

Abstract
For visual pigments, a covalent bond between the ligand (11-cis-retinal) and receptor (opsin) is crucial to spectral tuning and photoactivation. All photoreceptors have retinal bound via a Schiff base (SB) linkage, but only UV-sensitive cone pigments have this moiety unprotonated in the dark. We investigated the dynamics of mouse UV (MUV) photoactivation, focusing on SB protonation and the functional role of a highly conserved acidic residue (E108) in the third transmembrane helix. On illumination, wild-type MUV undergoes a series of conformational changes, batho → lumi → meta I, finally forming the active intermediate meta II. During the dark reactions, the SB becomes protonated transiently. In contrast, the MUV-E108Q mutant formed significantly less batho that did not decay through a protonated lumi. Rather, a transition to meta I occurred above ≈240 K, with a remarkable red shift (λmax ≈ 520 nm) accompanying SB protonation. The MUV-E108Q meta I → meta II transition appeared normal but the MUV-E108Q meta II decay to opsin and free retinal was dramatically delayed, resulting in increased transducin activation. These results suggest that there are two proton donors during the activation of UV pigments, the primary counterion E108 necessary for protonation of the SB during lumi formation and a second one necessary for protonation of meta I. Inactivation of meta II in SWS1 cone pigments is regulated by the primary counterion. Computational studies suggest that UV pigments adopt a switch to a more distant counterion, E176, during the lumi to meta I transition. The findings with MUV are in close analogy to rhodopsin and provides further support for the importance of the counterion switch in the photoactivation of both rod and cone visual pigments.
Visual pigments are seven transmembrane α-helical proteins that initiate the light transduction pathway in retinal photoreceptors. Whereas other G protein-coupled receptors interact with their ligands noncovalently, the visual pigments consist of 11-cis-retinal covalently attached to the apoprotein via a Schiff base (SB) linkage to a conserved lysine in transmembrane helix 7 (TM7) (Fig. 1). After absorption of light, the retinal chromophore isomerizes to the all-trans conformation and triggers a series of conformational changes that lead to the formation of the active state, R* or meta II. All-trans-retinal is eventually released from the vertebrate apoprotein, and the visual pigment can be regenerated with 11-cis-retinal.
Fig. 1.

The primary sequence of MUV (A, cytoplasmic side up) and a similarity-based model of the MUV binding site, with selected atoms of the retinal chromophore numbered (B, extracellular side up). The secondary structure elements (e.g., transmembrane segments) shown are based on the crystal structure of bovine rhodopsin. The five amino acids shown in larger yellow circles correspond to S85 in TM2; E108 in TM3, at the same position as the rhodopsin counterion (E113); E176 and S181 in β-strands 3 and 4 (EII, extracellular loop 2), respectively; and K291 in TM7, the retinal binding site (K296 in rhodopsin).
In dark-adapted rhodopsin, the SB pKa is extraordinarily high resulting in a protonated SB buried within the chromophore binding site (1). The protonation is important to prevent the spontaneous hydrolysis of the SB and contributes to the λmax (2, 3). The binding site of rhodopsin is neutral (4), and E113 serves as a counterion to the protonated SB (5–8). The stability of the salt bridge is enhanced by a single water molecule that interacts with the side chains of E113 and indirectly through a second water molecule that interacts with adjacent residues (9, 10). The SB pKa decreases to ≈6 in meta I and meta II (3). The deprotonation of the SB and apparent protonation of E113 on formation of meta II accompany the structural changes that allow interactions with transducin (11). The counterion is not necessary for SB protonation in rhodopsin, because mutants with neutral amino acid substitutions can be protonated by lowering the pH. Rather, the counterion is important for determining the SB pKa. Flash photolysis studies have shown that E113 is necessary for efficient deprotonation of the SB (12). Mutations in E113 lead to proteins that are able to activate transducin in the absence of the chromophore, suggesting that the counterion plays an important role in deactivation of R* (13).
