

Abstract
Motivation: The advent of high-throughput sequencing (HTS) technologies has made it affordable to sequence many individuals' genomes. Simultaneously the computational analysis of the large volumes of data generated by the new sequencing machines remains a challenge. While a plethora of tools are available to map the resulting reads to a reference genome, and to conduct primary analysis of the mappings, it is often necessary to visually examine the results and underlying data to confirm predictions and understand the functional effects, especially in the context of other datasets.
Results: We introduce Savant, the Sequence Annotation, Visualization and ANalysis Tool, a desktop visualization and analysis browser for genomic data. Savant was developed for visualizing and analyzing HTS data, with special care taken to enable dynamic visualization in the presence of gigabases of genomic reads and references the size of the human genome. Savant supports the visualization of genome-based sequence, point, interval and continuous datasets, and multiple visualization modes that enable easy identification of genomic variants (including single nucleotide polymorphisms, structural and copy number variants), and functional genomic information (e.g. peaks in ChIP-seq data) in the context of genomic annotations.
Availability: Savant is freely available at http://compbio.cs.toronto.edu/savant
Contact: savant@cs.toronto.edu
1 INTRODUCTION
The emergence of high-throughput (a.k.a. next-generation) sequencing technologies has made the acquisition of genomic data quicker and more affordable than ever before. Continued technological strides are being made to further improve throughput, cost and accuracy of the sequencing platforms, enabling large-scale studies of genomes, populations and diseases. Within the field of personalized genomics, ambitious sequencing projects such as the 1000 Genomes Project, the International Cancer Genome Consortium and the Autism Genome Project, are seeking to identify genomic variants among human genomes and to use this knowledge to determine the genetic underpinnings of human diseases by associating variants with symptoms (Hu-Lince et al., 2005; Via et al., 2010; http://www.icgc.org/). Many computational tools have been developed for single nucleotide polymorphism (SNP), insertion/deletion (indel) and other types of genetic variation discovery in individuals sequenced using high-throughput sequencing (HTS) platforms (Chiang et al., 2009; Hormozdiari et al., 2009; Li et al., 2008; also see Dalca and Brudno, 2010 and Medvedev et al., 2009 for reviews). The results of these analyses are being integrated into large-scale datasets, annotating each variant with the allele frequency in various populations, and enabling demographic and association studies.
Simultaneously, HTS has also revolutionized functional genomics, where the ability to sequence RNA and DNA to extremely high coverage has made possible RNA-seq and ChIP-seq (Pepke et al., 2009), methodologies that help discover rare RNA transcripts, identify the location of transcription factor binding sites on the genome, as well as discover the locations of nucleosomes. RNA-seq and ChIP-seq data also consist of many reads aligned to a reference genome, which are then binned and analyzed for peaks that indicate putative transcription factor binding sites and exons, respectively. Additionally, anomalously mapped reads or pairs from RNA-seq experiments can suggest alternative splicing or fusion transcripts.
The capacity of new sequencing technologies to generate huge volumes of raw sequence data has made its analysis a substantial informatics challenge. Sequencing machines typically output files containing a nucleotide sequence and quality values for each read, which can be tens to hundreds of gigabytes in size due to the sheer number of reads sequenced in a single run. These files can be easily parsed line-by-line by downstream computer programs because of their structured textual format, but are not intended for direct manual or visual analysis. While downstream analysis programs, such as read mappers, SNP callers and peak finders can identify regions of interest to the bench scientist, a visual analysis of the supporting data, as well as other, orthogonal, information is usually warranted before costly wetlab experiments are performed to confirm the biological validity of the prediction.
Visualization of genomic data gives researchers the benefit of looking at information in a more natural and interpretable way compared with a textual representation. There are many different tasks that are facilitated by visualization including: (i) integration of multiple related datasets into a single view, to gain insight into the interaction between genomic features, (ii) algorithm development, where visualization of many putative calls (e.g. genomic variants, promoter sites, intron–exon boundaries, etc.) helps with debugging and identification of true and false positives; and (iii) exploration of various genomic regions for specific signatures of functional sites that may be difficult to describe within a computer program, e.g. two closely spaced peaks in ChIP-seq data indicating adjacent binding sites. Without the convenience of a visualization tool, in each of these settings all regions of interest would have to be painstakingly considered via manual analysis of the supporting data.
