

Abstract
The gut microbiota refers to the trillions of microorganisms residing in the intestine and is integral in multiple physiological processes of the host. Recent research has shown that gut bacteria play a role in metabolic disorders such as obesity, diabetes, and cardiovascular diseases. The mechanisms by which the gut microbiota affects metabolic diseases are by two major routes: (1) the innate immune response to the structural components of bacteria (e.g., lipopolysaccharide) resulting in inflammation and (2) bacterial metabolites of dietary compounds (e.g., SCFA from fiber), which have biological activities that regulate host functions. Gut microbiota has evolved with humans as a mutualistic partner, but dysbiosis in a form of altered gut metagenome and collected microbial activities, in combination with classic genetic and environmental factors, may promote the development of metabolic disorders. This paper reviews the available literature about the gut microbiota and aforementioned metabolic disorders and reveals the gaps in knowledge for future study.
1. Introduction
Obesity results from the accumulation of excess adipose tissue; however, it is not a single disorder but a heterogeneous group of conditions with multiple causes. Major causes of the increasing prevalence of obesity include behavioral and environmental factors, such as excessive consumption of energy-dense foods and a sedentary lifestyle [1]. Still, it is now recognized that a series of underexplored physiological and environmental predispositions underlies the traditional risk factors for obesity and its associated metabolic disorders. In this regards, gut microbiota has recently been proposed as an environmental factor responsible for the weight gain and the altered energy metabolism that accompanies the obese state. Gut microbiota affects host metabolism by increased energy extraction, immune system modulation, and altered lipid metabolism. Furthermore, both the physical presence of bacteria and the metabolites from bacteria are responsible for these effects. An evaluation of the evidence indicating that the gut microbiota plays a role in energy balance and obesity-associated diseases such as diabetes and cardiovascular diseases (CVDs), as well as factors that mediate these hard endpoints, is presented in this paper.
2. Composition of the Microbiota in the Human Gastrointestinal Tract
Microbiota is a collection of microorganisms including bacteria, archaea, viruses, and some unicellular eukaryotes. Microbiota is associated with every multicellular organism on earth. In humans, it has been estimated that 1014 microorganisms reside in various parts of the body such as the surface of skin and in the gastrointestinal, genitourinary, and respiratory tracts. The gastrointestinal tract, which has the largest number of microorganisms in humans, is comprised of specialized compartments such as the mouth, esophagus, stomach, small intestine, large intestine (colon), rectum, and anus. Each of these compartments has unique physiological functions and anatomical structures. As a result, the chemical environment and habitable microorganisms differ tremendously in each compartment (Figure 1). In the colon, up to 1012 microorganisms/mL are reported. This is by far the highest density found in humans [2–4], and the vast majority of microorganisms belong to the phyla of Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria, with relatively lower numbers belonging to Fusobacteria, Verrucomicrobia, and TM7 [4–8]. Fungi and Archaea may also be resident [6, 9, 10], but comprise less than 1% of the total inhabitants. Altogether, a human gastrointestinal microbiota comprises more than 10,000 different phylotype s, most of which have not been characterized by a culturing technique or even sampled to date [8, 11]. However, this notion has been challenged by a recent finding by Goodman et al. who reported that 99% of the bacteria characterized in the phylum, class, and order level could also be found in the biomass from an anaerobic culture of original fecal samples [12].
Figure 1.

Association of the microbiota with humans. Microbiota presents in all parts of our body which has direct contact with external environment. The numbers of bacteria in the mouth (1010), on the skin (1012), and in the distal gut (1014) are presented in a relation to total number of parenchymal cells (1012). The composition of the microbiota in the digestive tract greatly differs in each of specialized compartments as illustrated [6, 13–15]. Physiological functions and chemical environment of each compartment are likely key factors influencing the bacterial habitants.
3. Gut Microbiota and Obesity
3.1. The Gut Microbiota Profile in Obesity
Several studies reported by members of the Gordon's lab showed that the gut microbiota differs at the phylum level depending on weight status [7, 16, 17]. In agreement with results from animal studies, it appears that human obesity is associated with a low abundance of intestinal Bacteroidetes and high abundance of Firmicutes. However, this result has been contradicted by other studies. Duncan et al. showed that no difference was found in the proportions of Bacteroidetes and Firmicutes in the feces of lean and obese subjects [18]. In another study, overweight and obese subjects had a ratio of Bacteroidetes to Firmicutes in favor of Bacteroidetes [19]. Recently, Jumpertz et al. applied the state-of-the-art pyrosequencing technique to examine the bacterial 16S rRNA genes and reported that no phylum level difference was found in between the obese and lean fecal microbiota [20]. Therefore, the phylum level difference of the gut microbiota between obese and lean individuals might not be universally true.
Compositional changes of the human gut microbiota in response to weight change have been examined by many groups. Ley et al. monitored the fecal gut microbiota in 12 obese subjects participating in a weight-loss program by consuming restricted diets for a year [7]. Following weight loss, the proportion of Bacteroidetes increased while the number of Firmicutes reciprocally decreased. Fecal microbiota compositions of overweight and obese adolescents also were determined in the EVASYON study group. After 10 weeks of energy restriction and exercise, participants who lost more than 4 kg body weight showed a significant increase in the population of Bacteroides fragilis group, as determined by a quantitative PCR technique [21]. Another EVASYON cohort study also reported that an increase in Bacteroides/Prevotella group positively correlated with the amount of weight loss [22].
To date, the most effective treatment for morbid obesity is gastric bypass surgery. The consequences of gastric bypass surgery in the composition of the fecal microbiota have been studied. Zhang et al. found that obesity was associated with an increase of family Prevotellaceae prior to the surgery, and following surgery Prevotellaceae was reduced to the level of lean individuals whereas other bacteria such as family Enterobacteriaceae and genus Akkermansia were enhanced [23]. In another clinical study, the number of fecal E. coli species was increased at 3 and 6 months after having gastric bypass surgery [24]. The impact of gastric bypass surgery on the gut microbiota profile in animals was an increase in the proportion of Enterobacter hormaechei by 200- and 42.8-fold at 2 and 8 weeks after the surgery, respectively [25]. Since both E. coli species and E. hormaechei belong to the family Enterobacteriaceae, a proportional increase of Enterobacteriaceae could be a common outcome of gastric bypass surgery. However, it is not known whether changes in Enterobacteriaceae populations are linked to the rapid weight loss and improvement of insulin sensitivity resulting from gastric bypass surgery.
3.2. Gut Microbiota as a Modulator of Obesity: Host Genome and Dietary Fats
3.2.1. Host Genome
Animal studies have shown that the host genome modulates gut microbiota composition. Obesity caused by a leptin mutation in mice (ob/ob) is associated with altered gut microbiota profiles [16, 17]. Ley et al. [16] analyzed more than 5000 bacterial 16S rRNA gene sequences from the cecal content of ob/ob mice, their lean ob/+ and +/+ siblings, and their ob/+ mothers. Homozygous ob/ob mutation coincided with 50% fewer Bacteroidetes and a proportional increase in Firmicutes in the gut. The gut microbiota profile of mice with a leptin receptor mutation (db/db) has recently been published [26]. In accordance to ob/ob mice, db/db mice cecal microbiota was characterized with higher abundance of phylum Firmicutes and lower abundance of phylum Bacteroidetes compared to the lean mice. Furthermore, certain genera such as Odoribacter, Prevotella, and Rikenella only present in the ceca of db/db mice, whereas Enterorhabdus presents in the ceca of lean mice. However, it is possible that the alterations in the bacterial composition in ob/ob and db/db mice are secondary to hyperphagia. A careful pair-feeding experiment would be needed to show if the amount of food intake contributes to the signature of gut microbiota associated with genetic obesity in mice.