Much less is known about the chromophore–protein interactions in cone pigments, which also have an acidic residue equivalent to RHO-E113. The Xenopus SWS1 cone pigment (VCOP, λmax ≈ 427 nm) has a protonated SB in the dark and remains so until formation of the active species (14–16). Neutralizing the conserved acidic residue (D108) in TM3 converts the violet-sensitive pigment into a UV-sensitive pigment (λmax ≈ 353 nm), consistent with a drop in the pKa and deprotonation of the SB (16). The VCOP-D108A mutant does not activate transducin in the dark but has a significantly prolonged R* lifetime. These data suggest that the counterion is necessary for the rapid production and decay of the active meta II in the SWS1 pigments.
The dark-adapted mouse SWS1 (MUV) visual pigment absorbs maximally in the UV (λmax ≈ 357 nm). Fourier transform infrared (FTIR) spectroscopic studies indicate that the dark-adapted form of the MUV pigment has an unprotonated SB (17). Thus, the sensitivity to UV light is accomplished by changing the pKa of the SB. Even though the MUV SB is neutral, there is still an acidic residue at the conserved position in TM3. Molecular modeling calculations predict that the conserved acidic residue is hydrogen bonded to the SB nitrogen in an arrangement similar to the binding site of rhodopsin but with the proton attached to the glutamic acid residue (14).
The photoactivation of MUV is remarkable in that the retinylidene SB is protonated during the formation of lumi and meta I (17). Subsequently, during the formation of the active species, meta II, the SB reverts to the unprotonated state. In this report, we investigate the role of the conserved counterion in TM3 in the photoactivation of the MUV visual pigment.
Materials and Methods
Preparation of Visual Pigments. Expression constructs and COS1 cell transfections were described previously (16). MUV samples had A357 ≈ 0.3–0.4 with an A280/Aλmax ratio of ≈4, and the E108Q mutant had A354 ≈ 0.3–0.4 and an A280/Aλmax ratio of ≈4.5.
Spectroscopy. Cryogenic. Low-temperature spectroscopy was performed as described (14, 17). Spectra of visual pigment samples (Aλmax ≈ 0.1) were collected in a solution containing 67% glycerol in buffer A (50 mM Hepes, pH 6.6/140 mM NaCl/3 mM MgCl2/0.05% N-dodecyl-β-d-maltoside). The spectra shown are the averages of four separate scans at each temperature, normalized to the absorbance at λmax of the initial dark sample. For studies below 230 K, samples were equilibrated at a specific temperature and then cooled to 75 K for spectra collection to avoid potential cryogenic artifacts. Spectra above 230 K were collected at the indicated temperature. Retinal oximes were extracted and analyzed as reported (17).
Acid denaturation. The E108Q pigment in buffer A was illuminated for 1 min at 22°C and subsequently acid-denatured by lowering the pH to 1.8. The experiments were performed under three different conditions: (i) buffer A; (ii) buffer B (10 mM Tris acetate, pH 7.0/100 mM NaCl/5 mM MgCl2/5 mM 2-mercaptoethanol/0.01% N-dodecyl-β-d-maltoside), equivalent to transducin assay buffer minus GTPγS; and (iii) buffer C (buffer A containing 5 mM 2-mercaptoethanol).
Transducin Activation Assays. Light-dependent activation of bovine transducin by using filter binding assays were carried out as described (16, 18).
Molecular Modeling. The conformational and spectroscopic properties of MUV were investigated by using mozyme/pm3 (19, 20), as implemented within mopac 2000 and MNDO-PSDCI (21, 22) theory. Calculations were based on the homology model of MUV reported in ref. 17, and unless noted otherwise, the backbone atoms in the transmembrane helices and the EII loop were fixed at the corresponding rhodopsin coordinates. Additional details of the modeling methods and results, as well as additional data on MUV-E108Q relating to the biochemical, photophysical, and spectroscopic properties of the photobleaching sequence, are provided in Supporting Methods, which is published as supporting information on the PNAS web site.
