

Abstract
In this report, we demonstrate the purification of DNA and RNA from a 10% serum sample using an oligonucleotide capture matrix. This approach provides a one-stage, completely aqueous system capable of purifying both RNA and DNA for downstream PCR amplification. The advantages of utilizing the polymer capture matrix method in place of the solid-phase extraction method is that the capture matrix eliminates both guanidine and the isopropanol wash that can inhibit downstream PCR and competition with proteins for the binding sites that can limit the capacity of the device. This method electrophoreses a biological sample (e.g. serum) containing the nucleic acid target through a polymer matrix with covalently bound oligonucleotides. These capture oligonucleotide selectively hybridize and retain the target nucleic acid, while the other biomolecules and reagents (e.g. SDS) pass through the matrix to waste. Following this purification step, the solution can be heated above the melting temperature of the capture sequence to release the target molecule, which is then electrophoresed to a recovery chamber for subsequent PCR amplification. We demonstrate that the device can be applied to purify both DNA and RNA from serum. The gag region of HIV at a starting concentration of 37.5 copies per microliter was successfully purified from a 10% serum sample demonstrating the applicability of this method to detect viruses present in low copy numbers.
Keywords: RNA purification, microfluidic, polymer matrix
1. Introduction
Purification of nucleic acids is required for most genetic analyses and nucleic acid assays for pathogen detection due to the inhibition of PCR by endogenous species (e.g. hemoglobin, heparin) [1, 2]. Additionally, it is one of the most time-consuming and labor-intensive diagnostic steps, so that increasing the speed and automation of this step can significantly reduce the time from raw sample to analytical output. Recent advances in lab-on-a-chip technology have demonstrated the ability to perform on a microfluidic platform the analytical steps for genetic analysis: nucleic acid purification, PCR amplification and detection [3, 4]. These devices for nucleic acid purification and analysis can offer advantages of reduced sample handling and contamination, a higher degree of automation, and faster turnover from raw sample to analytical output.
To date, the majority of reports on microfluidic purification have focused on purification of DNA via solid-phase extraction (SPE) [4-9]. However, relatively few reports of RNA purification on microfluidic devices have been published [8]. This is likely due to the additional challenge of RNA degradation by ubiquitous RNases. Thus far, there have been two basic approaches to RNA purification: capture using oligo-dTs and solid-phase extraction. Jiang et al. [10] and Satterfield et al. [11] used oligo-dTs bound to magnetic beads and a polymer monolith, respectively, to concentrate messenger RNA (mRNA) from total RNA (TRNA). While concentrating mRNA from TRNA is useful for applications such as creating a cDNA library, off-chip purification would be required to isolate the TRNA. Hong et al. lysed 1-100 bacterial cells and recovered the mRNA using oligo-dT beads [3], although this will likely recover less that 60% of bacterial RNA [12]. Additionally, compared to reports using SPE to isolate DNA or RNA, 100 cells will have less than 0.1% of the protein than 1 μl of human serum [8, 13].
Several reports have demonstrated the use of solid-phase extraction for the isolation TRNA from cell lysate [6, 14]. Witek et al. [6] utilized an array of photo-activated polycarbonate micropillars as the solid-phase, while Bhattacharyya et al. [14] and Hagan et al. [15] used silica beads. The Landers group pioneered the use of silica beads for DNA purification in microfluidic devices [5, 16]. However, two limitations to SPE have been that proteins bind strongly to the silica beads resulting in a low DNA binding capacity [17] and that residual guanidine or isopropanol can inhibit PCR when SPE and PCR are performed on a single chip [18]. Recent reports that have investigated methods to overcome these drawbacks, including a pH-induced DNA capture to remove the isopropanol wash [19] and a two-stage, dual-phase purification chip that increases DNA binding capacity and allows for a completely aqueous system [20]. While these reports are investigating methods to improve the SPE method, alternative purification approaches may be attractive for some applications, such as isolating nucleic acid targets that may be present in very low copy numbers.
In this report, we demonstrate a single-stage, completely aqueous system for the purification of RNA from serum using a polymer capture matrix. This approach has been demonstrated to purify ssDNA from a sub-microliter sample of a Sanger extension reaction prior to an electrophoretic separation [21]. Here, we demonstrate the ability to purify a specific target, the gag region of the HIV virus, from serum with a large sample volume for RT-PCR. The main challenges that were overcome in developing this method were preventing degradation of the RNA in serum, a low starting copy number, a large starting sample volume and the RNA component representing only a tiny fraction of the biological mass of a serum sample.