3.2.2. Dietary Fats
While the host genotype has been proven to affect microbiota, the effect of diet, specifically dietary fat, also plays an important role in determining bacterial composition. A high-fat-fed animal displays a significant shift in both bacterial and metagenomic profiles as compared to an animal on a normal, chow diet. Western diet-associated cecal microbiota is characterized with a reduction in the relative abundance of Bacteroidetes and an increase in the relative abundance of Firmicutes. In particular Mollicutes, a class of Firmicutes, has been found to be significantly more prevalent in CONV mice fed a Western-type diet [27]. An increase of genes involved in the import and processing of sugars in the gut metagenome was also found in Western diet-fed mice [27]. Examination of the role of the Western diet-associated cecal microbiota in facilitating weight gain has revealed that ex-GF mice receiving the Western diet-associated gut microbiota gained significantly more body fat than the mice receiving the chow diet-associated gut microbiota [27]. Murphy et al. reported that C57BL/6J mice responded to HF feeding with a progressive increase in abundance of Firmicutes over time [28]. Ravussin et al. also reported that in weight-matched animals, high-fat feeding was associated with an increase in the family of Lachnospiraceae and genera Bacteroides and Mucispirillum whereas low fat feeding was associated with an increase in genus Allobaculum [29].
The literature evidence further suggests that quality of the diet instead of the weight of animals is stronger modulator to the composition of the gut microbiota. It was found that diet explained 57% of the bacterial variation in the gut while genetic background only accounted for 12% of the variation in animals [30], which suggests the primacy of diet in determining the composition of the gut microbiota. Hildebrandt et al. reported a similar finding in that mice deficient in RELMβ gene had moderate impacts on the gut microbiota profile whereas high-fat feeding caused greater changes in the composition of the gut microbiota [31]. Metagenomic analysis of normal weight (RELMβ deficient) and obese mice (wildtype ) fed a high-fat diet found that the diet, rather than weight or genetic status, correlated with an increase in nutrient-transport genes and a decrease in amino acid and carbohydrate metabolism genes [31]. These data support the influence of the diet composition in the diversity and profiles of the gut microbiota in mice; however, the mechanism by which high-fat diets change the microbiota's functionality requires further study. Within the context of obesity, it appears that genetics may determine initial gut composition, but dietary fat is a potent modulator.
3.3. Mechanisms Linking the Gut Microbiota to Obesity
3.3.1. Inflammation
The interaction between the gut environment and diet (modulated by bacterial composition) may explain why genetically identical mice respond differently to a high-fat diet—some are prone and some resistant to weight gain. Sprague-Dawley male rats prone to weight gain exhibited ileal inflammation, decreased intestinal alkaline phosphatase activity (enzyme which detoxifies the bacterial component known to cause inflammation, lipopolysaccharide, or LPS), and increased innate immune system activation in the luminal wall as compared to the obesity-resistant rats [32]. Both obesity-prone and obesity-resistant rats had an overall decrease in bacteria on the high-fat diet; however, Enterobacteriales increased in the obesity-prone rats on a high-fat diet. In a study of genetically identical male rats, infusing a low level of LPS for 4 weeks caused the same amount of weight gain as a high-fat diet [33]. Rats that had a knockout of an immunoprotein (CD14), which is necessary to cause an inflammatory reaction to LPS, were immune to weight gain [33]. Together the data show that rats naturally prone to weight gain on a high-fat diet have intestinal inflammation, inflammation alone can cause weight gain in normal rats, and the absence of inflammation protects rats against weight gain from a high-fat diet. Therefore, it is hypothesized that the inflammatory mileu is integral in the development of obesity.
3.3.2. Fiaf
The mere presence of gut microbiota has recently been shown to contribute to obesity. A series of experiments comparing the germ-free (GF) to conventional (CV) mice concluded that the development of diet-induced obesity requires the colonization of complex gut microbiota [34–36]. Microbiota transplantation experiments showed that the accumulation of body fat depends on the type of the gut microbiota, which supports the role of the gut microbiota in the development of obesity [17, 27]. Mechanistically, the acceleration of fat mass gain in conventionalized mice can be partly explained by the suppression of hepatic de novo lipogenesis and by the inhibition of triglyceride storage in the white adipose tissue. The latter effect is thought to be caused by an excessive production of Fiaf (fasting-induced adipocyte factor or ANGPTL4) in the intestine of the GF mice [34]. Fiaf inhibits lipoprotein lipase (LPL) thereby blocking the disassociation of fatty acids from triglycerides for uptake into tissues and upregulating fatty acid oxidation and uncoupling proteins, potentially reducing the amount of fat storage in GF mice [37]. Fiaf also plays a role in the metabolic adaption to fasting via PPAR activation [38]. The importance of Fiaf as a mediator by which the gut microbiota regulates body weight was demonstrated in GF Fiaf-knockout mice. Unlike the GF wild-type animals, GF mice lacking Fiaf responded to a high-fat diet with excessive weight gains [39].
Although the function of Fiaf in blocking LPL activity is clear, the extent to which Fiaf from the intestine as compared to adipose tissue is causing this effect has not been elucidated. Initial studies characterizing Fiaf showed that it is located primarily in white and brown adipose tissue, as well as in the liver during fasting, and has a very low expression in small intestine [38]. Moreover, Fleissner et al. found an increase in intestinal Fiaf mRNA in GF mice but no increase in secreted plasma protein levels compared to CONV mice [40]. In a global Fiaf-knockout model, all Fiaf including that in the adipose tissue and in the intestine is eliminated, which diminishes the ability to determine the tissue specific contribution of Fiaf to the phenotype. Therefore, available evidence does not indicate that intestine-derived Fiaf has a significant impact on regulating the triglyceride storage in the adipose tissue, and perhaps the lack of weight gain in GF mice is due to other mechanisms. Determining the physiological contribution of intestinal Fiaf as well as its regulation by specific bacterial populations and metabolites warrants further investigation.
3.3.3. Energy Harvesting
Another mechanism by which the gut microbiota affects body weight is by increasing energy harvesting from dietary fibers. The intestinal microbiota breaks down indigestible polysaccharides (i.e., fiber) to short-chain fatty acids (SCFAs) providing 80 to 200 kcal per day or about 4–10% of daily energy intake in normal adults [41]. Metagenomic analysis of ob/ob cecal microbiota revealed that ob/ob-specific gut microbiota was enriched with bacterial genes capable of utilizing and fermenting dietary fibers [17]. The greater cecal SCFA concentrations of acetate and butyrate and lower fecal energy contents of ob/ob than in lean animals suggest additional absorption of SCFA was absorbed by the intestine of the ob/ob mice [17]. Murphy et al. independently evaluated whether energy harvesting is different between ob/ob and lean mice and found a similar result to that obtained by Turnbaugh et al. with 7-week-old mice but not at 15-weeks-old animals as older mice showed a similar amount of cecal SCFAs and fecal energy [28]. The concept of changing energy harvesting by the gut microbiota has also been tested in humans. Jumpertz et al. reported that the amount of stool energy in proportion of ingested calories was positively correlated with the abundance of phylum Bacteroidetes and negatively correlated with the abundance of phylum Firmicutes in the feces [20]. An estimate of 150 kcal difference can be achieved with a change of 20% relative abundance of Firmicutes and corresponding decrease of Bacteroidetes in the stool of lean individuals. Thus, excessive calories taken in the form of SCFAs from microbiota metabolism of fiber may be a contributing factor in the obese state.
3.3.4. Food Intake and Energy Expenditure
Food intake and energy expenditure are two key factors that determine energy balance in humans and animals. In humans, the role of gut microbiota in food intake has not been tested, and in animals the results are inconclusive. As nicely summarized by Wostmann, earlier work from the 1960s and 70s showed that food intake is lower in GF than in Conv rats [42]. Bäckhed et al. showed that GF C57BL/6J mice consumed more chow than Conv mice [35]; however, no relationship was found between chow intake and the presence or absence of the gut microbiota in C3H mice ([40] and personal communication with Professor Michael Blaut). On a Western diet, reported food intake in GF and Conv mice was similar, regardless of their genetic background ([34, 40] and personal communication with Professor Michael Blaut). On a semisynthetic high-fat diet, we observed that GF C57BL/6J mice consumed a less amount of food than Conv mice in a 10-week feeding experiment [36], but the same relationship was not observed in C3H mice [40]. Inconsistent findings of food intake in GF mice might be due to the species of animals (rat versus mouse), strain of mouse (C57BL6/J versus C3H), the quality of the diet (chow, Western, or high-fat diet), and the sample size in different studies.