Results
Formation of the Early MUV-E108Q Mutant Intermediates. Altering the conserved acidic residue in TM3 in MUV to a neutral residue (E108Q) causes a small blue shift in the λmax at room temperature, from 357 nm in MUV to 354 nm in MUV-E108Q. On cooling MUV-E108Q to 75 K, the absorbance spectrum exhibited a 6-nm red shift (λmax ≈ 360 nm). The MUV-E108Q pigment was illuminated with light at 305 nm (≈1 h) to produce a photostationary state (PSS305) (Fig. 2 and Fig. 7, which is published as supporting information on the PNAS web site). The PSS305 generated had a slightly red-shifted absorption maximum accompanied by a significant decrease of the protein band at ≈280 nm (Figs. 2 and 7D). Retinal oxime extractions revealed the isomeric mixture of the PSS305 of E108Q at 75 K as 68% 11-cis, 32% all-trans, and <1% 9-cis, significantly less all-trans and 9-cis than the MUV PSS305 (20% 11-cis, 72% all-trans, and 8% 9-cis), and PSS305 generated at 243 K (11% 11-cis, 83% all-trans, and 5% 9-cis, ±2%).
Fig. 2.

(A) Formation of the primary photostationary state of E108Q. The difference spectra between the photostationary state (PSS305) obtained after 15, 30, 45, and 60 min of illumination at 75 K minus the dark state spectrum are shown and labeled 15 m, 30 m, 45 m, and 60 m, respectively. (B) Thermal decay of the primary photostationary state of E108Q. The difference spectra calculated by subtracting the PSS305 formed at 75 K from the spectra equilibrated at 100, 160, 180, and 200 K are labeled accordingly. (C) Formation of the later intermediates of E108Q. Light minus dark difference spectra obtained after illumination at 243 K. The curve labeled PSS305 was the photostationary state obtained after illumination of the pigment with 305 nm light for 135 min. The remaining spectra were successive illumination of the PSS305 with 330 nm light for 15, 30, 60, 90, and 120 min (PSS330). (D) Thermal decay of the PSS330 generated at 243 K. Difference spectra were calculated by subtracting the PSS330 equilibrated at 253, 258, and 263 K from that equilibrated at 248 K plotted.
In contrast to wild-type MUV, raising the temperature of the MUV-E108Q PSS305 to 220 K does not result in any further spectral shift toward longer wavelengths. In fact, the PSS305 exhibits a spectral shift toward shorter wavelengths when warmed to 120 K and above (Fig. 2 and Fig. 8 A and B, which is published as supporting information on the PNAS web site). The aromatic region showed a decrease in absorbance as the temperature increased from 100 to 160 K. The absorbance recovered to that in the dark state by 200 K (Figs. 1B and 8).
RHO-E113Q exhibits a pH-dependent absorbance profile: at pH 8, the protein has λmax ≈ 390 nm at 75 K, indicating that the SB is unprotonated. On illumination at 340 nm, a slightly blue-shifted (compared to the dark) photostationary state (PSS340) is formed, reminiscent of the results of the E108Q mutant (Fig. 9A, which is published as supporting information on the PNAS web site). The PSS of the RHO-E113Q mutant at low pH, with a protonated SB, exhibits a pronounced red shift similar to that of MUV (Fig. 9B).
Formation of the Later MUV-E108Q Mutant Intermediates. The MUV-E108Q mutant has an absorption maximum at 350 nm at 243 K, identical to that at room temperature (Fig. 3). Illumination caused the formation of a λmax ≈ 520 nm absorbing meta I (Fig. 2 and Fig. 10A, which is published as supporting information on the PNAS web site) instead of ≈490 nm in wild-type MUV. On gradual warming, the MUV-E108Q PSS330 converts to a species that has an absorption maximum at ≈350 nm, similar to that of the dark state but slightly blue-shifted compared to wild-type meta II (Figs. 2 and 10C).
Fig. 3.

The pure electronic spectra of the MUV-E108Q intermediates obtained via the decomposition of the temperature-trapped photostationary state spectra are shown in the corresponding panels labeled with the intermediate and temperature at which it is stabilized and the absorption maximum. The spectra and absorption maxima of the wild-type MUV intermediates are shown in gray. Note that the E108Q mutant does not produce any lumi intermediate, whereas MUV does. The absorption maxima are indicated in nanometers and are accurate to ±2 nm.