2. Materials and Methods
2.1 Chip Fabrication
High-quality Optix® poly(methyl methacrylate) (PMMA) from Plaskolite, Inc. (Columbus, OH) was purchased with no surface treatments or coating that could interfere with the solvent bonding process. The purification cartridge was assembled from three PMMA layers: a bottom substrate, a middle layer with wells and channels, and a top layer with access ports. The layers were joined via solvent bonding, which dissolves the plastic at the interface and subsequently hardens to form transparent bond [22, 23]. The procedure, adapted from Lin et al. [23], used an azeotropic mixture of 1,2-dichloroethane (EDC) (J.T. Baker, Phillipsburg, NJ) and ethanol (EtOH) (Aaper Alcohol and Chemical, Co., Shelbyville, KY) (1:5 v/v EDC:EtOH) so that as the mixture evaporates during the bonding process, the composition remains unchanged. The EtOH serves as the diluent to prevent clogging of the microchannel during bonding. To bond two layers, the first layer is placed on an aluminum plate and several drops of EDC:EtOH solution are dispensed onto the PMMA. The second layer is immediately placed on top using the alignment pins to accurately position the two layers and a second aluminum plate is placed over the two PMMA sheets. Four c-clamps are used to apply uniform pressure across the aluminum plates for 8-9 minutes. The bonded PMMA layers are removed from the aluminum plates and sprayed with EtOH to remove any remaining bonding solution and dried. The third PMMA layer is solvent bonded similarly. An image of the purification cartridge is shown in Figure 1.
Figure 1.

Diagram of the cartridge middle layer with significant features labeled (left) and an image of the purification cartridge filled with colored solution (right): buffer and waste reservoirs (green), sample chamber (red), and capture chamber (blue). The red box around the capture chamber highlights the region where fluorescence images in Figure 2 were taken.
2.2 Polymer Synthesis
The polymer capture matrix and linear polyacrylamide (LPA) used in this study were synthesized via free-radical polymerization. The LPA was synthesized by dissolving acrylamide monomer (Amresco Inc., Solon, OH) at 3% w/v with 0.33% v/v isopropanol (Fisher Scientific, Pittsburgh, PA). This solution is bubbled with nitrogen for 45 minutes then initiated with 0.01% w/v 4,4’-azobis(4-cyanovaleric acid) (Sigma Aldrich, St. Louis, MO) and allowed to react for 4 hours at 50 °C in a jacketed reaction vessel. Following the reaction, the solution is allowed to cool and then poured into 100,000 Da molecular weight cut-off dialysis tubes (Spectrum Laboratories, Inc., Rancho Dominguez, CA). Following dialysis, the polymer solution is frozen, lyophilized and stored dry until use.
The polymer capture matrix was synthesized by dissolving 0.25 g of acrylamide in 5 mL of 0.5xTTE (25 mM Tris, 25 mM TAPS, 2 mM EDTA) with 6.25 nanomoles of acrydite-modified oligonucleotide (Operon Biotechnologies Inc, Huntsville, AL). The solution was bubbled for 30 minutes and initiated with 0.015% w/v ammonium persulfate and 0.015% w/v N, N, N’, N’-tetramethyethylenediamine (both from Sigma Aldrich). The reaction was allowed to proceed for 1 hour after which the solution was diluted to either 2.5 or 3% w/v with 0.5xTTE. The two capture sequences used in this study are:
SK462 capture sequence: 5′– GGC TGC TTG ATG TCC CCC CAC T
SK431 capture sequence: 5′– ATG TCA CTT CCC CTT GGT TCT CT
2.3 Electrophoresis System
Fluorescence imaging to visualize capture and release of target nucleic acids was performed on a Nikon TE200 inverted epifluorescence microscope (Nikon, Melville, NY) described previously [24]. Briefly, the light from a 100-W mercury lamp light source (Chiu Technical Corp, Kings Park, NY) was passed through a heat-absorbing filter and a standard FITC filter cube (Chroma Technology, Brattleboro, VT), then reflected off a dichroic mirror, and focused onto the cartridge with a Nikon 4x Plan Apo objective. The fluorescence is collected and directed into a VS4-1845 Generation 3 image intensifier (Videoscope International, Dulles, VA) and onto a 0.5 inch CCD, TM-6710-CL camera (JAI Pulnix, Sunnyvale, CA). Images were captured directly to a computer through a PIXCI control board (EPIX Inc., Buffalo Grove, IL) using XCAP-STD (EPIX Inc.) software. A power supply and software by Micronit Microfluidics BV(Enschede, The Netherlands) was used to control the voltages applied for electrophoresis. An in-house heating plate was used to release the target strand following capture. The Kapton flexible heater and surface thermocouple (both from Omega Engineering, Inc., Stamford, CT) were adhered to a thin copper plate and connected to a Digi-Sense temperature controller (Cole-Parmer Instrument Co., Vernon Hills, IL).
