

Abstract
The liver enzyme phenylalanine hydroxylase is responsible for conversion of excess phenylalanine in the diet to tyrosine. Phenylalanine hydroxylase is activated by phenylalanine; this activation is inhibited by the physiological reducing substrate tetrahydrobiopterin. Phosphorylation of Ser16 lowers the concentration of phenylalanine for activation. This review discusses the present understanding of the molecular details of the allosteric regulation of the enzyme.
Keywords: phenylalanine hydroxylase, allostery, regulation, phosphorylation, tetrahydrobiopterin
Phenylalanine hydroxylase (PheH) catalyzes the hydroxylation of phenylalanine to tyrosine, using tetrahydrobiopterin (BH4) as the source of electrons for the reaction (Scheme 1). The pterin substrate is oxidized to a 4a-hydroxypterin during the reaction; after loss of water, the resulting quinonoid dihydropterin is reduced by dihydropteridine reductase. PheH is a member of the family of aromatic amino acid hydroxylases [1]; the other members are tyrosine hydroxylase (TyrH) and tryptophan hydroxylase (TrpH). All three are non-heme iron enzymes involved in metabolic pathways critical for normal functioning of the nervous system. They have similar catalytic mechanisms but divergent regulatory properties [2–5]. PheH is found in the liver in humans, while rats also have low levels in the kidney [6, 7]. The lack of the enzyme results in phenylketonuria (PKU), a common genetic disease found in ~1/10,000 live births [8]. Individuals with PKU show high levels of phenylalanine in the blood [9], establishing that the physiological role of PheH is metabolism of excess phenylalanine.
Scheme 1.

PheH is highly conserved across mammals, with the sequence of the rat enzyme 92% identical to that of the human enzyme; rat PheH is one amino acid residue longer due to an additional serine at the C-terminus. PheH is found in Drosophila melanogaster as the henna protein [10], but the residues corresponding to the N-terminal 35 residues of the mammalian enzymes, including Ser16, are not conserved; thus, this is not a valid genetic system for studying the regulation of the enzyme. While the enzyme is not found in yeast, there are bacterial PheHs. These are monomeric proteins with structures similar to the catalytic core of eukaryotic PheHs, but lacking regulatory domains [11]. Consistent with the structure, their mechanism of catalysis is the same as liver PheH [12, 13], but there is no evidence for allosteric regulation of a bacterial PheH.
The short-term regulation of PheH is complex. The enzyme is activated by phenylalanine and inhibited by tetrahydrobiopterin (BH4); both phenomena were initially reported several decades ago [14, 15]. Activation of the enzyme by phosphorylation was similarly established in the 1970’s [16]. However, a complete understanding of the effects of substrates and phosphorylation on the activity of the enzyme and the structural changes responsible for the changes in activity are lacking. This review will attempt to describe the present understanding of the regulation of PheH. It will not discuss the catalytic mechanism of the enzyme, which is common to all the aromatic amino acid hydroxylases, since it has been reviewed elsewhere [17].
Effects of substrates on enzyme activity
The reaction catalyzed by PheH involves three substrates, two of which, the tetrahydropterin and oxygen, can react with one another in the absence of enzyme, making the assay itself technically difficult [18]. As discussed in more detail below, the order of addition of the substrates and the identity of the tetrahydropterin can have profound effects on the results. Moreover, the active site of the enzyme contains a non-heme iron that is intimately involved in catalysis. PheH is typically purified with the iron in the ferric state, so the iron must be reduced for catalytic activity [19, 20], a phenomenon also seen with TyrH [21]. Tetrahydropterins are effective reductants, so that simple addition of the ferric enzyme to an assay solution containing an excess of a tetrahydropterin will yield the active ferrous enzyme, but reduction can result in a lag of a few seconds in tyrosine formation. Reduction of the iron by tetrahydropterins was originally attributed to binding at a site other than the active or allosteric site [22]. It is more likely that the tetrahydropterin must still bind in some form in the active site to reduce the iron, but that the mode of binding is slightly different than occurs during turnover, as is the case with TyrH [21]. The enzyme is also reported to lose activity at longer assay times; this has been attributed to oxidative damage during turnover [22, 23].
PheH is activated by phenylalanine. For the rat enzyme with BH4 as the pterin substrate, pretreatment with phenylalanine is reported to increase the activity by 10- to 30-fold. In contrast, the enzyme is reported to be essentially fully active when a nonphysiological pterin such as 6-methyltetrahydropterin (6-MePH4) or 6,7-dimethyltetrahydropterin (6,7-Me2PH4) is used in assays [24, 25]. The effect of incubation with phenylalanine on the activity of the human enzyme is reported to be much smaller than the effect for the rat enzyme, only ~3 to 6-fold, due to the higher levels of activity of the unactivated human enzyme [26–29]. The reasons for the difference between the rat and human enzymes are not known. While the enzyme purified from rat liver is partially phosphorylated [30], the difference is also seen with the recombinant enzymes. The human enzyme is reported to be sensitive to proteolysis at the N-terminus [31], which would be expected to activate PheH, and deamidation, which can alter kinetic parameters but does not appear to activate the enzyme [32].
The physiological relevance of phenylalanine activation was established by two experiments. In the first, the initial rate of tyrosine formation by enzyme in lysates of rapid-frozen livers from rats was measured using BH4 in the assay [30]. The activity in the absence of phenylalanine pretreatment was <5% of that after preincubation, consistent with almost all of the enzyme in the liver being in the low activity form until excess phenylalanine in the diet was present to activate it. If the rats were given phenylalanine before being sacrificed, the activity in the absence of preincubation was ~1/3 that of the fully activated enzyme. In the second series of experiments, the conversion of 14C-phenylalanine to tyrosine and the amount of activity in the absence of phenylalanine pretreatment (activated enzyme) was determined in isolated livers treated with normal serum levels of phenylalanine (84 μM) or the higher levels expected after eating (310 μM). The amount of activated enzyme and the amount of tyrosine formed increased in parallel in livers exposed to the elevated level of phenylalanine.
Activation by phenylalanine has several effects on the kinetics of tyrosine formation, and different research groups have utilized different measures of activation. Nielsen [14] first reported that preincubation of rat PheH with phenylalanine resulted in a significant increase in the initial rate of tyrosine formation when enzyme was added to initiate the reaction. In the absence of such preincubation the reaction showed a distinct lag that increased with decreasing temperature and pH, consistent with the unactivated enzyme having low activity [14, 33]. Thus, the presence of a lag can be used as a measure of activation if BH4 is used as the pterin substrate [34]. If rat or human PheH is incubated for several minutes before adding enzyme and BH4 to start the reaction, varying the concentration of phenylalanine is generally reported to yield sigmoid kinetics if the concentration in the preincubation is the same as in the assays [25, 33, 35, 36] and hyperbolic kinetics if high concentrations (~1 mM) are used in the preincubation [31]. Based on this, the Hill coefficient for phenylalanine has also been used as a measure of the extent of activation [28, 37]. Finally, the relative activity with 6-MePH4 versus BH4 when the enzyme is not pre-treated with phenylalanine has been used as a simple measure of the extent of activation [38, 39].