Derivation of the Spectra of MUV Intermediates. Spectra of the pure intermediates were calculated by deconvolution using the HPLC results to assign the isomeric composition of PSS (Fig. 3) and assuming that the amount of the dark (11-cis) and the light (11-cis, all-trans, and 9-cis) states remained constant during the temperature ramping experiments. The spectral refinements for MUV-E108Q have a lower signal-to-noise ratio than wild type (17) because of the lower product yield. In contrast to the wild-type MUV, a lumi intermediate is not detected in E108Q. Meta I has a significantly red-shifted absorption compared to the wild-type intermediate. The absorption band of MUV-E108Q meta I shows evidence of an underlying feature at ≈430 nm, which had the same temperature-dependent formation and decay as the main meta I band (Fig. 2D). The meta II λmax (≈350 nm) indicates that the active intermediate has an unprotonated SB, as observed in other visual pigments.
Decay of the Active (Meta II) State of MUV-E108Q. The stability of E108Q meta II was determined by using an acid trapping assay. Pigments were illuminated to compare meta II stability under conditions used for spectroscopy and for biochemical assays. Most of the chromophore absorbance (λmax ≈ 354 nm in the dark) shifted to λmax ≈ 440 nm when the pH of illuminated samples was lowered to 1.8, even when pigments were incubated for 30 min at room temperature (Fig. 11, which is published as supporting information on the PNAS web site). These results show that very little free retinal was released. There was no difference in stability of the E108Q meta II in any of the buffers tested.
MUV and MUV-E108Q activated bovine transducin in a light-dependent manner (Fig. 4A). Activity was affected by continuous illumination (data not shown). The initial rates of activation were much faster for MUV-E108Q (≈168 mol GTPγS exchanged per mol pigment per min) compared to wild type (≈33 mol GTPγS exchanged per mol pigment per min). The time course of transducin activation was monophasic for MUV, completing in <20 sec. MUV-E108Q had biphasic kinetics, with the initial having the same time course as wild type, albeit with a higher efficiency and a slow phase extended for several minutes.
Fig. 4.

(A) Transducin activation by MUV and its counterion mutant, E108Q. The reaction was illuminated for 1 min, and then aliquots were removed and assayed for GTPγS bound to transducin. The solid lines represent the regression fit to the initial rate for each curve. (B) The decay of the active state of MUV and E108Q after illumination. The pigments were exposed to 1 min of illumination in the absence of transducin. Aliquots were added to transducin with a delay of 1–30 min and incubated for an additional 10 min in dark before assaying; a delay of 0 min was achieved by illuminating in the presence of transducin.
The MUV meta II state at 22°C decayed extremely fast (t1/2 < 30 sec) (Fig. 4B). Reducing the temperature did not slow down the decay of meta II significantly (data not shown). In contrast, MUV-E108Q had a slower meta II decay (t1/2 ≈ 6 min) (Fig. 4B). As seen in the acid-denaturation experiments, the stability of the meta II state of MUV-E108Q was enhanced, although the two assays differed in the magnitude of the effect.
Discussion
Structural Properties of the lumi and meta I Intermediates. The early portion of the MUV photoactivation pathway appears relatively intact in the MUV-E108Q mutant, although there are several distinct changes. E108Q forms a batho intermediate at 75 K that is slightly blue-shifted compared to wild type. The steady-state batho level (≈32% all-trans-retinal) is significantly less than in wild type (≈72% all-trans-retinal). This most likely reflects a more efficient reverse photochemical reaction in the mutant, as observed in the rhodopsin E113Q mutant (12). The minor changes in dark → batho spectra between the wild-type and E108Q mutant support the view that the batho shift is primarily due to conformational rather than electrostatic interactions with the chromophore (17). Presumably, the MUV batho intermediate has a twisted all-trans-retinal while the protein conformation remains the same as in the dark state, as in rhodopsin (23, 24). For both MUV (17) and E108Q, batho converts to BLI, with an accompanying blue shift in the absorbance spectra. Again, the E108Q BLI λmax is blue-shifted compared to wild type. BLI appears analogous to the previously observed intermediate in chicken blue opsin (25) and BSI in rhodopsin (26). However, there may be slight differences between the cone (BLI) and rod intermediates, because rhodopsin's BSI cannot be trapped at low temperature (26).