2.4 Purification Experiments
Prior to each purification experiment, the PMMA cartridge was flushed twice with 1 M nitric acid, filled with a 0.5% w/v hydroxypropyl methylcelluolose (Sigma Aldrich) solution for 5 minutes to coat the channels surfaces, then flushed with H2O. To load the cartridge, the buffer and waste wells were filled with a 3% LPA solution in 1xTTE (49 mM Tris, 49 mM TAPS, 2 mM EDTA). The capture chamber, shown as blue in Figure 1, was loaded with either LPA or the capture matrix. The sample was loaded using the two access ports connected to the sample well. Reservoirs were epoxied over the access port and filled with buffer. If any bubbles formed around the electrodes in these reservoirs, they were able to float to the surface and therefore did not impact the experiment.
Fluorescence imaging was used to determine the length of time to apply voltages to each of the access ports. The capture step in which the target was driven through the capture chamber required 46 minutes. The chip was then placed on a heating plate (75 °C for ssDNA experiments and 80 °C for RNA experiments) to release the target strands. Voltage was applied for 6.5 minutes to drive the target to the recovery well. The purification time required for the capture step is dependent on the cartridge dimensions and current limit of the power supply. Here, the goal was to purify a sample volume of 200 μl containing 20 μl of serum. This is a significantly larger sample volume when compared to reports using silica beads that typically purify 1 μl or less of a biological sample [4, 5, 15, 18]. Additionally, the power supply used for these experiments provides a maximum current of 1 mA. Given the large cross-sectional area of the purification channels (1.52 mm × 0.5 mm), the electrical resistance in the chip was low and electrophoresis conditions were selected to stay well below the 1 mA output. A different power supply capable of higher current output, or a chip designed with smaller channel cross-sectional areas for smaller biological sample volumes on the order of 1 μl, could significantly reduce the time required for the capture step.
The fluorescence imaging experiments used a FAM-labeled oligonucleotide, complementary to the SK462 capture sequence, to visualize nucleic acid capture and release. A sample of either 0.1xTTE or 10% serum with this oligonucleotide at 90 nM was imaged during electrophoresis using the system described above.
FAM-Labeled Target: 5’- FAM – AAA AGT GGG GGG ACA TCA AGC A
A 71-base oligonucleotide (Operon Biotechnologies, Inc.) complementary at one end to the SK462 capture matrix was used for purification experiments followed by PCR. The oligonucleotide was added to either a 0.1xTTE buffer sample or a 10% serum (Fisher Scientific) sample.
For purification of RNA, the target strand was the gag region of HIV-B prepared from armored RNA (Asuragen, Inc., Austin, TX). The RNA target sequence is shown in the supplemental information. This RNA sample provides a non-infectious target that is packaged in a protein coating to prevent degradation of the RNA prior to use so that the starting sample is at a known concentration. To prepare the RNA, the sample is heated to 75 °C for three minutes to remove the protein coating. The free RNA is then added to the sample solution, either 0.1xTTE or 10% serum with 0.5% w/v lithium dodecyl sulfate (Sigma Aldrich). The starting RNA concentration was 37.5 copies/μl. Given a sample chamber volume of 200 μl and a recovery well volume of approximately 150 μl, the maximum final RNA concentration is 50 copies/ μl. Since 5 μl of recovered solution were added to the PCR reaction, there is a maximum of 250 RNA copies in each PCR vial. All solutions used for the RNA purification experiments were made with 18 MΩ water treated with 0.1% v/v diethyl pyrocarbonate (Sigma Aldrich) and autoclaved.
This RNA target concentration was selected as it provided an amplicon peak well above the Agilent 2100 detector noise level. This minimized the impact of amplicon detection method selected on the percent deviation of the results. The limit-of-detection (LOD) of an assay depends both on the purification efficiency and the PCR efficiency. Future work that includes evaluating PCR efficiency can be used to determine a LOD for this assay.
2.5 PCR and Microchip Electrophoresis Detection
Samples recovered from the purification cartridge were amplified by PCR or RT-PCR using a conventional thermocycler (GeneAmp PCR System 2700, Applied Biosystems, Inc., Foster City, CA). Details on the PCR and RT-PCR conditions can be found in the supplemental information. PCR products were analyzed on the Agilent 2100 using the standard DNA 1000 kit (Agilent Technologies, Santa Clara, CA).
3. Results
A recent review by Wen et al. [8] states that there are relatively few demonstrations of RNA purification on microfluidic devices. This is likely due to the challenge of the reduced stability of RNA [8] in addition to the other constraints of developing a microfluidic purification platform (e.g. passivation of channel walls, precise movement of fluids and biomolecules, etc.). For this reason, the system was first validated using a ssDNA target and then used to purify RNA for RT-PCR
3.1 Fluorescence Imaging
Fluorescence imaging was used to establish the electrophoresis conditions necessary to drive the biomolecules through the capture matrix, as well as visually demonstrate capture and release of a target molecule. Figure 2 shows a series of fluorescence images that demonstrate that the target molecules will only be retained in the capture chamber if the capture matrix is loaded and that this does not occur if a LPA matrix is used.
