Disis ML, Schiffman K, Gooley TA, McNeel DG, Rinn K, Knutson KL. Delayed-type hypersensitivity response is a predictor of peripheral blood T-cell immunity after HER-2/neu peptide immunization. Clin Cancer Res. 2000;6:1347–50. [PubMed] [Google Scholar]
- 85.Schwaab T, Fisher JL, Meehan KR, Fadul CE, Givan AL, Ernstoff MS. Dye dilution proliferation assay: application of the DDPA to identify tumor-specific T cell precursor frequencies in clinical trials. Immunol Invest. 2007;36:649–64. doi: 10.1080/08820130701674760. [DOI] [PubMed] [Google Scholar]
- 86.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–72. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 87.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–6. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Okada H, Pollack IF. Do we need novel radiologic response criteria for brain tumor immunotherapy? Expert Rev Neurother. 2011;11:619–22. doi: 10.1586/ern.11.49. [DOI] [PubMed] [Google Scholar]
- 89.Kleijn A, Chen JW, Buhrman JS, et al. Distinguishing inflammation from tumor and peritumoral edema by myeloperoxidase magnetic resonance imaging. Clin Cancer Res. 2011;17:4484–93. doi: 10.1158/1078-0432.CCR-11-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lambert LA, Gibson GR, Maloney M, Durell B, Noelle RJ, Barth RJ., Jr Intranodal immunization with tumor lysate-pulsed dendritic cells enhances protective antitumor immunity. Cancer Res. 2001;61:641–6. [PubMed] [Google Scholar]
- 91.Bedrosian I, Mick R, Xu S, et al. Intranodal administration of peptide-pulsed mature dendritic cell vaccines results in superior CD8+ T-cell function in melanoma patients. J Clin Oncol. 2003;21:3826–35. doi: 10.1200/JCO.2003.04.042. [DOI] [PubMed] [Google Scholar]
- 92.Lipson EJ, Drake CG. Ipilimumab: an Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fakhrai H, Mantil JC, Liu L, et al. Phase I clinical trial of a TGF-beta antisense-modified tumor cell vaccine in patients with advanced glioma. Cancer Gene Ther. 2006;13:1052–60. doi: 10.1038/sj.cgt.7700975. [DOI] [PubMed] [Google Scholar]
- 94.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lallana ECH, WF, Fadul CE. The Neuroimmunology of Cancer. In: Rizvi SACPK, editor. Clinical Neuroimmunology: Multiple Sclerosis and Related Disorder, Current Clinical Neurology. Springer Scient+Business Media, LLC; 2011. pp. 233–54. [Google Scholar]
- 96.Rutkowski S, De Vleeschouwer S, Kaempgen E, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91:1656–62. doi: 10.1038/sj.bjc.6602195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu G, Akasaki Y, Khong HT, et al. Cytotoxic T cell targeting of TRP-2 sensitizes human malignant glioma for chemotherapy. AACR Meeting Abstracts 2005. 2005:1003-a-. doi: 10.1038/sj.onc.1208519. [DOI] [PubMed] [Google Scholar]