

Abstract

A concise formal synthesis of the cytotoxic macrolides (-)-salicylihalamides A and B is reported. Key features of the synthetic strategy include a chemoselective hydroboration, highly regio- and diastereoselective methyl cuprate addition, Pd-catalyzed formate reduction, and an E-selective ring-closing metathesis to construct the 12-membered macrocycle subunit. Overall, two routes have been developed from a readily prepared bicyclic phosphate (4-steps), a 13-step route and a more efficient 9-step sequence relying on regioselective esterification of a key diol.
I. Introduction
The cytotoxic macrolides salicylihalamides A (1a) and B (1b) were isolated from the Australian marine sponge Halicona sp. in 1997 by Boyd, Erickson, and co-workers (Figure 1).1 The structure and relative stereochemistry of the salicylihalamides (1a and 1b) were determined by NMR spectroscopic methods and Mosher ester analysis. These natural products possess a 12-membered unsaturated benzolactone core and an unusual enamide side chain with differing geometry about the C17-C18 double bond. In 2000, De Brabander and co-workers reassigned the absolute stereochemistry in the first total synthesis of 1a.2
Figure 1.
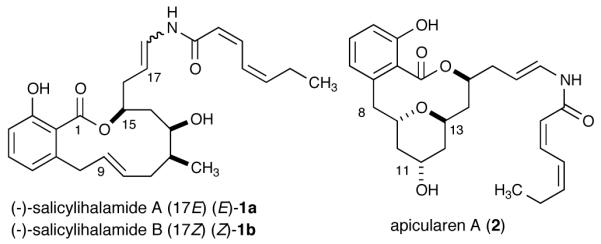
Structures of two important benzolactone enamide class compounds
When screened against the NCI’s 60 human tumor cell lines salicylihalamide A (1a) exhibited potent cytotoxicity with an average GI50 of 15 nM. In comparison to related benzolactone enamide natural products, e.g., apicularen A (2), salicylihalamide A (1a) exhibited the highest average sensitivity (GI50 = 7 nM) against melanoma cell lines.1 Furthermore, salicylihalamide A (1a) possesses selective inhibition of mammalian vacuolar type H+-ATPase (V-ATPase), with an IC50 value <1.0 nM against bovine brain V-ATPase.3 Salicilyhalamide A has attracted significant attention from the synthetic community due to its potent antitumor properties, structural features, and the limited availability of the material from natural sources.4
Previously, we have reported the synthesis of polyol synthons utilizing the concept of multivalent activation with temporary phosphate tethers whereby a number of chemo-, regio- and stereoselective transformations were realized.5 Herein, we disclose the formal total synthesis of salicylihalamide A in 13 steps from bicyclic phosphate 7 featuring the orthogonal protecting property of chiral aliphatic subunit 6 (overall, 17-step longest linear sequence (LLS)). A more efficient 9-step synthesis from 7 using regioselective esterification of diol 6 is also reported (overall, 13-step LLS), and is on par with the most efficient syntheses reported to date.4
Retrosynthetic analysis reveals the construction of the macrolactone (3 or 4) from the functionalized benzoic acid derivative 56 and the chiral, non-racemic subunit 6 via an E-selective ring-closing metathesis (RCM) using Grubbs catalyst (PCy3)2(Cl)2Ru=CHPh (cat-A)7 in both routes (Scheme 1). The key intermediate 6 can be derived from enantiomerically pure bicyclic phosphate (R,R,RP)-75a (derived via desymmetrization with Grubbs catalyst (IMesH2)(PCy3)(Cl)2Ru=CHPh (cat-B))8 involving a chemoselective hydroboration, highly regio- and diastereoselective cuprate addition, cross metathesis (CM) with the Hoveyda-Grubbs second generation catalyst (cat-C),9 and a Pd-catalyzed reductive allylic transposition using ammonium formate.5f
Scheme 1.

Retrosynthetic Analysis of (–)-Salicylihalamides
Results and Discussion
II. Construction of P-chiral, nonracemic bicyclo[4.3.1]phosphate 7
1,3-anti-diol 810 was desymmetrized using a phosphate tether-mediated RCM reaction to construct the P-chiral bicyclo[4.3.1]phosphate 7 (Scheme 2).5 In this strategy, pseudo-C2-symmetric phosphate triester 9 was synthesized from a 2-step sequential tripodal coupling5c of diol 8 and allyl alcohol with POCl3 or via a one-pot diol coupling with allyl tetraisopropylphosphorodiamidite followed by oxidation.5g The method is predicated on facile RCM occurring via the chair conformer bearing a cis-diaxial relationship in the reacting olefin pairs (allylphosphate ester cis to the terminal olefin) to yield the P-chiral, non-racemic bicyclo[4.3.1]-phosphate (R,R,Rp)-7.
Scheme 2.

Construction of P-chiral, nonracemic bicyclo[4.3.1]phosphate (R,R,Rp)-7
III. Synthesis of chiral subunit 6
Scheme 3 details the construction of advanced intermediate 6, which is the required intermediate in both routes from the bicycle (R,R,RP)-7. The primary alcohol 10 was obtained via a chemoselective hydroboration of the exocyclic olefin of (R,R,RP)-7, followed by oxidation under mild conditions (NaBO3•4H2O) developed by Burke and co-workers.11 Subsequent PMB-ether formation using the p-methoxybenzyl trichloroacetimidate derived from p-MeOPhCH2OH produced 11 in 92% yield. A regio- and diastereoselective SN2′ cuprate addition (CuCN•2LiCl, Me2Zn, dr = >95:5) to 11, followed by methylation (TMSCHN2 and MeOH) afforded monocyclic phosphate ester 12 in excellent overall yield (85%). The orthogonal alignment of the π* arbital (C=C) to the σ* orbital (C-O-PO) and concave nature of the bicycle 11 explains the regio and diastereoselectivity of the SN2′ reaction.5e,f Monocyclic phosphate 12 was subjected to cross metathesis with (Z)-diacetate 13 using 10 mol% of Hoveyda-Grubbs catalyst cat-C to generate CM adduct 14 in 83% yield.12 Pd-catalyzed, reductive allylic transposition [Pd(OAc)2, PPh3, HCOONH4] on CM adduct 14 occurred with excellent regioselectivity to afford the terminal olefin 15 in 94% yield.13 Removal of the phosphate ester in the presence of LiAlH4 provided diol 6 as a single diastereomer in excellent yield (98%).
Scheme 3.

Synthesis of Key Fragment 6
IV. Formal Total Synthesis of (–)-Salicylihalamide A & B in 13 steps from (R,R,Rp)-7
Initial efforts focused on the synthesis of benzolactone 3 from the key diol intermediate 6 utilizing protection with TIPSCl to differentiate the hydroxyl groups (Scheme 4). Diol 6 was selectively protected as a TIPS-ether 16 (86% yield),14 followed by MOM protection to furnish the fully protected triol 17 in 92% yield. Compound 17 was desilylated in 95% yield to afford the key subunit 18, which was ready for coupling with benzodioxinone 5. Alcohol 18 was treated with NaH, followed by addition of benzodioxinone 5, to provide ester 19 in 66% yield. Subsequent methylation resulted in the production of the known RCM precursor 20 in 90% yield.2a,4e To complete the formal synthesis, RCM reaction of 20 was carried out using condition developed by De Brabander and coworkers4e (10 mol% of cat-A)7 to furnish the known salicylihalamide macrolide 3 in 82% yield and with excellent E-selectivity (E/Z = 10:1). The physical and spectroscopic data of the synthetic sample (1H, 13C, IR, HRMS), as well as specific rotation, were all in full agreement with those reported in the literature.2a,4d,e
Scheme 4.