Knowledge of the number of binding sites for phenylalanine per PheH monomer is obviously critical for understanding the basis for activation, but this has been a matter of some controversy. In many cases methods used to analyze binding have been indirect, such as fluorescence or calorimetry, and would not necessarily have detected binding that did result in a change in the signal being monitored. In other cases the concentrations of the amino acid were likely too low to detect a site with a Kd value of 1 mM or above. The vast majority of binding analyses were carried out under conditions where the enzyme activation would occur during the titration. Using the large-zone gel filtration method of Hummel and Dreyer [40] and 100–200 μM phenylalanine, Shiman [41] found 0.8 binding sites per monomer. Parniak and Kaufman [37] and Shiman and coworkers [42] subsequently found ~1.5 phenylalanine sites per monomer using filter binding assays. When phenylalanine binding is assayed by isothermal titration calorimetry, human PheH is reported to bind only 1 molecule of phenylalanine/subunit, but the PheH from C. elegans to bind 2 [43]; however, concentrations of phenylalanine above 0.25 mM were not used in these analyses. Using a competition assay against a metal chelator in which only binding at the active site would be detected, a Kd value of ~ 1 mM has been measured. While this value is well above the Km value for phenylalanine, such a high Kd value would not be unusual if a tetrahydropterin must bind in the active site before phenylalanine during catalysis, as is the case with TyrH [44], and is suggested by the X-ray structures of PheH substrate complexes (see below).
The range of binding stoichiometries for phenylalanine suggests that the kinetics of binding to the regulatory and catalytic sites differ. Shiman et al. [45] reported that if PheH is incubated with a several-fold excess of 14C-phenylalanine and then 6-Me-PH4 added to start the reaction, one equivalent of the amino acid turns over much more slowly than the remainder. This is consistent with binding of phenylalanine at an allosteric site from which it is only slowly released and suggests not only that binding of phenylalanine to a regulatory site is tighter than binding to the active site, but also that binding and dissociation from the former are slow.
A number of other amino acids are also reported to be activators of PheH, although at much higher concentrations than phenylalanine [24, 46]. Tryptophan is an activator and a very slow substrate for PheH, with half-maximal activation reported to occur at 10 mM. A concentration of 5 mM tryptophan is reported to fully activate the enzyme for tyrosine formation and decrease the stoichiometry of phenylalanine binding from ~1.5 to 1 [46]; this has been attributed to binding of tryptophan at a regulatory site but not the active site under the experimental conditions.
Most binding assays have been carried out at 25 °C or higher and allowed binding and presumably activation to reach equilibrium. In contrast, Shiman et al. [42] reported that no binding of phenylalanine could be detected at times up to 3 min by filter-binding when the temperature is kept low to slow the conformational change accompanying activation, while at longer times and a higher temperature ~1.5 molecules of phenylalanine bound. This suggests that binding of the amino acid is coupled tightly to activation in that phenylalanine only binds tightly to the allosteric site after a conformational change has occurred. Kinetically, a slow conformational change followed by rapid and tight binding is difficult to distinguish from rapid binding that causes a slow conformational change. Overall, the data are consistent with a model in which PheH has an allosteric site that binds phenylalanine slowly and tightly (Kd < 0.1 mM), and an active site in the activated enzyme to which binding is looser (Kd ~ 1 mM).
Measurements of the stoichiometry of tetrahydropterin binding are consistent in finding one site per monomer [42, 47, 48]. In the absence of phenylalanine the binding is quite tight, with Kd values of ~ 0.1 μM and ~15 μM for BH4 and 6-MePH4, respectively, for both the rat and human enzymes. Measurement of the affinity for the activated enzyme is obviously more difficult, since turnover must be avoided unless the Km value is of interest. Shiman et al. [42], using a metal chelator to remove the active site iron and thereby inhibit the enzyme, reported a value of ~14 μM for binding of BH4 to the active enzyme. This was not done with 6-MePH4, but the binding of the air-stable analog 5-deaza-6-MePH4 was not affected by phenylalanine activation, while the binding of dihydrobiopterin (BH2) was [42], suggesting that the dihydroxypropyl side chain is required for tight binding of a tetrahydropterin to the unactivated enzyme. The reported Km values for BH4 and 6-MePH4 are generally in the range 20–50 μM for both pterins for the rat and human enzymes [26, 28, 49–51], in line with the reported Kd values for BH4 and 6-MePH4 for the activated enzyme. Mitnaul and Shiman [52] reported that a significant amount of biopterin in liver cells is retained by 30,000 Da cutoff filters, with the amount retained decreasing with increasing phenylalanine concentration, and attributed this to BH4 bound to unactivated PheH.
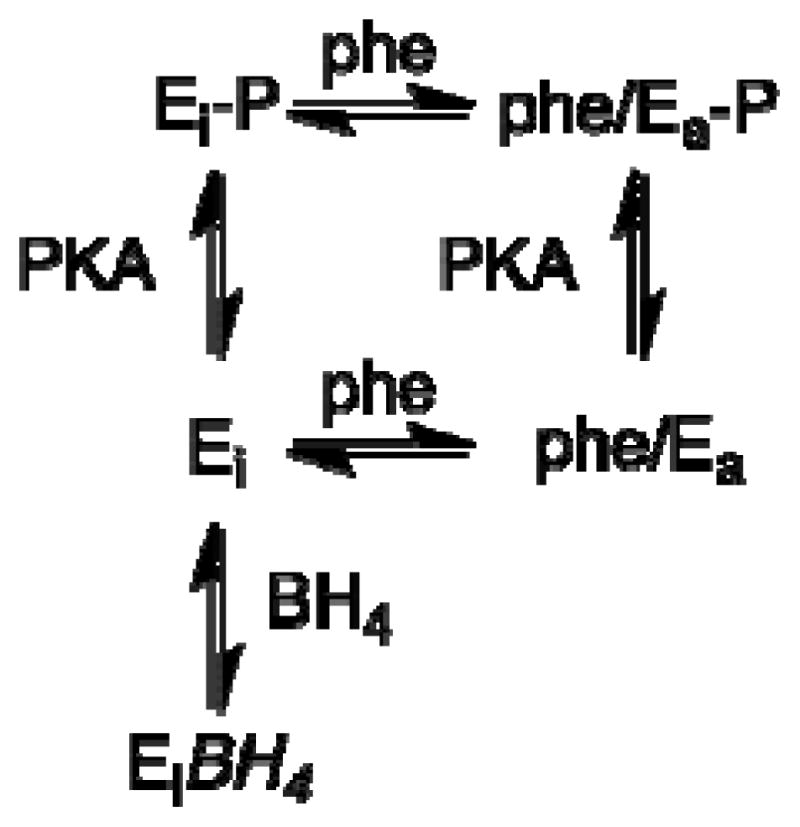
The most detailed mechanism for regulation of PheH was developed by Shiman and coworkers studying the enzyme from rat liver [53]. Key to the approach was the use of low temperature (5–10 °C) to slow the activation sufficiently for characterization of the unactivated enzyme and the use of metal chelators to probe binding to the active site without interference from binding at metal-free allosteric sites. The mechanism is shown in Scheme 2. Here, the resting form of the enzyme, Ei, is inactive. In the presence of phenylalanine, Ei converts slowly to an active form, Ea, in which there is an allosteric site that binds phenylalanine tightly. Only after the conformational change to Ea is the active site able to bind phenylalanine. BH4 will bind to Ei tightly, preventing activation by phenylalanine [42]. The binding site for tetrahydropterins in Ea differs structurally from that in Ei, but in both cases binding is to the enzyme active site. 6-MePH4 will also bind to Ei, but only very loosely, so that no effect is seen unless phenylalanine binds to trap the Ei/6-MePH4 complex. The phe/Ei/6-MePH4 complex can slowly convert to active enzyme.