Figure 2.
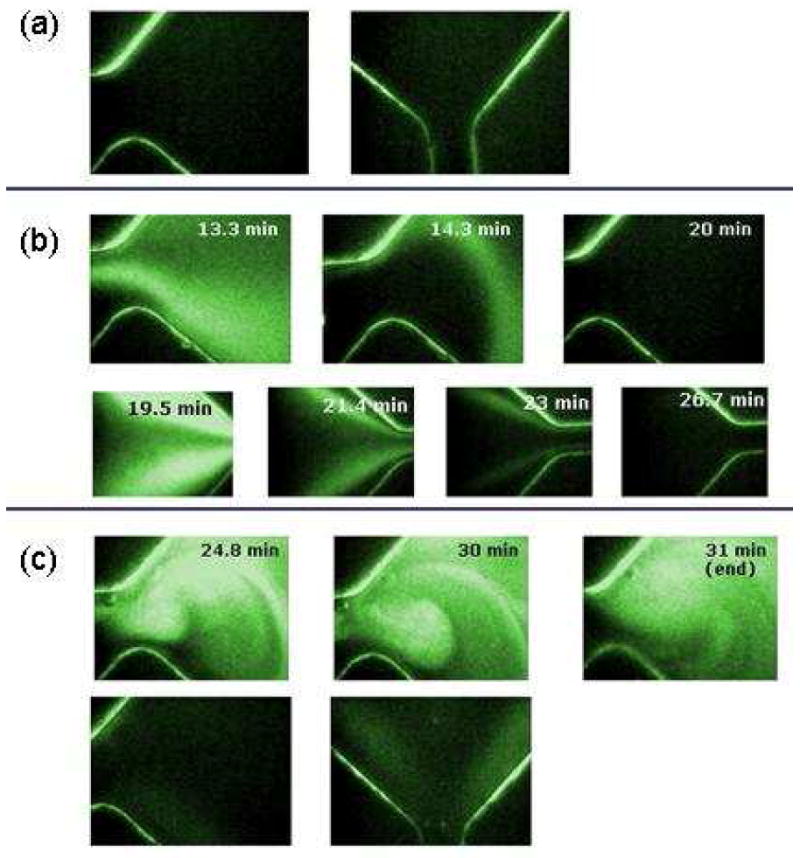
Fluorescence images to demonstrate capture of oligonucleotides described in detail in the text. (a) Image of the cartridge prior to the experiment. (b) FAM-labeled oligonucleotide being electrophoresed through the capture chamber when filled with LPA. Top row is the entrance to the capture chamber from the sample chamber. The bottom row is the exit from the capture chamber to the waste reservior (c) Top row is the FAM-labeled oligonucleotide being retained in the capture chamber by the SK462 matrix. Bottom row is following the dehybridation at 75 °C and electrophoresing the oligonucleotide to the recovery well so that the fluorescence returns to background.
Figure 2a shows the entrance to the capture chamber from the sample chamber and the exit of the capture chamber toward the recovery chamber prior to the experiment. This region is boxed in red in Figure 1. The orientation of the cartridge is the same in Figure 1 and Figure 2. These images show low background fluorescence from the chip except for the channel walls, which scatter light and therefore outline the features of the chip. Figure 2b shows the FAM-labeled olignucleotide being electrophoresed through the capture chamber when a LPA solution is loaded into the cartridge. The DNA molecules are being driven left-to-right from the sample well to the waste well. Since there are no capture oligonucleotide strands bound to the polymer chains, the target DNA is not retained and is completely driven out of the capture chamber to the waste well.
Figure 2c used the same conditions as in (b), except that the SK462 capture matrix was loaded into the capture chamber. Here, the fluorescence signal in the capture chamber remained strong throughout the capture step indicating successful hybridization of the target DNA. Upon heating the chip to 75 °C, the DNA was successfully driven to the recovery well resulting in a return to background fluorescence.
3.2 Purification of DNA oligonucleotides
Following the successful capture of fluorescently labeled oligonucleotides, we sought to recover and PCR amplify the target, with the PCR products analyzed on the Agilent 2100. A representative electropherogram of PCR products from a blank sample and a sample containing the target is shown in Figure 3. To combine the results of several experiments, the target peaks height was normalized to the upper DNA marker to account for chip-to-chip differences in absolute peak height. Differences in absolute peak height may arise due to slight differences in alignment of the chip with the laser and degradation of the gel dye over time.
Figure 3.