A number of tools have been developed for visualizing genome-based annotations. The UCSC and Ensembl genome browsers, for example, are popular online tools that have traditionally been used to display various biological datasets, such as genomic variants, expressed sequence tag (ESTs) and functional genomic data, in the context of high-quality, manually curated annotations (Hubbard et al., 2009; Kent et al., 2002). While both have been recently updated to support display of HTS data, their use for this purpose is onerous because: (i) a large amount of locally stored data must be uploaded to servers across the internet, (ii) once uploaded, the data being visualized cannot easily be manipulated or computed with; and (iii) server-side browsers are typically slow and non-interactive. Other visualization programs such as the Integrative Genomics Viewer, Artemis and Tablet (http://www.broadinstitute.org/igv; Milne et al., 2010; Rutherford et al., 2000) are designed to run on conventional desktop computers and thus make use of local storage capacity and computing resources to overcome the shortcomings of web-based browsers. While these popular browsers allow for interactive visualization of HTS data, they have limited analytic capability, and are not extensible through user-contributed modules. A substantial barrier for researchers who use genomic visualization tools, for all types of data, is the disconnect between the processes of visualization and computer-intensive analyses (Nielsen et al., 2010), a void which is caused by visualization tools being programmatically inaccessible.
Here, we introduce Savant, the Sequence Annotation Visualization and ANalysis Tool, which combines visualization of HTS and other genome-based data with powerful analytic tools. The Savant feature set (summarized in Table 1) was guided by three key design principles. (i) Ease of use: users can easily install the application, obtain and load data, and navigate to specific regions of interest. The general layout of data mirrors the standard genome browsers to shorten the learning curve. (ii) Speed and efficiency: the program quickly and dynamically sifts through very large datasets while maintaining a reasonable memory footprint. (iii) Access and extensibility: the underlying data is readily accessible from within the tool itself, and users can extend the application by adding any number of plug-ins for specific tasks.
Table 1.
Savant feature list
| Feature categories | Features |
|---|---|
| Data formats | FASTA, BED, SAM/BAM, WIG, GFF and any tab-delimited text file containing positional annotations |
| Speed and efficiency | Fast data access |
| Scalable to very large input files | |
| Small memory footprint | |
| Navigation | Zooming, panning and seeking using range controls or keyboard and mouse |
| Layout | Dockable modules supporting show/hide, rearrangement, maximize, float and close |
| Visualization | Compact view of sequence, point, interval and continuous tracks |
| Multiple display modes for specific track types | |
| Language | Implemented in Java |
| Operating systems | Works on Windows, Linux, Mac and any other platform having support for Java Virtual Machine |
| Extensibility | Plugin framework allows access to data, tracks and UI for extensive customization |
| Others | Bookmarking favorite locations |
| Locking overview tracks | |
| Novel representation of paired-end reads making structural variations easy to identify |
2 INTERFACE
Savant has a simple and intuitive interface, which is customizable through the use of a modular docking framework. Figure 1 shows a screenshot of Savant, and illustrates its various components, expanded upon in the following subsections.
Fig. 1.

Screenshot of Savant. (A) Range controls. Selection, zoom and pan controls for coarse navigation; text fields for fine navigation. Zooming and panning are also possible via keyboard and mouse commands. (B) Tracks. These represent the data in current range. Top: read alignments, with colored pixels representing differences between the reads and the reference. Bottom: color representation of the genome sequence. (C) Table View module, detached from the main interface. The table view module is displaying the mapped reads with SAM format fields. (D) Bookmarks module.
2.1 Navigation
There are several ways a user can specify the genomic region to be displayed by the viewer. Coarse navigation is made possible through a range selection panel whose horizontal length represents the length of the loaded genome, from which subranges can be chosen using the mouse. Alternatively, fine navigation is possible by entering the desired genomic range into text fields. Neighboring buttons enable zooming in and out and panning left and right. Each of these functions can also be engaged through mouse and keyboard shortcuts. Savant also uses a bookmarking framework to allow the user to switch between many regions of interest, as described lower.
2.2 Modules and docking framework
Savant uses a modular docking framework, similar to those used in most Integrated Development Environments (IDEs). Each module within the application appears as a separate window that can be shown, hidden, maximized, minimized, resized, closed or rearranged in any configuration the user desires. Modules can also be detached from the main interface and moved to a separate location, which is useful for maximizing screen usage on setups having multiple displays. Savant includes a number of modules which are described in subsections that follow. To demonstrate the power and utility of Savant's plug-in framework the Table View module was implemented as a plug-in.