Formal Total Synthesis of (–)-Salicylihalamides in 13 steps from (R,R,Rp)-7 (17-LLS).
V. Regioselective Esterification Studies on Key Fragment 6
To further streamline the process, we next explored the feasibility of a regioselective esterification of diol 6 in order to avoid the aforementioned orthogonal protection pathway. Scheme 5 highlights the key regioselective esterification studies on diol 6. As shown in entry one, use of NaH as the base yielded a readily separable mixture (1:1) of both isomers 21a (known) and 21b in 85% yield, while implementation of LiHMDS gave modest improvement of selectivity (2:1). However, implementation of NaHMDS as base resulted in a 3.6:1 mixture of the desired benzolactone 21a and its regioisomer 21b, which could be readily converted back to starting diol 6 (K2CO3, MeOH) for recycling.
Scheme 5.

Regioselective Esterification Studies on Key Fragment 6
VI. Formal Total Synthesis of (–)-Salicylihalamides A and B in 9 steps from (R,R,Rp)-7
Compound 21a was protected as the di-MOM ether 22 (86% yield), and subjected to RCM condition developed by De Brabander and coworkers4e (cat-A) to afford salicylihalamide macrolide 4 in 84% yield and with excellent E selectivity (E/Z = 9:1) (Scheme 6). The spectral data (1H, 13C, IR, HRMS) of 4 was in complete agreement with those reported in the literature along with specific rotation {[α]D = −29.6 (c 0.65, CHCl3)}.4e
Scheme 6.
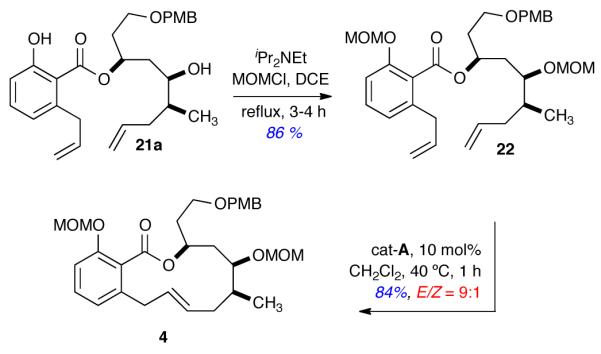
Formal Total Synthesis of (–)-Salicylihalamides A and B in 9 steps from (R,R,Rp)-7
Conclusion
In conclusion, the synthesis of key macrolactones 3 and 4 are reported representing formal syntheses of salicylihalamides A and B. Overall, a 13-step route (17-LLS) and a 9-step route (13-LLS) have been developed, from a common, readily prepared, chiral, nonracemic bicyclic phosphate (R,R,RP)-7, with an overall yield of 17.5% and 22.5%, respectively. Each route proceeds through the common diol subunit 6. The 13-step route requires differential protection of diol 6, while the more efficient 9-step sequence relies on a regioselective esterification of diol 6. The latter route has 13 steps in its longest linear sequence (LLS), which is on par with the most efficient syntheses of this key late stage subunit reported to date.
Experimental Section
General Methods
All reactions were carried out in oven- or flame-dried glassware, under argon atmosphere, using standard gas-tight syringes, cannulae, and septa. Stirring was achieved with oven-dried magnetic stir bars. Et2O, THF and CH2Cl2 were passed through a purification system employing activated Al2O3. Et3N was eluted through basic alumina and stored over KOH. Butyl lithium was titrated prior to use. All olefin metathesis catalysts were obtained commercially and used without further purification. All 1H and 13C NMR spectra were recorded in CDCl3 at 500 MHz, and 126 MHz instruments, respectively, and calibrated to the solvent peak. All mass spectra were obtained using electrospray ionization (ESI) (MeOH) coupled to high-resolution mass spectrometry (HRMS). Observed rotations at 589 nm were measured using an automatic polarimeter. Infrared spectra were obtained using a Fourier transform infrared (FTIR) spectrometer.
(1R,6R,8S)-8-(2-Hydroxyethyl)-2,9,10-trioxa-1-phosphabicyclo[4.3.1]dec-4-ene 1-oxide (10)
Bicyclic phosphate (R,R,Rp)-7 (1.50 g, 7.41 mmol) was dissolved in anhydrous THF (20 mL), followed by slow addition of 9-BBN (2.71 g, 22.2 mmol) in anhydrous THF (45 mL) under argon atmosphere. The solution was stirred at rt for 3 h. After completion of the reaction as monitored by TLC, the reaction mixture was cooled to 0 °C, and H2O (3.5 mL) was added dropwise, followed by addition of NaBO3•4H2O (10.26 g, 66.69 mmol) in one portion. After removing the ice bath, additional H2O (7 mL) was added, and the reaction mixture stirred at rt for 1 h. After complete oxidation as monitored by TLC, the crude solution was added dried (Na2SO4), filtered through pad of Celite® and washed with EtOAc. The filtrate was concentrated under reduced pressure and purified via flash column chromatography (10:1 EtOAc/MeOH) to provide alcohol 10 (1.30 g, 80%) as a white solid; [α]D = − 96.0 (c = 1.82, CHCl3); FTIR (neat) 3454, 3072, 2962, 2935, 2887, 1288, 1066, 975 cm−1; 1H NMR (500 MHz, CDCl3) δ 6.01 (dddd, J = 11.9, 6.7, 3.0, 2.2 Hz, 1H), 5.59 (ddd, J = 11.9, 3.9, 2.6 Hz, 1H), 5.18 (ddd, J = 24.6, 3.7, 1.9 Hz, 1H), 4.95 (dtd, J = 14.1, 5.6, 2.7 Hz, 1H), 4.86-4.74 (m, 1H), 4.35 (ddd, J = 27.9, 14.7, 6.7 Hz, 1H), 3.84-3.64 (m, 2H), 3.00 (s, 1H), 2.20 (ddd, J = 14.7, 12.0, 6.2 Hz, 1H), 1.97-1.89 (m, 1H), 1.86-1.77 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 129.7, 127.6, 77.4 (d, J = 6.6 Hz), 74.5 (d, J = 6.5 Hz), 63.0 (d, J = 6.4 Hz), 57.4, 38.1 (d, J = 9.2 Hz), 34.8; HRMS calcd. for C8H13O5PK (M+K)+ 259.0138; found 259.0138 (TOF MS ES+).