Scheme 2.

The observation of a lag in the formation of tyrosine when PheH is not pretreated with phenylalanine is central to the model of Scheme 2. In their initial studies, Shiman and Gray [33] found that the initial rate of tyrosine formation depended on the concentration of phenylalanine in the preincubation in a sigmoidal fashion and concluded that the resting enzyme had no activity until phenylalanine bound. The rate constant for activation in these studies was pH and temperature-dependent, so that lags were more obvious at and below pH 6.8 and 25 °C. Lysolecithin, another activator of PheH [35], had a similar effect on the activity of PheH. In the absence of preincubation, activation by lysolecithin was inhibited when BH4 was used in assays but not when 6-Me-PH4 was used. Conversely, longer lags were seen when the enzyme is preincubated with BH4 and assays were started with phenylalanine, consistent with the binding of BH4 to Ei inhibiting the conversion to Ea [53]. When phenylalanine at high concentrations (0.5 or 5 mM) was included with BH4 in a preincubation, less pterin was required to inhibit the activation; this suggested that the amino acid could trap the inactive Ei/BH4 complex. No inhibition could be detected with 6-MePH4 unless phenylalanine were also included in the preincubation. A significant complication in interpreting these experiments is the likelihood that the tetrahydropterin would oxidize to the dihydropterin during the aerobic preincubation, so that the inhibition is due to the oxidized cofactor. Consistent with this possibility, BH2 gave similar results to BH4.
Structural basis for regulation of phenylalanine hydroxylase
At present, there is no structure available of an intact PheH from a eukaryotic source. A variety of approaches have established that all three of the eukaryotic amino acid hydroxylases have three identifiable structural domains, an N-terminal regulatory domain that differs among the three enzymes, a homologous catalytic domain, and a homologous C-terminal domain involved in subunit-subunit interactions [1]. In both human and rat PheH, the regulatory domain extends to about residue 116 [34], while residue 425 is the beginning of a long alpha helix that forms a coiled-coil interaction with an adjacent subunit [63]. Most of the available structures lack the regulatory domain or the C-terminal helix or both. Still, it is possible to combine structures of the human enzyme lacking the regulatory domain [63] and the rat enzyme lacking the C-terminal 24 residues [64] to obtain a model for the intact enzyme. As shown in Figure 1A, the enzyme is a dimer of dimers. Figure 1B illustrates the interactions between individual domains within a dimer. In addition to extensive contacts between the two catalytic domains, each regulatory domain interacts with two catalytic domains but not with the other regulatory domain in the dimer. Figure 1C shows that in each monomer the N-terminal tail is over the active site. This observation led to the proposal that the N-terminus of the enzyme acts as an autoregulatory sequence [64], allowing limited substrate access unless the enzyme is activated by phenylalanine binding to the regulatory domain. Activation would result in the N-terminal 33 residues moving away from the active site. The proposal that the N-terminal ~30 residues is key to the activating effect of phenylalanine is supported by the results of site-directed mutagenesis. Removal of the entire regulatory domain from PheH results in an enzyme that has much higher activity in the absence of phenylalanine [36, 65] and does not exhibit a long lag in tyrosine formation when the assay is initiated by adding phenylalanine [34]. Removal of just the first 26 or 30 residues of rat PheH is sufficient to yield an enzyme that is mostly or fully activated [39, 66].
Figure 1.

Structure of PheH. A, Structure of the intact tetramer; B, structure of a dimer with the regulatory and catalytic domains on the same subunit colored different shades of red or blue; C, the structure of an individual monomer with the regulatory domain (blue) in cartoon mode and the active site iron shown as an orange sphere. Figures 1A and 1B were constructed by overlaying the dimeric structure of rat PheH lacking the C-terminal helix (pdb file 2PHM) and the tetrameric structure of human PheH lacking the regulatory domain (pdb file 2PAH). Figure C was constructed using pdb file 2PHM.
There is clear evidence for a significant structural change associated with phenylalanine binding but not with BH4 binding. Shiman et al. [67] reported that the enzyme was retained on a hydrophobic chromatography column if phenylalanine was present and eluted if the amino acid was removed. This observation was taken as evidence for a conformational change upon phenylalanine binding to PheH that both activated the enzyme and exposed a hydrophobic surface. This phenomenon is also the basis for purification of the enzyme. Phenylalanine binding results in an increase in the fluorescence of the enzyme, due mostly to an increase in the fluorescence of Trp120 [47, 68, 69]. The fluorescence is more easily quenched by sodium iodide in the presence of phenylalanine [47], suggesting that Trp120 becomes more solvent-exposed upon activation. Trp120 is located near the protein surface at the interface of the catalytic and regulatory domains within a subunit, so that the changes in its fluorescence when phenylalanine binds establish that the structural changes upon activation extend beyond the first 33 residues and likely involve altered domain-domain interfaces. In the absence of high-resolution information from crystal structures regarding the effects of phenylalanine on the PheH, the kinetics of deuterium incorporation into peptide bonds as monitored by mass spectrometry (HXMS) [70] have provided a slightly lower resolution picture [71]. In the presence of phenylalanine, there are extensive changes in deuterium incorporation at the interface between the regulatory and catalytic domains. There are also changes in the exchange kinetics of residues within the regulatory domain. The largest change is seen in the peptide containing residues 39–59. In all cases binding of phenylalanine results in increased deuterium incorporation, consistent with activation resulting in a more dynamic or open structure. Trp120 is located within the region showing extensive changes, although no peptide containing this residue was detected in the HXMS analyses. The HXMS analyses also show that the structures of several active site loops, including the Tyr138 loop, are altered in the presence of phenylalanine. This is consistent with conclusions drawn from the X-ray structures of the ferrous catalytic domain alone [72]. Since both the active site and the regulatory site for phenylalanine would have been occupied under the conditions of the HXMS analyses, it is not possible to unambiguously assign the loop movements to regulation or catalysis. Still, movement of the N-terminal tail away from the active site would be expected to give loops over the active site more freedom of motion and this would be reflected by increased deuterium incorporation from solvent.