2.3 Tracks
A track is a visual representation of one dataset. Each track shows data of a single type, such as a genome, read alignment, gene set or generic annotation. A user can specify the region of the loaded tracks to be displayed via the browser's many navigation controls. By default, multiple tracks are stacked on top of each other so that positions along their horizontal axes correspond to the same location of the genome, which is standard for genomic viewers (Gordon et al., 1998, Staden et al., 2000). Simultaneously, Savant enables the user to lock a certain track, while updating the others, as is described in Section 3.1
2.4 Bookmarks
It is often useful to make note of interesting regions while using the browser, or to load a set of such regions in order to quickly navigate between them. The Bookmarks module helps keep track of such locations. Users may add, remove or seek to a bookmarked region by using buttons within the module or keyboard shortcuts. Furthermore, bookmarks may be tagged with a description and exported for future use or for sharing among colleagues.
2.5 Table view
Finally, while Savant aims to provide the user with the ability to compute on the underlying data directly, through the plug-in framework described in Section 5, in many cases the user may wish to identify the underlying data elements for export to an external program, e.g. identifying the genomic sequence within a window to align against another genome, or downloading all of the supporting reads for a SNP to make sure they do not align elsewhere in the genome. In Savant, the user can display the underlying textual data from any loaded track within the Table View. This module displays records as rows and fields as columns in a spreadsheet. For each read mapping, for example, the Table View displays the read name, mapped position, CIGAR string and other SAM fields. The data can be sorted in either ascending or descending order with respect to any field. The spreadsheet can also be exported for further analyses.
3 VISUALIZATION
Savant retrieves and renders data every time a range change is requested by the user. Together, these processes happen quickly so as to confer seamless navigation around the genome. The renderer for each track is adaptive to both the display mode and the length of the viewed region chosen by the user.
3.1 Dynamic resolution changes
Savant dynamically adjusts its resolution—the amount of information it displays—to optimize both nucleotide- and genome-scale visualization of tracks. For example, read alignments can be visualized as a coverage track when the number of base pairs within a region is too large to enable the visualization of individual reads. Once the region is small enough, Savant seamlessly switches to a read-alignment view, as shown in Figure 2. In addition to presenting a more intuitive visualization, this feature also reduces the program's memory footprint and improves overall speed.
Fig. 2.

Read alignments, visualized at various resolutions and using two modes. (A) Chromosome-wide view of read mappings, showing the overall coverage (with no coverage in the centromere). (B) Regional view, still visualized as a coverage map, showing higher coverage in certain regions of the genome. (C) Local view, the reads are shown separately and differences between the reads and the reference genome are colored. Reads on the forward and reverse strand are shown with different shades of blue. (D) Matepair (arc) mode, showing the relative distance between the two reads of a pair. Taller arcs indicate larger distances between the pairs.
Individual tracks can also be locked to a particular range so that they are not updated until they are unlocked. Locked tracks can be used as overview profiles from which subregions can be selected to specify range changes for other tracks. Track locking also enables simultaneous viewing of high- and low-resolution profiles.
3.2 Display modes
Particular data types can be displayed in different modes. For example, interval annotations can be squished together on a single line or packed neatly so that none overlap (mimicking the squish and pack modes of the UCSC browser). The representation can be dynamically toggled within the browser, with each representation option emphasizing different aspects of the data. The variant and strand modes for read alignments, for instance, use colors to emphasize mismatches in reads and the strands to which reads are mapped, respectively. A novel mode for representing matepairs shows arcs between the mapped locations of paired reads, where the height of each arc is proportional to the inferred insert size. Arcs for anomalously mapped pairs, such as those suggestive of inversions or duplications, are colored differently. The various modes for read alignments are illustrated in Figure 2.
4 FILE FORMATTING AND PERFORMANCE
Savant supports a number of common text-based formats, which are described in Table 2. However, because text-files do not enable fast random access, Savant formats and saves each file so as to provide very efficient search operations at runtime. The speed with which Savant can sift through large datasets is enabled by the way in which it formats and indexes data. In particular, formatting involves converting text records into an indexed binary data structure specific to each data type. Sequence and continuous tracks are stored as fixed-width records, enabling direct lookup of records of interest. Annotation ranges (such as genes) are stored using a binning scheme similar to the one used in the UCSC Browser (Kent et al., 2002) and in BAM files (Li et al., 2009), so retrieving all ranges corresponding to some region usually requires only one, and at most O(log n) disk seeks. File formatting can be done directly through the application itself. Savant keeps its memory usage low by adjusting its sampling rate depending on the size of the visualized range.