(1R,6R,8S)-8-(2-((4-Methoxybenzyl)oxy)ethyl)-2,9,10-trioxa-1-phosphabicyclo[4.3.1]dec-4-ene 1-oxide (11)
To a stirring solution of NaH (142 mg, 5.91 mmol) in anhydrous Et2O (10 mL), under argon atmosphere, was slowly cannulated a solution of PMBOH (8.17 g, 59.09 mmol) in dry Et2O (30 mL) at rt. After stirring for 40 min, the solution was cooled to 0 °C, and Cl3CCN (8.54 g, 59.1 mmol) was slowly added via dropwise addition. After 5-10 min., the solution was removed from the ice bath and stirred for an additional hour. The reaction was quenched with saturated NaHCO3 (20 mL), and the layers separated. The aqueous layer was further extracted with Et2O (3 × 50 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude mixture was dissolved in dry CH2Cl2 (118 mL) and cannulated into a flask containing the phosphate-10 (2.6 g, 11.82 mmol), followed by the addition of PPTS (300 mg, 1.18 mmol) at rt under argon atmosphere. After stirring for 16 h, the reaction was quenched with saturated NaHCO3 (40 mL), and the layers separated. The aqueous layer was extracted with CH2Cl2 (3 × 50 mL), and the combined organic layers were washed with brine (100 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography (EtOAc) afforded PMB ether-11 (3.71 g, 92%) as a viscous, light yellow oil; [α]D = −64.66 (c = 2.40, CHCl3); FTIR (neat): 3055, 2933, 2866, 1612, 1512, 1298, 1093, 975, 738, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.24 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.02 (dddd, J = 11.9, 6.7, 3.1, 2.2 Hz, 1H), 5.57 (ddd, J = 11.9, 3.9, 2.6 Hz, 1H), 5.22-5.13 (m, 1H), 5.00 (ddt, J = 14.8, 8.9, 3.0 Hz, 1H), 4.81 (dddd, J = 10.5, 8.5, 4.1, 2.1 Hz, 1H), 4.43 (d, J = 2.9 Hz, 2H), 4.46-4.32 (m, 1H), 3.81 (s, 3H), 3.65-3.52 (m, 2H), 2.19 (ddd, J = 14.6, 12.0, 6.2 Hz, 1H), 2.02-1.84 (m, 2H), 1.74 (ddd, J = 14.6, 3.4, 2.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 159.2, 130.1, 129.8, 129.3, 127.8, 113.8, 77.2 (d, JCP = 6.3 Hz), 74.1 (d, JCP = 6.6 Hz), 72.8, 64.7, 62.9 (d, JCP = 6.4 Hz), 55.2, 35.9 (d, JCP = 9.4 Hz), 34.9 (d, JCP = 5.9 Hz); HRMS calcd. for C16H21O6PK (M+K)+ 379.0713; found 379.0706 (TOF MS ES+).
(4R,6S)-4-((S)-But-3-en-2-yl)-2-methoxy-6-(2-((4-methoxybenzyl)oxy)ethyl)-1,3,2-dioxaphosphinane 2-oxide (12)
Within a glovebox, CuCN (1.75 g, 19.51 mmol, dried overnight in a vacuum oven at 60 °C/0.3 mmHg and stored in a glovebox) and LiCl (1.65 g, 39.02 mmol, dried overnight in a vacuum oven at 60 °C/0.3 mmHg and stored in a glovebox) were added to a round bottom flask and sealed with a septa. The flask was removed from the glovebox and placed under a balloon of argon. Anhydrous THF (20 mL) was added to the mixture that was stirred for 20 min at rt then cooled to −30 °C. A solution of Me2Zn (16.2 mL, 1.2 M in toluene) was added rapidly via dropwise addition, and the solution was stirred for 30 min at −30 °C (solution turns deep green). After 30 min, phosphate 11 (1.32 g, 3.90 mmol) in anhydrous THF (6 mL) was cannulated dropwise (1 mL rinse) into the stirring reaction mixture, and the solution was immediately removed from the bath and stirred at rt for 2 h. Upon completion (monitored by TLC, baseline spot in EtOAc), the reaction was cooled to 0 °C and slowly quenched with 10% aqueous HCl (2 mL), followed by water (4 mL), and stirred at rt for 10 min, where pepper-colored salts formed. The solution was filtered through a pad of Celite and rinsed thoroughly with EtOAc. To the resulting biphasic solution was added 10% aqueous HCl (3 mL), and the layers were separated. The aqueous layer was extracted with EtOAc (2 × 25 mL), and the combined organic layers were concentrated under reduced pressure. The resulting oil was dissolved in MeOH (~10 mL), where TMSCHN2 (2 M in Et2O, ~5 mL) was added dropwise, resulting in a deep yellow solution. Excess TMSCHN2 was quenched via slow dropwise addition of glacial acetic acid (3-4 drops), and the solution was dried (Na2SO4), filtered, and concentrated under reduced pressure. Flash column chromatography (2:1 EtOAc/hexane) provided title compound 12 (1.23 g, 85%) as a clear oil, and as a ~1:1 mixture of diastereomers at phosphorus; FTIR (neat) 2925, 2852, 1265, 1093, 1033, 972, 749, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 8.3 Hz, 2H), 6.89 (d, J = 8.3 Hz, 2H), 5.85-5.74 (m, 1H), 5.15-5.05 (m, 2H), 4.82-4.63 (m, 1H), 4.44 (s, 2H), 4.49-4.30 (m, 1H), 3.81 (s, 3H), 3.78 (d, J = 6.6 Hz, 1.5H), 3.76 (d, J = 6.6 Hz, 1.5H), 3.69-3.52 (m, 2H), 2.57-2.36 (m, 1H), 2.23-2.05 (m, 2H), 1.92-1.80 (m, 2H), 1.10 (d, J = 6.9 Hz, 1.5 H), 1.06 (d, J = 6.9 Hz, 1.5 H); HRMS calcd. for C18H27O6PK (M+K)+ 409.1182; found 409.1188 (TOF MS ES+).
(4S,E)-4-((4R,6S)-2-Methoxy-6-(2-((4-methoxybenzyl)oxy)ethyl)-2-oxido-1,3,2-dioxaphosphinan-4-yl)pent-2-en-1-yl acetate (14)
The monocyclic phosphate 12 (1.0 g, 2.70 mmol) was weighed into a round bottom flask and dissolved in CH2Cl2 (degassed 10 min. with Ar, 27.0 mL), followed by the addition of diacetate 13 (0.56 g, 3.24 mmol) and cat-C (168 mg, 0.27 mmol) under argon at rt. The reaction mixture was heated at 45 °C for 1 h under continuous argon flow and, upon completion, as monitored by TLC, was concentrated under reduced pressure. Flash column chromatography (1:1 EtOAc/hexane) provided title compound 14 (0.99 g, 83%) as a clear oil, and as a ~1:1 mixture of diastereomers at phosphorus; FTIR (neat) 3053, 2954, 2927, 2852, 1737, 1265, 1247, 1093, 1031, 972, 1031, 972, 749, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.24 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.79-5.72 (m, 1H), 5.67-5.60 (m, 1H), 4.80-4.62 (m, 1H), 4.53 (t, J = 5.3 Hz, 2H), 4.47-4.29 (m, 3H), 3.81 (s, 3H), 3.77 (d, J = 7.4 Hz, 1.5 H), 3.75 (d, J = 7.4 Hz, 1.5 H), 3.68-3.53 (m, 2H), 2.59-3.38 (m, 1H), 2.23-2.04 (m, 2H), 2.07 (2s, 3H), 1.93-1.81 (m, 2H), 1.10 (d, J = 6.9 Hz, 1.5 H), 1.05 (d, J = 6.9 Hz, 1.5 H); HRMS calcd. for C21H31O8PK (M+K)+ 481.1394; found 481.1390 (TOF MS ES+).