In the absence of a structure of an intact PheH with phenylalanine bound, the location and even the presence of an allosteric site for phenylalanine in the regulatory domain has been a matter of some controversy. The regulatory domain of PheH has a similar fold to ACT domains; these are 70–80 residue domains often found in enzymes of purine and amino acid metabolism that are regulated by amino acid binding [73, 74]. Kobe et al. [64] suggested that PheH has a binding site for phenylalanine in the regulatory domain analogous to the allosteric site for serine in the ACT domain of 3-phosphoglycerate dehydrogenase [75]. The binding sites in ACT domains are generally on loops at the interfaces between domains, but the actual sites are poorly conserved [73]. If one assigns the binding site for phenylalanine in PheH to the same loop as is used in several other amino acid-binding ACT domains, that between the first beta strand and first helix [73, 76], the regulatory site is on the loop containing residues 42–47 (Figure 2). Alternatively, Flydal et al. [43], based on analogy to the structure of the ACT domain of prephenate dehydrogenase with phenylalanine bound, proposed that C. elegans PheH has a second phenylalanine binding site that would correspond to residues 46–48 and 65–69 in the regulatory domain of the human enzyme. Since these authors could only detect one binding site for phenylalanine in the human enzyme using calorimetry, the proposed site in the regulatory domain of the human enzyme was proposed to be nonfunctional. However, the PKU-associated mutations G46S, A47T, T63P/H64N, I65T, and R68S each decrease or eliminate binding of phenylalanine to a maltose binding protein-fusion of human PheH [77], suggesting that these residues in the regulatory domain are critical for a functional phenylalanine binding site. As noted above HXMS analyses of phenylalanine binding are similarly consistent with residues with a phenylalanine site within residues 39–59 [71]. Figure 2 shows the locations of residues 42–48 and 65–69 in the structure.
Figure 2.

Potential allosteric sites in the regulatory domain of PheH. Orange: binding site for BH4 based on structure of PCD/DCoH. Magenta: residues 42–48 and 63–69, potential binding sites for phenylalanine. Green: residues 19–31. Blue: residues 32–116. Red: catalytic domain, residues 117–426. The figure is based on pdb file 2PHM.
Based on a number of biophysical methods, Martinez and coworkers [28, 78–80] have argued that phenylalanine binds only at the active site of PheH and that the regulation involves homotropic interactions between different subunits. In this model, the role of the regulatory domain is to allow the local conformational change upon phenylalanine binding at one active site to be communicated to the active site in another subunit. The structures of ferrous human PheH lacking the regulatory domain have identified a number of structural differences between the protein with BH4 bound and the protein with both BH4 and an amino acid bound [72, 81]. In addition to movement of the BH4 closer to the iron and a change in the iron ligands (see below), several surface loops undergo significant motion. The loop showing the most dramatic movement is that containing Tyr138, which closes down over the active site to complete the binding site for the side chain of the amino acid substrate. Andersen et al. [72, 81] have similarly proposed that movements of surface loops are the critical conformational change responsible for activation, in that the Tyr138 loop and the N-terminal residues are adjacent in the intact structure, so that activation is due to binding of phenylalanine in the active site rather than at a regulatory site.
Very recently, NMR spectroscopy of the isolated regulatory domain of rat PheH has shown that the NMR shifts of a number of residues are perturbed in the presence of a stoichiometric amount of phenylalanine but not in the presence of the non-activating amino acid proline, providing strong evidence for a phenylalanine binding site in the regulatory domain [82]. Still, the location of such a site remains to be determined.
Conformational changes are seen in PheH lacking the regulatory domain upon binding of phenylalanine to the BH4-PheH complex, but they may involved in formation of an active site competent for a productive reaction with oxygen rather than with regulation. Spectroscopic studies of PheH and TyrH show that the ligands to the active site iron in both undergo a change from 6-coordinate to 5-coordinate when both the amino acid substrate and a tetrahydropterin are bound [83, 84]. In addition, the surface loop in TyrH that corresponds to the Tyr138 loop also shows significant motion upon substrate binding [85], although the regulatory mechanism and accompanying structural changes in that enzyme are different from those for PheH [3, 86, 87]. The structural changes detected by HXMS upon binding to rat PheH lacking the regulatory domain are much more localized than is the case for the intact enzyme [71], suggesting that binding to the regulatory site is required for large conformational changes. However, such a result would not be inconsistent with the role of the regulatory domain being only to communicate between different catalytic domains.
The crystal structure of the enzyme with an intact C-terminus is a tetramer, but PheH in solution appears to be a mixture of dimers and tetramers. This was first reported with enzyme isolated from rat liver using gel filtration and ultracentrifugation to measure the size [88, 89]. In the presence of phenylalanine there is a shift from predominantly dimer to predominantly tetramer [31, 49, 89]. For the rat enzyme the equilibrium between tetramer and dimer is pH-dependent, with tetramer predominating at pH 6 and dimer at pH 8 [49]. The equilibrium between the dimer and tetramer must be very slow or not occur at all, at least at the low (4–5 °C) temperature of these analyses, in that discrete peaks for dimer and tetramer are seen in gel filtration, and rechromatography of the isolated dimer still yields ~90% dimer under conditions where the initial chromatography shows the enzyme to be ~80% tetramer [36]. The structural basis for the isolable and non-equilibrating dimeric and tetrameric forms is unknown, but the contribution by Jaffe to this special issue [90] provides one possible explanation. The relevance of the phenomenon to the regulation has not been established, although Shiman et al. [45] concluded that the kinetics of activation by phenylalanine are more readily accommodated by activation of a tetrameric species. In addition, the reported rate constants for activation at low temperature (t1/2 ~ 14 min at 5 °C, [49]) appear to be too rapid to allow isolation of stable populations of dimers and tetramers.
Significantly, the active site iron in intact PheH appears to be unaffected when phenylalanine is bound to form the activated enzyme [91], suggesting that activation does not directly alter the active site structure. In contrast, binding of the amino acid substrate to a pterin-bound form of the enzyme lacking the regulatory domain does result in changes in the active site iron. The metal goes from hexacoordinate, with two histidines, a glutamate, and three water molecules as ligands to pentacoordinate, losing two water molecules and gaining the second glutamate carboxylate oxygen as a ligand [72, 84]. A similar change has been seen in TyrH [83]; in both enzymes it has been attributed to the need to open a site on the iron for oxygen.
The structural effects of BH4 binding differ from the effects of phenylalanine, consistent with the model of Scheme 2. Treatment of rat PheH with chymotrypsin will activate the enzyme, removing an N-terminal fragment of ~11,000 Mr, equivalent to almost the entire regulatory domain, and a C-terminal fragment of ~5,000 Mr [35, 92, 93]. Activation by chymotrypsin is slightly faster in the presence of phenylalanine, while it can be almost completely inhibited by BH4. These results suggest that binding of phenylalanine results in a more open, and chymotrypsin-accessible form and binding of BH4 shifts the equilibrium toward the chymotrypsin-resistant closed form. The fluorescence of PheH decreases in the presence of BH4, the opposite effect to phenylalanine [47, 69], again demonstrating that the two substrates have different effects on the structure.