Table 2.
Supported file formats
| File Format | Description |
|---|---|
| FASTA | Standard format for nucleotide sequences |
| BED | Format for describing coordinates of localized features on genomes, such as genes |
| SAM/BAM | Relatively standard format for large sets of nucleotide sequence alignments |
| WIG | Standard format for continuous-valued data. Useful for GC percent, probability scores and transcriptome data |
| GFF | General feature format for annotations of ranges |
| Tab delimited | Any tab-delimited file containing point, interval or continuous genome annotations |
The time and space requirements for the processes of data formatting and visualization were measured for a collection of human chromosome 1 datasets, including a genetic sequence, genes, SNPs, mammalian conservation and alignments of sequenced reads from an individual from the 1000 Genomes Project (∼40× coverage). The tests were performed on a Lenovo T61p laptop computer with an Intel Core 2 Duo CPU at 2.40 GHz and 3.0 GB of RAM. The results are summarized in Table 3. Formatting of the gene, SNP, sequence and conservation tracks took less than 10 min total, while the computation of a coverage track from a large set of read alignments took about an additional 40 min. The latter conversion is optional, and allows for dynamic switching between an alignment view and a coverage view as illustrated in Figure 2. Runtime performance was assessed by measuring the time taken to navigate to ranges of various sizes. Each measurement was performed on a newly started instance of Savant and the start location of the range was randomized. For seeking arbitrary ranges of sizes 10 million to 10K bp, Savant took less than a second to fetch and render data. The performance was worst for ranges just slightly shorter than 10K long, where the large number of BAM records that are displayed require 2 s to be fetched from disk. Savant renders virtually instantaneously for regions having sizes on the order of hundreds of base pairs, where most fine-scale visualization is done.
Table 3.
Time and space requirements for file formatting and visualization
| Data files |
Formatting |
Retrieval and visualization |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10M |
100K |
1K |
100 |
|||||||||
| File name | Size | Records | Out size | Time | #Recs. | Time (s) | #Recs. | Time (s) | #Recs. | Time (s) | #Recs. | Time (s) |
| chr1.genes.bed | 1.1 MB | 7.5K genes | 970 KB | 2 s | 423 | 0.18 | 10 | 0.05 | 7 | 0.03 | 5 | 0.00 |
| chr1.snps.point | 26 MB | 1.5M SNPs | 26 MB | 4 s | – | – | 559 | 0.01 | 12 | 0.00 | 6 | 0.00 |
| chr1.sequence.fasta | 242 MB | 249M nucs | 237 MB | 11 s | – | – | – | – | 1000 | 0.00 | 100 | 0.00 |
| chr1.conservation.wig | 1.2 GB | 249M vals | 1.2 GB | 6 m | 10Ma | 0.32 | 100Ka | 0.18 | 1000 | 0.02 | 100 | 0.01 |
| NA12878.chr1…bam | 25 GB | 262M reads | No formatting req. | Coverage shown at these ranges | 1101 | 0.05 | 150 | 0.04 | ||||
| NA12878.chr1…cov | Computed from BAM file | 1.2 GB | 42 m | 10Ma | 0.32 | 100Ka | 0.18 | Read alignments shown at these ranges | ||||
| TOTAL | 53 m | 0.82 s | 0.40 s | 0.10 s | 0.05 s | |||||||
The Data Files section describes input files. The Formatting section shows time required to format each input file and the resulting formatted file size. The Retrieval and visualization section shows the time taken to retrieve data from ranges of various sizes and the number of records retrieved and drawn. All operations require <50 MB of memory.
aTracks with continuous values are smoothed before rendering at large ranges. Sequence and point tracks are not rendered beyond certain ranges, denoted with a hyphen (–). Read alignments (BAM file) are replaced by coverage (precomputed from the BAM file) when visualizing longer regions.
5 PLUG-IN FRAMEWORK
Savant is able to integrate user-defined plug-ins, allowing one to accomplish very specific tasks. Each plug-in can be one of two general types. Interactive plug-ins are allocated dockable modules on which graphical user interface (GUI) elements such as buttons or text fields can be placed to respond to user input and retrieve data. Non-interactive plug-ins are not designated a GUI component but still have extensive access to the innards of the browser. A number of helper functions are provided to plug-ins which are summarized in Table 4. Plug-ins can be used, for example, to prototype a SNP finder by identifying variable columns currently in view, or for computing genome-wide statistics, such as the fraction of SNPs in exons.