Similar to food intake, the role of the gut microbiota on energy expenditure in humans is not known, and only limited information is available from animal models. Early work showed that basal metabolic rate, cardiac output, and body temperature of GF rats were lower than those of their Conv counterparts, indicating that microbiota may affect energy expenditure in animals [43]. Recent observations also indicated that the oxygen consumption was 25 to 40% less in GF than in Conv C57BL/6J mice [34]. This conclusion has been independently confirmed in GF C3H mice [40]. The comparison of energy expenditure in different gnotobiotic mice colonized with different gut microbiota would be needed to demonstrate the role of gut microbiota in energy expenditure.
4. Microbiota in Diabetes and Cardiovascular Diseases
4.1. Clinical Evidence
Few studies have reported the relationship between the gut microbiota composition and disease states, such as diabetes and cardiovascular diseases (CVDs), in humans. Larsen et al. showed that 16S rDNA sequences representing the class of Bacteroidetes were slightly higher in the diabetic subjects than in nondiabetic subjects, although the difference was not statistically significant [44]. In this study a lower proportion of class Clostridia and higher proportion of class Betaproteobacteria were associated with diabetes. Another study compared the fecal microbiota profile of three groups of subjects (lean control, obese diabetic, and obese nondiabetic) with a quantitative PCR technique and found diabetes was associated with a reduction of Faecalibacterium prausnitzii species [24]. A case-control study with 16 type 2 diabetics and 12 healthy controls found decreased B. vulgatus and Bifidobacterium in the diabetic group [45]. The interaction between the oral and gut microbiota and CVD, in particular atherosclerosis, has recently been discovered [46]. Interestingly, high abundance of certain bacteria was found in the atherosclerotic plaques and in the mouth microbiota, but no relationship was found between the plaque microbiota and the gut microbiota of affected patients [46].
4.2. Animal Studies
The role of the gut microbiota in type 1 and 2 diabetes has been researched in mouse models. Wen et al. showed that the development of type 1 diabetes in MyD88-deficient NOD mice, a model of type 1 diabetes, was dependent on the presence or absence of the gut microbiota [47]. Indeed, nearly all GF MyD88-deficient NOD mice developed diabetes, whereas the ex-GF mice with the same genetic background colonized with a consortium of 6 bacteria strains had much reduced incidence. This study demonstrates that bacterial presence is protective against development of type 1 diabetes.
Obesity and chronic low-grade inflammation are common aspects of both type 2 diabetes and CVD [48]. We and others postulated that gut microbiota could contribute to the onset of insulin resistance. In one study, a 2-week treatment with broad range antibiotics, norfloxacin and ampicillin, significantly reduced the number of the cecal microbiota in ob/ob mice, and the treated ob/ob mice exhibited marked reductions in fasting blood glucose levels and glucose intolerance [49]. Increase of liver glycogen and decrease of liver triglyceride stores were accompanied with improved glycemic control. This study showed that metabolic health was improved in ob/ob mice with reduced gut bacteria. To further examine if glucose tolerance of mice is affected by the presence or absence of the gut microbiota, we fed both GF and Conv C57BL/6J mice with a high-fat diet, and our results showed that GF mice did not develop diet-induced obesity and glucose intolerance [36]. Since all results based on comparisons between the GF and Conv mice cannot be extrapolated to represent normal physiology, evidence from microbiota transplantation studies will be needed to conclude if the gut microbiota composition predisposes the host to diet-induced glucose intolerance.
5. Possible Mechanisms: How the Gut Microbiota Affects Obesity-Related Metabolic Diseases
There is a reciprocal relationship between the host and its microbiome. Changes in the number of bacteria, proportion of certain phylotypes, and bacterial activities of the microbiome are sensed by the host. The main pathways by which the host and bacteria interact are when bacteria or the bacterial metabolites enter the host's circulation (Figure 2). There are multiple systems in our body to sense environmental cues, but the aforementioned bacterial components and metabolites have been implicated to the gut microbiota sensing with evidence related to the development of metabolic diseases.
Figure 2.
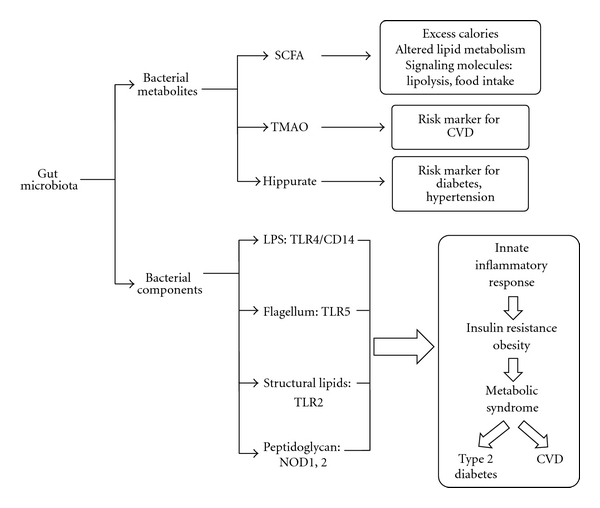
Gut microbiota can have an effect on the host's health via bacterial metabolites, that is, SCFA and TMAO, and immune responses to bacterial components, that is, TLR4 sensing of LPS. In general, bacterial metabolites have varied effects on metabolism and are markers of risk for disease, whereas bacterial components cause an innate inflammatory response resulting in insulin resistance leading to metabolic syndrome, type 2 diabetes, and CVD.
5.1. Bacterial Components
Our innate immune system is capable of sensing various type s of bacterial components via pattern recognition receptors (PRRs). In general, there are two type s of PRRs, toll-like receptors (TLRs) and Nod-like receptors (NLRs). TLRs are highly conserved transmembrane receptors, and each TLR recognizes specific ligands and is capable of activating inflammatory responses. In humans, some TLRs (TLR1, 2, 4, 5, 6, and 10) are expressed in the cell surface, but other TLRs (3, 7, 8 and 9) locate in the membrane of endolysosomal compartments [50]. Twenty-two NLRs have been identified in humans, but only two of them (NOD1 and NOD2) are well characterized [51]. In contrast to TLRs that are associated with membranes, NOD1 and NOD2 are located in cytoplasm [52]. PRRs recognize the structures of bacteria (e.g., LPS, lipoproteins, and peptidoglycan) in order to signal the immune system of a pathogen. PRRs not only engage in pathogen sensing, but are also implicated in the development of metabolic diseases such as insulin resistance and cardiovascular diseases. Therefore PRRs are perfect candidates for sensing the changes of the gut microbiota and more importantly mediating the subsequent inflammatory and metabolic responses.
5.1.1. LPS and TLR4 Sensing
Low-grade inflammation is a common comorbidity of type 2 diabetes and CVD. Although the cause of metabolic inflammation is unclear, there is evidence to suggest that LPS originating from Gram-negative bacteria in the gut induces low-grade inflammation and insulin resistance [48]. The presence of LPS is constantly monitored by the host via the TLR4 [53], which also recognizes compounds of nonmicrobial origin such as saturated fatty acids [54]. Evidence has suggested that metabolic endotoxemia (LPS, 50–100 pg/mL) can cause chronic low-grade inflammation and mild disturbances in energy metabolism implicated in the development of CVD and type 2 diabetes [55, 56]. A series of animal experiments has helped elucidate the relationship between gut microbiota, LPS, diet, and insulin sensitivity. First, when LPS was chronically infused to mice, it resulted in mild obesity and hepatic insulin resistance [33]. Mice deficient in TLR4 were protected from high-fat-diet-induced obesity and insulin resistance [57–59]. Second, the authors showed increased endotoxemia in mice consuming a high-fat diet [33, 60]. Finally, it was found that ob/ob mice or high-fat-diet-fed C57BL/6J mice treated with ampicillin and neomycin had altered gut microbiota composition and reduced endotoxemia with improved glucose tolerance [61]. Furthermore, a reduction of LPS by prebiotic (oligo-fructosaccharide) treatment significantly enhanced the whole body glucose tolerance and inflammatory markers in the liver and adipose tissue of mice fed a high-fat diet [62, 63]. Together, these data suggest that LPS enters circulation more readily while being on a high-fat diet, increased plasma LPS has a detrimental effect on glucose metabolism, and altering the microbiota can alleviate endotoxemia and insulin resistance. Therefore, available evidence suggests that LPS plays a critical role in the development of obesity-and obesity associated insulin resistance and low-grade inflammation.