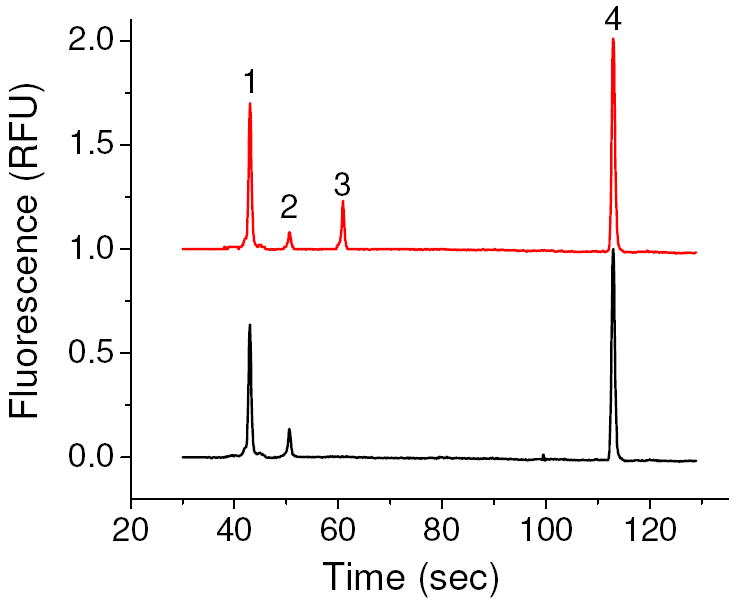
Representative elctropherograms of PCR products of a sample containing the target (top) and a blank sample (bottom). Peaks 1 and 4 are the lower and upper DNA markers, respectively, peak 2 is a primer-dimer and peak 3 is the amplified target.
To demonstrate that amplification of the target is due to the specific capture and recovery, two sets of control experiments were included with the capture and recovery experiments. The first control was to load the cartridge with a sample containing 1 pg/μl of the target oligonucleotide while loading a LPA matrix into the capture chamber. Lane 1 in Figure 4 shows that only one out of three runs resulted in a very small amount of PCR product. It is possible that either some oligonucleotide had adsorbed onto the surface and was released during the recovery step or that not all of the oligo had exited the capture chamber in this experiment. The next control was to load a sample with no target molecules with the SK462 capture matrix in the capture chamber. In this case, no PCR product was detected (Lane 2). These two experiments indicate that any detected PCR product is the result of specific capture and recovery of the target oligo.
Figure 4.
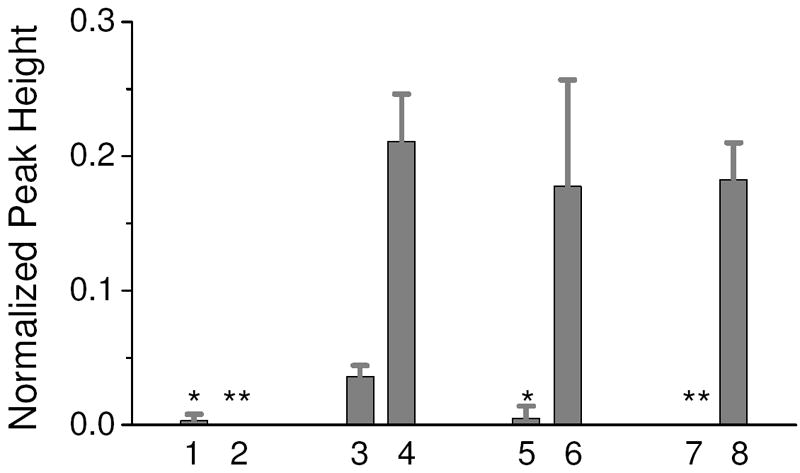
Normalized peak height results of ssDNA purification experiments. Lane 1 is a 1 pg/μl in 0.1xTTE using the LPA matrix. Lane 2 is a blank sample using the SK462 capture matrix. Lanes 3 and 4 are 50 fg/μl and 1 pg/ μl, respectively, in 0.1xTTE using the SK462 capture matrix. Lane 5 and 6 are 1 pg/ μl in 0.1xTTE + 10 mg/ml hemoglobin unpurified and purified, respectively. Lane 7 and 8 are 1 pg/ μl in 10% serum unpurified and purified, respectively. n=3 for all lanes. One PCR amplification per purified sample or control sample was performed. * - amplification detected in one out of the three PCR reactions. ** - no amplification detected in any PCR reaction.
Lanes 3 and 4 are the results of capturing and recovering the target oligonucleotide from 0.1xTTE solutions with starting concentrations of 50 fg/μl and 1 pg/μl, respectively. While this type of end-point detection for PCR does not provide a true quantitative measure of the starting oligo concentration, for these PCR conditions there is a reproducible difference in normalized peak height. Following successful capture and recovery of the target using the SK462 capture matrix, the capture matrix was challenged with more complex samples that have more real world application. The target was added to 0.1xTTE with 10 mg/ml hemoglobin solution and a 10% serum solution. Lanes 5 and 7 in Figure 4 shows that while the hemoglobin and serum inhibit PCR amplification, the purified sample recovered from the cartridge is readily amplifiable (Lanes 6 and 8).