In the model of Scheme 2, there is a BH4 regulatory site in addition to the active site, although it was proposed to be a modified form of the active site [42]. Kobe et al. [64], in their description of the structure of rat PheH catalytic and regulatory domains, noted that the regulatory domain had a similar fold to the bifunctional pterin-4a-carbinolamine dehydratase PCD/DCoH and suggested that the PheH regulatory domain also contained a similar pterin binding site. The pterin binding site in PCD/DCoH is made up mainly of residues on a loop at the interface between subunits [94]. Binding in an analogous fashion to PheH would result in BH4 binding at the interface of the catalytic and regulatory domains (Figure 2), where it would presumably block the conformational change required for activation. To date there is no direct structural evidence for a regulatory pterin binding site outside of the active site of PheH. Structures are available of PheH with BH4 or an analog bound, but these all lack the regulatory domain. The structures of the catalytic domain of ferrous PheH in the presence and absence of BH4 show only small differences [95], and many of these can be attributed to the higher resolution of the latter structure. In contrast, the binding of BH4 is different in the ternary complex of the ferrous catalytic domain with an amino acid bound [72] [Figure 3]. In the binary complex, the pterin ring forms hydrogen bonds to the protein backbone and there is a water-mediated hydrogen bond to Glu286, with the C4a of the pterin 5.9 Å from the iron. In the ternary complex, BH4 is shifted ~2.6 Å closer to Glu286 and the iron forming direct hydrogen bonds with that amino acid residue. Solstad et al. [96] have proposed that this motion is key to the inhibitory effects of BH4. If a structure of the catalytic domain of human PheH with bound pterin is superimposed on the structure of rat PheH containing both the regulatory and catalytic domain, as in Figure 1, the hydroxyl oxygens of the BH4 side chain are within hydrogen-bonding distance of Ser23 in the regulatory domain. When the ternary complex of the catalytic domain is utilized in the same superposition, the 2.6 Å displacement of the pterin eliminates the hydrogen-bond with Ser23. The binary complex with BH4 alone would correspond to the inhibited form, while the ternary complex would correspond to the activated form. This model explains why BH4 is inhibitory but not 6-MePH4, in that the latter lacks the dihydroxypropyl side chain and consequently cannot form hydrogen bonds with Ser23.
Figure 3.

Proposed interactions [96] between Ser23 in the regulatory domain and BH4 in the active site of the binary BH4 complex (magenta carbons and blue H-bonds) and the ternary BH4-amino acid complex (green carbons and H-bonds).
Knowledge of the structure of the combined catalytic and regulatory domains of rat PheH led Kobe et al. [64] to identify two possible hinge regions that are critical for the conformational changes involved in regulation (Figure 2). The first is residues 31–33; this is consistent with a rather simple motion in which the N-terminal 33 residues move away from the active site. The second is the loop from residues 111–117; movement around this region would allow for more drastic reorientation of the two domains with respect to one another. Based on the structure of the tetrameric catalytic domain of human PheH, Fusetti et al. [63] proposed that the tetramerization domain also contains two hinge regions, one containing residues 425–429 at the beginning of the long C-terminal helix and a second at Gly422 where there is a kink in the helix. The role of these hinges would be to allow the tetrameric structure to accommodate changes in the relative orientations of the subunits upon phenylalanine binding. However, the HXMS analyses of both the intact protein and the tetrameric catalytic domain did not find any detectable changes in the deuterium exchange kinetics of the peptides containing these residues in the presence of phenylalanine.
Effects of phosphorylation on phenylalanine hydroxylase
Glucagon treatment of rats results in increased phosphorylation of PheH, from a basal level of ~0.3 phosphates/monomer to 1 per monomer, and increases the basal activity of the enzyme [30, 54]. In vitro, several protein kinases, including cAMP-dependent protein kinase A (PKA), will phosphorylate the enzyme on Ser16 [16, 31, 55, 56]. PKA is likely to be the physiological kinase, since glucagon treatment of rats would activate PKA. Replacement of Ser16 with glutamate in either rat [57] or human PheH [27] replicates the effect of phosphorylation on the kinetics, confirming Ser16 as the phosphorylation site.
The effect of phosphorylation on the enzyme activity is much less than the effect of phenylalanine pretreatment. Instead of directly activating the enzyme, phosphorylation decreases the concentration of phenylalanine required to activate it. The effect is about 2-fold for both rat and human PheH [30, 38], and has been attributed to an increase in the rate constant for activation for the phosphorylated protein [30]. Phosphorylation is also reported to increase the Vmax value of the unactivated enzyme by ~1.4-fold for the human enzyme and ~3-fold for the rat enzyme [27, 57]. The enzyme from both sources still exhibits sigmoidal kinetics in the absence of phenylalanine treatment and is activated several-fold more by such treatment [27, 57].
The concentrations of the substrates affect the kinetics of phosphorylation, and this may play a role in mediating the activity of the enzyme in the liver. BH4 will decrease the rate of phosphorylation of rat PheH by PKA [58], with half-maximal inhibition occurring at 2 μM BH4 [59], several-fold less than the physiological concentration of BH4 (10–15 μM [52, 60, 61]). 6-MePH4 and 6,7-Me2PH4 have no effect even at substantially higher concentrations [58], in line with the general lack of effect of these substrates on the regulatory properties. Phenylalanine can increase the rate of phosphorylation up to 2-fold [58], with half-maximal activation occurring at ~ 50 μM [62], close to the typical concentration of phenylalanine in liver [30]. Higher concentrations of phenylalanine can overcome the effects of BH4, suggesting that modulation of the rate of phosphorylation provides an additional way of activating PheH.
The effects of phosphorylation on the activity of PheH suggest that the model of Scheme 2 is too simple. The data can be accommodated by adding phosphorylated forms of Ei and Ea to the scheme as shown in Scheme 3. The phosphorylated Ei would have low activity unless activated by phenylalanine, but activation would occur at lower concentrations of the amino acid. Binding of BH4 to Ei prevents phosphorylation, while the conformational change that occurs upon activation by phenylalanine increases the rate of phosphorylation, likely by making Ser16 more accessible.
Scheme 3.
The effects of phosphorylation on the structure of PheH are more subtle than the effects of phenylalanine binding, consistent with the less drastic effect on activity. The structure of the combined regulatory and catalytic domain of rat PheH after phosphorylation was also described in the original report by Kobe et al. [64] of the enzyme lacking only the C-terminal helix. Surprisingly, the structure of the phosphorylated enzyme was identical to that of the unphosphorylated enzyme. However, neither structure showed any electron density for the N-terminal 19 residues, including the phosphorylation site. There are a number of possible explanations for these results. The conformational change caused by phosphorylation could be limited to residues within the N-terminal 18 residues missing in the structures, with these residues being very mobile. Indeed, NMR spectroscopy [97] and HXMS [71] both show that the N-terminal residues missing from the X-ray structures are quite dynamic. Alternatively and less interesting, crystal packing forces may have overcome the conformational change caused by phosphorylation, or an adventitious protease may have removed the phosphorylation site from both samples. To address the effect of phosphorylation on the structure, Miranda et al. [27, 98] modeled the missing residues into the structure and then used molecular dynamics to determine the effects of phosphorylation of Ser16. These calculations indicated that the conformational change was limited to residues near Ser16, with an interaction between Arg15 and the serine-phosphate and repulsion between Glu280 at the opening to the active site being critical. Recent analyses of the effects of phosphorylation of Ser40 in the N-terminal regulatory domain of TyrH have shown that the effects of phosphorylation are also localized to the phosphorylation site and residues at the active site in that aromatic amino acid hydroxylase [87, 99]. A much more localized effect of phosphorylation on the structure compared to phenylalanine is consistent with the expanded model of Scheme 3.