Human studies have provided support for this hypothesis as well. In a cross-sectional study of 50 human subjects, fasting LPS levels in type 2 diabetics were significantly higher than the age, BMI, and sex-matched nondiabetic controls [64]. Furthermore, treatment of a subgroup of newly diagnosed type 2 diabetics with insulin-sensitizing drugs for 10 weeks decreased both endotoxin and insulin levels, such that the greater the change in endotoxin, the greater the change in insulin sensitivity [64]. Risk of incident diabetes was significantly associated with higher endotoxin (LPS) activity, such that those in the highest quartile of LPS activity had a 52% increased risk of having diabetes as compared to the lowest quartile [65]. Elevated LPS levels in type 1 diabetic and kidney vascular disease (IgAGN) patients were associated with higher serum triglycerides, earlier onset of diabetes, increased diastolic blood pressure, and elevated marker of inflammation, monocyte chemoattractant protein-1 [66]. In sum, Pussinen et al. conclude that “endotoxemia is a key player in the pathogenesis of diabetes and microbes may have a central role [65].”
Metabolic endotoxemia has been linked to the development of CVD as well. In a 5-year epidemiological study of 516 middle-aged men and women, those with plasma LPS levels over 50 pg/mL had a threefold increase in risk of developing atherosclerosis while the subpopulation of smokers or ex-smokers with the same level of LPS had a 13-fold increase [67]. In another study, endotoxin and TNFα were elevated systemically in those with acute heart failure as compared to stable heart failure or normal controls [68]. Nevertheless, intervention studies which lower LPS plasma levels and cause a subsequent decrease in CVD risk have not been conducted to our knowledge, and such result would clarify the importance of LPS in the etiology of CVD.
5.1.2. LPS Transport
The transport of LPS into blood can be through a number of pathways (Figure 3). Compromised intestinal tight junctions or “a leaky gut” enhances the possibility of bacteria translocation and uptake of the bacterial products. The bacterial products induce an inflammatory response, which is central to the pathophysiology of CVD and diabetes and provides a link between diseases of the gut and the vasculature. Indeed, individuals with inflammatory bowel disease were at a higher risk of developing coronary artery disease despite having lower rates of traditional risk factors (dyslipidemia, hypertension, obesity, diabetes) than their age-matched controls in a longitudinal cohort study [69]. In a study mentioned previously, obesity-prone rats were found to have increased gut permeability and higher plasma LPS as compared to obesity-resistant mice [32]. To further explore the mechanism by which leaky gut is related to the microbiome and diet, Cani et al. found that male C57BL6/J mice fed a high-fat diet had increased intestinal permeability and decreased expression of genes encoding for tight junction proteins, but administration of antibiotics with the high-fat diet effectively blunted these negative effects [33, 61]. In follow-up experiments, it was confirmed that the obese mouse controls had the highest levels of intestinal permeability; prebiotics were found to improve markers of intestinal permeability via glucagon-like peptide-2-mediated and cannabinoid-receptor-1-receptor-(CB1-) mediated pathways [70, 71].
Figure 3.
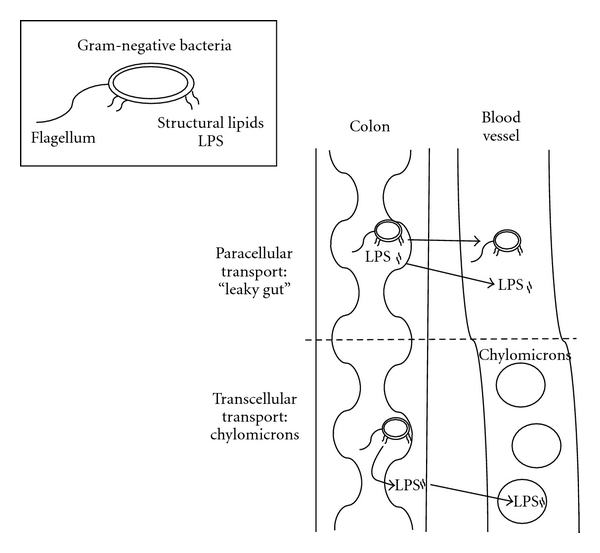
Diagram of the two primary methods of LPS entry into circulation: paracellular and transcellular. In paracellular transport or “leaky gut” either the entire bacteria or bacterial components such as LPS enter the circulation. In transcellular transport, chylomicrons take up LPS in circulation.
Another pathway by which LPS may enter the blood stream is its integration into chylomicrons; however, the mechanism has not been thoroughly elucidated. It is hypothesized that a certain amount of LPS is present within the enterocyte [72], attached to or within chylomicrons [73]. When chylomicron formation occurs during fat absorption, LPS is then transported into the lymph circulation with chylomicrons. It has been shown in humans that increased chylomicron formation due to a high-fat meal causes greater LPS transport postprandially as compared to a low-fat meal or no meal [60, 74]. Levels of sCD14 and IL-6, both markers of acute inflammation, were elevated after a mixed meal containing lipid in association with higher LPS levels [75]. In both animal and cellular models, chylomicron formation stimulated by a high-fat challenge significantly enhanced the transport of LPS from the intestinal lumen or enterocyte to circulation [73, 75]. This high-fat challenge did not affect the gut tight junctions in mice, and chemically inhibiting chylomicron formation in the presence of a high-fat stimulus effectively blocked LPS translocation. Together these data suggest that there is transcellular and chylomicron-dependent transport of LPS, which is induced by high-fat intake. While evidence for a role of LPS in chronic disease is intriguing, issues related to LPS measurement and LPS contamination make the study of this mechanism difficult; therefore, developing a fully quantitative LPS assay with a greater dynamic range should be considered for future study. Nevertheless, the role of the gut microbiota in pathogenesis of diabetes and CVD should not be overlooked and understanding the contribution of inflammation from gut microbiota may provide insight on new therapies to decrease disease risk.
5.1.3. Flagellum and TLR5; Structural Lipids and TLR2
TLR5 is highly expressed in epithelial cells of the intestinal mucosa [76] and is involved in mediating immune response through recognition of bacterial flagellin [77]. Vijay-Kumar et al. discovered that TLR5-knockout (T5KO) mice and wild-type littermates harboured different profiles of the gut microbiota. The changes of T5KO gut microbiota were mainly at the species level, which is in contrast with the ob/ob mouse model of obesity, where the alternation of the microbiota was characterized by a phylum-level shift [78]. In T5KO mice, altered gut microbiota profile was associated with metabolic syndrome characterized with increased visceral fat deposition, dyslipidemia, hypertension, and insulin resistance. The cause of metabolic syndrome by the T5KO gut microbiota was demonstrated with a transplantation study, and the results show that GF wild-type mice receiving the T5KO gut microbiota developed metabolic syndrome [78].
Similarly, TLR2 recognizes a wide array of molecules including structural lipids, lipoproteins, and lipopeptides found on the surface of bacteria [79]. Ligand-induced dimerisation of TLR2 with either TLR1 or TLR6 triggers a cascade of kinase activations and eventually activation of NFκB [80]. Several groups have shown that lacking TLR2 prevents mice from developing to high-fat-diet-induced obesity, hepatic steatosis, and insulin resistance [81, 82]. The liver, instead of skeletal muscle and white adipose tissue, is the major tissue with increased insulin sensitivity in TLR2-deficient mice [82]. However, disrupting TLR2 in the skeletal muscle and white adipose tissue with TLR2 antisense oligonucleotides also improved the whole-body insulin sensitivity of mice [83]. Together, data indicate several TLRs not only can sense the bacterial structures but also can mediate insulin resistance once activated.