(4S,6R)-2-Methoxy-4-(2-((4-methoxybenzyl)oxy)ethyl)-6-((S)-pent-4-en-2-yl)-1,3,2-dioxaphosphinane 2-oxide (15)
To a stirring solution of HCO2NH4 (63 mg, 1.00 mmol) in degassed DCE (4 mL) was added Pd(OAc)2 (12 mg, 0.05 mmol) and Ph3P (66 mg, 0.25 mmol) at rt, and the mixture was stirred for 15 min at rt under argon, at which point a solution of acetate 14 (220 mg, 0.50 mmol) in degassed DCE (2 mL) was added dropwise via cannula. The stirring solution was equipped with a reflux condenser and placed into an oil bath at 90 °C for 1 h. The reaction mixture was cooled to rt, washed with sat. NaHCO3 (6 mL) solution, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were rinsed with brine (10 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography (1:1 EtOAc/hexane) afforded the desired compound 15 (180 mg, 94 %) as a clear oil, and as a ~1:1 mixture of diastereomers at phosphorus; FTIR (neat): 3053, 2956, 2927, 2854, 12654, 1093, 1035, 970, 749, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 5.81-5.70 (m, 1H), 5.10-5.02 (m, 2H), 4.82-4.63 (m, 1H), 4.48-4.38 (m, 2H), 4.37-4.21 (m, 1H), 3.81 (s, 3H), 3.78 (d, J = 7.4, Hz, 1.5H), 3.76 (d, J = 7.7 Hz, 1.5 H), 3.69-3.54 (m, 2H), 2.42-2.27 (m, 1H), 2.22-1.76 (m, 6H), 0.90 (d, J = 6.8 Hz, 1.5H), 0.86 (d, J = 6.8 Hz, 1.5H); HRMS calcd. for C19H29O6PNa (M+Na)+ 407.1599; found 407.1612 (TOF MS ES+).
(3S,5R,6S)-1-((4-Methoxybenzyl)oxy)-6-methylnon-8-ene-3,5-diol (6)
To a suspension of LiAlH4 (53 mg, 1.10 mmol) in anhydrous Et2O (3 mL) was added dropwise a solution of 1:1 diastereomeric phosphate mixture 15 (170 mg, 0.45 mmol) in Et2O (2 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h, quenched via slow sequential addition of H2O (60 μL), 15% NaOH (60 μL), H2O (180 μL), and removed from the ice bath. After stirring for 30 minutes, 10% aqueous HCl (5 mL) was added to the reaction mixture, and the layers were separated. The aqueous layer was extracted with Et2O (3 × 10 mL), and the combined organic layers were rinsed with brine (1 × 10 mL)), dried (Na2SO4), filtered, and concentrated under reduced pressure. Flash column chromatography (1:1 EtOAc/Hexane) afforded diol 6 (125 mg, 98% yield) as a viscous oil; [α]D = +15.16 (c = 1.20, CHCl3); FTIR (neat) 3412, 2921, 2866,1298, 1093, 975, 740, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 8.6Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 5.82 (dddd, J = 16.9, 10.2, 7.6, 6.6 Hz, 1H), 5.07-4.99 (m, 2H), 4.46 (s, 2H), 4.20-4.12 (m, 1H), 3.81 (s, 3H), 3.77-3.71 (m, 1H), 3.72 (td, J = 9.3, 4.7 Hz, 1H), 3.65 (td, J = 9.2, 3.8 Hz, 1H), 3.59 (br. s, 1H), 2.97 (br. s, 1H), 2.35-2.28 (m, 1H), 1.97-1.88 (m, 2H), 1.71-1.55 (m, 4H), 0.87 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 159.3, 137.5, 129.7, 129.3, 115.9, 113.8, 73.0, 72.3, 69.9, 69.2, 55.2, 39.0, 38.6, 37.2, 36.1, 15.1; HRMS calcd. for C18H28O4Na (M+Na)+ 331.1885; found 331.1877 (TOF MS ES+).
(4S,5R,7S)-9-((4-Methoxybenzyl)oxy)-4-methyl-7-((triisopropylsilyl)oxy)non-1-en-5-ol (16)
To a stirring solution of diol 6 (50 mg, 0.16 mmol) in CH2Cl2 (4 mL) was added 2,6-lutidine (70 mg, 0.65 mmol), and the mixture was cooled to −78 °C. TIPSOTf (100 mg, 0.32 mmol) was added dropwise, and the reaction mixture was stirred for 2 h and then allowed to slowly warm to 0 °C. After completion of the reaction, as monitored by TLC, it was quenched with sat. NH4Cl, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 5 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography (1:10 EtOAc/Hexane) afforded the desired silyl ether 16 (64 mg, 86%) as a viscous oil; [α]D = +12.88 (c = 1.35, CHCl3); FTIR (neat) 3406, 2923, 2850, 1265, 1095, 740, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.24 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.78 (dddd, J = 16.9, 10.3, 7.5, 6.8 Hz, 1H), 5.06-4.95 (m, 2H), 4.41 (dd, J = 39.1, 11.6 Hz, 2H), 4.36-4.31 (m, 1H), 3.83 (d, J = 0.9 Hz, 1H), 3.81 (s, 3H), 3.80-3.76 (m, 1H), 3.50-3.39 (m, 2H), 2.25-2.17 (m, 1H), 2.10-1.93 (m, 2H), 1.89-1.80 (m, 1H), 1.76-1.65 (m, 1H), 1.64-1.50 (m, 2H), 1.08 (s, 12H), 1.06 (s, 9H), 0.83 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 159.2, 137.7, 130.3, 129.3, 129.2, 115.6, 113.7, 72.6, 71.6, 70.2, 66.3, 55.2, 38.9, 36.8, 36.3, 35.5, 18.1, 18.1, 17.7, 14.7, 12.3, 12.3; HRMS calcd. for C27H48O4SiNa (M+Na)+ 487.3220; found 487.3210 (TOF MS ES+).