Other modes of activation
PheH can be activated in other ways than by treatment with phenylalanine. Several phospholipids, including lysolecithin, will activate the enzyme in a similar matter to phenylalanine [47, 100], although there is no evidence that this is physiological. As noted above, proteases such as chymotrypsin activate the enzyme by removing the regulatory domain [35, 101]. Finally, the enzyme is activated by modification of Cys237 [37, 102]; the modified enzyme behaves similarly to enzyme activated by phenylalanine [37]. Mutagenesis of Cys237 also alters the regulatory properties of the enzyme [28]. Cys237 is located on the surface of the catalytic domain near the loop containing residues 69–75 in the regulatory domain of the other subunit in the same dimer, so that modifications of Cys237 would be expected to disrupt interactions between these domains. Consistent with a role of this interface in communicating the allosteric effect of phenylalanine between subunits, mutants of Arg68 also alter the regulatory properties of the enzyme [28].
Conclusions
The regulation of phenylalanine hydroxylase in the liver is obviously complex, with phosphorylation overlaid on the allosteric effects of both phenylalanine and BH4. The kinetic model of Scheme 2 developed by Shiman and coworkers remains the most comprehensive description of the effects of regulation. A key outstanding question is the structural basis for the allosteric effects of phenylalanine, including the location of the binding site responsible for regulation. There is a clear need for structures of the intact enzyme with only phenylalanine or BH4 bound.
Research Highlights.
Phenylalanine hydroxylase is activated by phenylalanine.
Phenylalanine activation is greatest with tetrahydrobiopterin as substrate.
Ser16 phosphorylation lowers the phenylalanine concentration needed for activation.
Activation is accompanied by altered interactions between domains.
Acknowledgments
This work was supported in part by NIH grants GM047291 and GM098140.
Abbreviations used
- PheH
phenylalanine hydroxylase
- TyrH
tyrosine hydroxylase
- BH4
tetrahydrobiopterin
- BH2
dihydrobiopterin
- 6-MePH4
6-methyltetrahydropterin
- 6,7-Me2PH4
6,7-dimethyltetrahydropterin
- HXMS
hydrogen/deuterium exchange mass spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fitzpatrick PF. Ann Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 2.Almas B, Le Bourdelles B, Flatmark T, Mallet J, Haavik J. Eur J Biochem. 1992;209:249–255. doi: 10.1111/j.1432-1033.1992.tb17283.x. [DOI] [PubMed] [Google Scholar]
- 3.Ramsey AJ, Fitzpatrick PF. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]
- 4.McKinney J, Knappskog PM, Haavik J. J Neurochem. 2005;92:311–320. doi: 10.1111/j.1471-4159.2004.02850.x. [DOI] [PubMed] [Google Scholar]
- 5.Daubner SC, Le T, Wang S. Arch Biochem Biophys. 2011;508:1–12. doi: 10.1016/j.abb.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGee MM, Greengard O, Knox WE. Biochem J. 1972;127:669–674. doi: 10.1042/bj1270669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh MC, Berry HK. J Exp Zool. 1979;208:161–167. doi: 10.1002/jez.1402080204. [DOI] [PubMed] [Google Scholar]
- 8.Konecki DS, Lichter-Konecki U. Hum Genet. 1991;87:377–388. doi: 10.1007/BF00197152. [DOI] [PubMed] [Google Scholar]
- 9.Williams RA, Mamotte CDS, Burnett JR. Clin Biochem Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]
- 10.Morales G, Requena J, Jimenez-Ruiz A, Lopez MC, Ugarte M, Alonso C. Gene. 1990;93:213–219. doi: 10.1016/0378-1119(90)90227-i. [DOI] [PubMed] [Google Scholar]
- 11.Erlandsen H, Kim JY, Patch MG, Han A, Volner A, Abu-Omar MM, Stevens RC. J Mol Biol. 2002;320:645–661. doi: 10.1016/s0022-2836(02)00496-5. [DOI] [PubMed] [Google Scholar]
- 12.Panay AJ, Fitzpatrick PF. Biochemistry. 2008;47:11118–11124. doi: 10.1021/bi801295w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panay AJ, Lee M, Krebs C, Bollinger JM, Jr, Fitzpatrick PF. Biochemistry. 2011;50:1928–1933. doi: 10.1021/bi1019868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nielsen KH. Eur J Biochem. 1969;7:360–369. doi: 10.1111/j.1432-1033.1969.tb19617.x. [DOI] [PubMed] [Google Scholar]
- 15.Bailey SW, Ayling J. J Biol Chem. 1978;253:1598–1605. [PubMed] [Google Scholar]
- 16.Abita JP, Milstien S, Chang N, Kaufman S. J Biol Chem. 1976;251:5310–5314. [PubMed] [Google Scholar]
- 17.Fitzpatrick PF. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearson AJ. Chem&Indus. 1974:233–239. [Google Scholar]
- 19.Wallick DE, Bloom LM, Gaffney BJ, Benkovic SJ. Biochemistry. 1984;23:1295–1302. doi: 10.1021/bi00301a043. [DOI] [PubMed] [Google Scholar]
- 20.Marota JJA, Shiman R. Biochemistry. 1984;23:1303–1311. doi: 10.1021/bi00301a044. [DOI] [PubMed] [Google Scholar]
- 21.Frantom PA, Seravalli J, Ragsdale SW, Fitzpatrick PF. Biochemistry. 2006;45:2372–2379. doi: 10.1021/bi052283j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiman R, Gray DW, Hill MA. J Biol Chem. 1994;269:24637–24646. [PubMed] [Google Scholar]
- 23.Fink RM, Elstner EF. Zeitschrift fuer Naturforschung. 1984;39C:734–737. doi: 10.1515/znc-1984-7-809. [DOI] [PubMed] [Google Scholar]
- 24.Phillips RS, Parniak MA, Kaufman S. J Biol Chem. 1984;259:271–277. [PubMed] [Google Scholar]