3.3 Purification of RNA
To inhibit RNA degradation in the starting serum sample and when first loaded into the cartridge, lithium dodecyl sulfate (LiDS) was added to the 10% serum sample at a concentration of 0.5% w/v. Lane 1 in Figure 5 show the normalized peak height of the recovered solution of blank samples with the SK426 capture matrix. Similar to the DNA experiments, the blank sample resulted in no amplification (purification experiments alternated between blank samples and samples containing RNA to demonstrate that RNA is not carried over from one experiment to the next). Lane 2 in Figure 5 shows the increase in RNA recovery using the SK426 capture matrix compared to the LPA matrix with no covalently bound capture oligonucleotides.
Figure 5.
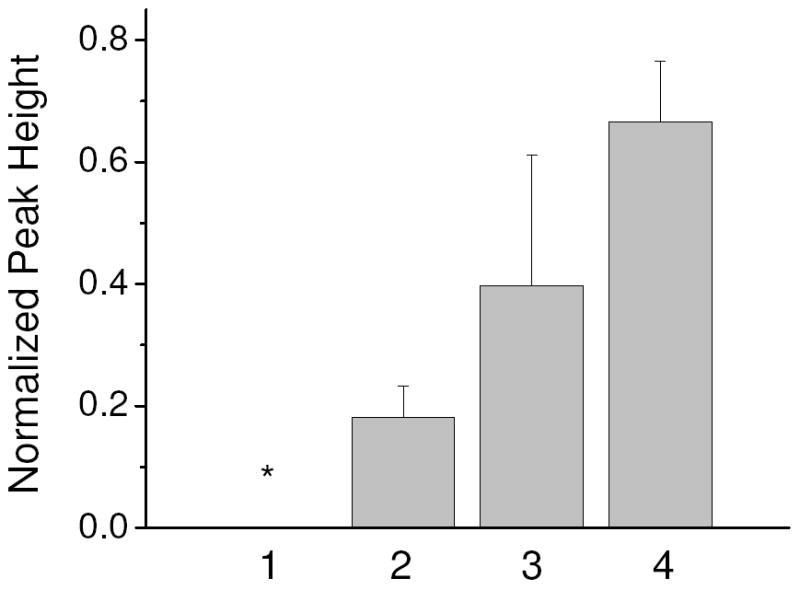
Normalized peak height results of the RNA purification experiments. The starting sample was 10% serum with 0.5% w/v LiDS. Lane 1 contained no RNA and the SK462 matrix in the capture chamber in which no product was detected. Lane 2-4 contained 37.5 RNA copies/μl with SK462 matrix (Lane 2) and SK431 matrix (Lane 3) in the capture chamber. Lane 4 is a positive template control containing 250 RNA copies. n=3. * - no amplification detected in any PCR reaction
An additional consideration when attempting to purify RNA via hybridization is the potential for RNA to form secondary structures that may interfere with hybridization and reduce capture efficiency. Therefore, the sequence of the HIV-B gag sample provided by Asuragen was put into Pfold, a program that predicts RNA secondary structure [25]. Pfold predicts that a six base region within the SK462 capture sequence is likely to be hybridized with another region of the gag sequence. However, the SK431 priming region is not predicted to contain any self-hybridizing sequences. Therefore, a capture matrix containing a portion of the SK431 primer region was tested. Lane 3 shows that significantly more RNA was recovered with the SK431 capture matrix than with the SK462 matrix. Another possibility for the improved recovery is the lower melting temperature of the SK431 capture sequence allows a more efficient recovery of the RNA. However, the temperature during the recovery step was more than 10 °C above the predicted melting temperature of the SK462 capture sequence, which should allow for complete denaturation.
While end-point detection is not quantitative, the normalized peak height of the RNA recovered using the SK431 capture matrix is 60% of the value of the positive template control with 250 copies. The positive template control was never introduced into serum and was PCR amplified immediately after heat lysing the armored RNA to ensure no RNA degradation. The reduced peak height for the samples purified by the cartridge may be due to several factors including some RNA degradation while in the serum and during electrophoretic purification, capture and recovery efficiencies of less than 100%, and residual PCR inhibitors (e.g. LiDS) reaching the recovery well. Therefore, while future work can progress toward evaluating and improving the recovery efficiency, this method appears to provide a valuable option for purifying RNA at starting concentrations below 50 copies per microliter.
4. Discussion
The work discussed here was aimed at providing an alternative method of nucleic acid purification that addresses several of the limitations of the current microfluidic purification approaches while purifying RNA, which is known to be challenging due to its susceptibility to degradation. Landers and coworkers have pioneered the use of silica beads for DNA purification in microchannels [5, 16, 17, 26], and have recently applied this method to purify RNA [15]. However, they have noted several limitations to the use of silica beads and have been working to overcome these drawbacks. Proteins, which constitute 89% of the mass of nucleic acids and proteins in a cell [8] compete for the binding sites on the silica bead surface, [20] and has limited extractions to less than 1 μl [5, 27]. Additionally, residual guanidine or isopropanol can inhibit PCR [18]. Recent investigations have worked to overcome these limitations with a pH-induced DNA capture and release method for a totally aqueous system [19] and a two-stage, dual-phase microchip to first remove proteins, then bind DNA [20]. The purification method demonstrated here is based on completely different means of retaining the target molecules and therefore is advantageous for certain targets and starting samples.