(5R,7S)-9,9-Diisopropyl-7-(2-((4-methoxybenzyl)oxy)ethyl)-10-methyl-5-((S)-pent-4-en-2-yl)-2,4,8-trioxa-9-silaundecane (17)
To a stirring solution of silyl ether 16 (60 mg, 0.129 mmol) in anhydrous CH2Cl2 (5 mL), under argon, was added iPr2NEt (167 mg, 1.292 mmol) and MOMCl (52 mg, 0.646 mmol) at 0 °C. The reaction was stirred at rt for 3-4 h. Upon completion (monitored by TLC), the reaction was diluted with CH2Cl2 (5 mL) followed by sat. NH4Cl solution (10 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were washed with brine (1 × 10 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude reaction mixture was purified through flash column chromatography (1:10 EtOAc/hexane) to afford MOM-ether 17 (60 mg, 92%) as a clear oil; [α]D = +9.12 (c = 1.08, CHCl3); FTIR (neat): 2923, 2850, 1460, 1265, 1097, 1039, 748, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.26 (d, J = 8.9 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 5.78 (dddd, J = 17.1, 10.2, 6.9, 6.4 Hz, 1H), 5.07-4.96 (m, 2H), 4.66 (dd, J = 10.4, 6.8 Hz, 2H), 4.43 (dd, J = 14.8, 11.5 Hz, 2H), 4.12-4.06 (m, 1H), 3.81 (s, 3H), 3.64 (ddd, J = 9.8, 6.6, 2.6 Hz, 1H), 3.57-3.52 (m, 2H), 3.36 (s, 3H), 2.15-2.08 (m, 1H), 1.94-1.80 (m, 3H), 1.67-1.60 (m, 2H), 1.52 (ddd, J = 14.2, 7.4, 3.4 Hz, 1H), 1.06 (br. s, 18H), 1.06-1.04 (m, 3H), 0.89 (d, J = 6.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 159.0, 137.5, 130.7, 129.2, 115.7, 113.7, 96.6, 79.9, 72.6, 68.1, 66.5, 55.7, 55.3, 38.3, 38.1, 37.1, 36.5, 18.3, 18.3, 14.2, 12.9; HRMS: calcd. for C29H52O5SiNa (M+Na)+ 531.3482; found 531.3502 (TOF MS ES+).
(3S,5R,6S)-1-((4-Methoxybenzyl)oxy)-5-(methoxymethoxy)-6-methylnon-8-en-3-ol (18)
A solution of protected triol 17 (52 mg, 0.110 mmol) in anhydrous THF (2 mL) was treated with TBAF (1 M in THF, 0.3 mL) at 0 °C and stirred for 2 h at rt. After completion of the reaction, as monitored by TLC, the reaction mixture was concentrated under reduced pressure. The crude product was purified through flash column chromatography (1:6 EtOAc/hexane) to afford alcohol 18 (34 mg, 95%) as a viscous oil; [α]D = +38.6 (c = 1.00, CHCl3); FTIR (neat) 3412, 2921, 2856,1298, 1093, 975, 749, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 8.6 Hz, 2H), 6.92-6.87 (d, J = 8.6 Hz, 2H), 5.78 (ddt, J = 17.0, 10.1, 7.0 Hz, 1H), 5.05-4.99 (m, 2H), 4.69 (dd, J = 10.1, 6.6 Hz, 2H), 4.45 (s, 2H), 4.05-3.96 (m, 1H), 3.81 (s, 3H), 3.75-3.60 (m, 3H), 3.41 (s, 3H), 3.31 (d, J = 3.4 Hz, 1H), 2.19-2.12 (m, 1H), 1.89-1.81 (m, 2H), 1.81-1.68 (m, 2H), 1.52 (dddd, J = 32.1, 14.2, 9.6, 2.8 Hz, 2H), 0.88 (d, J = 6.4 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 159.2, 137.2, 130.2, 129.3, 115.9, 113.8, 96.9, 79.0, 72.8, 68.3, 66.8, 55.9, 55.2, 37.6, 37.4, 37.0, 36.5, 14.2; HRMS calcd. for C20H32O5Na (M+Na)+ 375.2147; found 375.2146 (TOF MS ES+).
(3S,5R,6S)-1-((4-Methoxybenzyl)oxy)-5-(methoxymethoxy)-6-methylnon-8-en-3-yl 2-allyl-6-hydroxybenzoate (19)
To a suspension of NaH (24 mg, 0.852 mmol, 60% w/v dispersion in mineral oil) in anhydrous THF (2 mL), under argon, was added, dropwise, a solution of alcohol 18 (30 mg, 0.085) in anhydrous THF (1 mL) at 0 °C. The reaction mixture was stirred for 15 min at 0 °C. A solution of benzodioxinone 5 (37 mg, 0.170 mmol) in THF (1 mL) was added dropwise via cannula to the mixture, and the reaction was warmed to rt and stirred for 6 h. The reaction was quenched with saturated NH4Cl (5 mL), and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined organic layers were rinsed with brine (1×10 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Flash column chromatography (1:4 EtOAc/Hexane) afforded ester 19 (29 mg, 66%) as a viscous oil, along with recovered starting material (9 mg); [α]D = +12.3 (c = 1.00, CHCl3) FTIR (neat) 3435, 3053, 2956, 2925, 2854, 1646, 1265, 1033, 748, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 11.17 (s, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.22 (d, J = 8.8 Hz, 2H), 6.88 (dd, J = 8.3, 1.1 Hz, 1H), 6.82 (d, J = 8.8 Hz, 2H), 6.72 (dd, J = 7.4, 1.1 Hz, 1H), 5.97 (dddd, J = 16.9, 10.2, 6.1, 6.1 Hz, 1H), 5.72 (dddd, J = 17.0, 10.1, 7.0, 7.0 Hz, 1H), 5.62-5.56 (m, 1H), 5.03-4.90 (m, 4H), 4.63 (dd, J = 41.5, 6.9 Hz, 2H), 4.40 (s, 2H), 3.78 (s, 3H), 3.64 (ddd, J = 39.4, 15.7, 6.1 Hz, 2H), 3.58-3.51 (m, 3H), 3.37 (s, 3H), 2.11-1.98 (m, 3H), 1.97-1.88 (m, 1H), 1.88-1.70 (m, 3H), 0.89 (d, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 170.7, 162.3, 159.1, 142.4, 137.7, 136.9, 134.0, 130.1, 129.3, 122.3, 116.12, 116.0, 115.5, 113.7, 112.7, 112.6, 96.5, 78.0, 72.8, 71.8, 66.4, 55.9, 55.2, 39.9, 37.5, 36.0, 35.2, 34.8, 13.5; HRMS calcd. for C30H40O7Na (M+Na)+ 535.2672; found 535.2627 (TOF MS ES+).