5.1.4. Peptidoglycan and NOD1 and NOD2
Nucleotide oligomerization domain (NOD) 1 and 2 are intracellular sensors of bacterial peptidoglycan (PGN). NOD 1 preferentially responds to PGN fragment, containing meso-diaminopimelic acid (meso-DAP) [84, 85], which was found widely in Gram-negative but also in some Gram-positive bacteria, whereas NOD2 recognizes monosaccharide with a dipeptide stem such as muramyl dipeptide (MDP), which was found in both Gram-positive and Gram-negative bacteria [86, 87]. The role of NODs in controlling immune responses to bacterial ligand is reviewed by Clarke and Weisser, and these responses include the activation of innate defense, mediating antimicrobial peptide production, influencing phagocytosis of neutrophils and macrophages [52].
In addition to their role in regulating innate and adapted immunity, activation of NODs has been implicated in causing insulin resistance. In isolated human preadipocytes, a treatment with NOD1 ligand DAP led to an activation of NF-kB and increase of IL-6 secretion [88]. In 3T3-L1 adipocytes, DAP induces insulin resistance with a decrease in insulin-stimulated Akt phosphorylation (Ser 473 and Thr 308) and a reduced phosphorylated IRS-1 (Tyr 632) [89]. Impaired insulin signalling by DAP or a synthetic NOD1 ligand FK565 translated to a significant reduction of insulin stimulated glucose uptake in 3T3-L1 adipocytes [89, 90]. In vivo, the FK565 treatment resulted in whole-body insulin resistance with decreased glucose infusion rate, reduced glucose disposal rate, and diminished suppression of hepatic glucose production by insulin during a hyperinsulinemic euglycemic clamp [90]. The FK565-induced insulin resistance was absent in mice deficient in functional NOD1, which suggests the functional role of NOD1 activation in causing whole-body insulin resistance [90]. Similar to NOD1, activation of NOD2 also has been shown to induce insulin resistance. Tamrakar et al. reported that addition of NOD2 ligand MDP dose dependently suppressed basal and insulin-stimulated 2-DG glucose uptake in the cultured L6-GLUT4myc myotubes [91]. These data suggest that bacterial PGN can cause insulin resistance in tissues but each insulin-sensitive tissue responds to a different bacterial PGN. The liver and adipose tissue primarily react to NOD1 ligand whereas skeletal muscle preferentially reacts against NOD2 ligand. Together, available data suggest that bacterial PGN fragments can cause insulin resistance and PRRs such as NOD1 and NOD2 are responsible for triggering a cascade of events leading to inflammatory responses and insulin resistance.
5.2. Bacterial Metabolites
Bacterial metabolites encompass a group of molecules found in circulation that are a product of bacterial metabolism. The following bacterial metabolites: SCFA, TMAO, and hippurate, have been shown to affect diabetes or CVD risk.
5.2.1. SCFA Production and Bacterial Composition
Gut microbes ferment indigestible material in the colon to produce SCFAs, which is affected by the composition of the gut microbiota. Martin et al. applied 1HNMR spectroscopic analysis and examined the metabonomic profiles of feces from mice colonized with different gut microbiota [92]. Mice colonized with a human baby microbiota with a supplement of L. Paracasei differed from conventional and conventionalized mice with less fecal butyrate and propionate and more succinate. However, near identical metabonomic pattern was observed between the conventional and conventionalized mice. In another study, conventional rats were treated with penicillin and streptomycin, and the amounts of fecal acetate, n-butyrate, and propionate were greatly reduced by the treatment [93]. These results clearly demonstrated the changes of fecal SCFA concentrations as a result of gut microbiota modulation. Turnbaugh et al. have looked at the role gut microbiota profile plays in the production of SCFA at two levels: the capacity to produce SCFA and the type of SCFA produced. By looking at metagenomic differences between lean and obese mice, a higher concentration of butyrate and acetate in the caeca of obese mice was observed, while propionate was not significantly different between the two groups [17]. Although the increase in SCFA production capacity may be directly linked to the difference in microbiota profile between lean and obese Conv mice, other confounding factors such as calorie and fiber intake associated with the hyperphagic phenotype of ob/ob mice could be driving the upward changes in SCFA production and should be considered in future studies.
5.2.2. SCFA and Lipid Metabolism
As mentioned earlier, gut-derived SCFAs supply energy to the host. In addition, SCFAs are suggested to play a role in the regulation of blood lipids and therefore may have an effect on CVD risk. The three primary SCFAs produced by microbiota fermentation are acetate, propionate, and butyrate with the former two being absorbed into portal circulation and the latter used as an energy source for colonocytes. Acetate (or acetyl-CoA) is a substrate for cholesterol synthesis and is hypothesized to increase in plasma cholesterol, whereas propionate may decrease cholesterol by inhibiting acetyl-CoA synthetase activity, the enzyme that converts acetate to acetyl-CoA. Therefore, propionate is thought to inhibit the cholesterol-raising effect of acetate; however, the hypothesis is controversial.
Human studies have provided evidence to support the acetate: propionate hypothesis. In a cross-sectional study of normolipidemic men and women, the acetate: propionate ratio was positively associated with total and LDL cholesterol in men, but not women, even after adjusting for age and BMI [94]. Lactulose, a synthetic, nonabsorbable sugar, which is metabolized by microbiota to produce high levels of acetate, included in the diet of 6 healthy volunteers for 2 weeks, resulted in a significant increase in total and LDL cholesterol and apolipoprotein B with a trend towards increased serum acetate as compared to the control diet [95]. Furthermore, in a human comparative study using rectal infusions of acetate, propionate, or both, acetate increased serum cholesterol whereas propionate did not affect serum cholesterol, and the combination of the two did not cause an increase in serum cholesterol [96]. It should be noted that acetate is always produced to a greater extent than propionate and butyrate, and in vivo levels of propionate in the portal vein are quite low [97]; therefore, the optimal acetate:propionate ratio needed to lower serum cholesterol and how that translates to fiber intake in humans require further study.
Animal and in vitro studies have provided supporting evidence concerning acetate and propionate's role in cholesterol and fatty acid synthesis. The role of propionate in lipid metabolism and overall health is well reviewed by Hosseini et al. [97]. However, it is well known that there are many differences between the lipid metabolism of rodents and humans; therefore, more research must be done in models with similar lipid metabolism, such as hamsters.
5.2.3. SCFA as Signaling Molecules
In addition to their other physiological roles, SCFAs are signalling molecules which may help explain some mechanisms by which gut microbiota affects obesity and chronic diseases. Recently it has been discovered that SCFAs act as a ligand for G-protein-coupled receptors GPR41 (FFA3) and GPR43 (FFA2) [98–100]. FFA2 exhibits binding potency to SCFAs in the order of carbon chain length (C2 = C3 > C4 > C5 = C1), whereas FFA3 prefers to bind SCFAs with longer chain length (C3 = C4 = C5 > C2 = C1) [98–100]. FFA2 mRNA can be detected in various tissues, and the highest expression is found in immune cells such as neutrophils, monocytes, peripheral blood mononuclear cells, B cells, and polymorphonuclear cells [98–100]. Expression of FFA2 mRNA was also detected in the skeletal muscle [100], adipose tissue [101], distal ileum, and colon [102]. FFA3 is more widely distributed than FFA2. A high level of FFA3 expression can be found in the adipose tissue, pancreas, spleen, lymph nodes, bone marrow, blood mononuclear cells, and 3T3-L1 and 3T3-F442 preadipocytes [98, 99].
Biological functions of FFA2 and FFA3 in modulating lipid metabolism have been reported. Infusion of acetate reduced circulating free fatty acids in C57BL/6J and ob/ob mice, but the antilipolytic effect was abolished in FFA2-knockout mice [103], suggesting that FFA2 is partially responsible for the results of acetate infusion. Therefore, activation of FFA2 by short-chain fatty acids might be one of regulatory mechanisms controlling the basal lipolysis and circulating fatty acid concentrations.