The later steps in the photoactivation pathway are severely disturbed by the E108Q mutation. The batho or BLI intermediates do not decay to lumi above 180 K, as in wild type, but remain stable until ≈240 K. Apparently, placing a neutral residue at position 108 prevents stabilization of a protonated SB normally formed in wild-type lumi. The failure to form lumi in E108Q indicates that there may be a kinetic bottleneck, caused by the absence of a coordinated protonation of the chromophore and deprotonation of the counterion. The results presented here suggest the source of the proton is E108.
In addition to the change in pKa of the SB, there are structural changes that occur during MUV batho → lumi (17). The protein movements can be detected in the relaxation in the aromatic region of the absorbance spectrum. The data shown in Fig. 2 indicate the same changes occur, both in λmax and magnitude, in E108Q as is the wild-type pigment (17). This putative rearrangement of aromatic residue(s) during batho → lumi, perhaps involving TM3 and the SB environment in analogy to rhodopsin (27–30) could change the SB environment/pKa leading to protonation and a large red shift. Effectively, the chromophore and protein conformational changes on formation of lumi in MUV allow E108 to serve as a counterion. Results presented here suggest that the dark → lumi protein rearrangements occur, at least in part, in the E108Q mutant (Fig. 2) but that protonation of retinylidene SB does not.
The wild-type MUV meta I intermediate has a λmax (≈490 nm) comparable to the meta I of other visual pigments, including rhodopsin, which has a protonated retinylidene SB (12). In the E108Q pigment, the SB is unprotonated until meta I forms. What donates the proton in the E108Q meta I? One possibility is that it originates from solvent. Molecular models (Fig. 1) indicate that the SB is buried in the protein and probably not very accessible to solvent. Further, the energetics of lumi → meta I do not support protonation from solvent, because the lumi → meta I transition would not be expected to display the observed temperature dependence. Rather, we propose that a second counterion present within the binding site donates a proton to the chromophore. We explore this idea in the next section.
Secondary Counterion(s) in Mouse UV. We generated molecular models of wild-type MUV lumi and meta I retinal binding sites (Fig. 6). As discussed below, only E176 provides a satisfactory secondary counterion based on the energetics and spectroscopic properties of MUV meta I.
Fig. 6.

Schematic diagram of the key molecular changes in the mouse UV binding site in the dark state, batho, lumi, and meta I states. The arrows in A and C indicate the key conformational or chemical changes associated with transformation of the dark state to batho and lumi to meta I. The chromophore geometry in the batho state is all-trans, but the conformational details regarding the single bonds remains unknown. The relative location of the residues is approximate and does not reflect possible movement during the thermal relaxations (see Discussion). Molecular orbital (mozyme) calculations indicate that E176, when protonated (neutral), is freely rotating, and we use * to indicate that there are two tyrosine residues, Y187 and Y263, that alternate in hydrogen bonding to E176, depending on rotational state. Only in the meta I state do Y187 and Y263 both strongly hydrogen bond to E176 (D).
The total energy of MUV meta I is lower in energy by 4 kcal·mol–1 compared to MUV lumi when the first shell (all residues) within 5.6 Å of the chromophore are included (note that Fig. 5 A and B only contain a subset of these residues). When the backbone is released and a partial minimization is carried out, the relative energy of the meta I state drops below the lumi state by 9 kcal·mol–1 after 10 iterations. The primary stabilization of the meta I state is associated with the presence of two tyrosine residues, Y187 and Y263, which interact with E176 through highly effective hydrogen bonding (Fig. 6). The interactions of either of these residues with E176 were less stable in lumi. The estimated absorption maximum for both lumi and meta I are in qualitative agreement with the observed values (Fig. 5). Quantitatively, however, the calculated absorption maxima are overestimated for lumi by 10 nm 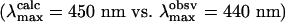 and meta I by 60 nm
and meta I by 60 nm 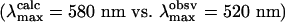 . This discrepancy could arise from improper positioning of the SB relative to the secondary counterion. For example, a shift of the SB nitrogen toward E176 would preferentially stabilize the ground state relative to the strongly allowed 1Bu*+-like π,π* state and lead to a blue-shift λmax.