- 25.Citron BA, Davis MD, Kaufman S. Protein Expression Purif. 1992;3:93–100. [PubMed] [Google Scholar]
- 26.Kowlessur D, Citron BA, Kaufman S. Arch Biochem Biophys. 1996;333:85–95. doi: 10.1006/abbi.1996.0367. [DOI] [PubMed] [Google Scholar]
- 27.Miranda FF, Teigen K, Thorolfsson M, Svebak RM, Knappskog PM, Flatmark T, Martinez A. J Biol Chem. 2002;277:40937–40943. doi: 10.1074/jbc.M112197200. [DOI] [PubMed] [Google Scholar]
- 28.Thorolfsson M, Teigen K, Martinez A. Biochemistry. 2003;42:3419–3428. doi: 10.1021/bi034021s. [DOI] [PubMed] [Google Scholar]
- 29.Carvalho RN, Solstad T, Bjorgo E, Barroso JF, Flatmark T. J Biol Chem. 2003;278:15142–15152. doi: 10.1074/jbc.M212180200. [DOI] [PubMed] [Google Scholar]
- 30.Shiman R, Mortimore GE, Schworer CM, Gray DW. J Biol Chem. 1982;257:11213–11216. [PubMed] [Google Scholar]
- 31.Martinez A, Knappskog PM, Olafsdottir S, Doskeland AP, Eiken HG, Svebak RM, Bozzini M, Apold J, Flatmark T. Biochem J. 1995;306:589–597. doi: 10.1042/bj3060589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solstad T, Flatmark T. Eur J Biochem. 2000;267:6302–6310. doi: 10.1046/j.1432-1327.2000.01715.x. [DOI] [PubMed] [Google Scholar]
- 33.Shiman R, Gray DW. J Biol Chem. 1980;255:4793–4800. [PubMed] [Google Scholar]
- 34.Daubner SC, Hillas PJ, Fitzpatrick PF. Arch Biochem Biophys. 1997;348:295–302. doi: 10.1006/abbi.1997.0435. [DOI] [PubMed] [Google Scholar]
- 35.Fisher DB, Kaufman S. J Biol Chem. 1973;248:4345–4353. [PubMed] [Google Scholar]
- 36.Knappskog PM, Flatmark T, Aarden JM, Haavik J, Martinez A. Eur J Biochem. 1996;242:813–821. doi: 10.1111/j.1432-1033.1996.0813r.x. [DOI] [PubMed] [Google Scholar]
- 37.Parniak MA, Kaufman S. J Biol Chem. 1981;256:6876–6882. [PubMed] [Google Scholar]
- 38.Doskeland AP, Martinez A, Knappskog PM, Flatmark T. Biochem J. 1996;313:409–414. doi: 10.1042/bj3130409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GA, Gu P, Kaufman S. Proc Natl Acad Sci USA. 2001;98:1537–1542. doi: 10.1073/pnas.031561698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hummel JP, Dreyer WJ. Biochim Biophys Acta. 1962;63:530–532. doi: 10.1016/0006-3002(62)90124-5. [DOI] [PubMed] [Google Scholar]
- 41.Shiman R. J Biol Chem. 1980;255 [PubMed] [Google Scholar]
- 42.Shiman R, Xia T, Hill MA, Gray DW. J Biol Chem. 1994;269:24647–24656. [PubMed] [Google Scholar]
- 43.Flydal MI, Mohn TC, Pey AL, Siltberg-Liberles J, Teigen K, Martinez A. Amino Acids. 2010;39:1463–1475. doi: 10.1007/s00726-010-0611-6. [DOI] [PubMed] [Google Scholar]
- 44.Fitzpatrick PF. Biochemistry. 1991;30:3658–3662. doi: 10.1021/bi00229a010. [DOI] [PubMed] [Google Scholar]
- 45.Shiman R, Jones SH, Gray DW. J Biol Chem. 1990;265:11633–11642. [PubMed] [Google Scholar]
- 46.Kaufman S, Mason K. J Biol Chem. 1982;257:14667–14678. [PubMed] [Google Scholar]
- 47.Phillips RS, Parniak MA, Kaufman S. Biochemistry. 1984;23:3836–3842. doi: 10.1021/bi00312a007. [DOI] [PubMed] [Google Scholar]
- 48.Pey AL, Thorolfsson M, Teigen K, Ugarte M, Martinez A. J Am Chem Soc. 2004;126:13670–13678. doi: 10.1021/ja047713s. [DOI] [PubMed] [Google Scholar]
- 49.Kappock TJ, Harkins PC, Friedenberg S, Caradonna JP. J Biol Chem. 1995;270:30532–30544. doi: 10.1074/jbc.270.51.30532. [DOI] [PubMed] [Google Scholar]
- 50.Haavik J, Doskeland AP, Flatmark T. Eur J Biochem. 1986;160:1–8. doi: 10.1111/j.1432-1033.1986.tb09932.x. [DOI] [PubMed] [Google Scholar]
- 51.Bjorgo E, de Carvalho RMN, Flatmark T. Eur J Biochem. 2001;268:997–1005. doi: 10.1046/j.1432-1327.2001.01958.x. [DOI] [PubMed] [Google Scholar]
- 52.Mitnaul LJ, Shiman R. Proc Natl Acad Sci USA. 1995;92:885–889. doi: 10.1073/pnas.92.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia T, Gray DW, Shiman R. J Biol Chem. 1994;269:24657–24665. [PubMed] [Google Scholar]
- 54.Donlon J, Kaufman S. J Biol Chem. 1978;253:6657–6659. [PubMed] [Google Scholar]
- 55.Wretborn M, Humble E, Ragnarsson U, Engstrom L. Biochem Biophys Res Commun. 1980;93:403–408. doi: 10.1016/0006-291x(80)91091-8. [DOI] [PubMed] [Google Scholar]
- 56.Doskeland AP, Schworer CM, Doskeland SO, Chrisman TD, Soderling TR, Corbin JD, Flatmark T. Eur J Biochem. 1984;145:31–37. doi: 10.1111/j.1432-1033.1984.tb08518.x. [DOI] [PubMed] [Google Scholar]
- 57.Kowlessur D, Yang XJ, Kaufman S. Proc Natl Acad Sci USA. 1995;92:4743–4747. doi: 10.1073/pnas.92.11.4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phillips RS, Kaufman S. J Biol Chem. 1984;259:2474–2479. [PubMed] [Google Scholar]
- 59.Doskeland AP, Haavik J, Flatmark T, Doskeland SO. Biochem J. 1987;242:867–874. doi: 10.1042/bj2420867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guerin T, Walsh GA, Donlon J, Kaufman S. Int J Biochem Cell Biol. 1998;30:1047–1054. doi: 10.1016/s1357-2725(98)00065-x. [DOI] [PubMed] [Google Scholar]
- 61.Rembold H, Buff K. Eur J Biochem. 1972;28:579–585. doi: 10.1111/j.1432-1033.1972.tb01946.x. [DOI] [PubMed] [Google Scholar]
- 62.Doskeland AP, Doskeland SO, Ogreid D, Flatmark T. J Biol Chem. 1984;259:11242–11248. [PubMed] [Google Scholar]
- 63.Fusetti F, Erlandsen H, Flatmark T, Stevens RC. J Biol Chem. 1998;273:16962–16967. doi: 10.1074/jbc.273.27.16962. [DOI] [PubMed] [Google Scholar]
- 64.Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, Stevens RC, Cotton RGH, Kemp BE. Nat Struct Biol. 1999;6:442–448. doi: 10.1038/8247. [DOI] [PubMed] [Google Scholar]