(3S,5R,6S)-1-((4-Methoxybenzyl)oxy)-5-(methoxymethoxy)-6-methylnon-8-en-3-yl 2-allyl-6-methoxybenzoate (20)
To a suspension of NaH (~2 mg, 0.078 mmol, 60% w/v dispersion in mineral oil) in anhydrous THF (1 mL) was added, dropwise, a solution of ester 19 (20 mg, 0.039) in anhydrous THF (2 mL). To this reaction mixture MeI (22 mg, 0.156 mmol) was added, and stirring was continued for 1 h at rt. The reaction mixture was quenched with cold water (2 mL), and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined organic layers were rinsed with brine (1 × 8 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Flash column chromatography (1:3 EtOAc/Hexane) afforded the methyl ether 20 (19 mg, 90%) as a viscous oil; [α]D = −1.6 (c = 0.50, CHCl3); FTIR (neat) 2952, 2925, 2852, 1641, 1265, 1069, 1033, 748, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.31-7.27 (m, 3H), 6.88 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 7.2 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 5.93 (dddd, J = 16.7, 10.2, 6.5, 6.5 Hz, 1H), 5.75 (dddd, J = 16.8, 10.2, 7.4, 6.5 Hz, 1H), 5.51-5.42 (m, 1H), 5.10-5.03 (m, 2H), 5.00-4.90 (m, 2H), 4.73 (dd, J = 11.4, 6.8 Hz, 2H), 4.46 (dd, J = 25.3, 11.3 Hz, 2H), 3.81 (s, 3H), 3.80 (s, 3H), 3.73-3.69 (m, 1H), 3.66-3.56 (m, 2H), 3.41 (s, 3H), 3.36 (d, J = 6.4 Hz, 2H), 2.11-1.98 (m, 3H), 1.97-1.89 (m, 1H), 1.88-1.79 (m, 1H), 1.77-1.67 (m, 2H), 0.91 (d, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.9, 159.1, 156.2, 138.1, 137.1, 136.4, 130.5, 130.2, 129.2, 124.1, 121.6, 116.4, 115.8, 113.7, 108.7, 97.0, 78.3, 72.7, 70.7, 55.8, 55.5, 55.3, 37.6, 37.2, 36.5, 35.3, 35.2, 13.7; HRMS calcd. for C31H42O7Na (M+Na)+ 549.2833; found 549.28631(TOF MS ES+).
(3S,5R,6S,E)-14-Methoxy-3-(2-((4-methoxybenzyl)oxy)ethyl)-5-(methoxymethoxy)-6-methyl-3,4,5,6,7,10-hexahydro-1H-benzo[c][1]oxacyclododecin-1-one (3)
Grubbs catalyst (Cy3P)2Cl2Ru=CHPh (~3 mg, 10 mol%, cat-A) was added to a solution of methyl ether 20 (14 mg, 0.026 mmol) in degassed, anhydrous CH2Cl2 (5 mL) at rt under argon. The stirring solution was equipped with a reflux condenser and placed into an oil bath at 40 °C for 1 h. After completion of the reaction, as monitored by TLC, the reaction mixture was concentrated under reduced pressure. Flash column chromatography (1:4 EtOAc/Hexane) afforded the major E-isomer 3 (11 mg, 82%) as a viscous oil (containing a small amount of Z-isomer, the E/Z ratio was 10:1 as determined by 1H NMR of the crude reaction); [α]D = −41.7 (c = 0.35, CHCl3); FTIR (neat) 2942, 2911, 2850, 1649, 1266, 1239, 1064, 1033, 908, 748, 702 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.8 Hz, 2H), 7.23 (t, J = 8.0 Hz, 1H), 6.88 (d, J = 8.8 Hz, 2H), 6.79 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 7.7 Hz, 1H), 5.53-5.45 (m, 2H), 5.35 (ddt, J = 15.2, 9.5, 2.1 Hz, 1H), 4.85 (dd, J = 46.3, 6.7 Hz, 2H), 4.48 (s, 2H), 4.16 (dd, J = 9.3, 3.6 Hz, 1H), 3.81 (s, 3H), 3.76-3.70 (m, 1H), 3.72 (s, 3H), 3.66 (t, J = 6.8 Hz, 2H), 3.44 (s, 3H), 3.33 (ddd, J = 14.0, 4.1, 2.1 Hz, 1H), 2.31 (d, J = 13.3 Hz, 1H), 2.13 (ddd, J = 18.8, 12.2, 6.2 Hz, 1H), 2.08-1.98 (m, 1H), 1.91 (dtd, J = 11.6, 7.4, 4.1 Hz, 1H), 1.82-1.65 (m, 2H), 1.46 (dd, J = 15.5, 9.4 Hz, 1H), 0.87 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 168.2, 159.1, 156.4, 139.1, 131.4, 130.7, 129.9, 129.0, 128.5, 124.5, 122.8, 113.7, 109.8, 96.8, 79.2, 72.6, 72.2, 66.6, 55.6, 55.3, 55.3, 37.7, 37.7, 36.4, 35.7, 34.0, 13.4; HRMS calcd. for C29H38O7Na (M+Na)+ 521.2515; found 521.2525 (TOF MS ES+).
(3S,5R,6S)-5-Hydroxy-1-((4-methoxybenzyl)oxy)-6-methylnon-8-en-3-yl 2-allyl-6-hydroxybenzoate (21a)
To a solution of diol 6 (50 mg, 0.16 mmol) in anhydrous THF (2 mL) was added, dropwise, NaHMDS (1 M in THF, 1.3 mL) at −20 °C, and the reaction mixture was stirred for 15 min at −20 °C. A solution of benzodioxinone 5 (42 mg, 0.14 mmol) in THF (1 mL) was added dropwise via cannula to the reaction mixture, and the combined mixture was warmed to 0 °C and stirred for 6 h. The reaction was quenched with saturated NH4Cl, and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 5 mL), and the combined organic layers were rinsed with brine (1 × 10 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Flash column chromatography (1:5 EtOAc/Hexane) yielded the both isomers 21a (32.8 mg) and 21b (9.2 mg) as viscous oils (65% overall yield); [α]D = − 10.0 (c = 0.25, CHCl ); FTIR (neat) 3435, 3053, 2956, 2925, 2854, 1656, 1265, 748, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 11.05 (s, 1H), 7.31(dd, J = 8.2, 7.6 Hz, 1H), 7.17 (d, J = 8.6, Hz, 2H), 6.87 (dd, J = 8.2, 1.1 Hz, 1H), 6.79 (d, J = 8.6 Hz, 2H), 6.71(dd, J = 7.5, 1.1 Hz, 1H), 5.95 (dddd, J = 16.9, 10.2, 6.0, 6.0 Hz, 1H), 5.76 (dddd, J = 16.9, 10.2, 7.6, 6.6 Hz, 1H), 5.66-5.60 (m, 1H), 5.01-4.93 (m, 3H), 4.89-4.83 (m, 1H), 4.38 (dd, J = 23.2, 11.5 Hz, 2H), 3.75 (s, 3H), 3.66 (dd, J = 15.7, 5.9 Hz, 1H), 3.55 (dd, J = 15.7, 5.9 Hz, 1H), 3.53-3.47 (m, 2H), 3.41-3.34 (m, 1H), 2.66 (d, J = 4.4 Hz, 1H), 2.27-2.21 (m, 1H), 2.10-1.94 (m, 2H), 1.91-1.78 (m, 2H), 1.71-1.64 (m, 1H), 1.62-1.57 (m, 1H), 0.84 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 171.4, 162.6, 159.8, 142.6, 137.7, 137.2, 134.4, 129.9, 129.4, 122.7, 116.4, 116.8, 115.4, 113.7, 112.2, 72.8, 71.8, 70.7, 66.7, 55.2, 40.0, 39.1, 38.6, 37.0, 35.1, 15.6; HRMS calcd. for C28H36O6Na (M+Na)+ 491.2410; found 491.2420 (TOF MS ES+).