Table 4.
Helper functions provided to plug-ins
| Category | Function descriptions |
|---|---|
| Range | Change range |
| Tracks | Add, remove, retrieve data, change display modes and resolutions |
| Bookmarks | Add, remove and seek |
| UI | Rearrange modules |
| Other | Take screenshot, export data, etc. |
Plug-in development is straight-forward and requires implementation of one Java interface. The Bookmark Intersection Plug-in, shown in Figure 3, is an example of an interactive plug-in. The plug-in allows the user to select two tracks, intersect them and load the intersecting regions into the list of bookmarks, enabling easy navigation to all of the regions of interest. Once developed, plug-ins can be shared among users through the Plugin section of the Savant web site.
Fig. 3.

Code used to make Bookmark Intersection Plug-in. The details of the UI that allows the user to select two tracks have been omitted. Once the two tracks are selected, the bookMarkTrackIntersections() method is run, which, for each interval of one track, finds overlapping intervals of the other, and saves intervals with overlap to the bookmark panel.
6 EXAMPLE USES
In this section, we demonstrate example usages of Savant to identify and visualize various polymorphisms between the reference human genome and a Yoruban genome (HapMap individual NA18507) sequenced with the Illumina platform (Bentley et al., 2008). The raw reads and MAQ mappings (in BAM format) were downloaded from the 1000 Genomes web site, while the conservation track (in WIG format), gene annotations and dbSNP data were obtained from the UCSC database. Figure 4A shows two likely SNP variants in the NA18507 genome. SNPs are typically supported by consistent mismatches in aligned reads with respect to the reference at the variant positions. The reads support the existence of a transition mutation (C to T) at a known SNP site, indicating a likely common variant in the human population. There is also considerable evidence (3/12 reads) for another transition (G to A) slightly upstream, indicating a putatively novel SNP. Both of these SNPs fall in an exon of a gene, as shown by the annotation, while the conservation appears to drop-off in the immediate vicinity of the SNPs, indicating weakened evolutionary constraint.
Fig. 4.
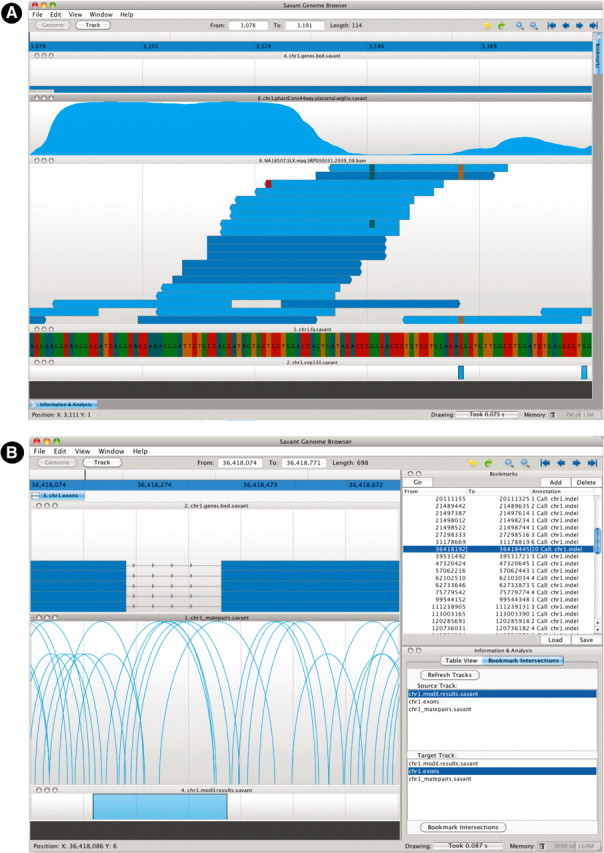
(A) Visualization of two SNP variants. The displayed tracks are, top to bottom, Conservation, Gene Models, Read Alignments, the Genome sequence and known SNPs from dbSNP. Two potential SNP variants are indicated by consistent mismatching colors within a column, with the downstream SNP previously known, while the upstream one, a heterozygous variant, not in dbSNP. (B) Identification and visualization of MoDIL indel variants that overlap exons via the plug-in framework. The visualized tracks are Gene Models, read mappings (visualized in the matepair mode) and MoDIL predictions. The panel on top right is the Bookmark module, while on the bottom right is the BookMarkIntersection plug-in described in Figure 3. To identify variants of interest, the user selects the two tracks in the BookMarkIntersection window, and those variants that overlap exons are added to the list of bookmarks. The user can then easily go through this list. In this particular case the indel likely occurs in the intron between the exons, but because of MoDIL's inability to accurately identify borders of variants, the variant is shown as overlapping.