SCFA signaling also affects pathways related to food intake. Leptin is an adipokine produced by the adipose tissue, and one of its functions is to negatively control the food intake. Recently, Xiong et al. demonstrated that SCFAs such as propionate and butyrate increased the expression of the leptin gene [104]. Conversely, knockdown of FFA3 by siRNA almost completely inhibited the ability of propionate to induce leptin gene expression [104]. FFA2 is expressed in peptide-YY-(PYY-) containing enteroendocrine cells, presumably L cells [102]. Since L cells are also responsible for the production and secretion of glucagon-like peptide (GLP-1), it is plausible that SCFAs may affect insulin secretion via multiple pathways and mechanisms. In support of this hypothesis, mice deficient in FFA2 showed reduced insulin secretion in an oral glucose tolerance test (OGTT), which further suggests the involvement of FFA2 in glucose-stimulated insulin secretion [105].
5.2.4. Trimethylamine-N-Oxide (TMAO)
Microbial metabolism of phosphatidylcholine may play a role in atheroprogression. Phosphatidylcholine is a phospholipid integral to cell membranes and present in higher-fat foods. Gut microbiota releases choline from dietary phosphatidylcholine where it is then metabolized to trimethylamine (TMA). TMA is transported to the liver via the portal vein where it is oxidized by flavin monooxygenase-3 to trimethylamine-N-oxide (TMAO). Wang et al. showed that increasing levels of plasma TMAO, choline, and betaine had dose-dependent relationships with the presence of CVD in a cohort of 1,876 men and women, after controlling for established risk factors and medication use [106]. The same group further demonstrated that atherosclerosis-prone mice fed either high-choline or TMAO diets had higher TMAO plasma levels and enhanced total aortic root atherosclerotic plaque area without any differences in plasma lipids or glucose. While the microbiota is necessary for methylamine production (GF mice do not excrete TMA), dietary interventions can modulate the metabolic outcomes. Conv mice fed high-fat diets had higher conversion of choline to methylamines as compared to those on low-fat diets [107]. Elevated plasma TMAO also is a signature biomarker of a high-meat diet as compared to low-meat and vegetarian diets in humans [108]. TMAO not only is a new biomarker for developing CVD but also a novel biomarker for food choice behavior.
5.2.5. Hippurate
Production of hippurate requires both microbial and mammalian metabolism. Low-molecular-weight aromatic compounds and polyphenols from the diet are metabolized by intestinal bacteria, resulting in a production of benzoic acid. Then, in the liver mitochondria, benzoic acid is conjugated with glycine to form hippurate, which subsequently is excreted into urine [109]. Therefore, urinary hippurate has been recognized as a marker of gut microbiota activity. Interestingly, urinary hippurate, acetate, and propionate were found lower in obese and insulin resistant Zucker (fa/fa) rats than in wild-type (-/-) or heterozygous mutation (fa/-) rats [110]. In humans, the amount of urinary hippurate discriminates the morbidly obese and insulin-resistant patients from age-matched control subjects [111]. In particular, obese urinary metabolic profile was characterized with a lower level of hippurate than that of lean controls. Recently, hippurate has been suggested as a biomarker discriminating people with elevated blood pressure from normotensive subjects. In the INTERMAP study, which collected 24-hour urine samples from 4630 participants in China, Japan, UK, and USA, an inverse association was found between the urinary hippurate and blood pressure [112]. Together, data suggest that metabolites of the gut microbiota origin hold a great promise as a diagnostic marker for people at the risk of obesity or cardiovascular diseases. However, since the intakes of dietary polyphenols found in fruits and vegetables can potentially affect the amount of urinary hippurate, both dietary habits and gut microbial metabolic activities should be considered when examining the association between urinary hippurate and metabolic diseases.
6. Conclusion
Obesity and its associated diseases such as type 2 diabetes and CVD are increasing at an alarming rate. During the past years, the discovery of the roles of the gut microbiota in energy homeostasis raised the question of whether commensal intestinal bacteria are friends or foes in maintaining a healthy weight. As summarized in this paper, published results so far do not offer a clear answer to this question. Factors specific to individuals, such as dynamic changes of microbiota and behavioral and genetic predispositions, likely work in a concert to determine the response of an individual to increased adipose stores. Thus, gut metagenome should be considered as a risk factor joining the classic factors such as host genetics and environmental facts for the development of metabolic diseases (Figure 4).
Figure 4.

New and old models describing the factors that contribute to the development of metabolic diseases. An old model is based on direct interactions between the environmental factors and genetic variations of individuals (nutrigenomic interactions). A new model includes the emerging discoveries related to the gut microbiota and the host. Environmental factors such as dietary fats affect the composition of the gut microbiota. Conversely, different profiles of gut microbiota regulate the production of short-chain fatty acids. Thus, the two way interactions can be described as nutri-metagenomic interactions. Crosstalks between the host and gut microbiota can also happen (host-metagenome interactions). Evidence that mutations of a host gene lead to alterations of gut microbiota profile and that mice colonized with different gut microbiota have different metabolic phenotype s supports the host-metagenome interactions.
Low-grade inflammation, increased oxidative stress, dyslipidemia, hypertension, and insulin resistance have all been linked with obesity. Thus far, many questions regarding the microbiota and obesity have been researched, such as do microbiota in the gut influence body weight, how do different gut microbiota profiles affect mucosal immune sensing, how is the production of bacterial metabolites regulated in the gut, and what are the impacts that bacterial metabolites have on the human body? However, most studies emphasize on the description of the gut microbiota in different populations and associations between bacterial phylotype s and certain metabolic outcomes. Therefore, it is paramount that the causality of metabolic diseases by disturbed gut microbiota be clearly demonstrated. High-priority questions that remain to be addressed are whether the gut microbiota should be considered as a pharmaceutical or nutritional treatment target for diabetes and CVD and what would be the ideal gut microbiota profile to prevent or to delay the onset of metabolic diseases.
For future study in this field, it is reasonable to rely on a classical approach to identify pathogens in the gut that could help explain metabolic diseases. However, taking a more global approach and regarding the gut microbiota as an ecosystem would be more appropriate. Specifically, research addressing the functionality rather than composition of the gut microbiota should be encouraged. Numerous human metagenome projects, such as MetaHIT (EU and China), MicrOBES (INRA, France), Meta-GUT (China), the Canadian Microbiome Initiative (Canada), and the NIH Human Gut Metagenome Initiative (USA), are currently on-going. Metaproteomics of human gut microbiota has been published [113], and nontargeted metabonomic profiling has recently been used to study the interactions between the gut microbiota and its host [92, 110]. Omics platforms are undoubtedly powerful tools to study the role of the gut microbiota in health and diseases. Hopefully, in the near future it will be possible to consolidate data from multiple omic platforms and identify whether certain profiles of gut microbiota or particular microbiota functionalities are a friend or foe of metabolic health. This would allow the design of proper diagnostic tools and therapeutic strategies to treat the consequences caused by dysbiosis between the gut microbiota and its host. In our opinion, gut microbiota may play an intriguing role in the development of obesity and obesity-related diseases, and, although microbiota seems strongly associated with obesity, a clear causal relationship remains to be established.
Acknowledgment
K. Harris is supported by the Nestle PhD, RD Training Fellowship, which is a competitive award funded by the Nestle Research Center for a nutritional science graduate student pursuing both degrees at the Pennsylvania State University.
References
- 1.World Health Organization. Obesity and Overweight. 2007.