. This discrepancy could arise from improper positioning of the SB relative to the secondary counterion. For example, a shift of the SB nitrogen toward E176 would preferentially stabilize the ground state relative to the strongly allowed 1Bu*+-like π,π* state and lead to a blue-shift λmax.
Fig. 5.

Molecular models of the lumi (A) and meta I (B) intermediates of mouse UV based on the assumption that a counterion switch occurs during the lumi (E108 counterion) to meta I (E176 counterion) transformation. The chromophore, binding site residues, and water were allowed to seek minimum energy, but the protein backbone and outer residues were fixed. The electronic spectra of the two intermediates are simulated by using MNDO-PSDCI molecular orbital theory in the C and D, and the calculated absorption band maxima are indicated in nanometers. The dipole moments of the binding site residues, μbs, are given in the upper right of A and B and the dipole moment vector is shown by using red-to-blue arrows. The MNDO-PSDCI calculations included all 36 single and 666 double excitations within the π electron manifold of the chromophore. The heights of the vertical bars in C and D are proportional to the oscillator strengths of the π* ← π transitions, where solid blue bars indicate Bu+-like states, dashed blue bars indicate Bu–-like states, solid red bars indicate Ag+-like states, and dashed red bars indicate Ag–-like states. The simulated chromophore spectra were generated assuming Gaussian profiles with full-widths at half-maxima of 4,000 cm–1.
Compared to meta I from other visual pigments, both MUV and E108Q meta I absorption bands are anomalous in shape because of inhomogeneous broadening. This may come about because the protonation occurs later in the photobleaching sequence than in other visual pigments. The protein may not be able to equilibrate to the presence of a charged chromophore at low temperatures. Hence, multiple protein configurations having different electrostatic stabilization properties may be present. Additional discussion of the modeling results shown in Fig. 5 may be found in Supporting Methods.
Nature and Implications of the Counterion Switch. Our results provide additional perspective on the counterion switch mechanism proposed for rhodopsin activation (31). It seems to us unlikely that nature would invoke a complicated SB → PSB(+):E108(–) → PSB(+):E176(–) → SB photoactivation sequence if it were not a key contributor to function. In fact, it appears that just such a mechanisms makes vision in the UV possible, because this activation strategy is operative for both protonated and unprotonated chromophores.
The model proposed by Yan et al. (31) assumes helix translocation during the dark reactions to form meta I such that the protonated SB moves toward E181. Previous studies have shown that there is significant motion as well as rotation of the transmembrane helices in meta II. In particular, movement of TM7, to which the chromophore is attached, is critical for coupling of the activated rhodopsin with transducin (32–36). In addition, there is evidence of significant movement of the β-ionylidene ring preceding the formation of lumi (27). There is no direct evidence concerning translocation of the SB toward the second counterion in MUV. However, the counterion switch is energetically favored whether the chromophore moves or stays fixed. The key amino acids acting in the proposed counterion switch (Fig. 6) are highly conserved in all of the visual pigments except M/LWS (medium/long wavelength sensitive; see ref. 37 for discussion). The MUV-E108Q mutant has altered photoactivation properties compared to the wild-type MUV pigment, highlighting the importance of the electrostatic interaction between E108 and the SB of UV opsins. The role of these interactions also extend to the inactivation of R*. Further work is needed to elucidate how the primary counterion participates in the hydrolysis of the unprotonated SB. We observed a discrepancy in the stability of the meta II state determined by transducin (t1/2 ≈ 6 min) versus acid denaturation (t1/2 > 30 min) assays. The difference could not be accounted for by differing buffer conditions (Fig. 11). It is possible that interaction with transducin causes a conformational change making the SB more labile to hydrolysis.