(4S,5R,7S)-7-Hydroxy-9-((4-methoxybenzyl)oxy)-4-methylnon-1-en-5-yl 2-allyl-6-hydroxybenzoate (21b)
[α]D = +2.0 (c = 0.25, CHCl3); FTIR (neat): 3439, 3046, 2950, 2931, 2867, 1661, 1243, 742, 729, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 11.26 (s, 1H), 7.35 (dd, J = 8.1, 7.6 Hz, 1H), 7.20 (d, J = 8.2 Hz, 2H), 6.91 (dd, J = 7.6, 1.1 Hz, 1H), 6.83 (d, J = 8.2 Hz, 2H), 6.76 (dd, J = 7.5, 1.1 Hz, 1H), 6.03-5.84 (m, 1H), 5.84-5.71 (m, 1H), 5.51-5.43 (m, 1H), 5.08-4.95 (m, 3H), 4.93-4.86 (m, 1H), 4.38 (dd, J = 21.1, 12.2 Hz, 2H), 3.79 (s, 3H), 3.78-3.76 (m, 1H), 3.68-3.50 (m, 3H), 3.31-3.27 (m, 1H), 2.31-2.23 (m, 1H), 2.04-1.90 (m, 2H), 1.87-1.65 (m, 4H), 0.97 (d, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 171.4, 162.9, 159.2, 142.4, 137.6, 136.1, 134.3, 132.5, 129.3, 122.6, 116.8, 116.3, 115.5, 113.8, 112.1, 76.6, 73.0, 68.0, 66.5, 55.6, 39.9, 38.5, 37.1, 36.9, 36.7, 15.2; HRMS calcd. for C28H36O6Na (M+Na)+ 491.2410; found 491.2415 (TOF MS ES+).
(3S,5R,6S)-1-((4-Methoxybenzyl)oxy)-5-(methoxymethoxy)-6-methylnon-8-en-3-yl 2-allyl-6-(methoxymethoxy)benzoate (22)
To a solution of ester 21a (25 mg, 0.053 mmol) in anhydrous DCE (5 mL), under argon, was added iPr2NEt (69 mg, 0.53 mmol) and MOMCl (43 mg g, 0.53 mmol) at rt. The stirring solution was equipped with a reflux condenser and placed into an oil bath at 90 °C for 3-4 h. Upon completion (monitored by TLC), the reaction was diluted with CH2Cl2 (5 mL), followed by saturated NH4Cl solution (6 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were washed with brine (1 × 10 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude reaction mixture was purified through flash column chromatography (1:4 EtOAc/hexane) to afford title compound 22 (25 mg, 86%) as a clear oil; [α]D = +5.27 (c = 0.55, CHCl3); FTIR (neat): 2952, 2925, 2852, 1641, 1265, 1033, 748, 703 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.30-7.24 (m, 3H), 7.04 (d, J = 7.9 Hz, 1H), 6.90-6.86 (m, 3H), 5.94 (dddd, J = 16.7, 10.2, 6.6, 6.6 Hz, 1H), 5.73 (dddd, J = 16.9, 10.2, 7.5, 6.6 Hz, 1H), 5.49-5.42 (m, 1H), 5.16 (dd, J = 26.3, 6.9 Hz, 2H), 5.10-5.03 (m, 2H), 4.99-4.90 (m, 2H), 4.72 (dd, J = 12.8, 6.9 Hz, 2H), 4.46 (dd, J = 21.6, 11.3 Hz, 2H), 3.81 (s, 3H), 3.70-3.57 (m, 3H), 3.45 (s, 3H), 3.41 (s, 3H), 3.37 (d, J = 6.5 Hz, 2H), 2.13-1.97 (m, 3H), 1.95-1.77 (m, 2H), 1.76-1.71 (m, 2H), 0.90 (d, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.7, 159.1, 153.8, 138.1, 137.1, 136.3, 130.45, 130.2, 129.2, 124.9, 122.6, 116.5, 115.9, 113.7, 112.2, 96.8, 94.4, 78.2, 72.7, 70.8, 66.5, 56.0, 55.8, 55.7, 37.6, 37.2, 36.5, 35.4, 35.1, 13.7; HRMS calcd. for C32H44O8K (M+K)+ 595.2673; found 595.2653 (TOF MS ES+).
(3S,5R,6S,E)-3-(2-((4-Methoxybenzyl)oxy)ethyl)-5,14-bis(methoxymethoxy)-6-methyl-3,4,5,6,7,10-hexahydro-1H-benzo[c][1]oxacyclododecin-1-one (4)
Grubbs catalyst (Cy3P)2Cl2Ru=CHPh (~3 mg, 10 mol%, cat-A) was added to a solution of compound 22 (15 mg, 0.027 mmol) in degassed, anhydrous CH2Cl2 (5 mL) at rt under argon. The stirring solution was equipped with a reflux condenser and placed into an oil bath at 40 °C for 1 h. After completion of the reaction, as monitored by TLC, the reaction mixture was concentrated under reduced pressure. Flash column chromatography (1:5 EtOAc/Hexane) afforded macrolide 4 (12 mg, 84%) as a viscous oil (a small amount of Z-isomer was observed, the E/Z ratio was 9:1 as determined by 1H NMR of crude reaction); [α]D = −29.6 (c = 0.65, CHCl3); FTIR (neat): 2952, 2921, 2850, 1639, 1263, 1249, 1064, 908, 736, 702 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.6 Hz, 2H), 7.21 (dd, J = 8.3, 7.7 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 7.6 Hz, 1H), 5.53-5.46 (m, 2H), 5.39-5.31 (m, 1H), 5.07 (s, 2H), 4.85 (dd, J = 44.1, 6.8 Hz, 2H), 4.47 (s, 2H), 4.14 (dd, J = 9.3, 3.6 Hz, 1H), 3.81 (s, 3H), 3.73 (dd, J = 16.4, 9.5 Hz, 1H), 3.69-3.64 (m, 2H), 3.44 (s, 3H), 3.40 (s, 3H), 3.34 (ddt, J = 16.4, 4.5, 2.3 Hz, 1H), 2.35-2.28 (m, 1H), 2.19-2.09 (m, 1H), 2.07-1.99 (m, 1H), 1.96-1.89 (m, 1H), 1.80-1.67 (m, 2H), 1.48 (dd, J = 15.4, 9.4 Hz, 1H), 0.88 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 168.0, 159.1, 154.2, 139.1, 131.3, 130.6, 129.9, 129.0, 128.5, 125.3, 123.9, 113.7, 112.8, 96.9, 94.4, 79.4, 72.6, 72.2, 66.5, 56.0, 55.6, 55.3., 37.7, 37.7, 36.4, 35.5, 34.0, 13.4; HRMS calcd. for C30H40O8Na (M+Na)+ 551.2621; found 551.2605 (TOF MS ES+).
Supplementary Material
Acknowledgement
This investigation was generously supported by funds provided by the National Institute of General Medical Sciences (NIH RO1 GM077309). The authors thank Dr. Justin Douglas and Sarah Neuenswander for assistance with NMR measurements and Dr. Todd Williams for HRMS analysis. The authors also thank Materia, Inc. for supplying metathesis catalyst and helpful suggestions.