- 2.Suau A, Bonnet R, Sutren M, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Applied and Environmental Microbiology. 1999;65(11):4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savage DC. Microbial ecology of the gastrointestinal tract. Annual Review of Microbiology. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 4.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3(7) doi: 10.1371/journal.pone.0002836. Article ID e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Current Opinion in Gastroenterology. 2008;24(1):4–10. doi: 10.1097/MOG.0b013e3282f2b0e8. [DOI] [PubMed] [Google Scholar]
- 6.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 8.Hattori M, Taylor TD. The human intestinal microbiome: a new frontier of human biology. DNA Research. 2009;16(1):1–12. doi: 10.1093/dnares/dsn033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller TL, Wolin MJ. Stability of Methanobrevibacter smithii populations in the microbial flora excreted from the human large bowel. Applied and Environmental Microbiology. 1983;45(1):317–318. doi: 10.1128/aem.45.1.317-318.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon GL, Gorbach SL. Intestinal flora in health and disease. Gastroenterology. 1984;86(1):174–193. [PubMed] [Google Scholar]
- 11.Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(15):6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(3):732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. Journal of Bacteriology. 2001;183(12):3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(12):4250–4255. doi: 10.1073/pnas.0306398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(31):11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 18.Duncan SH, Lobley GE, Holtrop G, et al. Human colonic microbiota associated with diet, obesity and weight loss. International Journal of Obesity. 2008;32(11):1720–1724. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 19.Schwiertz A, Taras D, Schäfer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18(1):190–195. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 20.Jumpertz R, Le DS, Turnbaugh PJ, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. American Journal of Clinical Nutrition. 2011;94(1):58–65. doi: 10.3945/ajcn.110.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santacruz A, Marcos A, Wärnberg J, et al. Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity. 2009;17(10):1906–1915. doi: 10.1038/oby.2009.112. [DOI] [PubMed] [Google Scholar]
- 22.Nadal I, Santacruz A, Marcos A, et al. Shifts in clostridia, bacteroides and immunoglobulin-coating fecal bacteria associated with weight loss in obese adolescents. International Journal of Obesity. 2009;33(7):758–767. doi: 10.1038/ijo.2008.260. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(7):2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furet JP, Kong LC, Tap J, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59(12):3049–3057. doi: 10.2337/db10-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li JV, Ashrafian H, Bueter M, et al. Metabolic surgery profoundly influences gut microbial—host metabolic cross-talk. Gut. 2011;60(9):1214–1223. doi: 10.1136/gut.2010.234708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geurts L, Lazarevic V, Derrien M, et al. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Frontiers in Microbiology. 2011;2, article 149 doi: 10.3389/fmicb.2011.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host and Microbe. 2008;3(4):213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy EF, Cotter PD, Healy S, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut. 2010;59(12):1635–1642. doi: 10.1136/gut.2010.215665. [DOI] [PubMed] [Google Scholar]
- 29.Ravussin Y, Koren O, Spor A, et al. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. doi: 10.1038/oby.2011.111. Obesity. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C, Zhang M, Wang S, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. The ISME Journal. 2010;4(2):232–241. doi: 10.1038/ismej.2009.112. [DOI] [PubMed] [Google Scholar]
- 31.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. 2009;137(5):1716–1716. doi: 10.1053/j.gastro.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de la Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. American Journal of Physiology. 2010;299(2):G440–G448. doi: 10.1152/ajpgi.00098.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 34.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabot S, Membrez M, Bruneau A, et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. The FASEB Journal. 2010;24(12):4948–4959. doi: 10.1096/fj.10-164921. [DOI] [PubMed] [Google Scholar]
- 37.Mandard S, Zandbergen F, Nguan ST, et al. The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. The Journal of Biological Chemistry. 2004;279(33):34411–34420. doi: 10.1074/jbc.M403058200. [DOI] [PubMed] [Google Scholar]
- 38.Kersten S, Mandard S, Tan NS, et al. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. The Journal of Biological Chemistry. 2000;275(37):28488–28493. doi: 10.1074/jbc.M004029200. [DOI] [PubMed] [Google Scholar]
- 39.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleissner CK, Huebel N, Abd El-Bary MM, Loh G, Klaus S, Blaut M. Absence of intestinal microbiota does not protect mice from diet-induced obesity. The British Journal of Nutrition. 2010;104(6):919–929. doi: 10.1017/S0007114510001303. [DOI] [PubMed] [Google Scholar]
- 41.Xu J, Gordon JI. Honor thy symbionts. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wostmann BS. Gernfree and Gnotobiotic Animal Models, Background and Applications. CRC; 1996. [Google Scholar]
- 43.Wostmann BS. Gernfree and Gnotobiotic Animal Models, Background and Applications. CRC; 1996. [Google Scholar]
- 44.Larsen N, Vogensen FK, van den Berg FWJ, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2) doi: 10.1371/journal.pone.0009085. Article ID e9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu X, Ma C, Han L, et al. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Current Microbiology. 2010;61(1):69–78. doi: 10.1007/s00284-010-9582-9. [DOI] [PubMed] [Google Scholar]
- 46.Koren O, Spor A, Felin J, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(supplement 1):4592–4598. doi: 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen L, Ley RE, Volchkov PY, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455(7216):1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. The Journal of Clinical Investigation. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Membrez M, Blancher F, Jaquet M, et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. The FASEB Journal. 2008;22(7):2416–2426. doi: 10.1096/fj.07-102723. [DOI] [PubMed] [Google Scholar]
- 50.Barton GM, Kagan JC. A cell biological view of toll-like receptor function: regulation through compartmentalization. Nature Reviews Immunology. 2009;9(8):535–541. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ting JPY, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28(3):285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clarke TB, Weiser JN. Intracellular sensors of extracellular bacteria. Immunological Reviews. 2011;243(1):9–25. doi: 10.1111/j.1600-065X.2011.01039.x. [DOI] [PubMed] [Google Scholar]
- 53.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 54.Lee JY, Hwang DH. The modulation of inflammatory gene expression by lipids: mediation through toll-like receptors. Molecules and Cells. 2006;21(2):174–185. [PubMed] [Google Scholar]
- 55.Feingold KR, Staprans I, Memon RA, et al. Endotoxin rapidly induces changes in lipid metabolism that produce hypertriglyceridemia: low doses stimulate hepatic triglyceride production while high doses inhibit clearance. Journal of Lipid Research. 1992;33(12):1765–1776. [PubMed] [Google Scholar]
- 56.Maitra U, Gan L, Chang S, Li L. Low-dose endotoxin induces inflammation by selectively removing nuclear receptors and activating ccaat/enhancer-binding protein δ. Journal of Immunology. 2011;186(7):4467–4473. doi: 10.4049/jimmunol.1003300. [DOI] [PubMed] [Google Scholar]
- 57.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of Clinical Investigation. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50(6):1267–1276. doi: 10.1007/s00125-007-0654-8. [DOI] [PubMed] [Google Scholar]
- 59.Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity. 2008;16(6):1248–1255. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- 60.Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. American Journal of Clinical Nutrition. 2007;86(5):1286–1292. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- 61.Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 62.Cani PD, Knauf C, Iglesias MA, Drucker DJ, Delzenne NM, Burcelin R. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes. 2006;55(5):1484–1490. doi: 10.2337/db05-1360. [DOI] [PubMed] [Google Scholar]