- 65.Daubner SC, Hillas PJ, Fitzpatrick PF. Biochemistry. 1997;36:11574–11582. doi: 10.1021/bi9711137. [DOI] [PubMed] [Google Scholar]
- 66.Jennings IG, Teh T, Kobe B. FEBS Lett. 2001;488:196–200. doi: 10.1016/s0014-5793(00)02426-1. [DOI] [PubMed] [Google Scholar]
- 67.Shiman R, Gray DW, Pater A. J Biol Chem. 1979;254:11300–11306. [PubMed] [Google Scholar]
- 68.Knappskog PM, Haavik J. Biochemistry. 1995;34:11790–11799. doi: 10.1021/bi00037a017. [DOI] [PubMed] [Google Scholar]
- 69.Koizumi S, Tanaka F, Kaneda N, Kano K, Nagatsu T. Biochemistry. 1988;27:640–646. doi: 10.1021/bi00402a022. [DOI] [PubMed] [Google Scholar]
- 70.Busenlehner LS, Armstrong RN. Arch Biochem Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 71.Li J, Dangott LJ, Fitzpatrick PF. Biochemistry. 2010;49:3327–3335. doi: 10.1021/bi1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andersen OA, Stokka AJ, Flatmark T, Hough E. J Mol Biol. 2003;333:747–757. doi: 10.1016/j.jmb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 73.Grant GA. J Biol Chem. 2006;281:33825–33829. doi: 10.1074/jbc.R600024200. [DOI] [PubMed] [Google Scholar]
- 74.Liberles JS, Thórólfsson M, Martínez A. Amino Acids. 2005;28:1–12. doi: 10.1007/s00726-004-0152-y. [DOI] [PubMed] [Google Scholar]
- 75.Schuller DJ, Grant GA, Banaszak LJ. Nature Struct Biol. 1995;2:69–76. doi: 10.1038/nsb0195-69. [DOI] [PubMed] [Google Scholar]
- 76.Siltberg-Liberles J, Steen IH, Svebak RM, Martinez A. Gene. 2008;427:86–92. doi: 10.1016/j.gene.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 77.Gjetting T, Petersen M, Guldberg P, Guttler F. Am J Hum Genet. 2001;68:1353–1360. doi: 10.1086/320604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martinez A, Haavik J, Flatmark T. Eur J Biochem. 1990;193:211–219. doi: 10.1111/j.1432-1033.1990.tb19325.x. [DOI] [PubMed] [Google Scholar]
- 79.Martinez A, Olafsdottir S, Flatmark T. Eur J Biochem. 1993;211:259–266. doi: 10.1111/j.1432-1033.1993.tb19894.x. [DOI] [PubMed] [Google Scholar]
- 80.Thorolfsson M, Ibarra-Molero B, Fojan P, Petersen SB, Sanchez-Ruiz JM, Martinez A. Biochemistry. 2002;41:7573–7585. doi: 10.1021/bi0160720. [DOI] [PubMed] [Google Scholar]
- 81.Andersen OA, Flatmark T, Hough E. J Mol Biol. 2002;320:1095–1108. doi: 10.1016/s0022-2836(02)00560-0. [DOI] [PubMed] [Google Scholar]
- 82.Li J, Ilangovan U, Daubner SC, Hinck AP, Fitzpatrick PF. Arch Biochem Biophys. 2011;505:250–255. doi: 10.1016/j.abb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chow MS, Eser BE, Wilson SA, Hodgson KO, Hedman B, Fitzpatrick PF, Solomon EI. J Amer Chem Soc. 2009;131:7685–7698. doi: 10.1021/ja810080c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wasinger EC, Miti3 N, Hedman B, Caradonna J, Solomon EI, Hodgson KO. Biochemistry. 2002;41:6211–6217. doi: 10.1021/bi0121510. [DOI] [PubMed] [Google Scholar]
- 85.Sura GR, Lasagna M, Gawandi V, Reinhart GD, Fitzpatrick PF. Biochemistry. 2006;45:9632–9638. doi: 10.1021/bi060754b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Daubner S, Lauriano C, Haycock J, Fitzpatrick P. J Biol Chem. 1992;267:12639–12646. [PubMed] [Google Scholar]
- 87.Wang S, Sura GR, Dangott LJ, Fitzpatrick PF. Biochemistry. 2009;48:4972–4979. doi: 10.1021/bi9004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaufman S, Fisher DB. J Biol Chem. 1970;245:4745–4750. [PubMed] [Google Scholar]
- 89.Tourian A. Biochim Biophys Acta. 1971;242:345–354. doi: 10.1016/0005-2744(71)90226-9. [DOI] [PubMed] [Google Scholar]
- 90.Selwood T, Jaffe EK. Arch Biochem Biophys. 2012 doi: 10.1016/j.abb.2011.11.020. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Loeb KE, Westre TE, Kappock TJ, Miti3 N, Glasfeld E, Caradonna JP, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:1901–1915. [Google Scholar]
- 92.Phillips RS, Iwaki M, Kaufman S. Biochem Biophys Res Commun. 1983;110:919–925. doi: 10.1016/0006-291x(83)91050-1. [DOI] [PubMed] [Google Scholar]
- 93.Iwaki M, Phillips RS, Kaufman S. J Biol Chem. 1986;261:2051–2056. [PubMed] [Google Scholar]
- 94.Cronk JD, Endrizzi JA, Alber T. Protein Sci. 1996;5:1963–1972. doi: 10.1002/pro.5560051002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Andersen OA, Flatmark T, Hough E. J Mol Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 96.Solstad T, Stokka AJ, Andersen OA, Flatmark T. Eur J Biochem. 2003;270:981–990. doi: 10.1046/j.1432-1033.2003.03471.x. [DOI] [PubMed] [Google Scholar]
- 97.Horne J, Jennings IG, Teh T, Gooley PR, Kobe B. Protein Sci. 2002;11:2041–2047. doi: 10.1110/ps.4560102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miranda FF, Thórólfsson M, Teigen K, Sanchez-Ruiz JM, Martínez A. Protein Sci. 2004;13:1219–1226. doi: 10.1110/ps.03595904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang S, Lasagna M, Daubner SC, Reinhart GD, Fitzpatrick PF. Biochemistry. 2011;50:2364–2370. doi: 10.1021/bi101844p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fisher DB, Kaufman S. J Biol Chem. 1972;247:2250–2252. [PubMed] [Google Scholar]
- 101.Abita JP, Parniak M, Kaufman S. J Biol Chem. 1984;259:14560–14566. [PubMed] [Google Scholar]
- 102.Gibbs BS, Benkovic SJ. Biochemistry. 1991;30:6795–6802. doi: 10.1021/bi00241a024. [DOI] [PubMed] [Google Scholar]