Footnotes
Supporting Information Available. Spectroscopic data of new compounds is available free of charge via the Internet at http://pubs.acs.org.
References
- (1)(a).Erickson KL, Beutler JA, Cardellina JH, II, Boyd MR. J. Org. Chem. 1997;62:8188–8192. doi: 10.1021/jo971556g. [DOI] [PubMed] [Google Scholar]; (b) Erickson KL, Beutler JA, Cardellina JH, II, Boyd MR. J. Org. Chem. 2001;66:1532. [Google Scholar]
- (2)(a).Wu Y, Esser L, De Brabander JK. Angew. Chem., Int. Ed. 2000;39:4308–4310. doi: 10.1002/1521-3773(20001201)39:23<4308::AID-ANIE4308>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Wu Y, Seguil OR, De Brabander JK. Org. Lett. 2000;2:4241–4244. doi: 10.1021/ol0068086. [DOI] [PubMed] [Google Scholar]
- (3).Boyd MR, Farina C, Belfiore P, Gagliardi S, Kim JW, Hayakawa Y, Beutler JA, McKee TC, Bowman BJ, Bowman EJ. J. Pharmacol. Exp. Ther. 2001;297:114–120. [PubMed] [Google Scholar]
- (4)(a).Smith AB, III, Zheng J. Synlett. 2001:1019–1023. [Google Scholar]; (b) Labrecque D, Charron S, Rej R, Blais C, Lamothe S. Tetrahedron Lett. 2001;42:2645–2648. [Google Scholar]; (c) Snider BB, Song F. Org. Lett. 2001;3:1817–1820. doi: 10.1021/ol015822v. [DOI] [PubMed] [Google Scholar]; (d) Fürstner A, Dierkes T, Thiel OR, Blanda G. Chem. Eur. J. 2001;7:5286–5298. doi: 10.1002/1521-3765(20011217)7:24<5286::aid-chem5286>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]; (e) Wu Y, Liao X, Wang R, Xie X-S, De Brabander JK. J. Am. Chem. Soc. 2002;124:3245–3253. doi: 10.1021/ja0177713. [DOI] [PubMed] [Google Scholar]; (f) Smith AB, III, Zheng J. Tetrahedron. 2002;58:6455–6471. [Google Scholar]; (g) Yang KL, Haack T, Blackman B, Diederich WE, Roy S, Pusuluri S, Georg GI. Org. Lett. 2003;5:4007–4009. doi: 10.1021/ol035630v. [DOI] [PubMed] [Google Scholar]; (h) Yadav JS, Srihari P. Tetrahedron:Asymmetry. 2004;15:81–89. [Google Scholar]; (i) Lebreton S, Xie X-S, Ferguson D, De Brabander JK. Tetrahedron. 2004;60:9635–9647. [Google Scholar]; (j) Herb C, Bayer A, Maier ME. Chem. Eur. J. 2004;10:5649–5660. doi: 10.1002/chem.200400617. [DOI] [PubMed] [Google Scholar]; (k) Holloway GA, Hügel HM, Rizzacasa MA. J. Org. Chem. 2003;68:2200–2204. doi: 10.1021/jo026798h. [DOI] [PubMed] [Google Scholar]; (l) Yang KL, Blackman B, Diederich W, Flaherty PT, Mossan CJ, Roy S, Ahn YM, Georg GI. J. Org. Chem. 2003;68:10030–10039. doi: 10.1021/jo0301550. [DOI] [PubMed] [Google Scholar]; (m) Herb C, Maier ME. J. Org. Chem. 2003;68:8129–8135. doi: 10.1021/jo035054g. [DOI] [PubMed] [Google Scholar]; (n) Yadav JS, Reddy PSR. Synthesis. 2007;7:1070–1076. [Google Scholar]; (o) Sugimoto Y, Konoki K, Murata M, Matsushita M, Kanazawa H, Oishi T. J. Med. Chem. 2009;52:798–806. doi: 10.1021/jm801265e. [DOI] [PubMed] [Google Scholar]; (p) Yadav JS, Rao NV, Rao PP, Reddy MS, Prasad A,R. Lett. Org. Chem. 2010;7:457–460. [Google Scholar]
- (5)(a).Whitehead A, McReynolds MD, Moore JD, Hanson PR. Org. Lett. 2005;7:3375–3378. doi: 10.1021/ol0512886. [DOI] [PubMed] [Google Scholar]; (b) Whitehead A, McParland JP, Hanson PR. Org. Lett. 2006;8:5025–5028. doi: 10.1021/ol061756r. [DOI] [PubMed] [Google Scholar]; (c) Waetzig JD, Hanson PR. Org. Lett. 2006;8:1673–1676. doi: 10.1021/ol0602809. [DOI] [PubMed] [Google Scholar]; (d) Waetzig JD, Hanson PR. Org. Lett. 2008;10:109–112. doi: 10.1021/ol7025944. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Whitehead A, Waetzig JD, Thomas CD, Hanson PR. Org. Lett. 2008;10:1421–1424. doi: 10.1021/ol8001865. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Thomas CD, McParland JP, Hanson PR. Eur. J. Org. Chem. 2009:5487–5500. doi: 10.1002/ejoc.200900560. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Venukadasula PKM, Chegondi R, Maitra S, Hanson PR. Org. Lett. 2010;12:1556–1559. doi: 10.1021/ol1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nicolaou KC, Kim DW, Baati R. Angew. Chem., Int. Ed. 2002;41:3701–3703. doi: 10.1002/1521-3773(20021004)41:19<3701::AID-ANIE3701>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- (7)(a).(PCy3)2(Cl)2Ru=CHPh (cat-A) Schwab P, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1996;118:100–110. Schwab P, France MB, Ziller JW, Grubbs RH. Angew. Chem., Int. Ed. 1995;34:2039–2041.
- (8).(IMesH2)(PCy3)(Cl)2Ru=CHPh (cat-B) Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q.
- (9).cat-C: Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179.
- (10)(a).Rychnovsky SD, Griesgraber G, Powers JP. Org. Synth. 1999;77:1–11. [Google Scholar]; (b) Davoille RJ, Rutherford DT, Christie SDR. Tetrahedron Lett. 2000;41:1255–1259. [Google Scholar]
- (11).Lucas BS, Luther LM, Burke SD. Org. Lett. 2004;6:2965–2968. doi: 10.1021/ol0488800. [DOI] [PubMed] [Google Scholar]
- (12)(a).Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Garber SB, Kingsbury JS, Harrity JPA. Org. Biol. Chem. 2004;2:8–23. doi: 10.1039/b311496c., and references cited therein; b) A study of the CM reaction using Hoveyda–Grubbs catalyst (cat-C) was reported by: Cossy J, BouzBouz S, Hoveyda AH. J. Organomet. Chem. 2001;624:327–332.
- (13).Tsuji J, Yamakawa T. Tetrahedron Lett. 1979:613–616. [Google Scholar]
- (14)(a).For selective silylation of similar 1,3-diols, see: Soltani O, DeBrabander JK. Org. Lett. 2005;7:2791–2793. doi: 10.1021/ol0510769. (b) See also, reference 5e.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.