The polymer capture matrix method described here provides a completely aqueous system in which competition with proteins for binding sites will not occur allowing a target-specific, one-stage purification. Since the system is based on specific hybridization of the target strand, it is compatible with many of the reagents used for cell lysis (e.g. surfactants). Moreover, this method should be scaleable to purify both large and small starting sample volumes. Here, the sample chamber was 200 μl with a 10% serum solution indicating it is possible to purify 20 μl, although higher serum concentrations may be possible (for comparison, the two-stage purification microchip was able to process up to 10 μl of whole blood [20]). Mathies and coworkers originally used the capture matrix approach to purify ssDNA for electrophoresis from a Sanger extension reaction from nanoliter volumes [21, 28]. While this starting sample is less complex than the serum sample from which ssDNA and RNA were purified here, it indicates the potential sample volume range over which this method may be used.
This method provides the potential to concentrate the target into a smaller volume for PCR. Here, the capture chamber concentrated the target into a volume approximately one-fifth of the starting sample volume. Redesigning the interface between the electrode and access ports (i.e. eliminating the additional reservoirs) can allow for at least another one-fifth reduction in volume concentrating the starting sample of 200 μl to 8 μl. It also has the potential to facilitate capturing more than one target at a time by either incorporating several capture strands into a polymer matrix, or by mixing several polymers that each have their own capture sequence. This would allow for multiplexed PCR amplification downstream to detect multiple targets of interest.
5. Conclusions
The work presented here demonstrates the purification of a target RNA strand from a large volume sample of 10% serum. This represents one of the first demonstrations of specific RNA purification in a microfluidic device from a complex starting sample. Analysis by microchip electrophoresis of the RT-PCR products of the recovered sample indicates successful RNA recovery from a starting concentration of only 37.5 copies per microliter. The use of oligonucleotides covalently bound to a polymer backbone is not limited by competitive binding of proteins and allows for a completely aqueous system. These advantages contribute to the recovery of the low number of RNA present in the background of serum. The capture oligonucleotide can be designed to purify any target of interest, and multiple oligonucleotide sequences can be incorporated into the polymer to purify multiple targets in a single run. Additionally, the PMMA cartridge can be flushed with nitric acid and recoated for multiple uses without sample carryover, but is also an inexpensive substrate that would allow the cartridges to be disposable. It should also be possible to purify from samples with higher serum concentrations. This would reduce the sample volume and should result in a shorter purification time and smaller recovery volume while maintaining the same number of total RNA copies in the starting sample. Additionally, redesigning the interface between the electrode and the recovery well will allow for concentration of the large starting sample into a smaller recovery volume. Successful recovery of the target RNA demonstrated here, coupled with a smaller recovery volume, could make this a valuable and versatile method for purifying RNA and DNA from serum for PCR in integrated microfluidic devices.
Supplementary Material
Acknowledgments
Financial support was provided by the NSF through the Northwestern University Nanoscale Science and Engineering Center (Award No. EEC-0647560) and the NIH (Grant No. 1 U01 AI061297-02). Brian Root was supported while on appointment as a U.S. Department of Homeland Security (DHS) Fellow under the DHS Scholarship and Fellowship Program, a program administered by the Oak Ridge Institute for Science and Education (ORISE) for DHS through an interagency agreement with the U.S Department of Energy (DOE). ORISE is managed by Oak Ridge Associated Universities under DOE contract number DE-AC05-06OR23100. All opinions expressed in this paper are the author’s and do not necessarily reflect the policies and views of DHS, DOE, NSF, NIH or ORISE. The authors would like to acknowledge Markus Data Services (Mundelein, IL) for machining the features of the PMMA cartridge.