Comments and Conclusions. We speculate that the protonation of the retinylidene SB linkage, the electrostatic interaction between the SB and the counterion, and the counterion switch that occurs during the photobleaching cascade are essential for the coordinated movement of helices as the protein becomes activated. This process ensures that the final active state, meta II, has the structural integrity necessary for transducin activation as well as rapid hydrolysis of the SB required for cone opsins. The SB becomes protonated in the lumi state through donation of a proton by E108, which takes on the secondary role of serving as a distant counterion to the protonated chromophore. Thus, the MUV-E108Q mutant does not form a lumi intermediate because glutamine cannot donate a proton. In the absence of the SB-counterion electrostatic interaction, the relative movement of the helices, especially TM3 and TM6, would take place in an uncoordinated fashion and, moreover, there would be no counterion-mediated protonation of the retinylidene SB during the batho to lumi transition. In addition, these studies support the importance of a counterion switch in the activation process of both the rhodopsins and the SWS1 cones. What remains to be understood is how the counterion switch mediates the structural changes that accompany formation of the meta II state.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grants to R.R.B. (GM-34548) and B.E.K. (EY-11256 and EY-12975), a National Science Foundation grant to R.R.B. (EIA-0129731), the Harold S. Schwenk Sr. Distinguished Chair funds for support of specialized instrumentation at the University of Connecticut, Research to Prevent Blindness (unrestricted grant to the Department of Ophthalmology, State University of New York, Upstate Medical University), and Lions of Central New York.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MUV, mouse SWS1 opsin; SB, Schiff base; TM, transmembrane helix.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1UV3).
References
- 1.Steinberg, G., Ottolenghi, M. & Sheves, M. (1993) Biophys. J. 64, 1499–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birge, R. R. (1993) Biophys. J. 64, 1371–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ebrey, T. (2000) Methods Enzymol. 315, 196–207. [DOI] [PubMed] [Google Scholar]
- 4.Birge, R. R., Murray, L. P., Pierce, B. M., Akita, H., Balogh-Nair, V., Findsen, L. A. & Nakanishi, K. (1985) Proc. Natl. Acad. Sci. USA 82, 4117–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakmar, T. P., Franke, R. R. & Khorana, H. G. (1989) Proc. Natl. Acad. Sci. USA 86, 8309–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhukovsky, E. A. & Oprian, D. D. (1989) Science 246, 928–931. [DOI] [PubMed] [Google Scholar]
- 7.Nathans, J. (1990) Biochemistry 29, 9746–9752. [DOI] [PubMed] [Google Scholar]
- 8.Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada, T., Stenkamp, R. E., et al. (2000) Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- 9.Okada, T., Terakita, A. & Shichida, Y. (2002) Tanpakushitsu Kakusan Koso 47, 1123–1130. [PubMed] [Google Scholar]
- 10.Stenkamp, R. E., Teller, D. C. & Palczewski, K. (2002) Chembiochem 3, 963–967. [DOI] [PubMed] [Google Scholar]
- 11.Jäger, F., Fahmy, K., Sakmar, T. P. & Siebert, F. (1994) Biochemistry 33, 10878–10882. [DOI] [PubMed] [Google Scholar]
- 12.Lewis, J. & Kliger, D. (2000) Methods Enzymol. 315, 164–178. [DOI] [PubMed] [Google Scholar]