- 63.Cani PD, Neyrinck AM, Fava F, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50(11):2374–2383. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- 64.Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. American Journal of Physiology. 2007;292(3):E740–E747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- 65.Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care. 2011;34(2):392–397. doi: 10.2337/dc10-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lassenius MI, Pietilainen KH, Kaartinen K, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34(8):1809–1815. doi: 10.2337/dc10-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiedermann CJ, Kiechl S, Dunzendorfer S, et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. Journal of the American College of Cardiology. 1999;34(7):1975–1981. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 68.Peschel T, Schönauer M, Thiele H, Anker S, Schuler G, Niebauer J. Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. European Journal of Heart Failure. 2003;5(5):609–614. doi: 10.1016/s1388-9842(03)00104-1. [DOI] [PubMed] [Google Scholar]
- 69.Yarur AJ, Deshpande AR, Pechman DM, Tamariz L, Abreu MT, Sussman DA. Inflammatory bowel disease is associated with an increased incidence of cardiovascular events. American Journal of Gastroenterology. 2011;106(4):741–747. doi: 10.1038/ajg.2011.63. [DOI] [PubMed] [Google Scholar]
- 70.Cani PD, Possemiers S, Van De Wiele T, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58(8):1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muccioli GG, Naslain D, Bäckhed F, et al. The endocannabinoid system links gut microbiota to adipogenesis. Molecular Systems Biology. 2010;6, article 392 doi: 10.1038/msb.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neal MD, Leaphart C, Levy R, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. Journal of Immunology. 2006;176(5):3070–3079. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- 73.Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. Journal of Lipid Research. 2009;50(1):90–97. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- 74.Ghanim H, Abuaysheh S, Sia CL, et al. Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. 2009;32(12):2281–2287. doi: 10.2337/dc09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laugerette F, Vors C, Géloën A, et al. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. Journal of Nutritional Biochemistry. 2011;22(1):53–59. doi: 10.1016/j.jnutbio.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 76.Muzio M, Bosisio D, Polentarutti N, et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. Journal of Immunology. 2000;164(11):5998–6004. doi: 10.4049/jimmunol.164.11.5998. [DOI] [PubMed] [Google Scholar]
- 77.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410(6832):1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 78.Vijay-Kumar M, Aitken JD, Carvalho FA, et al. Metabolie syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science. 2010;328(5975):228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clinical Microbiology Reviews. 2009;22(2):240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akira S. Mammalian Toll-like receptors. Current Opinion in Immunology. 2003;15(1):5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 81.Himes RW, Smith CW. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. The FASEB Journal. 2010;24(3):731–739. doi: 10.1096/fj.09-141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kuo LH, Tsai PJ, Jiang MJ, et al. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia. 2010;54(1):168–179. doi: 10.1007/s00125-010-1931-5. [DOI] [PubMed] [Google Scholar]
- 83.Caricilli AM, Nascimento PH, Pauli JR, et al. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. Journal of Endocrinology. 2008;199(3):399–406. doi: 10.1677/JOE-08-0354. [DOI] [PubMed] [Google Scholar]
- 84.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nature Medicine. 2010;16(2):228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Girardin SE, Boneca IG, Carneiro LAM, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 86.Girardin SE, Travassos LH, Hervé M, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. The Journal of Biological Chemistry. 2003;278(43):41702–41708. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- 87.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. The Journal of Biological Chemistry. 2003;278(11):8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 88.Stroh T, Batra A, Glauben R, et al. Nucleotide oligomerization domains 1 and 2: regulation of expression and function in preadipocytes. Journal of Immunology. 2008;181(5):3620–3627. doi: 10.4049/jimmunol.181.5.3620. [DOI] [PubMed] [Google Scholar]
- 89.Zhao L, Hu P, Zhou Y, Purohit J, Hwang D. NOD1 activation induces proinflammatory gene expression and insulin resistance in 3T3-L1 adipocytes. American Journal of Physiology. 2011;301(4):E587–E598. doi: 10.1152/ajpendo.00709.2010. [DOI] [PubMed] [Google Scholar]
- 90.Schertzer JD, Tamrakar AK, Magalhães JG, et al. NOD1 activators link innate immunity to insulin resistance. Diabetes. 2011;60(9):2206–2215. doi: 10.2337/db11-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tamrakar AK, Schertzer JD, Chiu TT, et al. NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology. 2010;151(12):5624–5637. doi: 10.1210/en.2010-0437. [DOI] [PubMed] [Google Scholar]
- 92.Martin FPJ, Sprenger N, Montoliu I, Rezzi S, Kochhar S, Nicholson JK. Dietary modulation of gut functional ecology studied by fecal metabonomics. Journal of Proteome Research. 2010;9(10):5284–5295. doi: 10.1021/pr100554m. [DOI] [PubMed] [Google Scholar]
- 93.Swann JR, Tuohy KM, Lindfors P, et al. Variation in antibiotic-induced microbial recolonization impacts on the host metabolic phenotypes of rats. Journal of Proteome Research. 2011;10(8):3590–3603. doi: 10.1021/pr200243t. [DOI] [PubMed] [Google Scholar]
- 94.Wolever TMS, Fernandes J, Rao AV. Serum acetate:propionate ratio is related to serum cholesterol in men but not women. Journal of Nutrition. 1996;126(11):2790–2797. doi: 10.1093/jn/126.11.2790. [DOI] [PubMed] [Google Scholar]
- 95.Jenkins DJA, Wolever TMS, Jenkins A, et al. Specific types of colonic fermentation may raise low-density-lipoprotein-cholesterol concentrations. American Journal of Clinical Nutrition. 1991;54(1):141–147. doi: 10.1093/ajcn/54.1.141. [DOI] [PubMed] [Google Scholar]
- 96.Wolever TMS, Spadafora P, Eshuis H. Interaction between colonic acetate and propionate in humans. American Journal of Clinical Nutrition. 1991;53(3):681–687. doi: 10.1093/ajcn/53.3.681. [DOI] [PubMed] [Google Scholar]
- 97.Hosseini E, Grootaert C, Verstraete W, Van de Wiele T. Propionate as a health-promoting microbial metabolite in the human gut. Nutrition Reviews. 2011;69(5):245–258. doi: 10.1111/j.1753-4887.2011.00388.x. [DOI] [PubMed] [Google Scholar]
- 98.Brown AJ, Goldsworthy SM, Barnes AA, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. The Journal of Biological Chemistry. 2003;278(13):11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 99.Le Poul E, Loison C, Struyf S, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. The Journal of Biological Chemistry. 2003;278(28):25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- 100.Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochemical and Biophysical Research Communications. 2003;303(4):1047–1052. doi: 10.1016/s0006-291x(03)00488-1. [DOI] [PubMed] [Google Scholar]
- 101.Hong YH, Nishimura Y, Hishikawa D, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146(12):5092–5099. doi: 10.1210/en.2005-0545. [DOI] [PubMed] [Google Scholar]
- 102.Karaki SI, Mitsui R, Hayashi H, et al. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell and Tissue Research. 2006;324(3):353–360. doi: 10.1007/s00441-005-0140-x. [DOI] [PubMed] [Google Scholar]
- 103.Ge H, Li X, Weiszmann J, et al. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology. 2008;149(9):4519–4526. doi: 10.1210/en.2008-0059. [DOI] [PubMed] [Google Scholar]
- 104.Xiong Y, Miyamoto N, Shibata K, et al. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(4):1045–1050. doi: 10.1073/pnas.2637002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bjursell M, Admyre T, Göransson M, et al. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. American Journal of Physiology. 2011;300(1):E211–E220. doi: 10.1152/ajpendo.00229.2010. [DOI] [PubMed] [Google Scholar]
- 106.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dumas ME, Barton RH, Toye A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(33):12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stella C, Beckwith-Hall B, Cloarec O, et al. Susceptibility of human metabolic phenotypes to dietary modulation. Journal of Proteome Research. 2006;5(10):2780–2788. doi: 10.1021/pr060265y. [DOI] [PubMed] [Google Scholar]
- 109.Nicholson JK, Holmes E, Wilson ID. Gut microorganisms, mammalian metabolism and personalized health care. Nature Reviews Microbiology. 2005;3(5):431–438. doi: 10.1038/nrmicro1152. [DOI] [PubMed] [Google Scholar]
- 110.Waldram A, Holmes E, Wang Y, et al. Top-down systems biology modeling of host metabotype-microbiome associations in obese rodents. Journal of Proteome Research. 2009;8(5):2361–2375. doi: 10.1021/pr8009885. [DOI] [PubMed] [Google Scholar]
- 111.Calvani R, Miccheli A, Capuani G, et al. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. International Journal of Obesity. 2010;34(6):1095–1098. doi: 10.1038/ijo.2010.44. [DOI] [PubMed] [Google Scholar]
- 112.Holmes E, Loo RL, Stamler J, et al. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature. 2008;453(7193):396–400. doi: 10.1038/nature06882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Verberkmoes NC, Russell AL, Shah M, et al. Shotgun metaproteomics of the human distal gut microbiota. The ISME Journal. 2009;3(2):179–189. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]