Abbreviations
- LPA
linear polyacrylamide
- PCR
polymerase chain reaction
- RT-PCR
reverse transcription polymerase chain reaction
- SPE
solid-phase extraction
- TRNA
total RNA
- mRNA
messenger RNA
- PMMA
poly(methyl methacrylate)
- EDC
1,2-dichloroethane
- EtOH
ethanol
- FAM
5 – carboxyfluorescein
- LiDS
lithium dodecylsulfate
References
- 1.de Mello AJ, Beard N. Lab on a Chip. 2003;3:11N–19N. doi: 10.1039/b301019h. [DOI] [PubMed] [Google Scholar]
- 2.Wilson IG. Applied and Environmental Microbiology. 1997;63:3741–3751. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong JW, Studer V, Hang G, Anderson WF, Quake SR. Nature Biotechnology. 2004;22:435–439. doi: 10.1038/nbt951. [DOI] [PubMed] [Google Scholar]
- 4.Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, et al. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19272–19277. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breadmore MC, Wolfe KA, Arcibal IG, Leung WK, et al. Analytical Chemistry. 2003;75:1880–1886. doi: 10.1021/ac0204855. [DOI] [PubMed] [Google Scholar]
- 6.Witek MA, Hupert ML, Park DSW, Fears K, et al. Analytical Chemistry. 2008;80:3483–3491. doi: 10.1021/ac8002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhattacharyya A, Klapperich CM. Analytical Chemistry. 2006;78:788–792. doi: 10.1021/ac051449j. [DOI] [PubMed] [Google Scholar]
- 8.Wen J, Legendre LA, Bienvenue JM, Landers JP. Analytical Chemistry. 2008;80:6472–6479. doi: 10.1021/ac8014998. [DOI] [PubMed] [Google Scholar]
- 9.Chung YC, Jan MS, Lin YC, Lin JH, et al. Lab on a Chip. 2004;4:141–147. doi: 10.1039/b310849j. [DOI] [PubMed] [Google Scholar]
- 10.Jiang GF, Harrison DJ. Analyst. 2000;125:2176–2179. doi: 10.1039/b005999o. [DOI] [PubMed] [Google Scholar]
- 11.Satterfield BC, Stern S, Caplan MR, Hukari KW, West JAA. Analytical Chemistry. 2007;79:6230–6235. doi: 10.1021/ac0709201. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar N. Microbiology-Uk. 1996;142:3125–3133. doi: 10.1099/13500872-142-11-3125. [DOI] [PubMed] [Google Scholar]
- 13.Betgovargez E, Knudsen V, Simonian MH. Journal of Biomolecular Techniques. 2005;16:306–310. [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya A, Klapperich CA. Sensors and Actuators B-Chemical. 2008;129:693–698. [Google Scholar]
- 15.Hagan KA, Bienvenue JM, Moskaluk CA, Landers JP. Analytical Chemistry. 2008 doi: 10.1021/ac8011945. [DOI] [PubMed] [Google Scholar]
- 16.Wolfe KA, Breadmore MC, Ferrance JP, Power ME, et al. Electrophoresis. 2002;23:727–733. doi: 10.1002/1522-2683(200203)23:5<727::AID-ELPS727>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 17.Tian HJ, Huhmer AFR, Landers JP. Analytical Biochemistry. 2000;283:175–191. doi: 10.1006/abio.2000.4577. [DOI] [PubMed] [Google Scholar]
- 18.Legendre LA, Bienvenue JM, Roper MG, Ferrance JP, Landers JP. Analytical Chemistry. 2006;78:1444–1451. doi: 10.1021/ac0516988. [DOI] [PubMed] [Google Scholar]
- 19.Cao WD, Easley CJ, Ferrance JP, Landers JP. Analytical Chemistry. 2006;78:7222–7228. doi: 10.1021/ac060391l. [DOI] [PubMed] [Google Scholar]
- 20.Wen J, Guillo C, Ferrance JP, Landers JP. Analytical Chemistry. 2007;79:6135–6142. doi: 10.1021/ac0703698. [DOI] [PubMed] [Google Scholar]
- 21.Paegel BM, Yeung SHI, Mathies RA. Analytical Chemistry. 2002;74:5092–5098. doi: 10.1021/ac0203645. [DOI] [PubMed] [Google Scholar]
- 22.Kelly RT, Li Y, Woolley AT. Analytical Chemistry. 2006;78:2565–2570. doi: 10.1021/ac0521394. [DOI] [PubMed] [Google Scholar]
- 23.Lin CH, Chao CH, Lan CW. Sensors and Actuators B-Chemical. 2007;121:698–705. [Google Scholar]
- 24.Chiesl T, Shi W, Barron AE. Analytical Chemistry. 2005;77:772–779. doi: 10.1021/ac049000y. [DOI] [PubMed] [Google Scholar]
- 25.Knudsen B, Hein J. Nucleic Acids Research. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu QR, Bienvenue JM, Hassan BJ, Kwok YC, et al. Analytical Chemistry. 2006;78:5704–5710. doi: 10.1021/ac060390t. [DOI] [PubMed] [Google Scholar]
- 27.Wen J, Guillo C, Ferrance JP, Landers JP. Analytical Chemistry. 2006;78:1673–1681. doi: 10.1021/ac051796t. [DOI] [PubMed] [Google Scholar]
- 28.Blazej RG, Kumaresan P, Cronier SA, Mathies RA. Analytical Chemistry. 2007;79:4499–4506. doi: 10.1021/ac070126f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.