- 13.Robinson, P., Cohen, G., Zhukovsky, E. & Oprian, D. (1992) Neuron 9, 719–725. [DOI] [PubMed] [Google Scholar]
- 14.Kusnetzow, A., Dukkipati, A., Babu, K. R., Singh, D., Vought, B. W., Knox, B. E. & Birge, R. R. (2001) Biochemistry 40, 7832–7844. [DOI] [PubMed] [Google Scholar]
- 15.Dukkipati, A., Vought, B. W., Singh, D., Birge, R. R. & Knox, B. E. (2001) Biochemistry 40, 15098–15108. [DOI] [PubMed] [Google Scholar]
- 16.Babu, K. R., Dukkipati, A., Birge, R. R. & Knox, B. E. (2001) Biochemistry 40, 13760–13766. [DOI] [PubMed] [Google Scholar]
- 17.Dukkipati, A., Kusnetzow, A., Babu, K. R., Ramos, L., Singh, D., Knox, B. E. & Birge, R. R. (2002) Biochemistry 41, 9842–9851. [DOI] [PubMed] [Google Scholar]
- 18.Max, M., Surya, A., Takahashi, J., Margolskee, R. & Knox, B. (1998) J. Biol. Chem. 273, 26820–26826. [DOI] [PubMed] [Google Scholar]
- 19.Stewart, J. J. P. (1996) Int. J. Quant. Chem. 58, 133–146. [Google Scholar]
- 20.Stewart, J. J. P. (1997) J. Mol. Struct. 401, 195–205. [Google Scholar]
- 21.Ren, L., Martin, C. H., Wise, K. J., Gillespie, N. B., Luecke, H., Lanyi, J. K., Spudich, J. L. & Birge, R. R. (2001) Biochemistry 40, 13906–13914. [DOI] [PubMed] [Google Scholar]
- 22.Martin, C. H. & Birge, R. R. (1998) J. Phys. Chem. A 102, 852–860. [Google Scholar]
- 23.Schoenlein, R. W., Peteanu, L. A., Mathies, R. A. & Shank, C. V. (1991) Science 254, 412–415. [DOI] [PubMed] [Google Scholar]
- 24.Stuart, J. A. & Birge, R. R. (1996) in Biomembranes, ed. Lee, A. G. (JAI Press, London), Vol. 2A, pp. 33–140. [Google Scholar]
- 25.Imai, H., Terakita, A., Tachibanaki, S., Imamoto, Y., Yoshizawa, T. & Shichida, Y. (1997) Biochemistry 36, 12773–12779. [DOI] [PubMed] [Google Scholar]
- 26.Hug, S. J., Lewis, J. W., Einterz, C. M., Thorgeirsson, T. E. & Kliger, D. S. (1990) Biochemistry 29, 1475–1485. [DOI] [PubMed] [Google Scholar]
- 27.Borhan, B., Souto, M., Imai, H., Shichida, Y. & Nakanishi, K. (2000) Science 288, 2209–2212. [DOI] [PubMed] [Google Scholar]
- 28.Ganter, U. M., Gärtner, W. & Siebert, F. (1988) Biochemistry 27, 7480–7488. [DOI] [PubMed] [Google Scholar]
- 29.Pan, D. & Mathies, R. A. (2001) Biochemistry 40, 7929–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada, T., Kandori, H., Shichida, Y., Yoshizawa, T., Denny, M., Zhang, B., Asato, A. E. & Liu, R. S. H. (1991) Biochemistry 30, 4796–4802. [DOI] [PubMed] [Google Scholar]
- 31.Yan, E. C. Y., Kasmi, M. A., Ganim, Z., Hou, J. M., Pan, D., Chang, B. S. W., Sakmar, T. P. & Mathies, R. A. (2003) Proc. Natl. Acad. Sci. USA 100, 9262–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farrens, D. L., Altenbach, C., Yang, K., Hubbell, W. L. & Khorana, H. G. (1996) Science 274, 768–770. [DOI] [PubMed] [Google Scholar]
- 33.Hubbell, W. L., Altenbach, C., Hubbell, C. M. & Khorana, H. G. (2003) Adv. Protein Chem. 63, 243–290. [DOI] [PubMed] [Google Scholar]
- 34.Hamm, H. (2001) Proc. Natl. Acad. Sci. USA 98, 4819–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdulaev, N. G. & Ridge, K. D. (1998) Proc. Natl. Acad. Sci. USA 95, 12854–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fritze, O., Filipek, S., Kuksa, V., Palczewski, K., Hofmann, K. P. & Ernst, O. P. (2003) Proc. Natl. Acad. Sci. USA 100, 2290–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birge, R. R. & Knox, B. E. (2003) Proc. Natl. Acad. Sci. USA 100, 9105–9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}