We also demonstrate the use of the plug-in framework to identify and investigate biologically interesting indel predictions made using an external tool. We used the dataset of predicted insertion and deletion (indel) polymorphisms predicted by MoDIL (Lee et al., 2009), which identifies these variants by analysis of matepair data. In particular, MoDIL identifies regions of the genome with a significant number of discordant matepairs (those with a distance between the mappings of the reads significantly different from the expected insert size of the library). We built a plug-in to identify those indels that overlap exons, the code of which is shown in Figure 3. Figure 4B displays the result of running this plug-in, together with one such region of interest. In this case, the deletion lies directly between two exons, and likely creates a genomic variant with a much shorter intron, or with the intron completely spliced out of the gene sequence. Taller arcs in the read visualization mode are indicative of ‘stretched’ matepairs that confirm the deletion, while the lack of read mappings within the deleted region indicates that the deletion is homozygous.
7 DISCUSSION AND FUTURE WORK
In this article, we introduce Savant, a genome browser customized for visualization of HTS data. In addition to dynamic visualization and support for multiple data types, Savant supports multiple novel features meant to simplify the analysis of genomic data, including a Bookmarks module to keep track of regions of interest, a Table View module to allow researchers access to the raw data underlying the visualizations and a plug-in framework that allows developers extensive access to the browser, enabling them to program novel analysis methods and extend the browser to support new visualization and data types.
In the near future, we plan to expand Savant by supporting additional file formats, while also allowing users to automatically download annotation tracks from various public resources, such as the UCSC Genome Browser and the 1000 Genomes Project. Additional functionality, for example, to save/restore user sessions, enabling novel visualization modes, such as superimposition of tracks, and combining the matepair and regular read alignment views in a single panel will also be added in subsequent releases. Savant is an open source tool, freely available at http://compbio.cs.toronto.edu/savant/. We are also working to build a user community, accessible through the same web site. The community can be used to share Savant plug-ins with other researchers, as well as to communicate with the development team, to report bugs and to suggest new functionality and improvements.
ACKNOWLEDGEMENTS
We would like to thank Misko Dzamba, Seunghak Lee and Paul Medvedev for assistance with this manuscript. We are also indebted to the scientists at The Centre for Applied Genomics (Toronto, Canada) for feature and design suggestions. MB is an Alfred P. Sloan Research Fellow.
Funding: CIHR Catalyst Grant (to M.B.); an NSERC Undergraduate Student Research Award for (M.F.).
Conflict of Interest: none declared.
REFERENCES
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalca AV, Brudno M. Genome variation discovery with high-throughput sequencing data. Brief. Bioinform. 2010;11:3–14. doi: 10.1093/bib/bbp058. [DOI] [PubMed] [Google Scholar]
- Gordon D, et al. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Hormozdiari F, et al. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lince D, et al. The autism genome project: goals and strategies. Am. J. Pharmacogenomics. 2005;5:233–246. doi: 10.2165/00129785-200505040-00004. [DOI] [PubMed] [Google Scholar]
- Hubbard T.JP, et al. Ensembl 2009. Nucleic Acids Res. 2009;37(Suppl. 1):D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, et al. Modil: detecting small indels from clone-end sequencing with mixtures of distributions. Nat. Methods. 2009;6:473–474. doi: 10.1038/nmeth.f.256. [DOI] [PubMed] [Google Scholar]
- Li H, et al. Mapping short dna sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, et al. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods. 2009;6:S13–S20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Milne I, et al. Tablet–next generation sequence assembly visualization. Bioinformatics. 2010;26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CB, et al. Visualizing genomes: techniques and challenges. Nat. Methods. 2010;7(Suppl. 3):S5–S15. doi: 10.1038/nmeth.1422. [DOI] [PubMed] [Google Scholar]
- Pepke S, et al. Computation for chip-seq and RNA-seq studies. Nat. Methods. 2009;6(Suppl. 11):S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Staden R, et al. The staden package, 1998. Methods Mol. Biol. 2000;132:115–130. doi: 10.1385/1-59259-192-2:115. [DOI] [PubMed] [Google Scholar]
- Via M, et al. The 1000 genomes project: new opportunities for research and social challenges. Genome Med. 2010;2:3. doi: 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]